
Abstract
The adult skin microbiome typically exhibits low microbial complexity, particularly on sebaceous sites, where lipophilic Cutibacterium and Malassezia spp. predominate. Current understanding of healthy skin microbiome is largely based on western, industrialized populations, with limited representation of diverse cultures and lifestyles. Here, we investigate the skin microbiome of a remote indigenous Yanomami community and reveal a complex microbial ecosystem comprising 115 previously unreported bacterial genomes. The Yanomami skin microbiome includes genera common to western populations alongside diverse environmental taxa that form multiplex interactions with the dominant eukaryote Malassezia globosa. Functional profiling indicates that this microbiome supports skin homeostasis by fortifying barrier integrity through lipid metabolism and acid production and mitigating oxidative stress. Longitudinal monitoring of western expeditioner’ skin demonstrates acquisition of the Yanomami microbiome following Amazonian immersion and its subsequent loss upon return to an industrialized setting. These findings reveal that diverse, environmentally enriched microbiota may confer skin benefits that are overlooked in current models of healthy skin.
Similar content being viewed by others

Introduction
The human skin serves as a protective external barrier and primary communication with the environment. Skin microbiota shape and maintain multiple physiological processes in this outermost layer of the human body1. The role of the skin microbiome includes maintenance of an acidic pH that protects against pathogen colonization2, establishing and modulating immune tolerance3,4,5,6, and fortifying skin barrier integrity7,8,9. Site-specific microenvironments are established by local pH, temperature, moisture, sebum content, topography, and other factors that influence community structure. Sebaceous (oily) surfaces are dominated by lipophilic microorganisms, specifically Cutibacterium spp and Malassezia spp, and differ from those found at more diverse and less stable dry or moist skin sites10,11,12. Site-specific interpersonal variation of skin microbiomes has also been linked with various host traits and habits, including age, sex, BMI, cohabitation, smoking, pet ownership, and antibiotic use12,13,14,15,16,17,18,19. Environment and lifestyle, specifically urbanization, also influence the skin microbiota, exemplified by the observation that individuals residing in rural communities harbor a more diverse and compositionally distinct microbiome than their urban-dweller counterparts18,20,21,22,23.
Perturbations to cutaneous microbial communities have been linked to various skin conditions, such as acne, rosacea, psoriasis, and atopic dermatitis24,25,26. These pathologies are considered industrialized comorbidities with less frequent occurrence in developing nations and unreported in hunter-gatherer populations27,28,29. The frequency of cutaneous disease inversely correlates with cutaneous microbial diversity observed along an urbanization gradient22,23. The least impacted populations are those of seminomadic hunter-gatherers with the highest skin microbial diversity22,23,30,31,32. Since the current understanding of the role of the skin microbiome in human health derives predominantly from studies of western, industrialized populations, it remains unclear if assumptions generated in western-biased populations apply across all cultures and lifestyles.
Here we investigate the skin microbiome of the Yanomami, one of the last remaining swidden horticulturalist and hunter-gatherer communities of the Amazon, minimally exposed to industrialization. We identify a complex microbial ecosystem comprising genera common to western skin alongside diverse environmental taxa that support skin homeostasis by fortifying barrier integrity and mitigating oxidative stress. These microbes are closely integrated with the surrounding environment and can be acquired by western skin during immersion in the Amazon but diminish upon return to industrialized settings. Our findings support the hypothesis that a skin microbiota enriched with environmental microbes confers benefits not captured by current models of healthy skin. Collectively, these results underscore lifestyle as a primary determinant of cutaneous microbiota composition, with industrialization likely driving the loss of beneficial environmental microbes.
Results
Study population and design
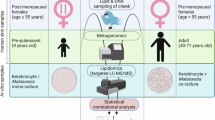
The Yanomami are people indigenous to the Amazon rainforests of Venezuela and Brazil, traditionally living a swidden horticulturalist, seminomadic hunter-gatherer lifestyle. Samples for this study were collected in one such community in Venezuela from two neighboring shabono (communal living structure—comprised of a naturally derived thatched roof and an open-aired shelter, with no dividing walls and a natural forest ground as the floor), situated within walking distance of a river where community members fish, bathe and collect water for drinking and cooking. To respect the Yanomami decision-making procedure that is followed for participation in the study, a community meeting was held where the field team explained each stage of sample collection, the goal, and the scope of the microbiome research project. To ensure an intercultural perspective, didactic posters and pictures were used with all the information translated into the Yanomami language. Community concurrence and free and prior informed verbal consent were obtained, after which sampling commenced.
A total of 94 skin samples were collected for metagenomic analysis from 17 Yanomami (50% female; 1–70 approximate years of age). During two expeditions, individuals from the same community were sampled at seven skin sites: retroauricular crease (RAC), face, upper back, scalp, volar arm, axilla, and toe web space (Supplementary Table 1), with no significant difference between expeditions in coverage of age groups or body sites sampled (Chi-square p = 0.319 and p = 0.443, respectively). The same body sites of a western adult male expeditioner were longitudinally sampled at various time points throughout the journey from the metropolitan United States to a remote Yanomami community and return, yielding a total of 45 skin samples (Supplementary Table 2). To compare indigenous skin microbiomes with those of western industrialized individuals, skin samples from the expeditioner (excluding those collected while in the Amazon) and 16 RAC samples (44% female; age range 18–40yo) from the Human Microbiome Project (HMP)33 data archive were included in the analysis. Median de-hosted metagenomic read depth was 2.17E7, 2.52E7 reads/sample for Yanomami skin samples and HMP samples, respectively. As proxy for sebaceous skin microbiome transcriptional activity, RNA extracted from chest swabs collected from Yanomami adults and westernized expeditioner (Supplementary Table 2) was profiled for microbial composition and functional analysis. To elucidate the relationship of Yanomami skin microbiota composition to the surrounding environment, samples of soil from an entrance trail to the shabono, water samples from a creek and river, and ash from the heath located in the shabono were collected and included in the study. Lastly, a Yanomami adult female (Yanomami traveler) accompanied the expeditioner on his return to the USA, and samples from her RAC, scalp, and volar arm were collected approximately two weeks into an extended stay in an industrialized setting, providing contrast with samples collected while in the native Amazon community (Supplementary Table 2).
Skin metagenomes of the Yanomami harbor previously unreported microbial genomes
We first assessed percentage of human reads across combined samples from the Yanomami RAC and face (n = 30), which were significantly lower (ANOVA of linear mixed-effects model (LME) p < 0.001) compared to the expeditioner westernized samples (USA samples, n = 10) collected from the same sebaceous body sites (Fig. 1A). This trend persisted across all body sites, although statistical significance was reached only for RAC, scalp, and arm (Supplementary Table 3), indicating possible reduced desquamation and potentially stronger skin barrier. Surprisingly, a lower percentage of bacterial (but not fungal) reads was detected in the Yanomami skin samples, resulting in a higher percentage of remaining unmapped reads (ANOVA of LME p < 0.001; Fig. 1A and Supplementary Table 3), which could represent bacterial reads not included in the databases used. This observation was consistent when the data were normalized to a read depth of 13 M, after removing human reads (Supplementary Fig. 1A). A portion of unmapped reads from Yanomami RAC/face samples presented as novel bacterial species previously unreported in existing databases, akin to observations of gut microbiomes reported for other hunter-gatherers34. Through reference-independent de novo assembly, reconstruction of bacterial metagenome-assembled genomes (MAGs) recovered 115 novel (≥50% completion and ≤5% contamination) bacterial genomes, constituting 59.0% of the total reconstructed prokaryotic MAGs obtained from the samples (Fig. 1B; Supplementary Table 4) when classified (≥90% average nucleotide identity) using The Genome Taxonomy Database (GTDB)35,36,37 or 155 novel (79.5%) MAGs (Supplementary Fig. 1B) when classified using The Skin Microbial Genome Collection (SMGC)38. The largest number of novel genomes were identified as Corynebacterium (12.3% and 13.3% of all MAGs, using GTDB or SMGC, respectively), with several genomes identified only to family (or higher), such as Dermatophilaceae, Micrococcaceae and Propionibacteriaceae. Additionally, 18 ( ≥ 50% BUSCO completion, dereplicated) fungal MAGs were recovered from the same samples, most of which were identified as Malassezia, including two potentially novel strains identified as Malasseziaceae (Supplementary Table 5). These results suggest that the Yanomami skin harbors an assortment of unique microbes yet to be identified and characterized.

A Percent of raw read distribution in combined RAC/face samples of Yanomami (n = 30) and western expeditioner (n = 10; ANOVA of LME, two-sided, p-value: ***<0.001; not shown >0.1; p = 0.00015, 2.48E-05, 0.7253, 5.47E-10, respectively). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR). B Summary of GTDB-based classification of bacterial MAGs reconstructed from Yanomami RAC/face (n = 30) samples. C Principal Coordinates Analysis (Bray-Curtis) of Yanomami (n = 94) bacterial microbiota across body sites and age groups. D Bacterial diversity (Shannon Index) of combined sebaceous samples (RAC/face/back/scalp; n = 53) across age groups. ANOVA of LME, two-sided, p = 0.0957, (Tukey multiple comparisons, two-sided: *p-value < 0.05, p = 0.023). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR). E Relative abundance of Cutibacterium in combined sebaceous samples (RAC/face/back/scalp) in Yanomami children (n = 14 samples) vs adults (n = 26 samples; ANOVA of LME, two-sided: p-value = 0.054). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR).
The Yanomami skin microbiome harbors diverse bacterial and fungal communities
We next assessed compositional and functional differences between Yanomami and western skin microbiomes. The microbial populations were analyzed by mapping to known microbial genomes using normalized 13 M reads after de-hosting. Similar to industrialized skin microbiomes, factors such as body site10,11,12 and age13,14,15,16,19 explained a portion of the variation in bacterial (Nested PERMANOVA estimated component of variation (ECV) = 20.4%, p = 0.0001 and ECV = 9.4%, p = 0.004, respectively Supplementary Table 6 and Fig. 1C) and fungal (Nested PERMANOVA ECV = 20.9%, p = 0.0001 and ECV = 10.1%, p = 0.024; Supplementary Table 6 and Supplementary Fig. 1C) Yanomami skin composition, where communities clustered primarily by microenvironment. The bacterial diversity of sebaceous skin (back, face, RAC and scalp) showed age-related variance, mirroring observations reported for industrialized skin. Specifically, bacterial diversity declined in adulthood (Fig. 1D, Tukey multiple comparisons p < 0.05), based on increased relative abundance of Cutibacterium spp. (Fig. 1E, ANOVA of LME p = 0.054; Supplementary Fig. 1D and Supplementary Fig. 2A). Fungal diversity was also lower in adult skin microbiota compared to that of children (Supplementary Fig. 2B, ANOVA of LME p < 0.05), corresponding to a higher relative abundance of Malassezia spp., (Supplementary Fig. 2C-D, ANOVA of LME p < 0.05). The decline in diversity in adult skin was not reflected in the richness of bacterial or fungal communities on combined sebaceous sites (data not shown), nor was it significant for dry skin (arm) samples from the same subjects (Supplementary Fig. 2E, F), indicating the community signature is primarily associated with restructuring rather than membership loss.
Compared to the western skin microbiome, bacterial communities of the Yanomami adult RAC were significantly richer (80% [IC range 76–87%]), more diverse (67% [IC range 60–74%], Wilcoxon, p = <0.001, Fig. 2A), and compositionally distinct (PERMANOVA R2 = 0.455, p = 0.001, Fig. 2B), with less dominance by Cutibacterium spp. (Fig. 2C). Higher bacterial diversity on Yanomami skin was not exclusive to RAC; it was also evident on other sebaceous body sites when contrasted with the westernized expeditioner samples (Supplementary Fig. 3A-C), as well as on the arm (dry skin site; Supplementary Fig. 3D). Bacterial diversity did not differ significantly in samples from the toe web (moist body site; Supplementary Fig. 3E), although composition remained distinct. The most abundant bacterial taxa detected on Yanomami skin were species of Corynebacterium (C. mucifaciens and C. ureicelerivorans), Bordetella (B. pertussis), Kocuria (K. indica and K. palustris), Micrococcus (M. aloeverae and M. luteus), Brachybacterium (B. paraconglomeratum), Microbacterium, and Moraxella (M. osloensis; Fig. 2C, D). At the genus level, a wide range of bacterial taxa were absent or only infrequently detected on western skin. These included species of Brachybacterium, Brevibacterium, Microbacterium, and Janibacter, amongst others (Fig. 2E), that were detected on Yanomami skin irrespective of the age of subjects. A subset of genera was frequently detected in both groups, including species of Corynebacterium and Cutibacterium (Fig. 2E), although in variable relative abundance (Supplementary Fig. 4A). The relative abundance of Staphylococcus, another shared genus, was similar for both groups (Wilcoxon with BH-correction q > 0.05). The repertoire of Staphylococcus spp. was greater on Yanomami skin, with significantly higher relative abundance of S. aureus, S. hominis, S. haemolyticus, S. saprophyticus, and S. arlettae (Fig. 2D), but no significant difference for S. epidermidis (Supplementary Fig. 4A). Notable, virulence factors of the Staphylococcus spp., expressed as a ratio of virulence genes to the relative abundance of species within the genus, trended lower for Yanomami (Supplementary Fig. 4B). Yanomami Staphylococcus virulence factors involved attachment and colonization, such as the surface protein SdrC-E, whereas virulence factors more common on western skin included antibiotic resistance and pathogenesis, namely ThyA and SePI (Supplementary Fig. 4C).

A Alpha Diversity of Yanomami (n = 5) and HMP (n = 16; Wilcoxon test, two-sided, p-value: ***<0.001, **<0.01; p = 0.00108 and 0.00039, respectively). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR). B Beta diversity of adult Yanomami (n = 5) and western HMP (n = 16; PERMANOVA two-sided p < 0.05, Bray-Curtis). C Mean relative abundance of dominant (≥2%) genera of adult Yanomami (n = 5) and western HMP (n = 16). D Most differential species (LEfSe, at >75% of subjects per group; Kruskal Wallis p < 0.05; pairwise Wilcoxon two-sided p < 0.05, LDA cutoff=2) on RAC of adult Yanomami (n = 5) and western HMP (n = 16). E Distribution of genera present at >80% (Yanomami RAC n = 11; western HMP n = 16; color indicates age group; Fisher’s exact test, FDR-adjusted: ****q < 0.0001, ***q < 0.001, **q < 0.01, *q < 0.05).
Relative abundance of bacteriophages reflected the bacterial profile (Supplementary Fig. 4D), with Propionibacterium phages significantly more abundant on western skin (Wilcoxon with Benjamini-Hochberg correction q < 0.05). The relative abundance of Propionibacterium phage correlated with the relative abundance of Cutibacterium (formerly Propionibacterium) on western (Spearman correlation, rho = 0.47, p = 0.016) but not Yanomami (rho = −0.14, p > 0.1) skin. A greater variety of Propionibacterium phages was detected on western skin, although the Yanomami skin microbiome harbored a higher relative abundance of the Propionibacterium phage PFR2 (Supplementary Fig. 4E).
Fungal communities of both groups were dominated by Malassezia spp. (Fig. 3A). However, the Yanomami mycobiota of the adult RAC samples were richer (Wilcoxon, p = <0.01, (Supplementary Fig. 5A) and compositionally distinct (PERMANOVA R2 = 0.376, p = 0.004, Supplementary Fig. 5B-C), but trended less diverse due to dominance by M. globosa (Fig. 3A, B and Supplementary Fig. 5D). Trending richer fungal communities were detected on all Yanomami sebaceous sites sampled (Supplementary Fig. 6A–C), though fungal diversity trends for those sites were mixed compared to that of the western expeditioner. No significant difference in alpha diversity matrices was observed for the arm (dry site; Supplementary Fig. 6D) and toe web mycobiota (moist site; Supplementary Fig. 6E), although all body sites were compositionally distinct, with reduced abundance of M. restricta (Supplementary Fig. 5D; Supplementary Fig. 6A–E and Fig. 3A). Other fungal species differentiating Yanomami RAC samples included M. japonica, M. furfur and Brettanomyces bruxellensis (Fig. 3B and Supplementary Fig. 5C).

A Mean relative abundance of dominant species (≥2%) in Yanomami (n = 5) vs western HMP (n = 16) individuals. B Most differential taxa (LEfSe;>50% of subjects/group; Kruskal Wallis p < 0.05; pairwise Wilcoxon two-sided p < 0.05, LDA cutoff = 2) in Yanomami (n = 5) vs western HMP (n = 16). Co-occurrence of dominant bacterial genera (80% prevalence) and all Malassezia species in the (C) western HMP (n = 16) and (D) Yanomami (n = 5) skin microbiomes (Spearman [abundance score], two-sided p < 0.05; where: Red—negative interactions; Green—positive interactions; thickness of lines indicates co-association strength).
The Yanomami skin microbiome features distinct interkingdom interactions and functional attributes
The interkingdom dynamics of the Yanomami skin microbiome were compared to western skin using network analysis of abundance scores, interactions of bacterial genera with members of the most dominant fungi, Malassezia, and interspecies interactions within this genus. The adult Yanomami RAC skin microbiome exhibited a more complex interactome than western skin. Whereas M. globosa was directly co-associated with Cutibacterium (negatively) and Staphylococcus (positively) on western skin (Fig. 3C), on Yanomami skin, M. globosa was co-associated with several bacterial genera, including Kocuria, Microbacterium, Neisseria, and Janibacter (Fig. 3D). Additionally, M. globosa was associated with M. slooffiae and indirectly with M.obtusa, forming a highly interactive hub (Fig. 3D). Conversely, M. restricta was observed to inversely correlate with Moraxella, Enhydrobacter, Paenibacillus, Rothia and directly with Cutibacterium (Fig. 3D). Relative abundance of M. globosa correlated positively with overall bacterial richness and diversity on the skin irrespective of lifestyle (Supplementary Fig. 7A–C), the inverse having been observed for M. restricta.
Reflective of compositional differences, the functional potential of the Yanomami skin microbiome was both greater than (Fig. 4A) and distinct from the western microbiome (PERMANOVA R2 = 0.47, p = 0.001, Fig. 4B). Functional pathways of the Yanomami skin microbiome that were most differential included lipid metabolism (biosynthesis and degradation), fermentation, metabolite activation, degradation of various amides/amines, and hormone metabolism (Fig. 4C). In contrast, western skin microbiome pathways included cofactor biosynthesis and degradation of carbohydrates and amino acids (Fig. 4C). To determine if these functional attributes were expressed on the skin, the Yanomami skin microbial transcriptome was compared to that of the western expeditioner, based on an additional sebaceous site (chest, exclusively utilized for RNA extraction and transcriptomics). The Yanomami transcriptional profile revealed richer, more diverse (Supplementary Fig. 8A) and compositionally distinct bacterial (Supplementary Fig. 8B-C) and fungal (Supplementary Fig. 8D-F) communities, with comparable membership (Supplementary Fig. 8C and Supplementary Fig. 8F) as observed from the metagenomic analyses, suggesting metabolically active microbes on Yanomami skin. Transcriptomics-based functional community output of the Yanomami microbiome was more complex and functionally divergent (Supplementary Fig. 8G-H) from that of the western microbiome. Active functional pathways of the Yanomami microbiome most differential were lipid biosynthesis, fatty acid and lipid degradation, fermentation, aromatic compound metabolism, degradation of amides/amines, hormone, and reactive oxygen species (Fig. 4D). Western skin microbial functional pathways included cofactor biosynthesis, amino acid metabolism, and carbohydrate degradation (Fig. 4D), suggesting physiologically divergent states of functional activity in these compositionally distinct microbial communities.

A Observed richness and diversity (Shannon index) of functional potential (3512 pathways assigned by MetaCyc) of the Yanomami (RAC, n = 5) and western HMP (RAC, n = 16) skin microbiome. (Wilcoxon test, two-sided, p-value: ***<0.001, **<0.01; p = 9.83E-05 and 0.00118, respectively). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR). B Functional potential composition (PERMANOVA two-sided p < 0.05 of Bray-Curtis dissimilarity; pathways assigned by MetaCyc at secondary pathway classes) of Yanomami (n = 5) and western HMP (n = 16) RAC samples. C Most differential functional secondary pathway classes in RAC samples (LEfSe; Kruskal Wallis p < 0.05; pairwise Wilcoxon two-sided p < 0.05, LDA cutoff=2). D Most differential functional secondary pathway classes of active microbiota in chest (RNA) samples of Yanomami adults (n = 4) and western expeditioner (n = 3) samples (LEfSe; Kruskal Wallis p < 0.05; pairwise Wilcoxon two-sided p < 0.05, LDA cutoff=2).
The Yanomami skin microbiome is integrated with their environment and diminished by industrialization
The Yanomami skin microbiome varied in bacterial and fungal composition (Supplementary Fig. 9A–H) across body sites (arm, axilla, back, face, RAC, and toe web), but unlike western skin10,11,12, greatest bacterial community richness was detected in the Yanomami face (sebaceous site) samples and the least in toe web (moist site, Supplementary Fig. 9A-B) samples, the latter also harboring the least rich fungal communities (Supplementary Fig. 9D-E). Arm (dry site) and axilla (moist site) microbiota shared significant community composition with sebaceous skin samples (Supplementary Fig. 9C and Supplementary Fig. 9F). Observations likely related to Yanomami’s lifestyle and the close connection to the environment.
The Yanomami live in communal shabono open to the environment and regularly swim in and use local water sources. Microbial exchange with their surroundings is likely a contributor to the microbial richness of the Yanomami skin microbiome. Yanomami skin microbiota across all body sites, clustered more closely with that of soil that had been sampled from an entrance trail to the shabono rather than that of the creek (Bray-Curtis distance matrix; Supplementary Fig. 10B), with toe web microbiota being most compositionally similar to that of soil. The more abundant cutaneous bacterial genera, namely Pseudomonas and Staphylococcus, were detected in both environmental profiles (Fig. 5A), Brachybacterium, Deinococcus, Janibacter, Microbacterium and Micrococcus as well as others (Fig. 5A; Supplementary Fig. 10A) were detected in soil. The mycobiome was reflective of creek microbial communities (Supplementary Fig. 10C-D) except toe web, unsurprisingly most similar in composition to soil, with M. restricta and Brettanomyces bruxellensis in creek and soil samples, respectively.

A Distribution of bacterial genera (≥2%, in at least three adult Yanomami) at various body sites (n = 5 individuals; n = 30 samples) that are co-detected in or exclusive to environmental samples (soil/creek). B Comparison of bacterial diversity (Bray-Curtis) of adult Yanomami (n = 9, combined sebaceous sites, n = 26 samples) and western expeditioner (n = 23 same body sites) at various timepoints; Pr- pre-expedition, Tr- in-transit, 1- Amazon, 2- Amazon, 3- Amazon, Po- post-expedition; ANOVA of LME, two-sided p = 0.02, (Tukey multiple comparisons, two-sided: ***p < 0.001; **p < 0.01; *p < 0.05). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR). C Similarity in bacterial composition (Bray-Curtis distance) between the western expeditioner (n = 23 combined sebaceous sites) and adult Yanomami (n = 26 same combined sites; repeated measures, non-parametric analysis by Friedman p < 0.05, followed by Dunn test [pair-wise, BH-corrected p < 0.025]). Boxplots: median, 1st- 3rd quartiles, whiskers min/max, outliers (1.5 × IQR). D Bacterial species present in ≥25% Yanomami adults (n = 9) at ≥0.1% relative abundance (all body sites n = 47) co-detected on the expeditioner at various time points (n = 54 samples).
Further evidence of environmental influence on the Yanomami skin microbiota was observed from the longitudinally sampled skin microbiome of the expeditioner, who adopted the local practices in the daily activities of the Yanomami community. An increase in microbial complexity was noted in samples from combined sebaceous body sites at all three time points of collection in the Amazon (approximately one week apart), with bacterial richness (Supplementary Fig. 10E), diversity (Fig. 5B), and composition closely resembling that of Yanomami microbiota (Fig. 5C, D). Apparent fungal richness gain for the Amazon samples did not reach statistical significance (Supplementary Fig. 10F), likely due to Malassezia dominance. The Yanomami cutaneous microbial community complexity diminished after integration into industrialized lifestyle, both in the microbiota of the expeditioner (Fig. 5D and Supplementary Fig. 10E-F) and a Yanomami family member who traveled out of the Amazon (Supplementary Fig. 11A–C), and was accompanied by loss of several bacterial genera namely Dietzia, Kocuria, Micrococcus, Brevibacterium, Brachybacterium, Deinococcus, Yimella and Janibacter, all prominent members of the Yanomami cutaneous microbiota (Fig. 5D and Supplementary Fig. 11C).
Discussion
The Yanomami represent one of the few indigenous groups in the Amazon remaining minimally impacted by industrialization, leading a seminomadic, swidden-horticulturalist, hunter-gatherer lifestyle. They experience few chronic cutaneous inflammatory conditions27,28,29, in contrast to an increasing frequency in industrialized populations globally. The Yanomami skin microbiome has been described (based on 16S rRNA biomarker sequencing) as significantly diverse compared to industrialized microbiome22,23,30,32, but inadequately characterized. This study aimed to elucidate the composition and functional attributes of the Yanomami skin metagenome and determine whether western-biased assumptions about the skin microbiome apply across cultures and lifestyles, especially as drastically different as the Yanomami, relatively unaffected by industrialization.
The Yanomami sebaceous skin metagenome was found to be complex, with a large component of total reads not mappable to human, fungal, bacterial, or viral genomes in the current literature and available databases, likely reflecting a connection to the diverse and relatively unexplored rainforest biome39 where the Yanomami reside. The Yanomami skin harbored a unique assortment of microbes with more than half of the reconstructed bacterial metagenome-assembled genomes (MAGs) belonging to previously unidentified microorganisms, including those co-evolved with mammalian and human skin40, classified only to family level, such as Dermatophilaceae, Micrococcaceae, Propionibacteriaceae and Malasseziaceae. The skin microbiome is reported as co-evolving in response to environmental and host cues, where microbial assemblages vary by skin site and are conditioned by the local cutaneous microenvironment, a relationship observed in the study reported here for Yanomami skin. Like that of industrialized skin10,11,12,13,14,15,16,19, the Yanomami skin bacterial and fungal community composition is influenced by site microenvironment and age of participants, with a bloom and gradual decrease in lipophilic Cutibacterium and Malassezia relative abundance with age. Community restructuring was reflected in a significant decline in microbial diversity in adults but not in bacterial richness and fungal assemblies on sebaceous sites, nor on dry skin for the same subjects, perhaps related to age-associated hormonal changes and increased sebum production in adulthood16.
Unlike industrialized sebaceous skin of relatively low microbial complexity due to dominance by Cutibacterium and Malassezia, where skin health correlates with less complex microbial communities41, the Yanomami adult skin microbiome was significantly more diverse. It was enriched for a wide range of environmental bacteria absent or infrequent on western skin, such as Brachybacterium, Brevibacterium, Microbacterium, and Janibacter spp. A subset of bacterial genera was common to both populations, such as Cutibacterium, Staphylococcus, and Corynebacterium spp., with the latter most abundant on Yanomami skin and representing novel MAGs. Corynebacterium spp. are key immunomodulatory members of the cutaneous microbiome42 and have previously been reported to support community diversity and evenness in species distribution within the modern skin microbiome43.
Staphylococcus was detected in both western and indigenous adult skin microbiomes. Although its relative abundance was indistinguishable, species diversity was greater on Yanomami skin, with a higher relative abundance of S. aureus, S. hominis, S. haemolyticus, S. gallinarum, and S. saprophyticus, with diminished virulence potential compared to western Staphylococcus. While indigenous Staphylococcus were putatively enriched for factors related to attachment and colonization, virulence attributes of western Staphylococcus included factors related to antibiotic resistance and enhanced pathogenesis, such as S. aureus thymidylate synthase44 and S. epidermidis pathogenicity islands45. Bacteriophage populations in this study proved unremarkable, possibly restricted by available bacteriophage sequences in public databases primarily derived from clinically relevant bacteria. Reflective of the bacterial profile, Propionibacterium phages were more abundant on the western skin, except for lytic phage, PFR2, previously isolated from P. freudenreichii, with reported ability to infect acne-associated C. acnes46 as a species abundant on the Yanomami skin.
The sebaceous adult skin mycobiota was dominated by the lipophilic yeast Malassezia. However, M. globosa and M. restricta dominated the skin mycobiome of the Yanomami and western skin, respectively. M. globosa was negatively co-associated with Cutibacterium in the western skin samples (as previously reported47) and positively with Staphylococcus. On the Yanomami skin, M. globosa formed a positive interaction hub, directly co-associating with several bacterial genera. The relative abundance of M. globosa correlated positively with overall bacterial richness and diversity on the skin of Yanomami and western subjects. Collectively, these observations indicate that M. globosa relative abundance may be a marker of greater bacterial complexity in sebaceous skin microbiota and, perhaps, as a gatekeeper of skin health. There is growing evidence that M. globosa may be beneficial to healthy skin in preventing colonization by pathogenic microorganisms48,49,50 and modulating inflammation via induction of immunoregulatory cytokine IL-1051. It may have a protective role in dermatitis, as suggested by the clinical observation of a lower M.restricta/globosa ratio on healthy skin compared to subjects with seborrheic dermatitis and dandruff, two conditions where Malassezia is implicated52.
Microbial diversity and interspecies interactions contribute to the functional output of microbial communities. Hence, it was unsurprising to observe a higher functional potential and more transcriptionally active microbial assemblages on Yanomami than western skin. Of the functional pathways that most differentiated the Yanomami microbiomes were those with the potential to contribute to maintaining skin homeostasis and fortifying skin barrier integrity. These included lipid metabolism and fermentation, contributing to pH and antimicrobial defensins regulation on the skin surface53,54. Degradation of secreted aromatic compounds and hormones by environmental bacteria55 abundant on the Yanomami skin may diffuse microbial virulence of pathogenic species56, whilst enhanced physiologic REDOX function renders additional protection against oxidative stress, such as that induced by UV exposure57,58. However, further mechanism-oriented studies on skin-microbiome interactions are needed to elucidate the specific contributions of these complex microbial communities to skin health and to explore their potential role in reducing the prevalence of skin disease in pre-industrialized communities.
Our findings suggest that similar to industrialized skin, where bacterial interconnections are observed with the indoor environment59,60 in which, on average, 90% of westerners spend their days61, the Yanomami skin microbiome is interconnected with their natural environment, with a suite of key differential cutaneous bacterial taxa co-detected across various environmental samples. Furthermore, longitudinal sampling of a western expeditioner skin microbiota throughout a journey to a remote Yanomami community provided evidence that the microbial complexity of the Yanomami skin may be acquired upon adoption of local practices. Temporary establishment of bacteria from contact with soil and aquatic environments has been reported on western skin62,63,64,65,66. Here, we conclude that prolonged and continuous exposure will result in incorporation of environmental taxa into an established skin microbiome. Our observation of the loss of the Yanomami skin bacterial complexity following adoption of a western lifestyle agrees with the report of bacterial diversity loss along an urbanization gradient in a cross-sectional assessment of the skin microbiome in the same geographic region22,23.
In summary, results of this study underscore lifestyle as a primary determinant in shaping the composition of cutaneous microbiota, with industrialization likely a pivotal factor driving the loss of environmental microbes such as those that enrich the Yanomami skin microbiome. The complex microbial communities observed in this study to comprise the indigenous skin microbiome challenge current western-centric and biased views of a healthy adult skin microbiome, whereby skin health is deemed correlating with less complex microbial communities. The findings reported here support the hypothesis that a diverse skin microbiota enriched with environmental microbes may confer benefits not captured by current models of healthy skin. Finally, we provide evidence suggesting that the adult human skin microbiome may be stably altered by prolonged microbial exposure, giving hope that continuous topical microbial supplementation can stabilize and substantially improve skin health by modulating the skin microbiome.
Limitations of the study
While the study represents the most comprehensive analysis of non-industrialized skin microbiome survey to date, it is limited to a relatively small sample size, a single Yanomami community and a western expeditioner, all of which was constrained by logistics, strict conformation to ethical practices for study of the last remaining minimally contacted hunter-gatherer population in the Amazonian rainforest, and lack of cutaneous metagenome studies of other populations maintaining a similar lifestyle for comparative reference. Other limitations of the study included the large number of uncharacterized genes, two-fold greater in Yanomami metagenomes and the lack of closely related homologs in currently available public databases, restricting putative functional prediction of genes in these communities. To address these limitations, studies involving deeply sequenced skin metagenomics of underrepresented non-westernized populations are needed for a more comprehensive understanding of the diversity and functional attributes of the human skin microbiome.
Methods
Inclusion and ethics statement
All samples used in this study were collected legally under valid permits from the Venezuelan government and ethically by The Yanomami Foundation (formally The Good Project) a nonprofit organization and was reviewed and approved by the Institutional Board of Directors, Venezuelan Institute of Scientific Research (IVIC; DIR-0021/1582/2017) and Advarra’s (formally IntegReview) Institutional Review Board (IORG0000689). Permission to collect samples was obtained from community leaders and each adult or parent of a child by verbal consent with the help of a translator and by video recording when possible. No written consent was acquired since this Yanomami community does not have a written form of communication. To minimize disturbance to the Yanomami way of life, sample collection was performed by a single Yanomami researcher, who was integrated into community’s daily activities. Western expeditioner provided a written informed consent.
This study was made possible by the dedication of local researchers at IVIC who enabled the acquisition of bioethical and legal permits for conducting research in the Yanomami territory of Venezuela and recruitment of the Yanomami participants in the study, as well as facilitated transport of the microbiome samples from Venezuela to the USA. David Good is a member of a Yanomami tribe and the executive director of the Yanomami Foundation, he personally collected the samples used in this study. The Yanomami Foundation is the custodian of the Yanomami Microbiome Project. Their mission is to equip the Yanomami communities with the tools, resources, and intercultural training necessary to safeguard the Yanomami way of life and preserve their Amazonian lifestyle and their microbiome. The Foundation does this partly by facilitating and advancing international, transdisciplinary collaborative research of the Yanomami microbiomes, believing this elucidates links between the unique Yanomami microbiome and their health and well-being. Importantly, this illuminates the vital importance of preserving the Amazonian rainforest and the intimate connection the Yanomami have with it. The Foundation sets a new precedent in bioethics by using community-based participatory approaches, exchanging intercultural skills and training, and ensuring equitable benefit sharing with the Yanomami communities in Venezuela and Brazil (for more detail of how the foundation supports local communities, visit www.yanomamifoundation.org).
Sample collection
Cutaneous microbiome samples were collected from arm, axilla, back, chest, face, retroauricular crease (RAC), scalp, and toe web using sterile swabs (Isohelix, Kent, UK) as described in the HMP sampling protocol67. Environmental samples were collected using swabs (hearth ash, soil, water) or Sterivex filters (water). All samples were preserved in DNA/RNA Shield (Zymo Research, Irvine, CA). Sample collection was performed by a single individual following IRB-approved protocols (detailed in Supplementary Methods).
Nucleic acid extraction and metagenomic sequencing
DNA was extracted from back swabs using ZymoBIOMICs MagBead DNA Kit (Zymo Research, Irvine, CA, USA), and ZymoBIOMICS Microprep kit (with Metapolyzyme enzymatic pre-treatment) was used for the other body site sample collection. Qiagen PowerSoil Pro kit (Qiagen, Germantown, MD, USA) was used to extract DNA from environmental samples. RNA was extracted from chest swabs and environmental samples (river, soil, and ash) using Qiagen RNeasy Plus kit (Qiagen, Germantown, MD, USA). DNA sequencing library was prepared using Nextera XT library prep kit (Illumina, San Diego, CA, USA), as previously described68. RNA sequencing library was prepared using the SMARTer Stranded RNA Total RNA Sample Prep Kit - Pico Input (Takara Bio USA, San Jose, CA, USA), as previously described69. All sequencing was done using 2 × 150 bp using Illumina HiSeq 4000 Instrument (Illumina Inc., San Diego, CA, USA).
Data analysis
Unassembled metagenomic sequencing reads from the dataset and HMP westerners were analyzed using CosmosID bioinformatics software package (CosmosID Inc., Rockville, MD, United States), as described previously in ref.70. Human reads were removed using genome assembly GRCh38_p6 and normalized by subsampling to 13 M reads using reformat.sh of the BBTools suite (http://bbtools.jgi.doe.gov). Processed reads were classified to taxa using CosmosID portal, employing k-mer mapping to unique genome regions for each taxon68,70 (details in Supplementary Methods). Reads were assigned to bacterial and archaeal, fungal, protist, viral (excluding phage), or bacteriophage lineages for taxonomic comparisons. Positive control taxa were removed from the dataset prior to analysis. R package Vegan was used to generate Shannon and Chao1 alpha-diversity from the strain-level abundance score matrices. Possible process contaminants were identified (Supplementary Table 7) as skin and environment in origin but not removed from the dataset. For functional gene classification, quality-filtered reads were translated and used to search the UniRef90 database71. Gene families were annotated using MetaCyc72, and normalized abundance values were calculated as copies per million (see Supplementary Methods for details). Shannon and Observed Richness alpha-diversity values for functional genes were calculated using R package Vegan.
Metagenome assembly and taxonomic assignment
Metagenome-assembled genomes (MAGs) from face and RAC samples were assembled with metaWRAP73. Adapters and host reads were removed, reads were assembled with metaSPAdes74 for prokaryotic MAGs, the remaining unassembled reads were assembled with MEGAHIT75, and contigs were aligned (SAMtools76 and BWA-MEM77) and binned (MetaBAT278, MaxBin 2.079, and CONCOCT80; see details in Supplemental Information). Following bin refinement, bacterial representative genomes (n = 195, with ≥50% completion and ≤5% contamination) were dereplicated (dRep)81 and classified with GTDB-tk82 (v2.10 07-RS207) as known (≥95% average nucleotide identity [ANI] to a genome from the GTDB36,37) or novel. The same ANI criteria were applied to bacterial MAGs compared against the Skin Microbial Genome Collection (SMGC)38.
Fungal contigs assembled with metaSPAdes74 were retrieved using EukRep83, indexed and aligned (SAMtools76 and BWA-MEM77), and binned (MetaBAT278). Fungal representative genomes (n = 18; ≥50% BUSCO84 completion) were dereplicated (dRep)81 and classified with taxator-tk85 and Blast+86 as known (≥95% ANI of NCBI assembly) or novel.
Statistical analyses
Univariate data comparisons between Yanomami and western individuals were performed using Wilcoxon rank test, Fisher’s Exact Test, or Analysis of variance (ANOVA) of linear mixed-effects (LME) models87 (lmerTerst package in R) to account for repeated measures, where appropriate. Univariate comparisons among Yanomami age groups were performed using Kruskal-Wallis, followed by Dunn test for multiple pairwise comparisons. Multiple comparisons p-values were adjusted using Benjamini-Hochberg88 (BH) correction (q-values). Multivariate data comparisons were performed using permutational analysis of variance (PERMANOVA89, adonis in R) on Bray-Curtis (BC) dissimilarity matrices (square root-transformed taxonomic data) and visualized using Principal Coordinates Analysis (PCoA). Linear discriminant analysis Effect Size90 (LEfSe; Kruskal Wallis p < 0.05 followed by pairwise Wilcoxon p < 0.05 and LDA cutoff of 2) algorithm provided between-group comparisons of taxa or functional gene relative abundances. Spearman correlation (Hmisc in R) of abundance scores was used to identify multi-kingdom co-occurrences. Malassezia species interactions to bacterial genera were plotted using igraph in R when p < 0.05, as previously described91. Differences across adult Yanomami body sites and similarities between environmental samples and adult Yanomami skin microbiota were evaluated by the Friedman test (group-wise comparison, to account for non-parametric, repeated measures) and the Dunn test. Longitudinal evaluation of the western expeditioner’s microbiota at combined sebaceous sites relative to the Yanomami was performed using LME models, followed by Tukey pairwise comparisons. Details for all analyses and analysis software used are provided in the Supplementary Methods.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw data from metagenomic and metatranscriptomic analyses generated in this study have been deposited in the NCBI under BioProject number PRJNA1083578.
References
Harris-Tryon, T. A. & Grice, E. A. Microbiota and maintenance of skin barrier function. Science 376, 940–945 (2022).
Nakatsuji, T. et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med 9, 1–12 (2017).
Scharschmidt, T. C. et al. Commensal Microbes and Hair Follicle Morphogenesis Coordinately Drive Treg Migration into Neonatal Skin. Cell Host Microbe 21, 467–477.e5 (2017).
Ito, Y. et al. Staphylococcus cohnii is a potentially biotherapeutic skin commensal alleviating skin inflammation. Cell Rep. 35, 109052 (2021).
Lunjani, N., Ahearn-Ford, S., Dube, F. S., Hlela, C. & O’Mahony, L. Mechanisms of microbe-immune system dialogue within the skin. Genes Immun. 22, 276–288 (2021).
Lebeer, S. et al. Selective targeting of skin pathobionts and inflammation with topically applied lactobacilli. Cell Rep. Med 3, 100521 (2022).
Kim, G. et al. Spermidine-induced recovery of human dermal structure and barrier function by skin microbiome. Commun Biol. 4, 369–375 (2021).
Uberoi, A. et al. Commensal microbiota regulates skin barrier function and repair via signaling through the aryl hydrocarbon receptor. Cell Host Microbe 29, 1235–1248.e8 (2021).
Zheng, Y. et al. Commensal Staphylococcus epidermidis contributes to skin barrier homeostasis by generating protective ceramides. Cell Host Microbe 30, 301–313.e9 (2022).
Oh, J. et al. Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64 (2014).
Oh, J., Byrd, A. L., Park, M., Kong, H. H. & Segre, J. A. Temporal Stability of the Human Skin Microbiome. Cell 165, 854–866 (2016).
Li, Z. et al. Characterization of the human skin resistome and identification of two microbiota cutotypes. Microbiome 9, 47 (2021).
Carrieri, A. P. et al. Explainable AI reveals changes in skin microbiome composition linked to phenotypic differences. Sci. Rep. 11, 4565 (2021).
Huang, S. et al. Human Skin, Oral, and Gut Microbiomes Predict Chronological Age. mSystems 5, e00630–19 (2020).
Howard, B. et al. Aging-Associated Changes in the Adult Human Skin Microbiome and the Host Factors that Affect Skin Microbiome Composition. J. Investig. Dermatol. https://doi.org/10.1016/j.jid.2021.11.029 (2022).
Park, J. et al. Shifts in the Skin Bacterial and Fungal Communities of Healthy Children Transitioning through Puberty. J. Investig. Dermatol. 142, 212–219 (2022).
Sharma, A. et al. Longitudinal homogenization of the microbiome between both occupants and the built environment in a cohort of United States Air Force Cadets. Microbiome 7, 70 (2019).
Moitinho-Silva, L. et al. Host traits, lifestyle and environment are associated with human skin bacteria. Br. J. Dermatol. 185, 573–584 (2021).
Kim, H. J. et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 9, 16748 (2019).
Kim, H.-J. et al. Fragile skin microbiomes in megacities are assembled by a predominantly niche-based process. Sci. Adv. 4, e1701581 (2018).
Lehtimäki, J. et al. Patterns in the skin microbiota differ in children and teenagers between rural and urban environments. Sci. Rep. 7, 45651 (2017).
McCall, L. I. et al. Home chemical and microbial transitions across urbanization. Nat. Microbiol. 5, 108–115 (2020).
Callewaert, C. et al. Skin Microbiome and its Interplay with the Environment. Am. J. Clin. Dermatol. 21, 4–11 (2020).
Fyhrquist, N. et al. Microbe-host interplay in atopic dermatitis and psoriasis. Nat. Commun. 10, 4703 (2019).
Chng, K. R. et al. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat. Microbiol. 1, 16106 (2016).
Rainer, B. M. et al. Characterization and Analysis of the Skin Microbiota in Rosacea: A Case–Control Study. Am. J. Clin. Dermatol. 21, 139–147 (2020).
Urban, K. et al. The global, regional, and national burden of atopic dermatitis in 195 countries and territories: An ecological study from the Global Burden of Disease Study 2017. JAAD Int. 2, 12–18 (2021).
Cordain, L. et al. Acne Vulgaris A Disease of Western Civilization. Arch. Dermatol. 138, 1584–1590 (2002).
Lynn, D., Umari, T., Dellavalle, R. & Dunnick, C. The epidemiology of acne vulgaris in late adolescence. Adolesc. Health Med. Ther. 13, https://doi.org/10.2147/ahmt.s55832 (2016).
Blaser, M. J. et al. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 7, 85–95 (2013).
Hospodsky, D. et al. Hand bacterial communities vary across two different human populations. Microbiol. (N. Y) 160, 1144–1152 (2014).
Clemente, J. C. et al. The microbiome of uncontacted Amerindians. Sci. Adv. 1, e1500183 (2015).
Peterson, J. et al. The NIH Human Microbiome Project. Genome Res. 19, 2317–2323 (2009).
Carter, M. M. et al. Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell https://doi.org/10.1016/j.cell.2023.05.046 (2023).
Wibowo, M. C. et al. Reconstruction of ancient microbial genomes from the human gut. Nature 594, 234–239 (2021).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996 (2018).
Saheb Kashaf, S. et al. Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Nat. Microbiol. 7, 169–179 (2022).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Ross, A. A., Rodrigues Hoffmann, A. & Neufeld, J. D. The skin microbiome of vertebrates. Microbiome 7, 79 (2019).
Myers, T. et al. A multi-study analysis enables identification of potential microbial features associated with skin aging signs. Front. Aging 4, 1304705 (2024).
Ridaura, V. K. et al. Contextual control of skin immunity and inflammation by Corynebacterium. J. Exp. Med. 215, 785–799 (2018).
Swaney, M. H., Sandstrom, S. & Kalan, L. R. Cobamide Sharing Is Predicted in the Human Skin Microbiome. mSystems 7, e0067722 (2022).
Kriegeskorte, A. et al. Inactivation of thyA in Staphylococcus aureus attenuates virulence and has a strong impact on metabolism and virulence gene expression. mBio 5, 1–15 (2014).
Banaszkiewicz, S. et al. Genetic Diversity of Composite Enterotoxigenic Staphylococcus epidermidis Pathogenicity Islands. Genome Biol. Evol. 11, 3498–3509 (2019).
Brown, T. L. et al. Dynamic interactions between prophages induce lysis in Propionibacterium acnes. Res Microbiol 168, 103–112 (2017).
Kim, J., Park, T., Kim, H. J., An, S. & Sul, W. J. Inferences in microbial structural signatures of acne microbiome and mycobiome. J. Microbiol. 59, 369–375 (2021).
Li, H. et al. Skin Commensal Malassezia globosa Secreted Protease Attenuates Staphylococcus aureus Biofilm Formation. J. Investig. Dermatol. 138, 1137–1145 (2018).
Goh, J. P. Z. et al. The human pathobiont Malassezia furfur secreted protease Mfsap1 regulates cell dispersal and exacerbates skin inflammation. Proc. Natl. Acad. Sci. USA 119, e2212533119 (2022).
Proctor, D. M. et al. Integrated genomic, epidemiologic investigation of Candida auris skin colonization in a skilled nursing facility. Nat. Med. 27, 1401–1409 (2021).
Donnarumma, G. et al. Analysis of the response of human keratinocytes to Malassezia globosa and restricta strains. Arch. Dermatol Res. 306, 763–768 (2014).
Tao, R., Li, R. & Wang, R. Skin microbiome alterations in seborrheic dermatitis and dandruff: A systematic review. Exp. Dermatol. 30, 1546–1553 (2021).
Kengmo Tchoupa, A., Kretschmer, D., Schittek, B. & Peschel, A. The epidermal lipid barrier in microbiome–skin interaction. Trends Microbiol. 31, 723–734 (2023).
Salgaonkar, N. et al. Glycerol fermentation by skin bacteria generates lactic acid and upregulates the expression levels of genes associated with the skin barrier function. Exp. Dermatol. 31, 1364–1372 (2022).
Chiang, Y. R., Wei, S. T. S., Wang, P. H., Wu, P. H. & Yu, C. P. Microbial degradation of steroid sex hormones: implications for environmental and ecological studies. Micro. Biotechnol. 13, 926–949 (2020).
Lesouhaitier, O. et al. Host Peptidic Hormones Affecting Bacterial Biofilm Formation and Virulence. J. Innate Immun. 11, 227–241 (2019).
Patra, V. et al. Ultraviolet exposure regulates skin metabolome based on the microbiome. Sci. Rep. 13, 7207 (2023).
Balasubramaniam, A. et al. Skin bacteria mediate glycerol fermentation to produce electricity and resist uv-b. Microorganisms 8, 1–11 (2020).
Lax, S. et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014).
Li, H., Zhou, S. Y. D., Neilson, R., An, X. L. & Su, J. Q. Skin microbiota interact with microbes on office surfaces. Environ. Int. 168, 107493 (2022).
Klepeis, N. E. et al. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J. Expo. Sci. Environ. Epidemiol. 11, 231–252 (2001).
Grönroos, M. et al. Short-term direct contact with soil and plant materials leads to an immediate increase in diversity of skin microbiota. Microbiologyopen 8, e00645 (2019).
Nielsen, M. C. & Jiang, S. C. Alterations of the human skin microbiome after ocean water exposure. Mar. Pollut. Bull. 145, 595–603 (2019).
Fuhrmeister, E. R. et al. Shared bacterial communities between soil, stored drinking water, and hands in rural Bangladeshi households. Water Res. X 9, 100056 (2020).
Roslund, M. I. et al. Biodiversity Intervention Enhances Immune Regulation and Health-Associated Commensal Microbiota among Daycare Children. Sci. Adv. 6, https://www.science.org (2020).
Mhuireach, G. et al. Temporary establishment of bacteria from indoor plant leaves and soil on human skin. Environ. Microbio. 17, 61 (2022).
Aagaard, K. et al. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 27, 1012–1022 (2013).
Hourigan, S. K. et al. Comparison of infant gut and skin microbiota, resistome and virulome between neonatal intensive care unit (NICU) environments. Front. Microbiol. 9, 1361 (2018).
Uritskiy, G. et al. Cellular life from the three domains and viruses are transcriptionally active in a hypersaline desert community. Environ. Microbiol. 23, 3401–3417 (2021).
Hasan, N. A. et al. Microbial community profiling of human saliva using shotgun metagenomic sequencing. PLoS One 9, e97699 (2014).
Bateman, A. et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 51, D523–D531 (2023).
Caspi, R. et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 36, D471–80 (2008).
Uritskiy, G. V., Diruggiero, J. & Taylor, J. MetaWRAP - A flexible pipeline for genome-resolved metagenomic data analysis 08 Information and Computing Sciences 0803 Computer Software 08 Information and Computing Sciences 0806 Information Systems. Microbiome 6, 158 (2018).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv 3, 13033997 (2013).
Kang, D. D. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ (2019).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. DRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927 (2020).
West, P. T., Probst, A. J., Grigoriev, I. V., Thomas, B. C. & Banfield, J. F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res 28, 569–580 (2018).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 1, e323 (2021).
Dröge, J., Gregor, I. & McHardy, A. C. Taxator-tk: Precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 31, 817–824 (2015).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 421 (2009).
Murphy, J. I., Weaver, N. E. & Hendricks, A. E. Accessible analysis of longitudinal data with linear mixed effects models. DMM Disease Models Mechan. 15, dmm048025 (2022).
Benjaminit, Y. & Hochberg, Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011).
Li, F. et al. Taxonomic assessment of rumen microbiota using total rna and targeted amplicon sequencing approaches. Front. Microbiol. 7, 987 (2016).
Acknowledgements
We are indebted to the Yanomami people for entrusting the stewardship of the Yanomami Microbiome Project to the Yanomami Foundation who enabled this study. The authors wish to acknowledge Monica Contreras (Instituto Venezolano de Investigaciones Científicas, IVIC) for support in the recruitment of Yanomami participants and navigating the permit process in Venezuela. Roger Cariban, coordinator of the Indigenous Health of Amazonas State (Ministerio del Poder Popular para la Salud, MPPS) for his medical oversite and assistance with sample collection. Orlana Lander (Instituto de Medicina Tropical, Universidad Central de Venezuela) for assistance with sample collection. Chris Callewaert (Ghent University) and Nayeim Khan (CosmosID) for assistance with exploratory analysis. As well as Martin Feelisch (University of Southampton), Howard Maibach (University of California, San Francisco), Emma Allen-Vercoe (University of Guelph), Carlos Bustamante (Stanford University; GalateaBio) for useful data discussions.
Author information
Authors and Affiliations
Contributions
J.D., Y.P., L.W. contributed to study conception and design. D.A.G. and H.C.A. managed bioethics and legal permit acquisition and served as cultural liaisons for the Yanomami people. D.A.G., H.C.A., J.D., Y.P. contributed to sample collection and management. J.D., Y.P., H.V., B.F., N.A.H., MD, J.O., A.D.K., T.L.D., R.R.C. contributed to data acquisition, analysis and interpretation; Y.P., H.V., B.F., J.D. performed data analysis. All authors have contributed to manuscript drafting and have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.D., Y.P., H.V. and L.W. report personal fees for employment with Weiss Biosciences Inc., a manufacturer of Symbiome™ Skincare products, outside of submitted work. The rest of the authors declare no relevant conflicts of interest. Weiss Biosciences Inc., has funded this study.
Peer review
Peer review information
Nature Communications thanks Astrid H. Lossius, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Durack, J., Piceno, Y., Vuong, H. et al. Yanomami skin microbiome complexity challenges prevailing concepts of healthy skin. Nat Commun 16, 5542 (2025). https://doi.org/10.1038/s41467-025-60131-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60131-7
