
Abstract
Atropisomers are sterically hindered molecules whose formation typically proceeds via atropisomeric intermediates and encumbered transition states. It is therefore largely accepted that the activation energy is higher for synthesis of atropisomers than for synthesis of similar, less sterically congested non-atropisomeric compounds. Here we show that atropisomer formation by nucleophilic aromatic substitution (SNAr) reactions can progress via non-atropisomeric intermediates and transition states. We put forth fast, mild, practical, regio- and chemoselective SNAr reactions that generate a diverse array of difficult-to-access heterobiaryl C─N atropisomers starting with readily available N─H heterocycles and aryl fluorides, as well as two catalytic methods employing N─SiR3 and N─H heterocycles for synthesis of title atropisomers in seconds. Products of SNAr are readily diversifiable, streamlining access to countless drug-like C─N atropisomers, including macrocycles, peptides, and analogs of achiral heterobiaryl pharmaceuticals. Supported by experimental and computational data, we discuss how steric repulsion is minimized in stereogenic axis-forming SNAr processes.
Similar content being viewed by others
Introduction
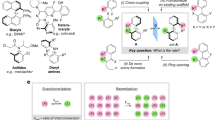
Atropisomeric molecules, or atropisomers, are chiral conformational isomers originating from restricted rotation around a sterically hindered single bond, namely a stereogenic axis1. The most stabilized conformations of atropisomers are intrinsically non-planar, for example in biaryl cases where the dihedral angle formed between the two aryl rings oscillates around 90°2. These unique conformational attributes impart the overall shape of molecules, leading to important implications in medicinal chemistry. Atropisomerism is introduced to minimize the entropic cost upon binding of a ligand to a biological receptor, often resulting in improved potency, selectivity and metabolic stability3. Atropisomerism has therefore become popular in drug discovery campaigns carried out both in industrial and academic settings. Crucial to such efforts are heterobiaryl-type atropisomers with a C─N stereogenic axis (Fig. 1a)4; prominent examples include marketed5,6 and lead drugs7,8,9,10, antibacterial atropopeptides11,12 recently isolated from natural sources and many other bioactive molecules13,14.

a Prominent bioactive heterocyclic C–N-based atropisomers are structurally diverse. b Desirable two-stage workflow involving (1) assembly and (2) diversification toward synthesis of libraries of atropisomers. c Atropisomer formation naturally proceeds via sterically congested transition states. Identifying reactions which proceed via less sterically demanding transition states may lead to faster processes. d Key features of a nucleophilic aromatic substitution strategy toward readily modifiable C–N atropisomers. FG, functional group; Z, CH or N; R, H or C-based group; Ea, energy of activation; SNAr, nucleophilic aromatic substitution.
Restricting motion on a heterobiaryl motif leads to particularly precise spatial positioning of substituents, which are placed on well-defined vectors with initial points at the atropisomeric core (Fig. 1b). Such preciseness may allow development of ligands that bind more strongly a given biological receptor or enable studies of intermolecular interactions within the receptor’s pocket. However, these studies are delayed due to a dearth of atropisomer synthesis strategies that are modular and allow programmable incorporation of groups at specific sites around atropisomeric biaryls. A desirable workflow for medicinal chemistry would consist of synthesizing atropisomeric molecules that could be modified sequentially by diversity-oriented synthesis15. This approach, involving assembly (phase 1) and diversification (phase 2), would enable elusive libraries of drug-like atropisomers to be produced with high control over the directionality of substituents in intrinsic tridimensional spaces16,17.
A direct way to synthesize heterobiaryl atropisomers is by metal-catalyzed cross-coupling reactions, gold standard for formation of C‒N and C‒C biaryl bonds18,19. However, such processes are not ideal for assembling diversifiable atropisomers. Cross-coupling reactions should preferably be employed for diversification but, for this to be possible, reactive aryl halides need to be left intact during atropisomer assembly. In the best possible scenario, considering C‒N bond formation, either an aryl iodide or a boronic acid, both sterically hindered, would have to react prior to less encumbered aryl bromides and chlorides. Even if catalysts were developed to overcome this challenge, the opportunity to use aryl iodides and boronates later for diversification would still be missed. In short, severe chemoselectivity issues arise if cross-coupling is employed for assembly and diversification. This discussion relates to a well-accepted notion that atropisomer formation involves strong repulsive steric interactions intrinsically, as stereogenic axis-forming reactions often progress via atropisomeric intermediates and encumbered transition states (Fig. 1c). Accordingly, we were keen to identify reactions which could assemble sterically hindered bonds via non-atropisomeric intermediates because, presumably, such processes would require lower energy of activation and enable synthesis of atropisomers more efficiently.
We entertained the idea that nucleophilic aromatic substitution (SNAr) would have potential for modular and direct synthesis of stereogenic axes without sacrificing future diversification initiatives. Our reasoning was based on the following: (1) aryl fluorides are more reactive than bromides and iodides in SNAr, an attribute which would facilitate the use of more reactive halides for cross-coupling in the modification stage; (2) a large number of 2,6-disubstituted aryl fluorides are readily available. A literature search reveals that a larger number of sterically hindered aryl fluorides are known, compared to analogous aryl bromides, iodides and boronates (see Supplementary Section 3 for details). (3) SNAr processes favor formation of C‒N vs C‒C bonds at large. This seemed a bonus in every way; not only N‒H heterocycles are readily available but heterobiaryl C‒N atropisomers are prominent and structurally diverse (Fig. 1a).
Synthesis of heterobiaryl C─N atropisomers through ipso substitution would benefit from reactions which, unlike cross-coupling20,21,22, would not be as sensitive to steric repulsions but highly sensitive to and activated by electronic effects. Reactions in which the more hindered reacting partner—with two large ortho substituents—would also be the more reactive one, are particularly desirable. Here, we show that SNAr reactions23,24,25 meet abovementioned demands of reactivity and structural diversity generation. Currently, methods for direct formation of a C─N stereogenic axis by SNAr are sparse and severely limited by the use of intricate chromium carbonyl complexes as electrophiles and harsh conditions26. We disclose a broadly applicable, modular SNAr strategy that is orthogonal to cross-coupling and other well-established methods, allowing for rapid synthesis of an assortment of atropisomers with drug-like structural features (Fig. 1d). The SNAr reactions shown herein are among the fastest and mildest atropisomer syntheses known to date.
Results
Reaction discovery
We identified that 2-methylindole undergoes reaction with ethyl 2-fluoro-3-nitrobenzoate in the presence of Cs2CO3 and DMSO to give atropisomer 1 in 97% yield, in one hour at room temperature (Fig. 2, grey box). The reaction was easily scalable and an aqueous workup sufficed to obtain 15.8 g of 1 as solid in >98% purity by 1H NMR. Atropisomer 1 is configurationally stable as no racemization was observed at 140 °C (ΔGrot‡ = 35.2 kcal/mol, measured at 165 °C in mesitylene solution). Apart from short reaction time and ambient temperature, conditions which are particularly uncommon to intermolecular atropisomer synthesis, several other reaction attributes are noteworthy. All reagents were commercially available and used as received (including reagent grade DMSO). The reaction was carried out with 1.0 equivalent of each SNAr reacting partner without precaution for exclusion of air and moisture and the total cost for synthesis of 1 was calculated to be <0.5 US dollars/mmol. Another distinct feature is the high selectivity (> 98:2, NMR detection limit) toward substitution at the indole N1 versus C3 position. As it will be shown herein, the SNAr protocol complements and can be used in combination with cross-coupling and other synthesis strategies for maximal diversification.

aAryl chloride instead of aryl fluoride employed; b70 °C; c2.0 equiv. 5,5-dimethylhydantoin used; d42 h. Z, CH or N; ΔGrot‡, energy barrier for C–N bond rotation (atropisomerism threshold at room temperature is 21 kcal/mol). Additional scope is shown in Supplementary Section 4, including electronically less activated aryl fluorides and six-membered nucleophiles.
Reaction scope (method A)
A key advantage is that a large number of N─H heterocycles and hindered aryl fluorides are commercial or easily synthesized. As a result, not only we synthesized a diverse set of C─N atropisomers, but each contained modifiable functional groups (Fig. 2, in color) and heteroaryl sites (e.g., indole C3). Minimal to no changes on the optimized protocol were required for obtaining high yields and chemoselectivities.
A broad range of aromatic heterocycles (H1-17) were incorporated (Fig. 2). Indole (1,6,13), benzimidazole (2,4,5), imidazole (8), indazole (9), tetrahydro-β-carboline (19), and carbazole (20) with various substituents afforded atropisomeric products readily. Heterocycles displaying structural similarities to purine nucleobases also reacted in high yields; examples include azaindole- (10,11), aminopurine- (12) and theophylline-containing (14) atropisomers. High selectivity was obtained toward N1 versus C3 for indole, N1 versus N3 for imidazole, N1 versus N2 for indazole, N9 versus N7 for aminopurine, and N7 versus N9 for theophylline (> 98:2 selectivity in all cases). For the imidazole and indazole derivatives, high regioselectivity was obtained on account of minimization of steric repulsion. Non-aromatic N-heterocycles were also suitable. For instance, reaction with unprotected 5,5-dimethylhydantoin resulted in synthesis of atropisomer 22 by substitution at N3 (85:15 selectivity), the most acidic position. Alternatively, in a case where N3 was substituted prior to the SNAr reaction, N1 reacted to generate compound 23. The method is not limited to 5-membered heterocycles; a pyrimidine nucleobase derivative, fluorouracil, delivered atropisomer 24 with full control of selectivity, favoring substitution at N1. It is currently challenging to employ many of such heterocycles in C─N forming cross-coupling reactions because, among other reasons, N─H heterocycles often strongly coordinate to Cu-, Ni-, and Pd-complexes, deactivating catalytic intermediates.
We gave preference to fluoroarenes (F1-19) with activating yet modifiable functional groups (Fig. 2). Compounds with nitro groups at ortho (1–5,8,10) and para positions (6,7), various esters (1,4,10,17,19,21,25,26), nitriles (8,15), sulfone (21), sulfonamide (23), chloro (2,5,11,12,14,23), bromo (3,4,6,8,10,13), iodo (9,10,17) groups reacted smoothly. Another advantage is that 6-membered heterocycles, many of which are commercial, are reactive electrophilic partners: synthesis of quinazoline- (16), quinoline- (17) and pyridine- containing (18,19) atropisomers proceeded in high yields. In two cases, toward 16 and 17, an aryl chloride was employed instead of aryl fluoride, underscoring other abundant SNAr active electrophiles are suitable. By and large, SNAr activators are polar functional groups in high oxidation state which are prevalent in pharmaceuticals and easily modifiable by well-established methods. Apart from the abovementioned, it merits attention that the scope includes heteroaryl NH2 (5,12), not only aryl halides but an example of an alkyl bromide (4), carbamate (19), methylketone (20), alkene (10) and click partners such as azide (25) as well as terminal and highly reactive strained alkynes (23,26). Additional scope is shown in Supplementary Section 4, including aryl fluorides that are less electronically activated and other six-membered nucleophiles.
Potential for Direct-to-Biology screenings
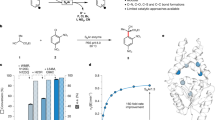
To further demonstrate practicality and potential for Direct-to-Biology (D2B) initiatives27 we selected a series of aryl fluorides (Fa-f) and heterocycles (Ha-f) which had not been used previously to react combinatorially (Fig. 3). Reactions were carried out with 2.0 equiv Cs2CO3 and 1.0 equiv of each reacting partner in DMSO-d6 as solvent for rapid yield quantification by 1H NMR. Structural characterization was carried out by NMR and mass spectrometry (MS) in low and high resolution modes (Supplementary Section 15 for details). Out of thirty-six reactions toward 27aa-ff, twenty-two afforded the desired product in > 70% yield, an excellent range for D2B. Other six reactions gave product in 40–70% and five in 10–40% yield. Only one reaction afforded product in less than 10% yield and two reactions failed completely. Successful reactions were observed in every column or row, but a few poor performing cases highlight the uniqueness of the combinatorial approach. For example, if one reacts Hd only with Fe, toward 27ed, one might believe Hd is a problematic substrate; however, this is not the case as Hd reacted successfully with four other fluoroarenes. It also merits mention that purification would be facile in most cases as reactions were clean and gave trace amounts of by-products. These results validate suitability for high-throughput synthesis followed by biological testing, which incidentally often employ DMSO solutions.

a Experimental attributes, such as the use of DMSO as solvent and resulting homogeneous solution after filtration, are ideal for D2B screenings. b Combinatorial experiments revealed that 60% of reactions generated product in greater than 70% yield, an excellent range for D2B approaches. Reactions were carried out with 1.0 equiv Fa-f, 1.0 equiv of Ha-f in DMSO-d6. Yields were measured by 1H NMR with an internal standard and characterization was carried out by NMR and MS. NMR nuclear magnetic resonance, MS mass spectrometry.
Configurational stability of atropisomers
Assessing atropisomer configurational stability is crucial in medicinal chemistry28. We measured experimentally the energy barrier to rotation around the stereogenic axis of representative compounds, allowing us to estimate barrier to rotation of all compounds (Supplementary Section 13 for details). Atropisomers with four substituents (other than H or a non-bonding electron pair) at the ortho position relative to the C─N stereogenic axis, the majority of the compounds, display high barrier to rotation and are therefore configurationally stable, class 3 atropisomers28 (ΔG‡438K (1) = 35.2 kcal/mol; ΔG‡438K (2) = 35.6 kcal/mol; and ΔG‡438K (23) > 35.6 kcal/mol). Most compounds with three substituents around the stereogenic axis are borderline class 2/3 atropisomers (ΔG‡353K (16) = 27.5 kcal/mol and ΔG‡383K (24) = 28.9 kcal/mol), and ones with the smallest substituents are class 2 (ΔG‡333K (7) = 24.1 kcal/mol and ΔG‡353K (18) = 25.8 kcal/mol). Most of the subsequent modifications performed in class 3 atropisomers would likely not affect enantiomeric excess, but would significantly lower enantiomeric ratio in class 2 compounds, especially modifications carried out above room temperature. Herein, we disclose a platform for discovery for which such matters are less relevant. In the context of medicinal chemistry, two aspects merit mention: (1) in early screenings, racemates are employed irrespective to the energy barrier for atropisomer interconversion. (2) Class 3 and borderline class 2/3 atropisomers are suitable for studies as single enantiomers upon separation by chiral chromatography or chemical methods.
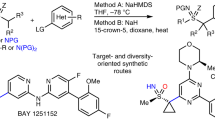
Catalytic SNAr starting with N‒SiR3 heterocycles (method B)
During our optimization studies, we observed that tertiary organic bases, for example DBU (1,8-Diazabicyclo[5.4.0]undec-7-ene), did not promote SNAr with indole derivatives as nucleophile. At the outset, we attributed the lack of reactivity to the fact that indole deprotonation, if feasible, would produce a tertiary ammonium cation, the proton of which could stabilize the nucleophile. Such findings led us to evaluate the possibility of generating negatively charged nucleophiles from N-silyl heterocycles by N‒Si cleavage with fluoride29. In this way, a proton would not be available and the SNAr could be rendered catalytic through intermediate Int-1 (Fig. 4b). We tested various fluoride sources. With 10 mol % TBAT (tetrabutylammonium difluorotriphenylsilicate), an anhydrous source of fluoride, H1-Si and F1a reacted in 3 min to give 1 in 95% yield in dry DMSO (Method B, Fig. 4a). With 10 mol % tetrabutylammonium fluoride, a lower yield (76%) was obtained, on account of competing hydrolysis of H1-Si to form H1. The fluoride-catalyzed process, which is milder and faster, improved several challenging cases. For example, reaction with 4-bromophthalimide was sluggish with Cs2CO3 (38% yield in 24 h), but with catalytic TBAT, we obtained 79% yield of 28 in 20 h (Fig. 4c). In some cases, more reactive esters were partially hydrolyzed under the Cs2CO3 protocol, but not in the fluoride-promoted reaction. Accordingly, atropisomer 29 was isolated in 61% yield after 2 h (vs <10% yield by Method A). Isatin ring opening was a major issue with Cs2CO3 in DMSO, in contrast, the catalytic reaction allowed us to secure 30 in 95% yield in 10 min. Another competing process in base-promoted SNAr reactions was undesired α-deprotonation followed by arylation, observed when 1-methylhydantoin was employed as nucleophile. Undesired arylation was not a problem in the catalytic variant, allowing atropisomeric product 31 to be synthesized in 45% yield (vs <2% by Method A).

a Fluoride-catalyzed reactions starting with H1-Si (Method B) or with H1 in presence of Me3SiCF3 (Method C) are remarkably fast. b Proposed catalytic cycles. c Method B enabled synthesis of atropisomers which could not be synthesized efficiently by Method A. d Method C enabled synthesis of atropisomers which could not be synthesized efficiently neither by Method A nor B. For additional scope by Method C, see Supplementary Section 5.3. e Catalytic methods foreshadow atroposelectivity, as shown by use of a benchmark chiral fluoride-based catalyst. TBAT tetrabutylammonium difluorotriphenylsilicate.
Catalytic SNAr starting with N‒H heterocycles (method C)
During the course of our studies on the abovementioned catalytic method, we failed on synthesis and isolation of labile silyl heterocycles, for example from benzimidazole derivatives among others. Mindful of the high reactivity of Int-1, and of reports on the reaction between fluoride anion with Me3SiCF330 to form trifluoromethyl anion, we envisioned a more direct catalytic process starting with N‒H heterocycles. We reacted H1 and F1a in the presence of 1.5 equivalents of Me3SiCF3 and 10 mol % TBAT (Fig. 4a). In the event, atropisomer 1 was synthesized in 94% yield in 30 s, after rapid and irreversible formation of fluoroform via Int-1 (Fig. 4b). En route to atropisomers 32 and 33 (Fig. 4d), we had observed several problems previously. Apart from abovementioned challenges in silylation, alcohols engaged in SNAr readily, leading to complex mixtures. Only by means of Method C the atropisomers 32 and 33 could be synthesized in high yields, 70 and 72%, respectively. This variant is likely to unlock atroposelective reactions. In a preliminary foray, with a benchmark chiral ammonium fluoride catalyst and our prototypical substrates, we observed low enantioselectivity (Fig. 4e). As this variant precludes background reactivity, we envision that catalyst development and fine-tuning of conditions will lead to high selectivity.
Mechanistic studies: SNAr for formation of stereogenic axis
We performed density functional theory (DFT) calculations and experiments to shed light on why SNAr reactions are efficient and fast toward a broad range of notoriously sterically hindered compounds, as well as to understand trends in reactivity (e.g., the influence of ortho substituents). Other crucial questions were: How does the nature of the nucleophile influence the SNAr reactions? What is the precise role of the base, and what is deprotonated, the heterocycle directly or a donor-acceptor complex? Does the cation play a role? Answering the above questions may lead to new and efficient atropisomer synthesis methods as well as to advances in substitution processes at large.
We started probing the fluoride-catalyzed reaction, a homogeneous process (Fig. 5). DFT indicates that forming an indole anion by reaction of H1-Si with fluoride is fast (via A-I and A-II with relative energies of 4.1 and 1.5 kcal/mol, respectively) and highly thermodynamically favored (A-III, −25.2 kcal/mol), due to formation of a strong F─Si bond and a tightly bound donor-acceptor complex involving 2-methylindole anion and F1a. Intermediate A-III reacts to form the Meisenheimer intermediate A-V via transition state A-IV. Intermediate A-V then undergoes rapid elimination to form 1 via transition state A-VI, regenerating the fluoride catalyst. The addition step, namely formation of the Meisenheimer complex, is turnover-limiting of the catalytic cycle (12.9 kcal/mol from A-III to A-IV versus 8.9 kcal/mol from A-V to A-VI). Alike the catalytic reaction, base-promoted processes proceed in a stepwise manner where the addition step is rate-determining (see Supplementary Section 14 for calculations on base-promoted reactions), consistent with previous studies with activated aryl fluorides31,32. Analysis of the transition state in the addition step reveals that the dihedral angle formed between the planes of the indole and the aryl ring is approximately 60°, closer to 80‒90° typical of atropisomers than to coplanarity. This arrangement places the ortho substituents—nitro, ester, and methyl groups—far from each other. The distance between the methyl group carbon (CMe) and the closest oxygen of the nitro (ONO2) and ester (Oest) groups are 3.5 and 3.8 Å, respectively (see Front and Lateral views). Another key feature is that minimal reorganization from transition state to product is required for atropisomer formation, unlike in cross-coupling reactions where steric interactions involving the ligand around the metal catalyst and ortho substituents on the substrates preclude the planes of aryl rings to be orthogonal19,33. We thus suggest that rates of SNAr reactions tend to be less sensitive to an increase in the number and size of ortho substituents. The SNAr reactions reported herein consist of atropisomer-forming processes that do not proceed via transition states with atropisomeric character in the rate-determining step. The energy for C‒N bond rotation in intermediate A-V was calculated to be 16.6 kcal/mol (Fig. 5), about 5.0 kcal/mol below the atropisomerism threshold of approximately 21 kcal/mol at room temperature34 and less than half the energy barrier to rotation measured for the same bond in product 1 (ΔG‡438K (1) = 35.2 kcal/mol, Fig. 2). The lower barrier in the Meisenheimer intermediate A-V is a result of the presence of a C(sp3)‒N versus a C(sp2)‒N bond typical of atropisomers such as in product 1. This discussion underscores the potential of SNAr for direct synthesis of stereogenic axis and other sterically congested bonds26,35. Our mechanistic analysis suggests that reactions toward atropisomers will benefit from electronic activation and non-atropisomeric intermediates and transition states (see Supplementary Section 14 for a detailed mechanistic discussion regarding the nature of nucleophile and its deprotonation, and Section 6 for the influence of ortho substituents).

The calculations support a stepwise mechanism in which the addition step is rate-determining (or turnover-limiting in the catalytic reactions) but it is not the atropisomer-forming step. This attribute is supported by the calculated energy barrier to rotation around the C–N bond in the Meisenheimer intermediate, which is 16.6 kcal/mol, therefore lower than the atropisomerism threshold (21 kcal/mol at room temperature) and less than half of that of the product (35.2 kcal/mol; see Supplementary Section 6,12, and 14 for mechanistic details). TS transition state; (X.X) = relative free energies in kcal/mol; DFT density functional theory, DFT method: PBE0-D3/def2-TZVP level in DMSO with CPCM solvation model.
Diversification and applications
Beyond the chemoselectivity observed with aryl halides, we discovered that (pinacol)boronate-containing 7-azaindole H23 reacts readily by Method A to afford atropisomer 34, which was then transformed to 35 by catalytic Chan-Lam reaction with piperidine (Fig. 6a)36. Realizing metal-catalyzed coupling while leaving aryl boronates intact is extremely challenging. A way to circumvent such challenge is to convert the C‒B bond before ipso substitution. However, this may be followed by other issues. For example, reaction between H23 with piperidine, under optimal Chan-Lam conditions, yielded traces of products because, in all likelihood, the 7-azaindole N‒H binds to Cu-based intermediates competitively and irreversibly. As such, a synthesis sequence involving Chan-Lam followed by SNAr toward 35 would be difficult to achieve, unlike the two-step sequence shown herein. Key to generation of libraries of atropisomers is diversification through selective and sequential cross-coupling reactions. One example entails selective N- versus C-substitution: fluorouracil-containing atropisomer 24 was modified by Chan-Lam reaction on the nitrogen or Sonogashira coupling on the aryl bromide site to generate 36 or 37, respectively (Fig. 6b). We also explored the innate reactivity of aryl halides in catalytic cross-coupling: aryl bromides react faster than aryl chlorides, allowing us to synthesize 38 and 39 sequentially (Fig. 6c). Likewise, an aryl iodide reacts prior to an aryl bromide, for example with atropisomer 9 as shown in Supplementary Section 4. The orthogonality with both aryl halide- and aryl boronate-based coupling reactions will allow for synthesis of countless hitherto inaccessible drug-like atropisomers. We diversified the atropisomers generated by SNAr in various other ways: by peptide- and cross-coupling reactions, click and biorthogonal chemistry and transformation of nitro, ester and other functional groups (see Supplementary Section 4 for strategic examples). Most diversification steps are orthogonal, enabling combinations and permutations which will give rise to structurally diverse and large libraries of potentially bioactive atropisomeric molecules.

a An aryl boronate is left intact under the standard assembly (SNAr) conditions, allowing for diversification by another C–N bond formation. Attempts to form firstly the C–N bond on the aryl boronate site were not productive. b In atropisomers with N–H and C–Br groups, selective C–N or C–C bond formations by cross-coupling are viable options. c An aryl bromide and an aryl chloride moieties, which remained intact during assembly, react sequentially and selectively.
Macrocycles are in the forefront of drug discovery (Fig. 1a). One way to bias macrocyclic conformation and in turn improve their pharmacological properties is to introduce atropisomeric moieties within the macrocyclic structure37. To synthesize modifiable atropisomeric macrocyclic platforms, we synthesized diene 40 by SNAr in 79% yield (Fig. 7a). Macrocyclization by ring-closing metathesis38 with a Ru-based catalyst gave a mixture of E and Z alkenes, 41 and 42, in 95% yield. The C‒C bond connecting the indole C2 and the aryl group is not a stereogenic axis but it becomes, after macrocyclization; the ring-closing strategy thus creates an additional stereogenic axis diastereoselectively. Reduction of the alkene and nitro groups in one pot gives a single atropisomeric macrocycle, bearing a modifiable indole and an aniline (43). Subsequent amide-coupling, indole C3 iodination and cross-coupling, en route to 45, showcase versatility and modularity.

a Synthesis of modifiable atropisomeric macrocycles by ring-closing metathesis. b Synthesis of linear and macrocyclic atropopeptides, inspired by naturally-occurring and antibacterial compounds such as cihunamide B (Fig. 1) and several others. c Inducing conformational changes (and chirality) by means of atropisomerism to heterobiaryl pharmaceuticals. This strategy is applicable to countless molecules in early drug discovery screenings.
Recently, several ribosomally synthesized and post-translationally modified (RiPPs) atropisomeric macrocyclic peptides have been isolated from natural sources and shown to possess antibacterial activity, including against resistant bacteria11,12. The vast majority of such intriguing molecules have a C─N stereogenic axis derived from tryptophan moieties (e.g., cihunamide B, Fig. 1). Inspired by the isolation and remarkable synthesis of the natural compounds39,40,41,42, we embarked on the synthesis of analogous, tryptophan-containing, atropopeptidic structures (Fig. 7b). SNAr with requisite tryptophan derivative and fluoroarene afforded a mixture of diastereomeric atropisomers 46 and 47 in 95% yield and 55:45 dr (diastereomeric ratio). Benzyl ester deprotection followed by amide coupling with H2N-Gly-Val-OtBu gave longer linear atropopeptides 48 and 49 in 72% yield over two steps and 58:42 dr. Global deprotection and peptide macrocyclization delivered 50 and 51 in 60% yield over two steps and 52:48 dr. Assignment of atropisomer stereochemistry was carried out by 1H‒1H ROESY in combination with computationally-determined conformational minima. The small changes in the dr throughout the synthesis, within the error of 1H NMR measurements, suggested high configurational stability. Indeed, the macrocycles 50 and 51 do not interconvert at 22 °C in CDCl3 solution over seven days, but epimerize slowly at 60 °C (ΔG‡333K (50 → 51) = 28.6 kcal/mol; (ΔG‡333K (51 → 50) = 29.0 kcal/mol). Nature provides one atropodiastereomer of RiPPs; we expect that our efforts, which afford two stable diastereomers, will unlock medicinal chemistry initiatives with stereochemically diverse set of atropopeptides with distinct conformation and tridimensional shapes.
We set out to introduce atropisomerism around C‒N-containing bioactive heterobiaryl compounds. We chose to demonstrate the concept with marketed pharmaceuticals, but the strategy also applies to drug leads or bioactive molecules in initial stages of medicinal chemistry efforts. Modifying conformation of biologically active molecules, especially to the extreme of introducing chirality, is crucial in structure-activity relationship studies and for understanding of ligand-receptor interactions. We employed commercially available reagents to introduce chirality to three bioactive molecules: anti-cancer nilutamide43 and nilotinib44, used for prostate cancer and leukemia, respectively, and the anti-inflammatory celecoxib (Fig. 7c)45 Nilutamide analogue 52, where a chloride was added and trifluoromethyl group moved toward the C‒N axis, was synthesized in 71% yield by Method A. Inspired by celecoxib, a sulfonamide group was placed at a hindered position to afford analogue 53, and a separable regioisomer—which is also atropisomeric—originated from substitution at N2 (versus N1 toward 53, see Supplementary Section 10.4 for details). For nilotinib, we modified the heterobiaryl portion to synthesize analogue 54 in 85% over two steps. It merits mention that the diarylaniline motif is present in most successful compounds of the same class (e.g., imatinib). We altered the portion of the molecule which differs across various analogues and is still under investigation.
Discussion
We disclosed a practical and broadly applicable SNAr strategy to form directly C─N stereogenic axis, unlocking synthesis of a diverse set of hitherto unknown heterocyclic atropisomers. More than hundred atropisomers were synthesized from readily available starting materials and adorned with functional groups that are prevalent in drugs and other bioactive molecules. Our strategy, when combined with cross-coupling and other established methods, enabled rapid access to compounds that would be challenging to synthesize otherwise. One advantage is that SNAr reactions require electron-withdrawing groups which, by being mostly in high oxidation states, can be modified easily, including to electron-donating ones by reduction; the opposite way by oxidation is more challenging and wasteful.
Development of catalytic SNAr is an active area of research46,47. We put forth two fluoride-catalyzed SNAr reactions, which were remarkably fast and broadened the scope. Catalytic reactions are less susceptible to undesired reactions such as hydrolysis, α−deprotonation or ring opening, and are amenable for atroposelective C‒N bond formation. By combining experimental and computational data, we demonstrated that atropisomer-forming reactions may proceed through unhindered transition states, challenging a common belief that atropisomer formation must involve strong repulsive steric interactions. SNAr reactions are unique as minimal reorganization is required from transition states to product formation, an attribute related to the high efficiency observed. More broadly, such aspects are to impact synthesis of compounds with sterically hindered bonds.
Inspired by naturally-occurring and synthetic bioactive molecules, we synthesized diversifiable macrocyclic atropisomers by ring-closing metathesis and peptide macrocyclization, including two enantio- and diastereomerically pure atropopeptides analogous to antibacterial RiPPs. Such molecules have been isolated as single atropodiastereomers from natural sources, thus our studies led to molecules with distinct structural features and untapped potential in medicinal chemistry. We provided a straightforward strategy for introduction of atropisomerism to achiral pharmaceuticals, highlighting a fine-tuning process for optimization of drug leads.
The assembly/diversification platform disclosed herein, which transforms readily available 2D building blocks into molecules that are inherently more 3D16,17, is ready for generation of structurally diverse libraries of potentially bioactive atropisomers. This work underscores how the development of chemoselective atropisomer synthesis methods may accelerate medicinal chemistry initiatives48.
Methods
General procedure for method A
A vial (8 mL, 15 × 50 mm) was charged with a stirring bar, 2-methylindole H1 (104.9 mg, 0.80 mmol), ethyl 2-fluoro-3-nitrobenzoate F1a (179.1 mg, 0.84 mmol) and grinded Cs2CO3 (521.3 mg, 1.60 mmol). Reagent grade DMSO (1.6 mL) was added by syringe, the vial was sealed with a plastic cap and the mixture was vigorously stirred for 1 h. Precautions for removal of air and moisture were not taken. After the reaction was complete, the reaction mixture was transferred with EtOAc (10 mL) and brine (10 mL) to a separatory funnel. The organic layer was washed with brine (2 × 10 mL) and the resulting aqueous layer was extracted with EtOAc (2 × 10 mL). The organic layers were combined, dried over Na2SO4 and filtered through cotton. The volatiles were removed under reduced pressure and the crude reaction mixture was purified by silica gel chromatography (petroleum ether:EtOAc 95:5 → 90:10, 2 × 10 cm silica) to afford 1 as yellow solid (238.7 mg, 92% yield). See Supplementary Section 5.4 for large-scale preparation of 1 in 97% yield.
General procedure for method B
A flame-dried Schlenk flask (10 mL) was charged with a stirring bar, N-TBS indole H1-Si (184.1 mg, 0.75 mmol), and ethyl 2-fluoro-3-nitrobenzoate F1a (106.6 mg, 0.5 mmol). The flask was evacuated for 10 min and backfilled with N2. Anhydrous DMSO (5.0 mL) and TBAT (54.0 mg, 0.1 mmol) were added under a positive flow of N2. Upon TBAT addition, the mixture turned from light yellowish to intense red. The mixture was stirred for 3 min, after which it was quenched with saturated NH4Cl solution (1.0 mL) and transferred with EtOAc (20 mL) and brine (20 mL) to a separatory funnel. The organic layer was washed with brine (2 × 20 mL) and the resulting aqueous layer was extracted with EtOAc (2 × 20 mL). The organic layers were combined, dried over MgSO4 and filtered. The volatiles were removed under reduced pressure and the crude reaction mixture was purified by silica gel chromatography (petroleum ether:EtOAc 95:5 → 90:10, 2 × 10 cm silica) to afford 1 as yellow solid (154.2 mg, 95% yield).
General procedure for method C
A flame-dried Schlenk flask (10 mL) was charged with a stirring bar, 2-methylindole H1 (98.3 mg, 0.75 mmol), ethyl 2-fluoro-3-nitrobenzoate F1a (106.6 mg, 0.5 mmol) and TBAT (54.0 mg, 0.1 mmol). The flask was evacuated for 10 min and backfilled with N2. Anhydrous DMSO (5.0 mL) and Me3SiCF3 (111 µL, 0.75 mmol) were added by syringe under a positive flow of N2. Upon Me3SiCF3 addition, the mixture turned from light yellowish to intense red. The mixture was stirred for 30 s, after which it was quenched with saturated NH4Cl solution (1.0 mL) and transferred with EtOAc (20 mL) and brine (20 mL) to a separatory funnel. The organic layer was washed with brine (2 × 20 mL) and the resulting aqueous layer was extracted with EtOAc (2 × 20 mL). The organic layers were combined, dried over MgSO4 and filtered. The volatiles were removed under reduced pressure and the crude reaction mixture was purified by silica gel chromatography (petroleum ether:EtOAc 95:5 → 90:10, 2 × 10 cm silica) to afford 1 as yellow solid (151.8 mg, 94% yield).
Data availability
All data in support of the findings of this study are available within the Article and its Supplementary Information. Source data are provided with this paper. Data are also available from the corresponding authors upon request. Source data are provided with this paper.
References
Clayden, J., Moran, W. J., Edwards, P. J. & LaPlante, S. R. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed. 48, 6398–6401 (2009).
Toenjes, S. T. & Gustafson, J. L. Atropisomerism in medicinal chemistry: challenges and opportunities. Future Med. Chem. 10, 409–422 (2018).
Basilaia, M., Chen, M. H., Secka, J. & Gustafson, J. L. Atropisomerism in pharmaceutically relevant realm. Acc. Chem. Res. 55, 2904–2919 (2022).
Glunz, P. W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett. 28, 53–60 (2018).
Lanman, B. A., Parsons, A. T. & Zech, S. G. Addressing atropisomerism in the development of Sotorasib, a covalent inhibitor of KRAS G12C: structural, analytical, and synthetic considerations. Acc. Chem. Res. 55, 2892–2903 (2022).
Wang, J. et al. Discovery and assessment of atropisomers of (±)-Lesinurad. ACS Med. Chem. Lett. 8, 299–303 (2017).
Chandrasekhar, J. Atropisomerism by design: discovery of a selective and stable phosphoinositide 3-kinase (PI3K) β inhibitor. J. Med. Chem. 61, 6858–6868 (2018).
Rojas, C. et al. 663P safety and preliminary efficacy of the KRAS G12C inhibitor MK-1084 in solid tumors and in combination with Pembrolizumab in NSCLC. Ann. Oncol.34, S466–S467 (2023).
Yoon, D. S. et al. Atropisomerism observed in galactose-based monosaccharide inhibitors of galectin-3 comprising 2-methyl-4-phenyl-2,4-dihydro-3H-1,2,4-triazole-3-thione. J. Med. Chem. 67, 14184–14199 (2024).
Watterson, S. H. et al. Discovery of BMS-986142: a reversible inhibitor of Bruton’s tyrosine kinase (BTK) conformationally constrained by two locked atropisomers. J. Med. Chem. 59, 9173–9200 (2016).
An, J. S. et al. Discovery and biosynthesis of Cihunamides, macrocyclic antibacterial RiPPs with a unique C − N linkage formed by CYP450. Catal. Angew. Chem. Int. Ed. 62, e202300998 (2023).
Nanudorn, P., et al. Atropopeptides are a novel family of ribosomally synthesized and posttranslationally modified peptides with a complex molecular shape. Angew. Chem. Int. Ed. 61, e202208361 (2022).
Bringmann, G., Gulder, T., Gulder, T. A. M. & Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 111, 563–639 (2011).
Smyth, J. E., Butler, N. M. & Keller, P. A. A twist of Nature – the significance of atropisomers in biological systems. Nat. Prod. Rep. 32, 1562–1583 (2015).
Schreiber, S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964–1969 (2000).
Hung, A. W. et al. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. USA 108, 6799–6804 (2011).
Meyers, J., Carter, M., Mok, N. Y. & Brown, N. On the origins of three-dimensionality in drug-like molecules. Future Med. Chem. 8, 1753–1767 (2016).
Rodríguez-Salamanca, P., Fernández, R., Hornillos, V. & Lassaletta, J. M. Asymmetric synthesis of axially chiral C − N atropisomers. Chem. Eur. J. 28, e202282861 (2022).
Hedouin, G., Hazra, S., Gallou, F. & Handa, S. The catalytic formation of atropisomers and stereocenters via asymmetric Suzuki−Miyaura couplings. ACS Catal. 12, 4918–4937 (2022).
Frey, J. et al. Enantioselective synthesis of N−C axially chiral compounds by Cu-catalyzed atroposelective aryl amination. Angew. Chem. Int. Ed. 59, 8844–8848 (2020).
Ishida, M. et al. First atroposelective Chan–Lam coupling for the synthesis of C–N linked biaryls. Chem. Commun. 60, 678–691 (2024).
Thönnißen, V., Westphäling, J., Atodiresei, I. L. & Patureau, F. W. Atroposelective Chan–Evans–Lam amination. Chem. Eur. J. 30, e202304378 (2024).
Guo, F., Fang, S., He, J., Su, Z. & Wang, T. Enantioselective organocatalytic synthesis of axially chiral aldehyde-containing styrenes via SNAr reaction-guided dynamic kinetic resolution. Nat. Commun. 14, 5050 (2023).
Armstrong, R. J. & Smith, M. D. Catalytic enantioselective synthesis of atropisomeric biaryls: a cation-directed nucleophilic aromatic substitution reaction. Angew. Chem. Int. Ed. 53, 12822–12826 (2014).
Cardenas, M. M., Toenjes, S. T., Nalbandian, C. J. & Gustafson, J. L. Enantioselective synthesis of pyrrolopyrimidine scaffolds through cation-directed nucleophilic aromatic substitution. Org. Lett. 20, 2037–2041 (2018).
Kamikawa, K., Kinoshita, S., Matsuzaka, H. & Uemura, M. Stereoselective synthesis of axially chiral N−C bonds in N-aryl indoles. Org. Lett. 8, 1097–1100 (2006).
Thomas, R. P. et al. A direct-to-biology high-throughput chemistry approach to reactive fragment screening. Chem. Sci. 12, 12098–12106 (2021).
LaPlante, S. R. et al. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 54, 7005–7022 (2011).
Sekino, K. et al. Fluoride-ion-catalyzed synthesis of ladder-type conjugated benzobisbenzofurans via intramolecular nucleophilic aromatic substitution reaction under metal-free and mild conditions. Org. Lett. 22, 2892–2896 (2020).
Liu, X., Xu, C., Wang, M. & Liu, Q. Trifluoromethyltrimethylsilane: nucleophilic trifluoromethylation and beyond. Chem. Rev. 115, 683–730 (2015).
Rohrbach, S. et al. Concerted nucleophilic aromatic substitution reactions. Angew. Chem. Int. Ed. 58, 16368–16388 (2019).
Kwan, E. E., Zeng, Y., Besser, H. A. & Jacobsen, E. N. Concerted nucleophilic aromatic substitutions. Nat. Chem. 10, 917–923 (2018).
Wencel-Delord, J., Panossian, A., Leroux, F. R. & Colobert, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 44, 3418–3430 (2015).
O̅ki, M. In recent advances in atropisomerism. In Topics in Stereochemistry Vol. 14 (eds Eliel, E. L., Wilen, S. H. & Allinger, N. L.) 1−81 (John Wiley and Sons, New York, 2007).
Shindo, M., Koga, K. & Tomioka, K. A. Catalytic method for asymmetric nucleophilic aromatic substitution giving binaphthyls. J. Am. Chem. Soc. 114, 8732–8733 (1992).
Vantourout, J. C., Miras, H. N., Isidro-Llobet, A., Sproules, S. & Watson, A. J. B. Spectroscopic studies of the Chan–Lam amination: a mechanism-inspired solution to boronic ester reactivity. J. Am. Chem. Soc. 139, 4769–4779 (2017).
Nishio, S. et al. Axially chiral macrocyclic E-alkene bearing bisazole component formed by sequential C‒H homocoupling and ring-closing metathesis. Org. Lett. 14, 2476–2479 (2012).
Hoveyda, A. H. Evolution of catalytic stereoselective olefin metathesis: from ancillary transformation to purveyor of stereochemical identity. J. Org. Chem. 79, 4763–4792 (2014).
Reisberg, S. H. et al. Total synthesis reveals atypical atropisomerism in a small-molecule natural product, Tryptorubin A. Science 367, 458–463 (2020).
Kin, Y. C. et al. Atroposelective total synthesis of Darobactin A. J. Am. Chem. Soc. 144, 14458–14462 (2022).
Nesic, M. et al. Total synthesis of Darobactin A. J. Am. Chem. Soc. 144, 14026–14030 (2022).
Yu, L., Nagata, Y. & Nakamura, H. Atroposelective synthesis of Cihunamide B. J. Am. Chem. Soc. 146, 2549–2555 (2024).
Decensi, A. U. et al. Monotherapy with Nilutamide, a pure nonsteroidal antiandrogen, in untreated patients with metastatic carcinoma of the prostate. J. Urol. 146, 377–381 (1991).
Weisberg, E. et al. AMN107 (Nilotinib): a novel and selective Inhibitor of BCR-ABL. Br. J. Cancer 94, 1765–1769 (2006).
Saxena, P., Sharma, P. K. & Puhorit, P. A journey of Celecoxib from pain to cancer. Prostaglandins Other Lipid Mediat. 147, 106379 (2020).
Shigeno et al. Catalytic concerted SNAr reactions of fluoroarenes by an organic superbase. J. Am. Chem. Soc. 146, 32452–32462 (2024).
Pistritto, V. A., Schutzbach-Horton, M. E. & Nicewicz, D. A. Nucleophilic aromatic substitution of unactivated fluoroarenes enabled by organic photoredox catalysis. J. Am. Chem. Soc. 142, 17187–17194 (2020).
Brown, D. G. & Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone?. J. Med. Chem. 59, 4443–4458 (2016).
Acknowledgements
P.H.S.P. thanks financial support from the Czech Science Foundation (GAČR Junior Star 24-11562 M), European Research Council (ERC Starting Grant, 101160990) and IOCB start-up funds. M.S. thanks IOCB for a postdoctoral fellowship. We are indebted to our colleagues from IOCB for their generosity with chemicals, instruments and computational resources. We acknowledge Professors Amir Hoveyda, Filippo Romiti, and Stella Gonsales, as well as Dr. Juan del Pozo and Shawn Ng for helpful suggestions.
Author information
Authors and Affiliations
Contributions
P.H.S.P. conceived the project, directed the investigations and composed the manuscript with revisions provided by authors. P.D., M.S., P.H.S.P. designed and carried out nucleophilic aromatic substitutions. M.S. designed the catalytic reactions and M.S., P.D. and J.E. performed catalytic experiments. P.D., M.S. and V.B. carried out the combinatorial experiments and V.B. synthesized the atropopeptides. M.S., K.M. and P.H.S.P. planned and carried out diversification reactions and synthesis of conformationally-modified bioactive compounds. M.D. carried out NMR experiments for characterization of atropisomers. D.B., P.H.S.P. and M.S. conceived and performed the mechanistic investigations. D.B. performed computational studies.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Šimek, M., Dey, P., Blahout, V. et al. Nucleophilic aromatic substitutions enable diversity-oriented synthesis of heterocyclic atropisomers via non-atropisomeric intermediates. Nat Commun 16, 4856 (2025). https://doi.org/10.1038/s41467-025-60101-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60101-z
