
Abstract
Transcriptional regulators (TRs) are master controllers of gene expression and play a critical role in both normal tissue development and disease progression. However, existing computational methods for identification of TRs regulating specific biological processes have significant limitations, such as relying on distance on a linear chromosome or binding motifs that have low specificity. Many also use statistical tests in ways that lack interpretability and rigorous confidence measures. We introduce BIT, a Bayesian hierarchical model for in-silico TR identification. Leveraging a comprehensive library of TR ChIP-seq data, BIT offers a fully integrated Bayesian approach to assess genome-wide consistency between user-provided epigenomic profiling data and the TR binding library, enabling the identification of critical TRs while quantifying uncertainty. It avoids estimation and inference in a sequential manner or numerous isolated statistical tests, thereby enhancing accuracy and interpretability. BIT successfully identifies perturbed TRs in perturbation experiments, functionally essential TRs in various cancer types, and cell-type-specific TRs within heterogeneous cell populations, offering deeper biological insights into transcriptional regulation.
Similar content being viewed by others

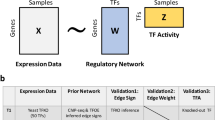
Introduction
Transcriptional regulators (TRs), comprising transcription factors, cofactors, chromatin modulators, and various regulatory molecules, play a key role in regulating gene transcription1,2,3. TRs are essential in diverse cellular processes, including growth, differentiation, morphogenesis, and apoptosis4,5,6. Dysfunction of TRs can lead to various diseases such as cancers7,8,9, diabetes10,11, and heart diseases12,13. Thus, gaining insights into the identity and function of TRs that regulate specific biological processes is essential in a broad range of biological and medical research, particularly in finding biomarkers and therapeutic targets14,15,16. Because traditional wet lab experiments to identify TRs are labor-intensive and time-consuming, researchers are increasingly turning to efficient statistical and machine learning methods for TR identification, especially those leveraging next-generation sequencing (NGS) data.
The in-silico identification of TRs regulating a specific biological process typically requires two key components. The first component is input data that are often generated from cost-effective techniques, such as a set of genes identified from transcriptomics techniques (e.g., microarray, RNA-seq) or a set of accessible chromatin regions derived from epigenomic profiling techniques17 (e.g., ATAC-seq). These input data typically reflect the downstream effects of key TR activity or provide epigenomic features closely associated with TR binding sites. For example, differential gene expression between diseased and normal samples is often attributed to the change in key TR activity18,19,20.
In our recent comprehensive review21, we discussed the limitations of using gene sets as input. This approach requires assigning each gene a representative chromatin location, typically the transcription start site (TSS). The association between a TR and a gene is then assessed based on the linear distance between the binding sites of the TR and the TSS of this gene, which often underestimates long-range interactions, such as distant enhancers that locate thousands of base pairs away from their target genes22. In contrast, alternative input such as chromatin accessibility profiling data directly correlate with TR binding activity23,24,25. As accessible chromatin is a prerequisite for TR binding, functional TRs can be inferred as those with binding sites enriched in the input accessible chromatin regions, thereby avoiding the use of linear distance. In addition, epigenomic profiling techniques such as ATAC-seq have gained increasing popularity over the past decade. The abundance of such datasets has stimulated the development of computational methods to uncover insights into transcriptional regulation.
The second component of in-silico TR identification is a reference library consisting of TR ChIP-seq data or TR binding motifs. TRs bind to specific cis-regulatory elements to exert their regulatory function2, and thus, data that accurately capture TR binding sites serve as a valuable reference. TR ChIP-seq provides an accurate representation of in vivo TR binding profile26. However, each ChIP-seq dataset can only decipher one TR’s binding profile under a specific condition. It is not feasible to conduct thousands of ChIP-seq experiments for one single condition to measure every TR’s binding profile. Therefore, researchers often rely on computational methods to infer functional TRs based on a library of binding profiles collected from thousands of existing ChIP-seq experiments that cover many known TRs. In addition, TR binding sites can also be inferred by TR binding motifs, which are DNA sequence patterns that reveal TR binding preference.
Despite tremendous successes in advancing biomedical research, existing computational methods can suffer from various limitations, regardless of the reference type. (i) For methods (e.g., WhichTF27 and HOMER28) that leverage TR binding motifs as reference, the highly similar motifs shared among many TRs make accurate identification challenging29. In addition, motifs alone cannot characterize any context-specific binding pattern, and the lack of well-defined motifs for some TRs further reduces the reliability of using motifs for TR identification2,30. (ii) For methods using TR ChIP-seq data (e.g., BART31 and ChIP-Atlas32), each TR’s binding profile can vary in different cell types or environmental conditions. However, published methods have no rigorous treatment of the within-TR heterogeneity when dealing with multiple binding profiles from the same TR. Further, TRs with more datasets can benefit from randomness and have a greater chance of being ranked higher in the final output, leading to biased results21. (iii) Most existing methods use frequentist approaches for measuring TR statistical significance, lacking coherent probabilistic interpretation or uncertainty estimation. For example, ChIP-Atlas32 ranks TRs using p values from Fisher’s exact tests, which assess significance under a fixed-parameter null hypothesis (e.g., odds ratio = 1) but fail to capture epistemic uncertainty, especially when experiments cannot be identically replicated. Given the dynamic nature of TR binding activity, the ability to quantify uncertainty is not just beneficial but essential.
To address these limitations, we propose a Bayesian hierarchical model for TR identification, named BIT (Bayesian Identification of Transcriptional regulators from epigenomics-based query regions sets). The input of BIT is a set of user-provided epigenomic regions containing the chromosome numbers and coordinates of the regions, typically referred to as peaks, often derived from genome-wide epigenomic profiling of specific biological processes. Compared to existing methods, BIT offers the following advantages.
First, BIT leverages over 10,000 TR ChIP-seq datasets gathered from previous studies covering a large collection of TRs. This approach bypasses the need for computationally predicted motifs, which can lead to unreliable TR predictions in motif-based methods28,33,34. In addition, the context-specific binding patterns captured by TR ChIP-seq will not be neglected.
Second, through a unique hierarchical model setup, BIT integrates information across multiple TRs and across multiple ChIP-seq datasets of the same TRs, appropriately accounting for both between- and within-TR heterogeneity in binding profiles. This formal model-based approach enables BIT to estimate the uncertainty associated with varying context-specific binding patterns existing within different ChIP-seq datasets for the same TR, leading to a more reliable and robust prediction, as shown in the analysis results of this study. By contrast, many existing methods need to conduct thousands of separate ad-hoc statistical tests (e.g., BART31 and ChIP-Atlas32), which often give higher rankings to TRs with more ChIP-seq datasets21.
Third, BIT employs a Bayesian framework to handle its modeling and computational needs. Besides information pooling for improved prediction, a Bayesian approach is inherently attractive, due to the following: (i) It enables straightforward quantification of estimation uncertainty by deriving 95% credible intervals from posterior sampling, representing the range within which the true value of an unknown quantity (e.g., a parameter of interest or a function of parameters) likely falls. (ii) In situations when meaningful prior knowledge is available, it enables researchers to formally incorporate such knowledge through prior specification for improved results. In all other situations, with the default non-informative prior setting, it becomes a fully automated procedure that does not require any tuning parameter. In contrast, none of the existing methods can offer all these advantages altogether (Table 1). The R package of BIT is available on GitHub (https://github.com/ZeyuL01/BIT) along with a detailed manual. We also provide an easy-to-use online portal (http://43.135.174.109:8080/).
Results
The integration of epigenomic profiling data has proven to be a feasible approach for identifying TRs in biomedical research24,35,36. It is well recognized that the binding sites of TRs critical to a specific biological process are primarily enriched in active regulatory regions of downstream genes that drive the process37. These epigenomic regions can be profiled by sequencing techniques17 (e.g., ATAC-seq), which offer a “snapshot” of genome-wide TR activity, akin to distinct “fingerprints” at a crime scene, providing clues about individuals involved. To link this TR fingerprints snapshot to specific TRs, we leverage TR ChIP-seq data. In humans and mice, there are ~1600 known TRs regulating a wide variety of biological processes, and tens of thousands of TR ChIP-seq experiments have been conducted, each creating a unique reference fingerprint for an individual TR (under some specific condition). To use this wealth of data, we pre-processed a substantial collection of TR ChIP-seq datasets, creating a comprehensive library of TR binding profiles (distinctive fingerprints for individual TRs) in human and mouse genomes, respectively. We utilized 10,140 human TR ChIP-seq datasets covering 988 TRs and 5681 mouse TR ChIP-seq datasets covering 607 TRs. These datasets were sourced from GTRD38, one of the most comprehensive databases in epigenomic research (Supplementary Fig. 1). Each library serves as a powerful reference. Users can compare their own snapshots of TR fingerprints (from ATAC-seq, etc.) with those in the corresponding library to identify TRs whose binding patterns show the highest consistency, indicating their functional relevance in the biological context under study.
BIT operates based on two key biological principles: (1) If a TR is truly involved in regulating a given biological process, its binding pattern across the genome should align more closely with the user-provided input (active regulatory regions) than a TR that is irrelevant. A stronger correlation between TR binding sites and these input regions suggests a greater likelihood that the TR regulates key genes, thus significantly impacting the biological process under investigation. (2) Each TR possesses a distinct binding pattern, enabling its identification even in the presence of some variation. This means that the binding patterns of a specific TR will be more similar across different conditions, compared to the binding patterns of other TRs (Supplementary Fig. 2). This inherent specificity allows for accurate recognition and differentiation of TRs within complex biological systems.
These principles, also used in other bioinformatics tools27,31,39, underscore that an active TR involved in a cellular process will exhibit frequent DNA interactions, leading to captured epigenomic regions near its binding sites. In addition, while variations exist in ChIP-seq data for the same TR due to factors like measurement errors, data quality, tissue specificity, and cell-type specificity, the overall consistency stemming from TR binding preferences allows for unique binding characteristics to emerge. This uniqueness, when compared to input epigenomic data, enables the differentiation of functional TRs from non-functional ones.
BIT only requires a set of epigenomic regions containing the chromosome numbers and coordinates of the regions as input (e.g., Peaks called by MACS2 with the common format such as bed/narrowPeak) and consists of two main steps: (i) data preprocessing and (ii) Bayesian computation and posterior inference with a Gibbs sampler (see “Methods” and Supplementary Note 1). First, BIT transforms the coordinates of processed peak files to fixed-length binary vectors with 0s and 1s. Second, BIT formulates a Bayesian hierarchical model to integrate user-provided information with TR binding information from the reference library and then uses Markov Chain Monte Carlo (MCMC) to sample from the joint posterior distribution. Finally, BIT assigns each TR a BIT score based on posterior draws (Fig. 1). Based on the assumptions stated above, the BIT score can be used as the overall importance measure of the TR being functional. Alternatively, it can be interpreted as the Jaccard index that quantifies the average similarity between the binary vectors of the TR’s binding profiles and the binary vector of the input epigenomic regions.

a Transcription regulators (TRs), such as transcription factors, cofactors, and chromatin modulators, mostly bind to accessible chromatin regions to regulate gene transcription. NGS techniques such as TR ChIP-seq and ATAC-seq can acquire information on TR binding sites and accessible regions, respectively. Input regions and collected TR ChIP-seq peaks are transformed into binary vectors, where the proportion of the matching cases among all informative cases serves as the response variable. b BIT is a Bayesian hierarchical model, combined with Pólya-Gamma data augmentation, to integrate information from multiple TRs and multiple ChIP-seq datasets. c A Gibbs sampler is used to draw posterior samples and estimate key model parameters. d TR-level BIT scores are generated and ranked to output a TR ranking list. This figure has elements created in BioRender. Lu, Z. (2025) https://BioRender.com/4cshxif.
To verify BIT’s performance, we first conducted simulations to examine the efficiency of its posterior estimation. As BIT is built on a unique hierarchical model with meaningful parameters, we used a naïve method as the baseline for comparison, which estimates the parameters based on straightforward summary statistics as described in “Methods.” Second, we examined BIT’s performance with three different applications: (i) We identified TRs using differentially accessible regions (DARs) derived from TR perturbation experiments. Perturbed TRs should be responsible for the DARs, and therefore, these DARs can serve as input with known ground truth. (ii) We tested BIT in TR identification using cancer-type-specific accessible epigenomic regions. The identified TRs were cross-validated by findings in previous publications and results derived from CRISPR/Cas9 loss-of-function analysis. (iii) Given the rapid development of single-cell techniques, cell-type-specific accessible regions can now be derived from scATAC-seq data. We further applied BIT to two sets of cell-type-specific accessible regions and validated the BIT predictions. We also benchmarked results from BIT against other state-of-the-art methods in the three applications.
BIT can estimate model parameters and TR rankings with high accuracy
We explored the performance of BIT through extensive simulations. As will be detailed in the “Methods” section, our BIT model separates candidate TRs in the reference library into two distinct groups: (1) TRs with binding profiles generated from multiple TR ChIP-seq datasets and (2) TRs with only one binding profile generated from a single TR ChIP-seq dataset. We use \(i\) and \(i^{\prime}\) to index TRs in the first and second groups, respectively. To cover potential scenarios, we first varied the global mean parameters \(\mu\) and variance \({\tau }^{2}\) to control the distribution of \({\theta }_{i}\). The \({\theta }_{i}\) denotes the mean (logit-scale) proportion of matching between the input binary vector representing the user-provided set of epigenomic regions and those representing binding profiles of the \(i{{\rm{th}}}\) TR in the reference library, measuring its importance relevant to the biological process under study. Thus, μ represents the global mean (logit-scale) proportion of matching across all TRs while \({\tau }^{2}\) captures the between-TR variability. We also varied \({\sigma }_{0}^{2}\), the common variance of \({\theta }_{i{\prime} 1}\) within the second TR group, where \({\theta }_{i{\prime} 1}\) is the logit-scale proportion of matching between the input binary vector and the reference vector representing the single binding profile of the \(i^{\prime}{{\rm{th}}}\) TR. In our simulation, we examined the effect of one such parameter at a time while keeping the other two fixed at their default values (\(\mu=-4\), \({\tau }^{2}=0.75\), and \({\sigma }_{0}^{2}=1.5\)), determined based on an exploratory data analysis of TR perturbation experiments in the “Methods” Section. The range of the varied parameters was also decided based on the exploratory analysis to cover various real scenarios (\(\mu \in \{-5,-4.5,\ldots,-2.5\}\),\({\tau }^{2}\in \{{{\mathrm{0.5,0.7}}},\ldots,1.5\}\), and \({\sigma }_{0}^{2}\in \left\{{{\mathrm{1,1.2}}},\ldots,2\right\}\)). Other parameters were then generated from Gaussian distributions with pre-determined global parameters according to our BIT model. Finally, the number of input matching and informative bins was generated from a binomial distribution with logit−1 transformed \({\theta }_{{ij}}\) (\({\theta }_{{i}^{{\prime} }1}\)). We repeated the above steps and generated 100 replicate datasets for each combination of \(\mu\), \({\tau }^{2}\), and \({\sigma }_{0}^{2}\).
We further investigated the effect of adjusting the number of candidate TRs (\(I\)) included in our analysis. Our human reference library covers 988 TRs, and our mouse reference library covers 607 TRs. Both libraries are expected to rapidly grow as more and more TR ChIP-seq datasets are being generated. Thus, we considered \(I={{\mathrm{500,1000,1500}}}\) in our simulation. In addition, the count of TR ChIP-seq datasets for the \(i\) th TR was simulated randomly from a distribution fitted using the actual counts within the first group in the human reference library.
We evaluated BIT in three aspects: (i) the mean squared errors (MSEs) for estimating global parameters \(\mu,{\tau }^{2},{\sigma }_{0}^{2}\) and TR-level importance parameters (\({\theta }_{i}\)’s and \({\theta }_{i{\prime} }\)’s); (ii) the ability to recover the original ranks of TRs according to their importance, measured by Spearman’s rho. (iii) the robustness of BIT to the normality assumption of the BIT model. Comparison was made with a baseline method based on naïve estimation, as detailed in the “Methods” section.
Overall, BIT estimates all global parameters with high efficiency (Fig. 2a). First, it is noted that the MSEs of BIT for estimating \(\mu\), \({\tau }^{2}\), and \({\sigma }_{0}^{2}\) are consistently lower than that of the baseline method and are close to zero in all cases. Second, a lower number of candidate TRs (i.e., smaller \(I\)) can result in relatively higher MSE, but the difference is much less obvious for BIT. Third, the MSE for the estimation of \({\tau }^{2}\) tends to increase as the value of \({\tau }^{2}\) goes up. This is reasonable as the increased variability would lead to greater estimation uncertainty. Nevertheless, BIT still significantly outperforms the baseline method in estimating \({\tau }^{2}\).

a Mean squared errors for estimation of global parameters using 100 replicate datasets in each simulation setting. b The average of MSEs for estimation of importance parameters (averaged over different TRs) over 100 replicates. c Box plots of Spearman’s rho correlation over 100 replicates between ranks from BIT or the baseline method and the true ranks of simulated TRs. Box plots show the median (center line), 25th and 75th percentiles (box edges), and whiskers extending to the largest or smallest value that is within 1.5 times of the interquartile range, and points beyond this range are plotted as outliers. d The distributions of simulated vs. real numbers of ChIP-seq datasets of individual TRs. e Data generated in our sensitivity analysis from t and gamma distributions, to examine whether heavy-tailedness and skewness would significantly affect the performance of BIT. f Box plots of Spearman correlation over 100 replicates under t and gamma distributions, with baseline under normal distribution. As in (c), box plots show the median (center line), 25th and 75th percentiles (box edges), and whiskers extending to the largest or smallest value that is within 1.5 times of the interquartile range, and points beyond this range are plotted as outliers. Source data are provided as a Source Data file.
BIT offers improved performance in estimating TR-level importance parameters as well, as evidenced by much lower average MSEs (averaged over the different TRs) in various settings (Fig. 2b). Accurate estimation of these parameters is crucial for determining precise TR rankings. We examined Spearman’s rho correlation, which is the Pearson correlation between the rank values of the estimated and true importance parameters of the candidate TRs. We find that for both methods, larger \({\tau }^{2}\) yields higher correlation while larger \({\sigma }_{0}^{2}\) does the opposite (Fig. 2c). As \({\tau }^{2}\) controls the between-TR heterogeneity, larger \({\tau }^{2}\) would make candidate TRs more easily distinguishable and so lead to better ranking performance (i.e., higher Spearman’s \(\rho\)). On the contrary, \({\sigma }_{0}^{2}\) controls the noise level (i.e., how \({\theta }_{i{\prime} 1}\) differs from \({\theta }_{i{\prime} }\)) for TRs in the second group. Thus, increasing \({\sigma }_{0}^{2}\) would make the estimation of \({\theta }_{i{\prime} }\) more difficult, reducing the ranking performance.
The number of datasets associated with each TR in the first group was simulated using a lognormal distribution with right-skewness and heavy-tailedness, right truncated at 1000, which closely mimics the actual count distribution observed in the human reference library (Fig. 2d). We also checked the robustness of BIT to the normality assumption in the BIT model. We used two types of non-Gaussian distributions, the Student’s t-distributions (with degrees of freedom 3, 5, and 10) and gamma distributions (shape parameter \(\alpha\) = 3, 5, 10, scale parameter \(\beta\)= 1), to generate \({\theta }_{i}\)’s and \({\theta }_{i{\prime} }\)’s. The overall shapes of these generated distributions are different from Gaussian distributions (Fig. 2e). The t-distributions with only a few degrees of freedom are known for heavy-tailedness, and gamma distributions for skewness. The simulated data are then shifted and scaled to recover the same mean and variability for comparison while keeping the heavy-tailedness or skewness. The ranking performance of both methods appears to decline as the deviation from normality becomes larger. Nevertheless, BIT is much less sensitive, and the advantages over the baseline method persist (Fig. 2f).
In summary, through simulations in settings mimicking various real scenarios, we showed BIT’s superior performance in accurately estimating parameters and recovering original TR ranks. It consistently outperformed the baseline method, demonstrating high efficiency and robustness even when normality is violated.
BIT can identify perturbed TRs from differentially accessible regions (DARs)
DARs derived from differential accessibility analysis between two biological conditions are critical in understanding the activity of TRs. Here, we leverage DARs from TR perturbation experiments to validate BIT’s capacity to pinpoint key TRs. The targeted nature of these experiments provides a ground truth, allowing us to assess BIT’s ability to identify the perturbed TRs.
The first experiment is an acute depletion of CTCF in the MLL-rearranged human B cell lymphoblastic leukemia (B-ALL) cell line SEM40. The second is a ZBTB7A-knockout experiment in the HUDEP-2 cell line41. The third involves a FOXA2-knockout in the pancreatic progenitors 1 differentiation stage of human pluripotent stem cells42. BIT has successfully identified each perturbed TR as one of the top-ranked TRs in all three experiments, and it has also correctly identified several other TRs that contribute to the biological process involved (Fig. 3a and see Supplementary Data 1 for full lists).

a The top 10 TRs identified by BIT using DARs from each of the three TR perturbation experiments. Posterior estimates of BIT scores are reported with error bars indicating the upper and lower bounds of 95% credible intervals. b Protein-protein interaction of CTCF derived from STRING. c BIT-identified TRs have binding patterns consistent with the differentially accessible regions observed in TR perturbation experiments. d The top 5 terms ranked by gene ontology enrichment analysis of top TRs in the output list of three cases. The enrichment analysis uses one-sided Fisher’s exact tests. GO terms are ranked by Benjamini–Hochberg adjusted p values. e The scatterplot of TRs by using BIT scores from gain accessible regions versus loss accessible regions. Each dot represents a TR. f Mean reciprocal rank (MRR) using five perturbed TRs to compare BIT and five state-of-the-art methods (BART, ChIP-Atlas, i-cisTarget, WhichTF, and HOMER). Source data are provided as a Source Data file.
Using DARs from the first experiment as the input regions, the perturbed TR, CTCF, was ranked first. Other top-ranked TRs include RAD21, SMC3, and SMC1A, which are all components of the cohesin complex. SMC proteins and RAD21 are core proteins that form the ring-shaped cohesin complex (cohesin ring)43,44. CTCF and the cohesin ring can be crucial in forming chromatin loops and topologically associating domains. These two structures are critical in regulating gene transcription45,46. Previous studies have confirmed that both CTCF and the cohesin ring are essential for the maintenance of the two structures when using acute depletion assays to handle proteins47,48, which can explain why BIT also identified the other three TRs in this study. The result was also validated by the protein-protein interactions of CTCF retrieved from the STRING database49, which shows strong associations among CTCF, SMC3, SMC1A, and RAD21 (Fig. 3b).
In the second experiment, in addition to the top-ranked ZBTB7A (the perturbed TR), GATA1 and KLF1 were also ranked in the top 10, which have binding patterns consistent with the DARs derived from the experiment (Fig. 3c). Early studies have revealed that erythroid differentiation and mutation can be controlled by GATA1-dependent transcription50,51, and that ZBTB7A is a cofactor of GATA1 and plays an essential anti-apoptotic role during terminal erythroid differentiation52. This explains why GATA1 was also ranked at the top after the knockout of ZBTB7A. In addition, KLF1 has a multifunctional role in erythropoiesis and acts as one of the master regulators53.
In the third experiment, FOXA2 ranked third, but notably, other known pancreatic regulators (PDX1, GATA6, FOXA1, and GATA4) were also identified. This is consistent with their established roles in pancreatic development and disease54,55,56,57. For example, the pioneer factors FOXA1/2 could initiate chromatin accessibility58, allowing subsequent binding and transcriptional regulation by PDX1, GATA6, and GATA4.
The results of these three experiments were also validated using Gene Ontology (GO) enrichment analysis, where we used the top TRs identified by BIT to examine enriched biological processes (Fig. 3d). The count in this figure indicates the number of TRs associated with each process, and the p value was calculated using hypergeometric tests to evaluate whether the association of TRs with a biological process is higher than expected by chance. We observed that the top five enriched biological processes, ranked by p values, include those involving sister chromatid cohesion formation for the first experiment, erythrocyte differentiation and homeostasis for the second experiment, and pancreas differentiation and development for the third experiment. This further supports that BIT was able to identify TRs from DARs.
We further categorized the DARs into two groups based on whether these regions showed increased (gained) or decreased (lost) accessibility after the perturbation of TRs. We separately applied BIT to the two groups and noticed that the perturbed TRs were ranked differently according to their BIT scores (Fig. 3e). For example, CTCF and FOXA2 were ranked higher in the group of gained accessibility regions, while ZBTB7A was ranked higher in the group of lost accessibility regions. This provides insights into how the perturbation of these TRs affects chromatin accessibility and demonstrates that BIT can offer additional understanding of the functional role of TRs from the DAR analysis.
We also applied BIT to two more datasets derived from TR knockout in mouse thymocytes to study its performance on data from animal models59 and validated the results with GO enrichment analysis (Supplementary Fig. 3). BIT not only ranks the perturbed TRs in top positions (no. 3 and no. 2, respectively) but also identifies other experimentally validated TRs strongly associated with perturbed TRs or biological processes involved.
In addition to bulk TR perturbation data, we leveraged a single-cell TR perturbation dataset60 for validation, which screened 40 TRs with the CRISPR technique in K562 (a well-known leukemia cell line). Among them, GATA1 knockdown exhibited the strongest target perturbation, consistent with the finding from the original study60. In contrast, the perturbation effects of the remaining TRs were weaker and difficult to distinguish from each other using UMAP projection (Supplementary Fig. 4a). This may partly result from the essential role of GATA1 in erythrocyte differentiation and development61. As anticipated, BIT accurately ranked GATA1 as the top TR when provided with DARs generated by comparing GATA1 knockdown cells with negative control-treated cells (Supplementary Fig. 4b). In addition, GO enrichment analysis of the top-ranked TRs identified by BIT in GATA1 knockdown cells also reveals significant enrichment for terms related to leukemia (Supplementary Fig. 4c).
Finally, to compare BIT with other state-of-the-art methods (BART, ChIP-Atlas, i-cisTarget, WhichTF, and HOMER), we calculated the mean reciprocal rank (MRR) for each method using five perturbed TRs in bulk datasets (Fig. 3f) and 40 TRs in the single-cell perturbation dataset (Supplementary Fig. 4d). MRR is a commonly used metric to evaluate a method’s ability to rank relevant terms at top positions62. Among the six methods, BIT achieved the highest MRR for both bulk and single-cell perturbation data. Additionally, BIT identified the most perturbed TRs within the top 10 and top 50 lists from the single-cell perturbations (Supplementary Fig. 4e).
BIT can identify cancer-type-specific TRs
To further validate BIT’s efficacy, we leveraged cancer-type-specific accessible regions obtained from The Cancer Genome Atlas (TCGA) database63. TRs are well-established drivers of crucial cellular processes in various cancers. However, a significant portion of accessible regions recur across multiple cancer types. Including possible non-specific regions can hinder the identification of critical TRs specific to one cancer type. Therefore, employing cancer-type-specific accessible regions offered a more focused approach. We used regions specific to cancers originating from nine tissue types to validate BIT’s performance: breast, bladder, colon, lung, liver, mesothelium, prostate, squamous cell, and testis. More specifically, the ATAC-seq samples used to generate these regions encompassed a broad range of carcinomas, including breast invasive carcinoma (BRCA), bladder urothelial carcinoma (BLCA), and others (see Supplementary Note 2 for an extended discussion). The identified TRs were cross-validated through existing publications and CRISPR/Cas9 screening data from The Cancer Dependency Map Consortium (DepMap)64, which evaluates cancer cell viability after gene knockout.
First of all, many of BIT’s top-ranked TRs are well-known regulators of normal cell processes such as differentiation and proliferation, and have also been implicated in tumor development. A total of 32 TRs (listed by cancer type in Fig. 4a) were cross-validated with existing literature (Supplementary Data 2), with some already serving as biomarkers or therapeutic targets in cancer treatment65,66. For instance, top-ranked TRs in breast cancer included FOXA1, ESR1, and PGR, while prostate cancer revealed PIAS1, AR, and HOXB13, and testicular cancer revealed NANOG, SOX17, and POU5F1 (see Supplementary Data 3 for full lists). Beyond TR identification using estimated BIT scores, BIT also gauges estimation uncertainty. For example, PIAS1 and SOX13 were identified in multiple cancer types, but with higher uncertainty in their BIT score compared to other TRs in the same cancer (Fig. 4b). This suggests their potential roles as pan-cancer regulators.

a Thirty-two TRs identified by BIT from TCGA cancer-type-specific accessible regions validated by existing literature. b BIT scores of top 10 TRs. Posterior estimates of BIT scores are reported with error bars indicating upper and lower bounds of 95% credible intervals. The center is estimated BIT score. c Jaccard index compares cancer-specific accessible regions between any two cancer types (lower triangle) and the sets of top TRs identified by BIT between any two cancer types (upper triangle). d Cumulative number of BIT-identified top 10 and top 50 TRs with minimum Chronos scores of ≤−2 to >0. e Box plots for the Pearson correlation coefficients between Chronos scores and BIT scores for the top 50 TRs identified by BIT across 209 cell lines from nine cancer types. Here, box plots show the median (center line), 25th and 75th percentiles (box edges), and whiskers extending to the largest or smallest value that is within 1.5 times of the interquartile range, and points beyond this range are plotted as outliers. f Numbers of functionally essential TRs identified in the top 10 and 50 TRs by BIT and state-of-the-art methods (BART, ChIP-Atlas, HOMER, i-cisTarget, WhichTF). g The results of gene ontology enrichment analysis of top 20 TRs identified by BIT from breast cancer. The enrichment analysis uses one-sided Fisher’s exact tests. GO terms are ranked by Benjamini–Hochberg adjusted p values. Source data are provided as a Source Data file. This figure has elements created in BioRender. Lu, Z. (2025) https://BioRender.com/4cshxif.
Second, we investigated the specificity of BIT-identified TRs across different cancer types. The low Jaccard similarity coefficients (0.004–0.150) of the input binary vectors indicated minimal overlap between cancer types (Fig. 4c, lower triangle), confirming that retrieved accessible regions are generally specific to their corresponding cancer type. Furthermore, the top-ranked TRs identified by BIT varied significantly across cancer types (Fig. 4b), consistent with the generally low Jaccard similarity (Fig. 4c, lower triangle). This highlights BIT’s ability to identify key TRs specific to each cancer type.
Third, it is noted that several top-ranked TRs, such as NCOA3 and TET2 in breast cancer, lack known motifs2. This absence means these TRs cannot be detected using motif-based methods, despite solid experimental evidence supporting their significant roles in tumor development67,68. In contrast, BIT uses TR ChIP-seq data to prioritize these TRs, irrespective of motif information. This highlights BIT’s advantage over traditional motif-based methods, especially when known motifs are ambiguous or absent.
Fourth, given the critical roles of many TRs in cellular viability7,69,70, we hypothesized a significant overlap between BIT’s top-ranked TRs and functionally essential TRs (i.e., TRs strongly correlated with cancer cell viability) identified in corresponding cancer types via CRISPR/Cas9 knockout screenings. Indeed, approximately half of the BIT-identified TRs were functionally essential TRs (Fig. 4d): 52 out of 90 top 10 TRs and 232 out of 450 top 50 TRs across various cancer types. Here, functionally essential TRs were defined as those with a minimum Chronos (minC) score64,71 (across all cell lines) less than −0.4; a more negative minC indicates greater relevance. Furthermore, we computed Pearson correlation coefficients between Chronos and BIT scores of the top 50 TRs identified by BIT in 209 cell lines across the nine cancer types. Box plots showed in seven out of nine cases, the two scores of most cell lines are negatively correlated, which indicates that higher-ranked TRs generally exhibited lower Chronos scores, thus having a greater impact on cancer cell viability after gene knockout (Fig. 4e).
Last but not least, we benchmarked BIT against state-of-the-art methods (e.g., BART, ChIP-Atlas, i-cisTarget, WhichTF, and HOMER) by comparing their top-ranked TRs with functionally essential TRs from CRISPR/Cas9 knockout data. We observed that BIT identified the highest total number of functionally essential TRs, which was verified by DepMap in the top 10 and top 50 TRs separately, by summing the results from nine cancer types (Fig. 4f). While cellular viability is not the sole determinant of TR functionality in cancer, BIT’s robust performance highlights its efficacy in identifying crucial TRs from cancer-type-specific accessible regions, even amidst the multifaceted nature of TR functionality.
In addition to the cross-validation with CRISPR/Cas9 knockout analysis, we conducted several more analyses using various resources: (1) GO enrichment analysis using BIT’s top TRs identified from breast cancer-specific accessible regions revealed enriched GO terms strongly associated with breast cancer (Fig. 4g). (2) We retrieved super-enhancer-targeted genes from Cistrome72 for six available cancer types, which rank genes based on adjacent super-enhancer activities. We noticed many BIT-identified TRs are also highly ranked by Cistrome (Supplementary Fig. 5a). (3) Kaplan–Meier plotter73, a public analysis tool that has collected data from over 30,000 cancer samples, is used to investigate the correlation between gene expression levels and patient survival time. It is observed that multiple BIT-identified TRs show a strong correlation with patient survival time (Supplementary Fig. 5b).
BIT can identify cell-type-specific TRs
The rise of single-cell omics technologies has created a need for computational tools capable of analyzing transcriptional regulation at the individual cell level. scATAC-seq, a powerful technique for profiling chromatin accessibility in single cells, often involves clustering cells and annotating them to specific cell types to overcome the challenge of sparse data. An important task in downstream analyses of scATAC-seq data is to identify key TRs associated with accessible regulatory regions in specific cell types.
In this study, we investigated the application of BIT for identifying crucial TRs using cell-type-specific accessible regions derived from scATAC-seq data. Two datasets, including 10K peripheral blood mononuclear cells (PBMCs) and primary liver cancer samples, were used, where the cell types were previously annotated based on the dimension reduction results and marker genes. UMAP projections show a clear separation of different cell types in each dataset (Fig. 5a). SnapATAC274 was applied to generate cell-type-specific accessible regions. We then applied BIT to accessible regions from various cell types in the PBMC and liver cancer datasets and compared the results (Supplementary Data 4) with those from SnapATAC274, ArchR75, and scBasset76. These tools integrate multiple steps for processing and analyzing scATAC-seq data, with motif enrichment analysis being a common approach for inferring TR activity in each annotated cell type. Since these tools are widely used with scATAC-seq analysis, we specifically evaluated BIT’s performance in comparison to its motif enrichment analysis functions from a practical perspective. The results were thoroughly validated by multiple approaches, as described below.

a UMAP projection of human Peripheral blood mononuclear cells (PBMCs) and primary liver cancer cells. b Top 20 TRs identified by BIT using PBMC B cell-specific and liver malignant cell-specific accessible regions, verified by existing literature and Human Protein Atlas (HPA). Posterior estimates of BIT scores are reported with error bars indicating upper and lower bounds of 95% credible intervals. c UMAP visualization of gene activity for PAX5, EBF1, and POU2F1 in PBMCs and HNF4A, FOXA2, and HNF4G in liver cancer samples, each identified by BIT with high activity in B or malignant cells. d Venn plots of top 20 TRs identified by BIT, ArchR, SnapATAC2, and scBasset from PBMC B cells and liver malignant cells. e Gene ontology enrichment results of top 20 TRs identified by BIT, ArchR, SnapATAC2, and scBasset from PBMC B cells. The analysis uses one-sided Fisher exact tests. GO terms are ranked by Benjamini–Hochberg adjusted p values. f Kaplan–Meier Plots of HLF, HNF4A, NFIA, PHOX2B (potential tumor suppressors or differentiation regulators), and CDX2, ETV5, HNF1A, NR1H3 (potential oncogenes) in liver cancer samples. g Numbers of BIT, ArchR, SnapATAC2, and scBasset identified top 10 to 50 TRs from liver malignant cells with minimum Chronos score ≤−0.4. Source data are provided as a Source Data file.
First, we evaluated the credibility of the top 20 TRs identified by BIT in the two investigated cell types (PBMC B cells and liver malignant cells; Fig. 5b) based on existing publications and by examining whether each identified TR has a significantly elevated protein expression in the Human Protein Atlas (HPA) for the respective cell type (see Supplementary Data 5 for details). Notably, BIT successfully captured several TRs critical for B cell development. These include PAX5, EBF1, SPIB, IRF4, IKZF1, and BCL11A, which were previously reported for their roles in maintaining B cell identity and verified by HPA as lineage-specific TRs. In addition, POU2AF1, MEF2B, BCL6, BACH2, POU2F2, BATF, IKZF3, REL, and PBX1, though not present in the HPA-verified list, serve as pivotal regulators in B cell proliferation, differentiation, the germinal center transcriptional program, and immune response, as supported by the literature (Supplementary Data 5).
For liver cancer malignant cells, six of the top 20 TRs (HNF4A, FOXA2, NR2F6, HNF1A, HLF, and AR) were verified as tissue-specific by HPA and reported in the literature. Furthermore, the top TRs identified from scATAC-seq (Fig. 5b) showed strong consistency with those from bulk ATAC-seq (Fig. 4b). Seven of the top 10 TRs identified from bulk data (SOX13, HNF4A, NFIL3, NR2F6, HNF4G, HLF, and NFIA) also appeared in the list from single-cell data. While bulk analysis used accessible regions from all cells, the current study leveraged single-cell data specific to malignant cells. This consistency supports the robustness of BIT in identifying critical TRs across data types.
Second, gene activity plots were generated to demonstrate the high specificity of the identified TRs in both PBMC B cells and liver malignant cells (Fig. 5c; left panel for B cells and right panel for malignant cells). Gene activity was assessed based on the number of Tn5 insertions within the gene’s regulatory regions, a standard approach in scATAC-seq analysis74,77. Multiple BIT-identified top TRs exhibited significantly higher gene activity in the studied cell types than other cell types, including PAX5, EBF1, and POU2F1 in PBMC B cells and HNF4A, HNF4G, and FOXA2 in liver cancer malignant cells.
Third, for PBMC B cells and liver malignant cells, we compared the top 20 TRs identified by BIT with those from motif enrichment analysis in ArchR, scBasset, and SnapATAC2. The four sets showed little to no overlap (Fig. 5d). A similar pattern was observed across all HPA-available cell types in the PBMC dataset (Supplementary Fig. 6a). As mentioned in the introduction, the similarity of binding motifs can hinder the differentiation between TRs from the same protein family. This is evident in the top-ranked TRs identified using ArchR and SnapATAC2, where results of PBMC B cells include the POU and IRF families, and results of liver cancer malignant cells exhibit enrichment exclusively for the FOX and ZNF families. It is important to note that not all members within these families hold equal importance. Their inclusion in the top rankings solely reflects the inability of motif enrichment analysis to identify which specific TR within the family directly interacts with the motif site in the accessible chromatin regions. In contrast, BIT’s ability to pinpoint specific TRs, even within families with similar binding motifs, offers a distinct advantage in understanding the precise regulatory mechanisms at play in different cell types. We further compared the number of HPA-verified TRs among the top 20 for the six HPA-available cell types from PBMCs using these four approaches. BIT identified the most HPA-verified TRs in four cell types and had the highest total count overall (Supplementary Fig. 6b).
Fourth, we performed GO enrichment analysis for the top TRs in PBMC B cells identified by BIT and by motif enrichment, separately. The results revealed strong enrichment for biological processes relevant to B cells or mononuclear cells in the BIT-identified TRs. Conversely, the motif-enrichment-identified TRs displayed enrichment for less specific biological processes (Fig. 5e).
Fifth, we explored the potential of BIT-identified TRs from liver malignant cells as prognostic biomarkers. Kaplan–Meier survival analysis revealed statistically significant associations between their expression levels and patient survival (Fig. 5f). While some TRs correlated with worse prognosis, others were linked to improved survival, suggesting diverse roles in tumor progression and differentiation. TRs correlated with better prognosis may reflect transcriptional programs that constrain malignant potential or promote tumor differentiation, while those linked to worse prognosis may drive tumor progression and survival under selective pressures. These findings highlight the functional relevance of BIT-identified TRs in shaping tumor behavior and their prognostic value in liver cancer. In addition, CRISPR/Cas9 knockout experiments in liver cancer cell lines revealed that BIT identified more functionally essential TRs compared to TRs identified by ArchR, SnapATAC2, and scBasset, respectively (Fig. 5g).
For an unbiased analysis, we present results for the eight remaining PBMC cell types (Supplementary Fig. 7) and six remaining primary liver cancer cell types (Supplementary Fig. 8) besides those for PBMC B cells and liver malignant cells. These results align with observations from Fig. 5. For example, GO enrichment analysis of BIT-identified TRs across PBMC cell types (Supplementary Fig. 7) revealed significant enrichment of biological processes directly linked to their core functions (e.g., T cell differentiation, myeloid cell differentiation). In the liver cancer dataset, we identified additional context-specific pathways (Supplementary Fig. 8c), such as positive regulation of defense response in TAMs and lymphocyte activation in immune response in B cells. These findings highlight the tumor microenvironment’s influence on TR activity, where proximity to malignant cells may perturb canonical regulatory programs through microenvironmental crosstalk or oncogenic signaling. In addition, gene activity plots (Supplementary Figs. 7b and 8b) demonstrate that top-ranked TRs exhibit cell-type-specific activity patterns consistent with their functional roles. Collectively, our findings demonstrate the feasibility of using BIT for the downstream analysis of cell-type-specific accessible regions in scATAC-seq data. Beyond feasibility, BIT offers valuable insights into potential regulatory mechanisms. This makes it a compelling alternative to the current dominant motif-based enrichment methods used in scATAC-seq analysis.
Discussion
We proposed a Bayesian hierarchical approach, called BIT, to identify TRs using user-provided epigenomic regions (peaks), often derived from genome-wide epigenomic profiling data. Using various simulation and application studies, we demonstrated the effectiveness of BIT and its advantages over existing state-of-the-art methods.
Though BIT demonstrated superior performance consistently in various applications, BIT can suffer from the biological complexities of TR regulation, which can reduce the method’s performance. TR binding is inherently dynamic, changing across cell types and time in response to diverse signals and environmental factors78. Chromatin structure and epigenetic modification can further complicate this by affecting the binding affinity and specificity23,79. In addition, cooperative binding can also change binding patterns as TRs frequently cooperate to regulate gene expression80. A large number of interactions between TRs, DNA, and other cellular components challenge accurate prediction and functional interpretation, while the currently gathered context-specific binding profiles from ChIP-seq experiments only represent a very tiny part of all possible experiments.
Similar to other NGS-based computational methods, data quality is a critical issue that can affect BIT’s performance. Multiple types of sequencing biases, such as GC content bias or PCR amplification bias, can potentially reduce the reliability of the input epigenomic region set and binding profiles generated by NGS experiments81. For instance, the Illumina platform is known for a strong GC bias, wherein epigenomic regions with high or low GC content may be over- or under-represented in the sequencing data82. Preferential amplification of certain sequences can also lead to the overrepresentation of specific genomic regions83. Therefore, the performance of BIT can depend on the quality of the NGS data used.
In the manuscript, we primarily used ATAC-seq datasets that profile chromatin accessibility as input data. However, other techniques, such as DNase-seq, can also profile the chromatin accessibility and serve as input. In addition, alternative epigenomic profiling techniques are available that also correlate with TR binding activity. For example, H3K27ac ChIP-seq data can mark active promoters and enhancers84, which are potential TR binding sites. Therefore, these datasets can also be employed to identify TRs regulating the biological processes of interest.
To assess the similarity of top-ranked TRs across methods, we computed Jaccard similarity indices of the top 10, 50, and 100 TRs identified by BIT, BART2, ChIP-Atlas, i-cisTarget, HOMER, and WhichTF in our studies of TR perturbation-derived DARs and cancer-type-specific accessible regions. Overall, Jaccard similarity among these approaches is generally low (Supplementary Fig. 9). However, ChIP-seq-based methods (BIT, BART2, and ChIP-Atlas) exhibit relatively higher similarity compared to motif-based methods, likely due to their reliance on overlapping reference databases derived from publicly available TR ChIP-seq datasets. Since TR ChIP-seq provides the most accurate information for TR binding, this shared foundation contributes to the observed similarities. It is worth mentioning that the highest similarity indices often occur between BIT and another approach, suggesting BIT’s strong ability to capture true signals.
BIT implements a Gibbs sampling scheme to draw posterior samples for thousands of parameters defined within the model. As shown in Supplementary Fig. 10a, its computational efficiency depends on two main factors: (1) the number of parameters and (2) the length of the MCMC chain. The number of parameters is determined by the available TRs and associated datasets in each reference library.
We compared the computational efficiency of BIT with the other methods using bulk TR perturbation data. For BIT, we measured the time to convergence (Supplementary Fig. 10b), while for other methods, we recorded the time to obtain results. BIT’s speed is slightly slower than BART but significantly faster than the other four methods (Supplementary Fig. 10c).
With the growing availability of ChIP-seq experiments, the number of parameters for BIT to infer is expected to increase significantly. In addition, it is common for researchers to generate dozens or even hundreds of epigenomic profiling datasets in a single project. Analyzing these datasets using the Gibbs sampler can become increasingly time-consuming. Therefore, alternative approaches such as variational inference85,86 may be considered for improving computational efficiency.
In summary, BIT offers valuable insights into transcriptional regulation by providing accurate TR identification through a rigorous Bayesian hierarchical model. This approach leverages the wealth of accumulated TR ChIP-seq data, representing the most accurate in vivo context-specific binding patterns. With the increasing popularity of epigenomic profiling techniques, particularly bulk and single-cell ATAC-seq, BIT addresses a critical need for effective tools to analyze this rapidly increasing omics data.
Methods
Preprocessing epigenomic NGS data
Bulk data
In the TR perturbation-derived DAR analysis, raw ATAC-seq sequence reads were first cleaned using Trim-galore (v0.6.10). Read quality was checked by FastQC (v0.12.1). Paired-end reads were mapped to the human genome (hg38) or mouse genome (mm10) using bowtie2 (v2.5.2) applying parameters: bowtie2 --very-sensitive -X 2000. SAMtools (v1.16.1) was used for the following steps: (i) convert SAM files to BAM files, (ii) remove unmapped reads and duplicates, (iii) generate index files for BAM files. Peaks were called using MACS2 (v2.2.9.1). BAM files, index files, and peak files were used for DAR analysis using DiffBind (v3.10.1). Reference TR ChIP-seq datasets were retrieved from GTRD (v21.12), datasets with too low number peaks ( < 500) and individual peaks with a low q-value (<10) were removed. Cancer-type-specific accessible regions were directly retrieved from TCGA.
Single-cell data
The single-cell TR perturbation dataset was retrieved from the GEO archive. We obtained DARs for 40 perturbed TRs using the pipeline provided by the data authors (https://github.com/GreenleafLab/SpearATAC_MS_2021), by comparing cells treated with single guide RNA (sgRNA) targeting one specific TR against control cells treated with non-targeting single guide RNA (sgNT). 10K PBMCs and primary liver cancer scATAC-seq fragment files were retrieved from the 10XGenomics and GEO archive. The cell-type-specific accessible regions were generated from each dataset using SnapATAC2 (v2.6.0). When applicable, the cell type annotations were also retrieved and applied. For comparative benchmarking with established single-cell data analysis methods, in addition to SnapATAC2, we used ArchR (v1.0.2) and scBasset (https://github.com/calico/scBasset).
Region mapping
BIT only requires user-provided epigenomic profiling data as input, while leveraging a comprehensive reference library of TR binding profiles from a large collection of TR ChIP-seq datasets. All binding sites (peaks) from a single dataset are mapped to disjoint, consecutive, pre-defined bins of 1000 bps over the entire genome. Each bin is assigned either 0 or 1 according to whether the summit of the peak falls into the bin (in case that the summit information of a peak is missing, the middle point is used instead). Such mapping is also done to the user-provided epigenomic regions. Thus, all data (input and references) are transformed into binary vectors of the same length. We then compare the binary vector from the input with that from each of the TR ChIP-seq datasets, where we define three conditions for any given bin: (i) matching if (1,1); (2) mismatching if (1,0) or (0,1); and (3) non-informative if (0,0). In all input vs. reference comparisons, the first two cases (informative bins) are much fewer than the third case (non-informative bins), and we drop the third case to mitigate the sparsity while not losing much useful information. Note that the proportion of matching bins among all informative bins is the Jaccard Index, a measure of similarity between two binary vectors. To assess the impact of bin width on BIT’s performance, we analyzed bulk TR perturbation datasets using bin widths of 200 and 500 bps. The results indicate that our default 1000 bps setting performed well, with no significant effect observed from varying bin width (Supplementary Fig. 11).
The data model of BIT
BIT is the first model-based method that formally distinguishes TRs with multiple datasets (or binding profiles) from those with only a single dataset (or binding profile) in the reference database, to account for both within-TR and between-TR heterogeneity and allow for information borrowing across different TRs and datasets via a hierarchical setup. Our modeling choices are made according to the principle of parsimony in statistics. Thus, BIT strikes a balance between clarity and effectiveness in addressing complex biological questions. It aims to reduce the risk of overfitting while preserving the key insights needed for accurate predictions. An exploratory analysis using the three human TR perturbation datasets reveals the following: (1) When grouped by the same TR, the distribution of the log odds (for matching among informative bins) often exhibits a roughly bell shape, with different groups (TRs) showing different centers and spreads. (2) When calculating the (average) log odds for each TR and categorizing these quantities based on whether the TRs have single or multiple ChIP-seq datasets, the distributions of the two categories exhibit similar centers but different variabilities (Fig. 6). It is worth mentioning that we also examined how factors, including the number of reference datasets available for a TR and number of peaks in a reference dataset, affect the within-TR variability of the log odds. No significant patterns were observed (Supplementary Fig. 12).

a The distributions of log odds of matching bins among informative bins for the 30 TRs with the highest numbers of TR ChIP-seq datasets available in the reference database. b The distributions of the (average) log odds for all TRs, where TRs are categorized into two groups based on whether each TR has single or multiple TR ChIP-seq datasets. Source data are provided as a Source Data file.
We index TRs with multiple reference datasets by \(i\), \(i={{\mathrm{1,2}}},\ldots,M\), each with \(j={{\mathrm{1,2}}},...,{J}_{i}\) datasets. TRs with a single reference dataset are indexed by \(i^{\prime}={{\mathrm{1,2}}},...,{M}^{c}\), all with \(j\equiv 1\) dataset. Let \({x}_{{ij}}\) \(\left({x}_{i{\prime} 1}\right)\) be the number of matching bins out of \({n}_{{ij}}\) \(\left({n}_{i{\prime} 1}\right)\) informative bins by comparing the vectors between the user input and the \(j\) th reference dataset of the \(i{{\rm{th}}}\) TR (or the only dataset of the \({i}^{{\prime}}{{\rm{th}}}\) TR). The probability of a bin being matching given it is informative is denoted by \({p}_{{ij}}\) \(\left({p}_{i{\prime} 1}\right)\) such that \({x}_{{ij}}\sim {\mbox{Binom}}({n}_{{ij}},{p}_{{ij}})\) and \({x}_{{i}^{{\prime} }1}\sim {\mbox{Binom}}\left({n}_{{i}^{{\prime} }1},{p}_{{i}^{{\prime} }1}\right)\). Let \({\theta }_{{ij}}\equiv {\mbox{logit}}({p}_{{ij}})\left({\theta }_{{i}^{{\prime} }1}\equiv {\mbox{logit}}\left({p}_{{i}^{{\prime} }1}\right)\right)\) be the log odds of matching among informative bins, which can be interpreted as the Jaccard similarity index (or simply, the importance score) at the dataset level. We further model \({\theta }_{{ij}}({\theta }_{{i}^{{\prime} }1})\) by \({\theta }_{{ij}}\sim N\left({\theta }_{i},{\sigma }_{i}^{2}\right)\), \({\theta }_{{i}^{{\prime} }1}\sim N\left({\theta }_{{i}^{{\prime} }},{\sigma }_{0}^{2}\right)\), and all \({\theta }_{i},{\theta }_{i{\prime} }\sim N\left(\mu,{\tau }^{2}\right)\), where \(\mu\) and \({\tau }^{2}\) represent the global mean and variability of TR-level importance scores. In this way, for each TR with multiple datasets, we model its importance scores by a normal distribution with distinctive mean \({\theta }_{i}\) and variance \({\sigma }_{i}^{2}\), reflecting its overall importance and within-TR heterogeneity, respectively (Fig. 6a); all TRs with only one dataset share a common variance \({\sigma }_{0}^{2}\) for information pooling; and all TRs, no matter which group they are in, share a common mean \(\mu\) (Fig. 6b).
We emphasize that \({\tau }^{2}\) controls the between-TR heterogeneity, while each \({\sigma }_{i}^{2}\) governs the noise levels of the datasets associated with the same TR. Additionally, \({\sigma }_{0}^{2}\) accounts for the noise levels for TRs with only a single dataset. These parameters are critical as they balance the estimates between TR-specific data and global population mean, reducing overfitting through regularization.
It turns out that the entire hierarchical model (Fig. 1b) is simple yet effective, as shown in our simulation and applications:
Unlike many existing algorithms31,32,39, BIT inherently integrates information from reference datasets of different TRs to assign one final rank of each TR based on the TR-level importance score \({\theta }_{i}\left({\theta }_{i{\prime} }\right)\) rather than conducting thousands of separate ad-hoc statistical tests.
The (hyper)parameters of our BIT model are collectively denoted by \(\Theta=\{\mu,{\tau }^{2},{\sigma }_{0}^{2},{\left\{{\theta }_{i}\right\}}_{i=1}^{M},{\left\{{\theta }_{{i}^{{\prime} }}\right\}}_{{i}^{{\prime} }=1}^{{M}_{c}},{\left\{{\sigma }_{i}^{2}\right\}}_{i=1}^{M},{\left\{{\theta }_{{ij}}\right\}}_{i=1,j=1}^{M,{J}_{i}},{\left\{{\theta }_{{i}^{{\prime} }1}\right\}}_{{i}^{{\prime} }=1}^{{M}_{c}}\}\) and observed data are \(X=\left\{{\left\{{x}_{{ij}}\right\}}_{i=1,j=1}^{M,{J}_{i}},{\left\{{x}_{{i}^{{\prime} }1}\right\}}_{{i}^{{\prime} }=1}^{{M}_{c}}\right\}\). Then the joint distribution of \(\left({{{\rm{X}}}},\Theta \right)\) can be written as
where
Prior specification
The global location parameter \(\mu\) and all variance parameters, including \({\tau }^{2}\), \({\sigma }_{0}^{2}\), and \({\left\{{\sigma }_{i}^{2}\right\}}_{i=1}^{M}\) are assumed a priori independent. For \(\mu\), we consider the non-informative flat prior \(\mu \sim {\mbox{uniform}}({L}_{\mu },{U}_{\mu })\). The default values are \({L}_{\mu }=-10\) and \({U}_{\mu }=10\), which correspond to a proportion of matching cases among informative cases between 0.00005 and 0.99995, providing a sufficiently wide coverage for all plausible values of \(\mu\) suggested by the data. Detailed discussion about how to set up the upper and lower bounds can be found in Jia et al.87. For each variance parameter involved, we specify an inverse gamma prior distribution \({\mbox{IG}}(a,b)\), where \(a\) and \(b\) are tiny positive values to make prior very vague and diffuse (e.g., \(a=b=0.01\)).
Pólya-Gamma data augmentation and Gibbs sampler
BIT uses a Gibbs sampler, an efficient Monte Carlo Markov chain (MCMC) algorithm to generate posterior samples from the joint probability distribution \(p\left(\varTheta {|X}\right)\) that is proportional to \(p\left(X,\varTheta \right)\). We proceed to derive the full conditionals to set up the Gibbs sampler efficiently. The full conditional posterior distribution of each parameter can be written as \(p\left(\theta {|X},\varTheta \setminus \theta \right)\), where \(\varTheta \setminus \theta\) denotes the set of parameters after removing \({{{\rm{\theta }}}}\) from \(\varTheta\). For \(\mu,{\tau }^{2},{\sigma }_{0}^{2},\) and \({\sigma }_{i}^{2}\), the conditional posterior distributions are known distributions which can be directly sampled, given below:
However, it is difficult to sample from the conditional posteriors of \(p\left({\theta }_{{ij}}{|X},\varTheta \setminus {\theta }_{{ij}}\right)\) and \(p\left({\theta }_{i{\prime} 1}{|X},\varTheta \setminus {\theta }_{i{\prime} 1}\right)\) as these are intractable. We employ a data augmentation strategy based on Pólya-Gamma (PG) latent variables88. Specifically, we define auxiliary variables \({\lambda }_{{ij}}\) and \({\lambda }_{i{\prime} 1}\), which follow the PG distribution.
Then the conditionals given the defined auxiliary parameters can be easily written in the form of a known distribution.
where
Similarly, we can derive the conditional posterior of \({\theta }_{i{\prime} 1}\). Given that all conditionals are now known distributions, we can design an efficient Gibbs sampler in which all quantities are drawn sequentially and generated readily without using any built-in sampling algorithms (such as Metropolis-Hastings or Acceptance/Rejection sampling) that can greatly slow down the computation.
Bayesian inference and TR rankings
Inference about TR-level importance parameters can be made by marginalizing over posterior samples generated from the Gibbs sampler. Let \({\theta }_{i}^{(t)}\) and \({\theta }_{{i}^{{\prime} }}^{(t)}\) be the posterior draw of \({\theta }_{i}\) and \({\theta }_{{i}^{{\prime} }}\) in the \(t{{\rm{th}}}\) iteration of MCMC after the burn-in period, where \(t=1,\ldots,T\). Then \({\theta }_{i}\) and \({\theta }_{{i}^{{\prime} }}\) are estimated by \({\widetilde{\theta }}_{i}={\sum }_{t=1}^{T}{\theta }_{i}^{(t)}/T\) and \({\widetilde{\theta }}_{{i}^{{\prime} }}={\sum }_{t=1}^{T}{\theta }_{{i}^{{\prime} }}^{(t)}/T\). Then, these logit-transformed probabilities \({\left\{{\widetilde{\theta }}_{i}\right\}}_{i=1}^{M}\) and \({\left\{{\widetilde{\theta }}_{{i}^{{\prime} }}\right\}}_{{i}^{{\prime} }=1}^{{M}_{c}}\) can be used to produce a final ranked TR list or to be further transformed back to the 0-1 scale. To gauge prediction uncertainty, credible intervals of any parameter of interest can be easily obtained from percentiles of draws or as highest posterior density intervals.
The naïve method as a baseline for comparison in simulation
To compare the performance of BIT in estimating model parameters, we used a naïve method as the baseline, where \(\mu\) is estimated by
\({\tau }^{2}\) is estimated by
\({\sigma }_{0}^{2}\) is estimated by
and \({\theta }_{i}\) and \({\theta }_{{i}^{{\prime} }}\) are estimated by
In the unlikely case that \(\frac{{x}_{{ij}}}{{n}_{{ij}}}=0\) or \(\frac{{x}_{{i}^{{\prime} }1}}{{n}_{{i}^{{\prime} }1}}=0\), we add a tiny positive value 0.0001 to the zeros.
BIT validation analyses
Computation of MRR
Let the rank of the perturbed TR in the \(i{{\mathrm{th}}}\) output list generated by a method be denoted as \({R}_{i}\), the MRR of the five perturbed TRs (CTCF, ZBTB7A, FOXA2, RUNX1, BCL11B) was computed as:
We computed the metric separately for the six methods (BIT, BART, ChIP-Atlas, i-cisTarget, WhichTF, and HOMER). We similarly computed the metric in a single-cell Perturbation study.
Cross-validation with CRISPR/Cas9 screening data
Data regarding TR knockout effects on the viability of cell lines were retrieved from DepMap. TRs not found in the BIT database or DepMap data were filtered out. Cell lines of the same lineage were grouped to find TRs with a Chronos score lower than −0.4, classifying them as functionally essential TRs. We then examined top-ranked TRs to determine how many were functionally essential in each cancer type.
To compare BIT with state-of-the-art methods, the original implementations of BART (v2.0), HOMER (v4.11.1), and WhichTF (v0.2) were retrieved from https://github.com/zanglab/bart2, http://homer.ucsd.edu/homer/, and https://bitbucket.org/bejerano/whichtf. For ChIP-Atlas and i-cisTarget, we used online portals. The same epigenomic profiling datasets given to BIT were used as input.
Gene ontology enrichment analysis
We used the R package clusterProfiler (v4.14.4) to conduct GO enrichment analysis. The top-ranked TRs in the output list of BIT were first converted to their ensemble IDs, next by contrasting them to the universe of all available genes. We computed the significance of the enrichment of the top TRs in the GO terms. The top 10 TRs in TR perturbation experiments, and the top 20 TRs in the remaining cases were used. We used a q-value cutoff of 0.05 and listed the top 5 or 10 terms in the final outputs.
Statistics and reproducibility
Analyses were mainly performed in R (4.3.0 and 4.4.1) and Python (3.10). The GO enrichment analysis used one-sided Fisher exact tests with p values adjusted by the Benjamini–Hochberg Procedure to control for false discovery rate. Correlation between ranked lists was quantified using Spearman’s rho, while Pearson’s correlation was used for paired numeric data. For rank-based evaluations, rank 1 indicates the highest TR importance, and overall method performance in identifying correct TRs was evaluated using the MRR.
As this was not a clinical trial, no statistical method was used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment. All eligible ChIP-seq datasets from the GTRD database were used, following the removal of datasets with insufficient peaks and low-quality peaks as detailed in the “Methods” section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Only public datasets were used in this study. The reference ChIP-seq datasets are available at GTRD (v21.12) [http://gtrd.biouml.org:8888/downloads/current/]. HPA-verified TRs lists are available at Human Protein Atlas (v24.0) [https://www.proteinatlas.org/]. The TR perturbation experiment data are available at GEO under the accession codes GSE15323740 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153237], GSE17341641 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173416], GSE11410242 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114102], GSE23433159 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE234331], and GSE16885160 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168851]. Cancer-type specific accessible regions are available at TCGA63 (GDC 2019 v18.0) [https://gdc.cancer.gov/about-data/publications/ATACseq-AWG]. CRISPR/Cas9 knockout screening data are available at DepMap64 [https://depmap.org/portal/]. PBMCs scATAC-seq data are available at 10XGenomics [https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/1.0.0/pbmc_granulocyte_sorted_10k]. Liver cancer scATAC-seq data are available at GEO under accession code GSE22726589 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227265]. Processed data are available at Zenodo [https://zenodo.org/records/14231098]. Source data are provided with this paper.
Code availability
BIT90 software is available at GitHub [https://github.com/ZeyuL01/BIT] with a GPL-3.0 license. BIT is also available as a web portal for online analysis [http://43.135.174.109:8080/].
References
Casamassimi, A. & Ciccodicola, A. Transcriptional regulation: molecules, involved mechanisms, and misregulation. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20061281 (2019).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Andersson, R. & Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21, 71–87 (2020).
Mullen, A. C. et al. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576 (2011).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Hwang, J. Y. & Zukin, R. S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 48, 193–200 (2018).
Bradner, J. E., Hnisz, D. & Young, R. A. Transcriptional addiction in cancer. Cell 168, 629–643 (2017).
Chen, Y., Xu, L., Lin, R. Y., Müschen, M. & Koeffler, H. P. Core transcriptional regulatory circuitries in cancer. Oncogene 39, 6633–6646 (2020).
Pei, H., Guo, W., Peng, Y., Xiong, H. & Chen, Y. Targeting key proteins involved in transcriptional regulation for cancer therapy: Current strategies and future prospective. Med. Res. Rev. 42, 1607–1660 (2022).
Rottner, A. K. et al. A genome-wide CRISPR screen identifies CALCOCO2 as a regulator of beta cell function influencing type 2 diabetes risk. Nat. Genet. 55, 54–65 (2023).
Balakrishnan, S., Dhavamani, S. & Prahalathan, C. beta-Cell specific transcription factors in the context of diabetes mellitus and beta-cell regeneration. Mech. Dev. 163, 103634 (2020).
Hong, J. H. & Zhang, H. G. Transcription factors involved in the development and prognosis of cardiac remodeling. Front. Pharmacol. 13, 828549 (2022).
Bauer, A. J. & Martin, K. A. Coordinating regulation of gene expression in cardiovascular disease: interactions between chromatin modifiers and transcription factors. Front. Cardiovasc. Med. 4, 19 (2017).
Bhagwat, A. S. & Vakoc, C. R. Targeting transcription factors in cancer. Trends Cancer 1, 53–65 (2015).
Bushweller, J. H. Targeting transcription factors in cancer - from undruggable to reality. Nat. Rev. Cancer 19, 611–624 (2019).
Darnell, J. E. Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2, 740–749 (2002).
Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 21, 22 (2020).
Libermann, T. A. & Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell Biol. 10, 2327–2334 (1990).
Gilley, J., Coffer, P. J. & Ham, J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162, 613–622 (2003).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 (2003).
Lu, Z., Xiao, X., Zheng, Q., Wang, X. & Xu, L. Assessing next-generation sequencing-based computational methods for predicting transcriptional regulators with query gene sets. Brief Bioinform. 25, https://doi.org/10.1093/bib/bbae366 (2024).
Kulaeva, O. I., Nizovtseva, E. V., Polikanov, Y. S., Ulianov, S. V. & Studitsky, V. M. Distant activation of transcription: mechanisms of enhancer action. Mol. Cell. Biol. 32, 4892–4897 (2012).
Tsompana, M. & Buck, M. J. Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7, 33 (2014).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20, 207–220 (2019).
Minnoye, L. et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Prim. 1, 10 (2021).
Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 (2009).
Tanigawa, Y., Dyer, E. S. & Bejerano, G. WhichTF is functionally important in your open chromatin data?. PLoS Comput. Biol. 18, e1010378 (2022).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013).
Slattery, M. et al. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 39, 381–399 (2014).
Wang, Z. et al. BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 34, 2867–2869 (2018).
Oki, S. et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 19, https://doi.org/10.15252/embr.201846255 (2018).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Tang, F. et al. Chromatin profiles classify castration-resistant prostate cancers suggesting therapeutic targets. Science376, eabe1505 (2022).
Wang, Z., Zhang, Y. & Zang, C. BART3D: inferring transcriptional regulators associated with differential chromatin interactions from Hi-C data. Bioinformatics 37, 3075–3078 (2021).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Kolmykov, S. et al. GTRD: an integrated view of transcription regulation. Nucleic Acids Res. 49, D104–D111 (2021).
Qin, Q. et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 21, 32 (2020).
Xu, B. et al. Acute depletion of CTCF rewires genome-wide chromatin accessibility. Genome Biol. 22, 244 (2021).
King, A. J. et al. Reactivation of a developmentally silenced embryonic globin gene. Nat. Commun. 12, 4439 (2021).
Lee, K. et al. FOXA2 is required for enhancer priming during pancreatic differentiation. Cell Rep. 28, 382–393.e387 (2019).
Gruber, S., Haering, C. H. & Nasmyth, K. Chromosomal cohesin forms a ring. Cell 112, 765–777 (2003).
Haering, C. H., Löwe, J., Hochwagen, A. & Nasmyth, K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell 9, 773–788 (2002).
Kadauke, S. & Blobel, G. A. Chromatin loops in gene regulation. Biochim. Biophys. Acta 1789, 17–25 (2009).
Hansen, A. S., Cattoglio, C., Darzacq, X. & Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9, 20–32 (2018).
Nora, E. P. et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e922 (2017).
Rao, S. S. P. et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e324 (2017).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Leonard, M., Brice, M., Engel, J. D. & Papayannopoulou, T. Dynamics of GATA transcription factor expression during erythroid differentiation. Blood 82, 1071–1079 (1993).
Cheng, Y. et al. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 19, 2172–2184 (2009).
Maeda, T. et al. LRF is an essential downstream target of GATA1 in erythroid development and regulates BIM-dependent apoptosis. Dev. Cell 17, 527–540 (2009).
Siatecka, M. & Bieker, J. J. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 118, 2044–2054 (2011).
Wang, H. et al. Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J. Biol. Chem. 276, 25279–25286 (2001).
Watt, A. J., Zhao, R., Li, J. & Duncan, S. A. Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev. Biol. 7, 37 (2007).
Fujimoto, K. & Polonsky, K. S. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 11, 30–37 (2009).
Allen, H. L. et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 44, 20–22 (2011).
Iwafuchi-Doi, M. & Zaret, K. S. Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692 (2014).
Gamble, N. et al. PU.1 and BCL11B sequentially cooperate with RUNX1 to anchor mSWI/SNF to poise the T cell effector landscape. Nat. Immunol. 25, 860–872 (2024).
Pierce, S. E., Granja, J. M. & Greenleaf, W. J. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat. Commun. 12, 2969 (2021).
Granja, J. M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 37, 1458–1465 (2019).
Craswell, N. in Encyclopedia of Database Systems (eds L. Liu & M. Tamer ÖZsu) 1703 (Springer US, 2009).
Corces, M. R. et al. The chromatin accessibility landscape of primary human cancers. Science 362, https://doi.org/10.1126/science.aav1898 (2018).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017).
Watson, P. A., Arora, V. K. & Sawyers, C. L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711 (2015).
Patel, H. K. & Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 186, 1–24 (2018).
Anzick, S. L. et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277, 965–968 (1997).
Kim, M. R., Wu, M. J., Zhang, Y., Yang, J. Y. & Chang, C. J. TET2 directs mammary luminal cell differentiation and endocrine response. Nat. Commun. 11, 4642 (2020).
Durbin, A. D. et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 50, 1240–1246 (2018).
Gryder, B. E. et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 51, 1714–1722 (2019).
Reddy, J. et al. Predicting master transcription factors from pan-cancer expression data. Sci. Adv. 7, eabf6123 (2021).
Liu, T. et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12, R83 (2011).
Győrffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation5, 100625 (2024).
Zhang, K., Zemke, N. R., Armand, E. J. & Ren, B. A fast, scalable and versatile tool for analysis of single-cell omics data. Nat. Methods 21, 217–227 (2024).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Yuan, H. & Kelley, D. R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Gertz, J. et al. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell 52, 25–36 (2013).
Zhang, Y. & Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 (2001).
Jolma, A. et al. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388 (2015).
Meyer, C. A. & Liu, X. S. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat. Rev. Genet. 15, 709–721 (2014).
Benjamini, Y. & Speed, T. P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40, e72 (2012).
Aird, D. et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 12, R18 (2011).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936 (2010).
Zhang, C., Bütepage, J., Kjellström, H. & Mandt, S. Advances in variational inference. IEEE Trans. Pattern Anal. Mach. Intell. 41, 2008–2026 (2019).
Blei, D. M., Kucukelbir, A. & McAuliffe, J. D. Variational inference: a review for statisticians. J. Am. Stat. Assoc. 112, 859–877 (2017).
Jia, G. et al. RCRnorm: an integrated system of random-coefficient hierarchical regression models for normalizing NanoString nCounter data. Ann. Appl. Stat. 13, 1617–1647 (2019).
Polson, N. G., Scott, J. G. & Windle, J. Bayesian inference for logistic models using Pólya–Gamma latent variables. J. Am. Stat. Assoc. 108, 1339–1349 (2013).
Craig, A. J. et al. Genome-wide profiling of transcription factor activity in primary liver cancer using single-cell ATAC sequencing. Cell Rep. 42, 113446 (2023).
Lu, Z., Xu, L. & Wang, X. BIT: Bayesian Identification of Transcriptional regulators from epigenomics-based query region sets v. v1.0. Zenodo, https://github.com/ZeyuL01/BIT (2025).
Acknowledgements
This work was supported by the following funding: the Rally Foundation, Sam Day Foundation, Children’s Cancer Fund (Dallas), the Cancer Prevention and Research Institute of Texas (RP180319, RP200103, RP220032, RP170152 and RP180805), and the National Institutes of Health funds (R21CA259771, P30CA142543, R01HG011996, and R01HL144969 to L.X.) (R01CA258584 and R15GM131390 to X.W.). Figures 1 and 4 have elements created using BioRender (BioRender.com) by Lu, Z. (2025), available at https://BioRender.com/4cshxif.
Author information
Authors and Affiliations
Contributions
Lu, Z. conducted data processing, coding, simulation, and validation. Lu, Z. and Wang, X. developed BIT’s model and methodology and designed the algorithm and numerical studies. Xu, L. and Wang, X. conceived the ideas, supervised the study, and acquired the funding. All members have read, revised, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Elisa Salviato and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Z., Xu, L. & Wang, X. BIT: Bayesian Identification of Transcriptional regulators from epigenomics-based query region sets. Nat Commun 16, 4966 (2025). https://doi.org/10.1038/s41467-025-60269-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60269-4
