
Abstract
Metabolites in plasma form biosignatures of a range of common complex human diseases. Discovering variants with pleiotropic effects across metabolites can reveal underlying biological mechanisms. We therefore performed uni- and multivariate genome-wide association studies (GWAS) on 249 circulating metabolic markers across 328,006 UK Biobank and Estonian Biobank participants. We investigated rare variation through whole exome sequencing gene burden tests, analysed the role of body mass index through Mendelian randomization, and performed genome-wide interaction analyses with sex. We discovered 15,585 loci summed over the univariate GWAS, with high pleiotropy across markers, linked to a wide range of disorders. Findings from common and rare variant gene tests converged on lipid homeostasis pathways. 31 loci interacted with sex, mapped to genes involved in cholesterol processing. The findings offer insights into the genetic architecture of circulating metabolites, revealing pleiotropic loci, highlighting the role of rare variation, and uncovering sex-specific molecular mechanisms of lipid metabolism.
Similar content being viewed by others


Introduction
Lipoproteins, fatty acids, amino acids and ketone bodies are circulating markers of metabolic processes essential for human health. Reliable quantification of absolute concentrations of these metabolites can now be achieved through high-throughput nuclear magnetic resonance (NMR) spectroscopy1. Metabolomics data in large population samples such as the UK Biobank (UKB), coupled to national health records, has allowed researchers to identify numerous associations between patterns of metabolite concentrations and a wide range of common medical conditions2. These metabolites hold potential for precision medicine as they have been shown to predict long term outcomes3, and could aid in combatting key public health issues, including the adverse effects of the worldwide obesity epidemic4.
Charting the pleiotropic genetic architecture of metabolic biomarkers, through the effects of common and rare variants, is key to understanding interindividual differences in metabolic processes. Genome-wide association studies (GWAS) of metabolomics data have confirmed there is a substantial genetic component to these metabolite concentrations and have identified hundreds of genetic variants associated with individual metabolites5,6,7. The sets of metabolites included in metabolomics panels are strongly genetically correlated to each other8; joint analysis through a multivariate approach may improve discovery of variants with widespread effects by leveraging shared genetic signal across the metabolites9. Additionally, characterizing the influence of rare variants on metabolites through whole exome sequencing (WES) data complements previous GWAS efforts, as rare variants are likely to be particularly impactful and point towards promising drug targets10,11.
Obesity and sex are likely important moderators of the relation between an individual’s genetic make-up and metabolic health. As obesity and its downstream medical conditions co-occur with changes in metabolite concentrations12; disentangling the causal role of obesity in determining these levels can aid in devising treatment strategies. Biological sex is a further important determinant of metabolic activity13, yet there is little knowledge about sex-dependent genetic influences. Males and females differ substantially in basal metabolic activity, as well as in their propensity to develop prominent metabolic conditions, such as obesity, coronary artery disease (CAD) and type 2 diabetes (T2D)14. Previous studies have shown that there is a genetic basis for sex differences in metabolism, beyond the impact of gonadal hormones15.
Here, we take advantage of the latest generation of targeted metabolomics technology available in the UKB and Estonian Biobank (EstBB), to perform a large GWAS of circulating metabolic traits, leveraging NMR spectroscopy data from over 300,000 individuals. We expand on previous work on this data by employing a multivariate approach to boost discovery of variants with widespread shared effects across metabolites, and perform quantification of the global genetic architecture. We further incorporate WES data to increase knowledge about the impact of both common and rare variants. Lastly, we identify widespread sex-specific effects and estimate the influence of obesity (indexed by body mass index, BMI) to provide insight into the influence of individual, clinically relevant factors.
Results
We conducted GWAS of 249 circulating metabolites from the Nightingale NMR metabolomic platform, charting their shared and specific genetic architectures. This panel encompasses 228 lipids, lipoproteins and fatty acids, and 21 non-lipids, including amino acids, ketone bodies, fluid balance, glycolysis- and inflammation-related metabolites. See Supplementary Data 1 for an overview of these circulating metabolites, their categories, and sample sizes. For the main analyses we used data from UKB, including 207,836 White British participants, with a mean age of 57.4 years (standard deviation (SD) 8.0 years), 53.7% female. Additionally, there were data on 27,509 non-White British UKB participants, with a mean age of 54.5 years (SD = 8.4 years), 54.3% female. From EstBB, we included 92,661 unrelated White European participants, with a mean age of 50.9 years (SD = 16.2 years), 65.7% female, which we used to test for generalization of the discovered loci across different populations. For each of these subsets, identical analyses were carried out, covarying for age, sex, and the first twenty genetic principal components to control for population stratification16.
Univariate GWAS
We estimated the effective number of independent traits in our analyses to be 96, based on matrix spectral decomposition17 of the phenotypic correlation between all 249 metabolite concentrations. We therefore set the univariate GWAS significance threshold at α = 5 × 10−8/96 = 5.2 × 10−10. The GWAS of all individual 249 metabolites revealed a median of 63 loci discovered per metabolite (range 8 to 98), for a total of 15,585 loci when summing over the individual univariate GWAS, as shown in Fig. 1a. Accounting for locus boundary overlap across the univariate GWAS, there were 465 unique genomic regions involved, suggesting high numbers of shared genetic variants across the metabolites. Of these, 166 regions were novel, in that they did not overlap with the 276 regions identified by the previously largest GWAS of 233 metabolites of the Nightingale metabolomics panel5. The most significant novel loci were rs4760682, mapped to the PFKM gene, and rs7584089, mapped to PDK1. Both showed strongest association with pyruvate levels (B = −0.11, p = 2.6 × 10−187; B = −0.09, p = 8.9 × 10−117). This befits the central role that the mapped genes play in glycolysis and fatty acid metabolism, with their overexpression previously coupled to T2D18, cancer19, and Alzheimer’s disease20. Supplementary Data 1 lists the number of significant loci and lead SNPs for each of the 249 individual metabolites. Supplementary Datas 2 and 3 list information on all discovered regions (per metabolite and aggregated), and whether they are novel. All Manhattan plots are provided in Supplementary Data 19.

a Scatterplot displaying the effect sizes (y-axis) of all 15,585 locus lead variants identified through univariate genome-wide association study (GWAS) of 249 metabolites, ordered by their minor allele frequency (x-axis) and colour coded by metabolite category. The two loci with the strongest effects on individual GWAS are demarcated by vertical dashed lines and annotated. On the right side of the figure, the local genomic regions of these two loci are depicted, with b showing rs7412 mapped to APOE and c showing rs1047891 mapped to CPRS1, both of which were also the only fine-mapped variants in these regions. The chromosomal location is on the x-axis and -log10(p-value) on the y-axis, with colours reflecting the linkage disequilibrium between the lead and surrounding variants, as indicated in the legend. The bottom panel shows the genes located in these regions. The vertical dashed lines highlight the position of the lead variants in their respective genes.
We applied a combination of PolyFun and FINEMAP, Bayesian fine-mapping procedures bundled in the SAFFARI pipeline21, to each of the univariate GWAS summary statistics, to obtain a set of high-confidence causal variants and genes. Out of the original 15,585 loci, we retained 2629 variants that had a posterior probability >0.95 of being part of a credible set. We then mapped these variants to 2498 protein-coding genes using OpenTargets22. The two fine-mapped loci with the largest effect sizes (rs7412, APOE, and rs1047891, CSP1) had strongly divergent patterns of associations, being highly pleiotropic (influencing nearly all lipid measures) versus one individual association (with glycine). APOE is well-known for playing a central role in lipid homeostasis, and variation in its gene has been associated with a wide variety of traits23. CPS1 on the other hand is an enzyme involved in a specific pathway degrading choline to urea, with variation in its gene linked to blood pressure and CAD through its strong effects on glycine levels24. Figure 1b and c illustrates the mapping of these genomic regions. Supplementary Data 4 lists the fine-mapping results in more details, including all mapped genes and their coupling to individual metabolites.
We checked cross-population generalization of the effects of fine-mapped variants in the White EstBB cohort and in the non-White British UKB subset. For the EstBB replication set (n = 92,645 individuals), 99.0% of the 2,019 available variants showed the same direction of effect, and 91.3% of these effects were nominally significant. In the additional UKB subset (n = 27,509 individuals), we found that 95.9% of all 2,207 available fine-mapped variants showed the same direction of effects, and 75.3% were nominally significant. Thus, our results suggest cross-population generalization of the discovered genetic associations. Supplementary Fig. 1 shows the relationship between the number of discovered loci in UKB and replicated loci in EstBB, per metabolite.
Multivariate GWAS
Genetic variants are likely to have pleiotropic effects across the metabolites, given these metabolites are components of the same biological system, as also indicated by the univariate GWAS findings. We therefore jointly analysed all measures with the Multivariate Omnibus Statistical Test (MOSTest)9, which prioritizes the identification of pleiotropic variants by leveraging shared genetic signal across the univariate measures, yielding a multivariate association with each genetic variant.
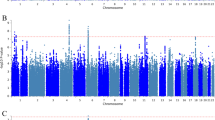
For the primary sample, MOSTest revealed 12,216 independent significant SNPs and 2690 lead SNPs across all metabolites, for a total of 534 loci covering 8.3% of the genome, see Fig. 2a. The lead SNPs of 96 of these loci did not show genome-wide significant effects on any of the individual metabolites, i.e. they were detected only through MOSTest due to their distributed signal across the metabolites. Supplementary Fig. 2 summarizes the significance of the locus lead SNPs across all metabolites, illustrating the pervasive pleiotropy of most discovered variants. Indeed, 48 of these SNPs showed a genome-wide significant association with more than 100 metabolites, as summarized in Supplementary Data 5. This table also lists the comparison to the previous GWAS, showing that 274 of these 276 regions overlap with the MOSTest-discovered loci, while MOSTest uncovered another 241 loci not reported in this previous GWAS.

a Manhattan plot of the output of the multivariate genome-wide association study (GWAS) on all 249 metabolites, with the observed -log10(p-value) for each variant shown on the y-axis. The x-axis shows the relative genomic location, grouped by chromosome, and the red dashed line indicates the genome-wide significance threshold of 5 × 10−8. The colour coding represents the number of genome-wide significant associations of each variant with metabolites at the univariate level, ranging from 0 (in black) to 214 (in red), illustrating the extent of pleiotropy. Loci with p < 1 × 10−300 have been annotated with mapped gene names. b heatmap showing 30 multivariate GWAS-identified loci with the highest number of Bonferroni-corrected significant associations with diseases, based on published GWAS. On the x-axis are the locus lead variants with mapped genes, and on the y-axis the categories of diseases as compiled by OpenTargets. The cell colouring indicates the -log10(p-value) of the variant-disease association, as indicated by the legend on the right.
We performed phenome-wide association studies (pheWAS) of each of the 534 loci identified through MOSTest, querying GWAS Catalog and FinnGen GWAS summary statistics through the ‘otargen’ R package, leveraging the OpenTargets ‘diseases’ categorization in order to determine clinical relevance25. There were a total of 1253 Bonferroni-corrected significant associations, with 341 unique traits across 372 studies. The results, fully listed in Supplementary Data 6, show that many of the discovered variants are associated with cardiovascular diseases, as expected, as well as commonly comorbid conditions. Specific variants also show high pleiotropy across diseases, as illustrated in Fig. 2b. For instance, the pleiotropic variant rs3184504 was mapped to SH2B3 on chromosome 12, which is a key regulator of signalling pathways involved in inflammatory responses26. Among the ten most pleiotropic variants was also a novel locus at chromosome 7 mapped to IRF5, which encodes a transcription factor that induces proinflammatory cytokines, and has been named a potential therapeutic target for a wide range of autoimmune diseases27. These examples reflect the well-known coupling of low-grade inflammation to metabolic dysregulation, contributing to patterns of comorbidity28.
Gene-based analyses
Next, we ran gene-based analyses to identify the most significant genes and their enrichment among specific biological pathways. Aggregating across all common variants within 17,849 protein-coding genes, we found 1921 significant genes based on the 249 individual univariate GWAS summary statistics, and 2590 genes from the multivariate GWAS. Tests of tissue-specificity, covarying for mean expression across all tissues, revealed differential gene expression in the liver for nearly all metabolites (243 out of 249), in line with its central role in metabolism of both lipids and amino acids. Differential expression in other tissues was more specific to a metabolite category, as can be seen for the spleen, summarized in Fig. 3a. Competitive gene-set analysis for each individual metabolite GWAS, testing for 7522 Gene Ontology (GO) biological processes, primarily uncovered associations with lipoprotein particle modification, organization, and homeostasis, see Fig. 3b. Supplementary Data 7 lists all identified genes, and Table 8 contains the complete results of the gene-set analyses for each individual metabolite.

a Stacked bar plot summarizing the output of tests of tissue-specific gene expression, with the top 15 tissues on the x-axis. b Competitive gene-set analysis of Gene Ontology biological processes, with top 15 pathways listed on the x-axis. For both plots, the number of significant associations with metabolites is shown on the y-axis and the colours indicate metabolite categories. c Venn diagram of the number of genes identified through gene-based tests of the multivariate GWAS, univariate GWAS and rare variant WES data.
Rare variant data
We ran SKAT-O29 gene burden tests on the UKB WES data (N = 200,330), restricted to intragenic variants with MAF < 0.005, to characterize the impact of rare exonic variation on metabolites. There were 338 protein-coding genes with a significant burden, see Supplementary Data 9. Figure 4 lists the top genes identified, showcasing the widespread impact of apolipoprotein genes, well-known for their association with obesity and Alzheimer’s disease (AD), on the lipid metabolites. Many of the additional identified highly pleiotropic genes with impactful rare variants also play roles in metabolic conditions and healthy ageing through interrelated pathways30,31,32, directly or indirectly. For instance, ZPR1 codes for a zinc finger protein that regulates axonal growth, which has been coupled to neuronal cell death following a high-fat diet33. As impactful rare variation signals potential for manipulation of these pathways, we queried the drug-gene interaction database (DGIdb v5.0.6)34 and found significant enrichment for targets of nine drugs. These include three lipid-lowering agents, three antibacterial drugs, a drug used to treat leukemia, an anticonvulsant, and a platelet aggregation inhibitor (Supplementary Data 10).

a Stacked bar plot showing the number of significant associations (x-axis) with genes identified through whole-exome sequencing-based gene burden tests (y-axis), coloured by metabolite category. b Grouped scatterplot, showing the -log10(p-value) of Bonferroni-corrected significant genes on the y-axis, grouped by category on the x-axis, with the dot colours also reflecting category. The top three most significant genes per category are annotated.
See Fig. 3c for overlapping and unique components of the identified sets of genes through the three different gene-level analyses, capturing genes with effects driven by rare, common, and/or pleiotropic variants. The unique and shared sets of genes were coupled to the GWAS Catalog through hypergeometric tests, with results summarized in Supplementary Data 11.
Global genetic architecture
We determined the SNP-based heritability, h2, as well as the polygenicity and average magnitude of non-null effects (‘discoverability’)35,36 for each metabolite. Overall, the output showed that the metabolites vary widely in their global genetic architecture; h2 ranged from 02 (acetoacetate, standard error (SE) = 0.002) to 0.21 (triglycerides to total lipids in very large HDL, SE = 0.001), all p-values < 1.1 × 10−21. Polygenicity estimates spread across two orders of magnitude, from oligogenic (phenylalanine with an estimated 17 causal variants) to moderately polygenic (creatinine with an estimated 1529 causal variants). Similarly, discoverability ranged from 1.2 × 10−4 (lactate) to 2.4 × 10−3 (omega-3%). Supplementary Fig.. 3 depicts the estimated proportion of h2 explained as a function of sample size, for 37 metabolites validated for clinical use1. This showed that over 70% of h2 for omega-3 and omega-6 fatty acid concentrations is explained by the currently discovered variants. All estimates, for each of the 249 metabolites, are listed in Supplementary Data 1.
Analyses of BMI and sex
Given BMI and sex have been associated with substantial interindividual variation in metabolic activity12,13, we sought to determine the phenotypic, causal and genetic relation of these individual determinants with the metabolites. First, we conducted linear regression analyses, regressing each metabolite onto BMI, sex, BMI*sex, and age. These models produced highly significant associations with BMI, sex, and their interaction across nearly all metabolites, as summarized in Supplementary Data 12. This underscores the importance of sex and BMI as individual-level influences when investigating the biological underpinnings of metabolic processes. Notably, there was a very high correlation between the coefficients of BMI and sex (r = 0.87, p = 4.1 × 10−79), indicating that these factors share mechanisms that in turn impact metabolites.
As BMI is a modifiable factor, we next sought to estimate the causal nature of the identified relationships between BMI and metabolites. We ran bidirectional two-sample Mendelian randomization (MR), combining inverse variance weighted (IVW) MR with the weighted median and MR Egger approach37. There were no instances where the metabolites had a significant causal effect on BMI consistently across the different MR methods. BMI had a multiple comparisons-corrected significant (p < 0.05/96) causal effect on 79 metabolites for the IVW and weighted median approach. However, when further thresholded by the MR Egger approach, sensitive to horizontal pleiotropy, the causal effect of BMI on only six metabolites remained: albumin, phenylalanine, average diameter for LDL particles, cholesterol % in small LDL, tyrosine, and valine, see Fig. 5. The full results, in both directions, are provided in Supplementary Datas 13–15.

Plot listing coefficients from two-sample Mendelian randomization (MR) analyses on the x-axis, and the 6 different metabolites that showed a significant influence of BMI on the y-axis. The dots and lines represent the point estimates with their standard error around the mean, colour-coded by the MR method used.
Sex-specific genetic influences
Our identification of significant interactions between BMI and sex on metabolite concentrations underlines the need for sex-specific research into metabolic health. We therefore first ran univariate GWAS within both sexes separately, to compare the overall genetic architecture between men (n = 96,281) and women (n = 111,560). Through paired t-tests applied to sex-specific LDSC heritability estimates, we found that the mean h2 was significantly higher for women than for men (h2 = 0.148 vs. .132, t = 12.8, p < 1 × 10−16). Men’s h2 was still higher than that of the overall GWAS (h2 = 0.132 vs. 0.128, t = 7.5, p = 9 × 10−13), suggesting heritability estimates may be lowered by combining two subsamples (men and women) with differing genetic influences. We further calculated genetic correlations between the two sets of sex-specific GWAS and found that these ranged between 0.85 and 1. While these correlations were high, the majority differed significantly from 1, as reported in Supplementary Data 16.
Given the identification of sex-specific genetic components through LDSC, we ran multivariate GWAS with an interaction term between sex and each genetic variant, to discover individual variants with sex-specific effects. We found 31 loci with a genome-wide significant interaction effect. Of these, 8 loci had no whole-genome significant interaction effect on any individual metabolite. Next, we mapped the loci to 29 genes through OpenTargets, see Fig. 6a. Functional annotation of the 29 mapped genes revealed tissue-specific upregulation in kidney, liver, and heart tissues based on GTEx v8 data, and enrichment for GO pathways involved primarily in cholesterol regulation. Coupling these 29 genes to the GWAS Catalog showed enrichment among gene lists reported for metabolic syndrome, CAD, T2D, and steatotic liver disease, which are well-known for having sex differences in prevalence and etiology14.

a Manhattan plot of the multivariate genome-wide association study with an interaction term with sex on all 249 metabolites, with the observed −log10(p-value) of each interaction shown on the y-axis. The x-axis shows the relative genomic location, grouped by chromosome, and the red dashed line indicates the genome-wide significance threshold of 5 × 10−8. The y-axis is clipped at -log10(p)=150. Loci have been annotated with mapped gene names. b Illustration of an identified significant cross-over interaction between sex and rs1065853 on chromosome 19, showing opposite effects on phospholipid concentrations (y-axis) in men and women (x-axis). c An interaction effect of rs964184 on chromosome 11, illustrating effects on very low density lipoprotein (VLDL) cholesterol concentrations only in women. In both plots, the line colors indicate genotypes, and error bars represent standard error of the mean. Male n = 96 281, female n = 111 560.
Follow-up in the univariate summary statistics showed that the interaction effects were often present for numerous metabolites, with one interaction effect (rs1065853, APOE) being genome-wide significant across 110 metabolites. Figure 6b provides an example of univariate cross-over interaction between sex and the rs1065853 genetic variant on lipid levels, with highly significant effects in females (B = 0.145, p < 1 x 10-16), but not males (B = −0.002, p = 0.84). Figure 6c shows another significant sex*gene variant interaction effect of rs964184 (ZPR1), which also influences cholesterol levels only in females (B = 0.110, p = 1 x 10-16) and not males (B = 0.008, p = 0.21). In total, there were 496 univariate genome-wide significant interactions. In EstBB, the concordance rate was 99.3%, and 113 out of 158 (71.5%) of the available lead variants was nominally significant. In the non-White UKB subset, the concordance rate was 90.7%, with 268 out of the 496 lead variants being nominally significant (54.0%). The lists of all multivariate and univariate loci with significant interactions are provided in Supplementary Data 17 and 18.
Discussion
Here, we reported results from a large-scale GWAS of circulating metabolite concentrations. This led to the identification of the largest number of discovered genetic determinants across these metabolites to date, mapped to genes with roles in lipid homeostasis. Using a multivariate approach, our findings emphasized the pervasive pleiotropy across metabolic measures that underscore and expand the findings from other GWAS using this data7. We further went beyond previous studies by integrating WES data in our analyses to uncover a sizeable role for rare variants. We identified the causal effect of BMI on specific amino acids, indicating obesity as a primary target for improving metabolic health. Last, we discovered sex-specific genetic effects on metabolite concentrations, which may explain the substantial sex differences in metabolic health.
Locus discovery was high, in line with the estimated genetic architecture. The complementary univariate and multivariate GWAS approaches employed in this study particularly emphasized the pervasive pleiotropy across the set of included metabolites, in accordance with previous findings8. Joint analyses of these interrelated measures are essential to boost discovery of variants with small, yet distributed effects. The clinical relevance of this discovery is underscored by the results of the pheWAS analyses, showing the association of many of these pleiotropic variants with medical conditions across domains. This likely contributes to the extensive comorbidity across complex medical conditions with a cardiometabolic component38,39, which is an important determinant of clinical outcomes39,40.
The gene-based analyses illustrated the relative contributions of common and rare variation, with extensive pleiotropy, to determining metabolite levels. The WES gene burden tests, aggregating across rare variants, identified 335 genes with widespread associations across both lipid and non-lipid metabolite categories. Among the most pleiotropic were apolipoprotein genes, well-known for their involvement in diabetes and CAD as well as in brain disorders41. Particularly notable in this context is the identification of BACE1 on chromosome 11 among the most influenced genes, the protein product of which is central to the generation of amyloid-B peptides in neurons and a key enzyme in the pathophysiology of AD42. Overall, this rare variant data confirms the presence of impactful rare variants with high potential for druggability, as confirmed by the coupling to DGIdb. The generated data on the specificity of these genetic effects on metabolites is important information for research into comorbidities and for predicting utility as a biomarker and drug target.
The findings of the gene-by-sex interaction analyses underscore the substantial differences between males and females in metabolism13. This is likely to be a strong explanatory factor of sex differences in the prevalence of a wide array of cardiometabolic conditions14, advocating for the investigation of sex-specific mechanisms. The notoriously low power of interaction effects43 is counteracted by our multivariate approach. MOSTest is insensitive to differences in the directions of these interactions across the univariate measures, which would hamper other approaches to aggregation across measures. The identification of the widespread sex-dependent effects of rs1065853 showcases the potential of these interaction terms to identify variants that explain interindividual variation beyond their main effects. This SNP, located in a known enhancer of APOE, is well known for its association with numerous metabolic and clinical outcomes, including AD and CAD44,45. The identification of such non-linear effects represent a new frontier in genomics, which needs to be explored in order to further resolve interindividual heterogeneity. Our findings particularly suggest value of additional sex-specific research into obesity and metabolic health.
The MR analyses provided evidence for the causal effect of BMI, as a proxy of obesity, on circulating metabolic biomarkers, emphasizing the importance of obesity as a primary target for treatment of cardiometabolic conditions. In accordance with previous findings in smaller samples, we show that BMI has a significant causal effect on levels of several metabolites46, primarily amino acids, while there was no evidence of effects of any metabolites on BMI. Obesity therefore appears to drive changes in these amino acids, which may then cause complications47. Branched chain amino acids, including valine, have been robustly associated with an increased risk of T2D48, which may be driven by higher BMI and insulin resistance49. The importance of targeting BMI is further underscored by our finding that higher BMI lowers albumin levels, which is a key marker of liver function and general nutritional status, as well as a predictor of a wide range of cardiovascular outcomes50. Notably, the use of MR methods that safeguard against horizontal pleiotropy substantially reduced the number of causal relationships identified with lipid-related measures. This suggests a sizeable role for pleiotropic effects complicating the relationship between genetically mediated obesity and these measures, in line with our GWAS findings. It speaks, for instance, to the complex role of the GLP-1 secretory system, currently hailed as a highly promising therapeutic target for treatment of obesity, with divergent findings across both human and animal studies51. A better understanding of the role of genetic susceptibility and sources of interindividual variation is needed to optimize individual outcomes.
Strengths of this study are the large sample size and the use of high quality, accurately measured metabolomics data. We further complemented the analysis of common variants influencing individual metabolites with a multivariate approach for greater discovery of pleiotropic variants and inclusion of WES data to uncover the role of rare variants. While this allowed for greater insight into the overall genetic architecture of metabolism, focused follow-up studies are needed to generate a deeper understanding of the specific determinants of subsets of metabolites. Genetic discovery was based on a single large cohort, with a relatively homogeneous population of White British individuals. We included two replication samples with varying genetic ancestry, enabling estimation of generalization of the findings. However, given known ethnic differences in the association between obesity and metabolic conditions such as T2D52, the role of ethnicity should be investigated in further detail. It should also be noted that the data collection was not done under fasting conditions, which has been shown to obscure associations between genetic variation and metabolites5. Ideally, future studies include gene-by-time interaction analyses to further increase our understanding of the genetic regulation of metabolite concentrations.
To conclude, metabolic health is central to the most prevalent and impactful medical conditions in our society, indicating a strong need for new therapeutic targets. Knowledge about causal individual-level determinants is central to develop effective strategies that optimally treat the individual. Here, we showed that accurate NMR-derived circulating metabolite concentrations share genetic influences that can be leveraged to boost discovery of pleiotropic variants of high relevance for cardiometabolic diseases. The summary statistics made freely available can be used by follow-up studies to further enhance our understanding of metabolism and related diseases, identify potential drug targets for these diseases, and contribute to the development of more effective interventions by identifying individual-level determinants.
Methods
The conducted research complies with all relevant ethical regulations. It has been approved by the UK’s National Health Service National Research Ethics Service (ref. 11/NW/0382) and the Estonian Council on Bioethics and Human Research (24 March 2020, nr 1.1-12/624). The study design and conduct complied with all relevant regulations regarding the use of human study participants and was conducted in accordance to the criteria set by the Declaration of Helsinki.
Participants
For the UKB, we obtained data under accession number 27412. The composition, set-up, and data gathering protocols of the UKB have been extensively described elsewhere53. It has obtained informed consent from its participants. For the primary analyses, we selected unrelated White Europeans (KING cut-off 0.05)54 that had the Nightingale metabolomics data, as well as genetic and complete covariate data available (N = 207,836, mean age 57.4 years (SD = 8.0), 53.7 % female). BMI was taken from UKB field 21001, with a mean of 27.4 (SD = 4.8). For the generalization analyses, we made use of data from non-White European UKB participants (N = 27,509, mean age 54.5 years (SD = 8.4), 54.3 % female). Ethnicity was based on self-report confirmed by genetics (UKB field 22006).
EstBB is a volunteer-based biobank composed of ~213,000 individuals with data available on genotype, phenotype and electronic health records55. All EstBB participants have signed an informed consent form. All analyses were conducted using data according to release S60 from EstBB. Specifically, individuals were selected under conditions identical to those used for the UKB data for filtering and quality control, resulting in 92,661 unrelated White European participants, with a mean age of 50.9 years (SD = 16.2 years), 65.7% female. BMI values (mean 26.1, SD = 5.3) were either calculated at the time of recruitment and blood donation or referenced from EHR within a year from enrollment.
Data collection and pre-processing
We included all 249 metabolites from the Nightingale NMR metabolomics panel, encompassing 228 lipids, lipoproteins or fatty acids and 21 non-lipid traits, namely amino acids, ketone bodies, fluid balance, glycolysis-, and inflammation-related metabolites, as QC’ed and released by UKB2. We applied additional pre-processing through the ‘ukbnmr’ R package, to remove sources of technical noise56.
We applied rank-based inverse normal transformation57 to each measure, leading to normally distributed measures as input for the GWAS.
Univariate GWAS and univariate interaction GWAS
We made use of the UKB v3 imputed data, which has undergone extensive quality control procedures as described by the UKB genetics team58. After converting the BGEN format to PLINK binary format59, we set a minor allele frequency threshold of 0.005, leaving 11,144,506 SNPs.
We carried out univariate GWAS on each of the 249 metabolites through PLINK2, which were then combined into a multivariate GWAS through the freely available MOSTest software (https://github.com/precimed/mostest). Details about the procedure and its extensive validation have been described previously9. GWAS on each of the normalized measures were carried out using the standard additive model of linear association between genotype vector, \({g}_{j}\), and phenotype vector, \(y\). In all analyses we covaried for mean-centered age and twenty genetic principal components. We additionally covaried for biological sex, except in the sex-specific analyses.
Association of genotype*sex interaction with each of 249 metabolites was tested with PLINK2, including genotype, sex, mean-centered age and 20 genetic principal components as covariates. Produced univariate GWASs were then combined into multivariate MOSTest analysis. Calibration of the null distribution for the MOSTest analysis was performed permuting both genotypes and sex independently.
Clumping
For both univariate and multivariate GWAS, independent significant variants and genomic loci were identified in accordance with the Psychiatric Genomics Consortium locus definition60. First, we selected a subset of variants that passed genome-wide significance threshold, and used PLINK to perform a clumping procedure at LD r2 = 0.6 to identify the list of independent significant variants. Second, we queried the reference panel for all candidate variants in LD r2 of 0.1 or higher with any independent significant variant. Further, for each independent significant variant, its corresponding genomic loci were defined as a contiguous region of the independent significant variants’ chromosome, containing all candidate variants in r2 = 0.1 or higher LD with the independent significant variant. Adjacent genomic loci were merged if separated by less than 250 KB. A subset of independent significant variants with LD r2 < 0.1 was selected as lead variants (with potentially more than one lead variant per locus). Finally for each locus the most significant among all lead variants was defined as the locus lead variant. Allele LD correlations were computed from a random subset of 10% of the study population to lower computational burden. The number of unique significant loci across all univariate GWAS was determined through the min-P approach61.
Gene mapping
We used the Variant-to-Gene (V2G) pipeline from Open Targets Genetics, to map lead variants to genes based on the strongest evidence from quantitative trait loci (QTL) experiments, chromatin interaction experiments, in silico functional prediction, and proximity of each variant to the canonical transcription start site of genes22.
PheWAS
We used the ‘otargen’ R package to conduct the pheWAS analyses on each of the 534 MOSTest-identified locus lead SNPs. We restricted the analyses to the FinnGen and GWAS Catalog study sources, and selected only traits that had the term ‘disease’ in the trait category. The results were thresholded to associations of each of the locus lead SNP at p < 0.05 divided by the unique number of traits included (n = 7684).
Fine-mapping procedure
We used the SAFFARI pipeline to perform statistical and functional fine mapping21. This consisted of applying PolyFun+FINEMAP to each of the GWAS in order to identify sets of functionally-informed highly credible causal variants, selecting those that were part of a credible set with a posterior probability >0.95 prioritizing these for follow-up. By default, SAFFARI excludes the major histocompatibility complex (MHC) region on chromosome 6 (28–34 Mb).
WES gene burden tests
We used Regenie (v3.1.1) to perform omnibus SKAT-O tests to combine variance component tests and burden tests for each of the 249 metabolites, with age, sex and 20 genetic principal components as covariates. We merged the genotype data of chromosome 1 to 22 into a single PLINK file, lifted the genomic build from GRCh37 to GRCh38, and filtered with PLINK (--maf 0.01 --mac 20 --geno 0.1 --hwe 1e-15 --mind 0.1) to select 591,260 SNPs for step 1. Step 2 variants were rare (MAF < 0.005) with the following annotation masks: LoF, missense (0/5), missense (5/5), missense (>=1/5), and synonymous. We used relevant annotation files described elsewhere: https://biobank.ctsu.ox.ac.uk/crystal/refer.cgi?id=916. We included the same set of protein coding genes and multiple comparisons correction as used for the MAGMA gene-based analyses. The analyses were conducted on the Research Analysis Platform (https://ukbiobank.dnanexus.com).
Gene-set analyses
We carried out gene-based analyses using MAGMA v1.08 with default settings, which entails the application of a SNP-wide mean model62. We used a randomly selected set of 10,000 white British UKB participants as reference panel. Gene-set analyses were done in a similar manner, restricting the sets under investigation to those that are part of the Gene Ontology biological processes subset (n = 7522), as listed in the Molecular Signatures Database (MsigdB; c5.bp.v7.1).
For tissue-specificity analyses, we applied MAGMA gene-property analyses to test relationships between tissue-specific gene expression profiles and the identified gene associations. This encompassed running one-sided tests for each of 30 general tissue types, testing whether the association between each tissue’s known gene expression levels and the gene-based Z-scores is greater than 0, corrected for the average expression across all tissue types and a set of technical confounders. We used preprocessed and normalized GTEx v8 tissue expression values63 as provided through FUMA’s downloads (https://fuma.ctglab.nl/).
Multiple comparison’s correction for these analyses consisted of a Bonferroni correction for the number of protein-coding genes, with α = 0.05/17,849 = 2.8 x 10-6
Drug enrichment analysis
The Drug Gene Interaction Database (DGIdb, (https://www.dgidb.org/) v.5.0.6 (04/04/2024)34 was used to identify drug-gene interactions among the genes identified from the WES gene burden tests. The DGIdb provides information on drugfig
-gene interactions from 28 diverse sources that are aggregated and normalized. The database collects drug-gene interactions based on information about therapeutic targets and their corresponding drugs, knowledge from clinical trials, as well as potentially clinically actionable drug-gene associations based on metadata such as molecule structure and molecular weight34. Gene-set enrichment analysis (GSEA) was performed to test if the genes identified from the WES gene burden tests were significantly (FDR < 0.05) enriched for targets of specific drugs.
LDSC
We applied univariate64 and cross-trait65 LDSC to estimate narrow-sense heritability and genetic correlations, respectively. For this, we formatted the GWAS summary statistics using our standardized pipeline, including ‘munging’ and removal of all variants in the extended MHC region (chr6:26–34 Mb), in accordance with recommendations (https://github.com/precimed/python_convert/blob/master/sumstats.py).
MiXeR analysis
We applied a causal mixture model35,36 to each of the main univariate GWAS summary statistics, with the extended MHC region excluded, to estimate the percentage of variance explained by genome-wide significant SNPs as a function of sample size. For each SNP, \(i\), MiXeR models its additive genetic effect of allele substitution,\({\beta }_{i}\), as a point-normal mixture, \({\beta }_{i}=\left(1-{\pi }_{1}\right)N\left({\mathrm{0,0}}\right)+{\pi }_{1}N\left(0,{\sigma }_{\beta }^{2}\right)\), where \({\pi }_{1}\) represents the proportion of non-null SNPs (‘polygenicity‘) and \({\sigma }_{\beta }^{2}\) represents the variance of effect sizes of non-null SNPs (‘discoverability‘). Then, for each SNP, \(j\), MiXeR incorporates LD information and allele frequencies for 9,997,231 SNPs extracted from the EUR population of the 1000 Genomes Phase3 data to estimate the expected probability distribution of the signed test statistic, \({z}_{j}={\delta }_{j}+{\epsilon }_{j}=N{\sum }_{i}\sqrt{{H}_{i}}{r}_{{ij}}{\beta }_{i}+{\epsilon }_{j}\), where \(N\) is the sample size, \({H}_{i}\) indicates heterozygosity of i-th SNP, \({r}_{{ij}}\) indicates an allelic correlation between i-th and j-th SNPs, and \({\epsilon }_{j}\sim N\left(0,{\sigma }_{0}^{2}\right)\) is the residual variance. Further, the three parameters, \({\pi }_{1},{\sigma }_{\beta }^{2},{\sigma }_{0}^{2}\), are fitted by direct maximization of the likelihood function. Finally, given the estimated parameters of the model, the power curve\(S\left(N\right)\) is then calculated from the posterior distribution \(p\left({\delta }_{j}|{z}_{j},N\right)\).
For quality control of the MiXeR results, we used the Akaike Information Criterion (AIC), comparing the Gaussian mixture model fit to that of the infinitesimal model. In this study, the AIC values of all 249 metabolites were positive, i.e. the Gaussian mixture had better model fit, warranting the inclusion of the results.
Mendelian randomization
We ran bidirectional MR, investigating the causal relationships between BMI and the 249 metabolites, with the TwoSampleMR R package. For this, we combined the BMI GWAS summary statistics from the GIANT consortium with no UKB participants (N = 339 224)66, to prevent sample overlap, with the metabolomics GWAS summary statistics generated in this study. We selected only genome wide significant variants for the analysis, clumped using PLINK with clump_p = 1, clump_r2 = 0.001, clump_kb = 10000 against the 1000 Genomes Phase3 503 EUR samples keeping other settings default. We calculated MR regression coefficients using the inverse variance weighted method and the weighted median method. To create robust findings, we only selected findings that showed a multiple comparisons-significance (p < .05/96) across both these methods. As an additional check, we ran MR-Egger and selected those relationships with nominal significance on this test.
Statistical analyses
All pre-processing steps and analyses performed outside the above-mentioned tools and software, e.g. formatting the data and creating the graphs, were carried out in R, v4.2.
Sensitivity analyses
We ran two sets of variations on the primary GWAS, to investigate the role of medication and of the preprocessing pipeline. First, we re-ran the primary GWAS controlling for insulin, blood pressure, and cholesterol-lowering medication. Second, we re-ran without the ‘ukbnmr’ pre-processing pipeline, directly on the originally released metabolomics data. For both variations, the produced summary statistics were highly comparable with the primary GWAS, with median genetic correlations of 0.992 and 0.998 across the metabolites, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data incorporated in this work were gathered from public resources. All input data described for the main analyses are available through UK Biobank, subject to approval from the UK Biobank access committee. See https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access for further details. The output data is provided in the Supplementary Data tables, as referenced in the Results. GWAS summary statistics have been uploaded to Zenodo, with the following DOIs: [main UKB], [meta-analysed UKB and EstBB], [sex-interaction], [male-specific], [female-specific], [MOSTest main and sex-interaction]. Source data are provided with this paper.
Code availability
The MOSTest and MiXeR code is available via https://github.com/precimed (GPLv3 license).
References
Würtz, P. et al. Quantitative Serum Nuclear Magnetic Resonance Metabolomics in Large-Scale Epidemiology: A Primer on -Omic Technologies. Am. J. Epidemiol. 186, 1084–1096 (2017).
Julkunen, H. et al. Atlas of plasma NMR biomarkers for health and disease in 118,461 individuals from the UK Biobank. Nat. Commun. 14, 604 (2023).
Group, N. H. B. C. et al. Metabolomic and genomic prediction of common diseases in 477,706 participants in three national biobanks. medRxiv https://doi.org/10.1101/2023.06.09.23291213 (2023).
Saklayen, M. G. The global epidemic of the metabolic syndrome. Curr. Hypertens. Rep. 20, 1–8 (2018).
Karjalainen, M. K. et al. Genome-wide characterization of circulating metabolic biomarkers. Nature https://doi.org/10.1038/s41586-024-07148-y (2024).
Lotta, L. A. et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet. 53, 54–64 (2021).
Tambets, R. et al. Genome-wide association study for circulating metabolites in 619,372 individuals. medRxiv https://doi.org/10.1101/2024.10.15.24315557 (2024).
Smith, C. J. et al. Integrative analysis of metabolite GWAS illuminates the molecular basis of pleiotropy and genetic correlation. Elife 11, e79348 (2022).
van der Meer, D. et al. Understanding the genetic determinants of the brain with MOSTest. Nat. Commun. 11, 3512 (2020).
Bomba, L. et al. Whole-exome sequencing identifies rare genetic variants associated with human plasma metabolites. Am. J. Hum. Genet. 109, 1038–1054 (2022).
Nag, A. et al. Assessing the contribution of rare-to-common protein-coding variants to circulating metabolic biomarker levels via 412,394 UK Biobank exome sequences. medRxiv https://doi.org/10.1101/2021.12.24.21268381 (2021).
Park, S., Sadanala, K. C. & Kim, E.-K. A metabolomic approach to understanding the metabolic link between obesity and diabetes. Mol. Cells 38, 587–596 (2015).
Varlamov, O., Bethea, C. L. & Roberts, C. T. Jr. Sex-specific differences in lipid and glucose metabolism. Front. Endocrinol. 5, 241 (2015).
Gerdts, E. & Regitz-Zagrosek, V. Sex differences in cardiometabolic disorders. Nat. Med. 25, 1657–1666 (2019).
Link, J. C. & Reue, K. Genetic Basis for Sex Differences in Obesity and Lipid Metabolism. Annu. Rev. Nutr. 37, 225–245 (2017).
Privé, F., Luu, K., Blum, M. G. B., McGrath, J. J. & Vilhjálmsson, B. J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 36, 4449–4457 (2020).
Li, J. & Ji, L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95, 221–227 (2005).
Keildson, S. et al. Expression of Phosphofructokinase in Skeletal Muscle Is Influenced by Genetic Variation and Associated With Insulin Sensitivity. Diabetes 63, 1154–1165 (2014).
Ahsan, H. et al. A genome-wide association study of early-onset breast cancer identifies PFKM as a novel breast cancer gene and supports a common genetic spectrum for breast cancer at any age. Cancer Epidemiol. Biomark. Prev. 23, 658–669 (2014).
Newington, J. T. et al. Overexpression of pyruvate dehydrogenase kinase 1 and lactate dehydrogenase A in nerve cells confers resistance to amyloid β and other toxins by decreasing mitochondrial respiration and reactive oxygen species production. J. Biol. Chem. 287, 37245–37258 (2012).
Koromina, M. et al. Fine-mapping genomic loci refines bipolar disorder risk genes. medRxiv https://doi.org/10.1101/2024.02.12.24302716 (2024).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021).
Martínez-Martínez, A. B. et al. Beyond the CNS: the many peripheral roles of APOE. Neurobiol. Dis. 138, 104809 (2020).
Jia, Q. et al. Genetic determinants of circulating glycine levels and risk of coronary artery disease. J. Am. Heart Assoc. 8, e011922 (2019).
Pendergrass, S. et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet. Epidemiol. 35, 410–422 (2011).
Devallière, J. & Charreau, B. The adaptor Lnk (SH2B3): an emerging regulator in vascular cells and a link between immune and inflammatory signaling. Biochem. Pharmacol. 82, 1391–1402 (2011).
Almuttaqi, H. & Udalova, I. A. Advances and challenges in targeting IRF5, a key regulator of inflammation. FEBS J. 286, 1624–1637 (2019).
Silveira Rossi, J. L. et al. Metabolic syndrome and cardiovascular diseases: Going beyond traditional risk factors. Diabetes Metab. Res. Rev. 38, e3502 (2022).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
McDade, E. et al. The case for low-level BACE1 inhibition for the prevention of Alzheimer disease. Nat. Rev. Neurol. 17, 703–714 (2021).
Aragam, K. G. et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 54, 1803–1815 (2022).
Zhou, S. et al. Association study of BUD13-ZNF259 gene rs964184 polymorphism and hemorrhagic stroke risk. Int. J. Clin. Exp. Med. 8, 22503 (2015).
Chittilla, M., Akimbekov, N. S. & Razzaque, M. S. High-fat diet-associated cognitive decline: Is zinc finger protein 1 (ZPR1) the molecular connection?. Curr. Res. Physiol. 4, 223–228 (2021).
Cannon, M. et al. DGIdb 5.0: rebuilding the drug–gene interaction database for precision medicine and drug discovery platforms. Nucleic Acids Res. 52, D1227–D1235 (2024).
Holland, D. et al. Beyond SNP Heritability: Polygenicity and Discoverability of Phenotypes Estimated with a Univariate Gaussian Mixture Model. bioRxiv https://doi.org/10.1101/498550 (2019).
Frei, O. et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat. Commun. 10, 2417 (2019).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389 (2017).
Kivimäki, M. et al. Overweight, obesity, and risk of cardiometabolic multimorbidity: pooled analysis of individual-level data for 120 813 adults from 16 cohort studies from the USA and Europe. Lancet Public Health 2, e277–e285 (2017).
Correll, C. U. et al. Cardiometabolic comorbidities, readmission, and costs in schizophrenia and bipolar disorder: a real-world analysis. Ann. Gen. Psychiatry 16, 1–8 (2017).
Canoy, D. et al. Association between cardiometabolic disease multimorbidity and all-cause mortality in 2 million women and men registered in UK general practices. BMC Med 19, 1–10 (2021).
Elliott, D. A., Weickert, C. S. & Garner, B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin. Lipidol. 5, 555–573 (2010).
Cole, S. L. & Vassar, R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2, 1–25 (2007).
Clayton, D. G. Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet 5, e1000540–e1000540 (2009).
Strawbridge, R. J. et al. Carotid intima-media thickness: novel loci, sex-specific effects, and genetic correlations with obesity and glucometabolic traits in UK Biobank. Arterioscler. Thromb. Vasc. Biol. 40, 446–461 (2020).
Lutz, M. W. & Chiba-Falek, O. Bioinformatics pipeline to guide late-onset Alzheimer’s disease (LOAD) post-GWAS studies: Prioritizing transcription regulatory variants within LOAD-associated regions. Alzheimers Dement. Transl. Res. Clin. Interv. 8, e12244 (2022).
Bell, J. A. et al. Effects of general and central adiposity on circulating lipoprotein, lipid, and metabolite levels in UK Biobank: a multivariable Mendelian randomization study. Lancet Reg. Heal. 21, 100457 (2022).
Marott, S. C., Nordestgaard, B. G., Tybjærg-Hansen, A. & Benn, M. Causal associations in type 2 diabetes development. J. Clin. Endocrinol. Metab. 104, 1313–1324 (2019).
Lotta, L. A. et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a Mendelian randomisation analysis. PLoS Med. 13, e1002179 (2016).
Mahendran, Y. et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia 60, 873–878 (2017).
Arques, S. Human serum albumin in cardiovascular diseases. Eur. J. Intern. Med. 52, 8–12 (2018).
Hira, T., Pinyo, J. & Hara, H. What is GLP-1 really doing in obesity?. Trends Endocrinol. Metab. 31, 71–80 (2020).
Chiu, M., Austin, P. C., Manuel, D. G., Shah, B. R. & Tu, J. V. Deriving ethnic-specific BMI cutoff points for assessing diabetes risk. Diabetes Care 34, 1741–1748 (2011).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779–e1001779 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Leitsalu, L. et al. Cohort profile: Estonian biobank of the Estonian genome center, university of Tartu. Int. J. Epidemiol. 44, 1137–1147 (2015).
Ritchie, S. C. et al. Quality control and removal of technical variation of NMR metabolic biomarker data in ~120,000 UK Biobank participants. Sci. Data 10, 64–64 (2023).
Beasley, T. M., Erickson, S. & Allison, D. B. Rank-based inverse normal transformations are increasingly used, but are they merited?. Behav. Genet. 39, 580–595 (2009).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7–7 (2015).
Watanabe, K., Taskesen, E., Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826–1826 (2017).
Van der Sluis, S., Posthuma, D. & Dolan, C. V. TATES: efficient multivariate genotype-phenotype analysis for genome-wide association studies. PLoS Genet 9, e1003235–e1003235 (2013).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219–e1004219 (2015).
Consortium, G. te et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236 (2015).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Acknowledgements
This work was partly performed on the TSD (Tjeneste for Sensitive Data) facilities, owned by the University of Oslo, operated and developed by the TSD service group at the University of Oslo, IT-Department (USIT). (tsd-drift@usit.uio.no). Computations were also performed on resources provided by UNINETT Sigma2 - the National Infrastructure for High-Performance Computing and Data Storage in Norway. Computation of Estonian Biobank data was carried out in part in the High-Performance Computing Center of University of Tartu. The Estonian Biobank Research Team, responsible for data collection, genotyping, QC, and imputation, comprises Andres Metspalu, Tõnu Esko, Reedik Mägi, Mait Metspalu, Mari Nelis Georgi Hudjashov, Priit Palta, Nele Taba, Erik Abner, Jaanika Kronberg, Urmo Võsa. The authors were funded by the Research Council of Norway (273291 O.A.A., 273446 O.A.A., 296030 O.A.A., 324252 O.A.A., 324499 O.A.A., 326813 O.A.A.), Estonian Research Council (grant PSG615 K.L., PSG1028 M.A.), The University of Tartu grant (PLTGI24925 M.A.), European Union’s Horizon 2020 research and innovation program (847776 CoMorMent O.A.A., 964874 RealMent O.A.A.), and Marie Skłodowska-Curie grant 801133 (P.P.), National Institutes of Health (U24DA041123 A.M.D., R01AG076838 A.M.D., U24DA055330 A.M.D., OT2HL161847, A.M.D.).
Author information
Authors and Affiliations
Consortia
Contributions
D.v.d.M., Z.R., A.S., S.D., and O.A.A. conceived the study; D.v.d.M., Z.R., A.O., G.K., and A.S. pre-processed the data. D.v.d.M., Z.R. A.O., E.K. and A.S. performed all analyses, with conceptual input from J.R., S.S., P.P., S.D. and O.A.A.; D.v.d.M. drafted the manuscript. Z.R., A.O., P.P., G.K., S.E.S., M.K., J.R., I.E.S., N.P., M.T., G.H., L.N.R., E.K. E.B.R.T., N.E.S., J.P.B., K.S.O., O.B.S., O.F., A.M.D., S.D., K.L., M.A., L.M., A.A.S. and O.A.A. critically revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.M.D. is Founding Director, holds equity in CorTechs Labs, Inc. (DBA Cortechs.ai), and serves on its Board of Directors and Scientific Advisory Board. A.M.D. is the President of J. Craig Venter Institute (JCVI) and is a member of the Board of Trustees of JCVI. He is an unpaid consultant for Oslo University Hospital. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. O.F. is a consultant to Precision Health. O.A.A. has received speaker fees from Lundbeck, Janssen, Otsuka, Lilly, and Sunovion and is a consultant to Cortechs.ai. and Precision Health. All other authors report no potential conflicts of interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
van der Meer, D., Rahman, Z., Ottas, A. et al. Pleiotropic and sex-specific genetic mechanisms of circulating metabolic markers. Nat Commun 16, 4961 (2025). https://doi.org/10.1038/s41467-025-60058-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60058-z
This article is cited by
-
Genetic architecture of plasma metabolome in 254,825 individuals
Nature Communications (2025)
-
A genetic map of human metabolism across the allele frequency spectrum
Nature Genetics (2025)
