
Abstract
The human mitochondrial helicase Twinkle is essential for mitochondrial DNA (mtDNA) replication and integrity. Using biochemical and single-molecule techniques, we investigated Twinkle’s real-time kinetics, including DNA loading, unwinding, and rewinding, and their regulation by its N-terminal Zinc-binding domain (ZBD), C-terminal tail, and mitochondrial SSB protein (mtSSB). Our results indicate that Twinkle rapidly scans dsDNA to locate the fork, where specific interactions halt diffusion. During unwinding, ZBD-DNA interactions and C-terminal tail control of ATPase activity downregulate kinetics, slowing down the helicase. Binding of mtSSB to DNA likely outcompetes ZBD-DNA interactions, alleviating the downregulatory effects of this domain. Furthermore, we show that ZBD-DNA interactions and ATP binding also regulate rewinding kinetics following helicase stalling. Our findings reveal that ZBD and C-terminal tail play a major role in regulation of Twinkle´s real-time kinetics. Their interplay constitutes an auto-regulatory mechanism that may be relevant for coordinating the mtDNA maintenance activities of the helicase.
Similar content being viewed by others

Introduction
Mitochondrial DNA (mtDNA) is maintained and replicated differently from its nuclear counterpart. Twinkle is the sole replicative helicase found in human mitochondria1,2. It presents strand separation activity needed for leading strand replication. In addition, Twinkle exhibits strand annealing, strand-exchange and branch migration activities and is essential for organization of mitochondrial RNA granules3,4,5,6,7, suggesting a multi-contextual role in mitochondrial nucleic acid metabolism. The essential function of Twinkle is underscored by disease variants of the helicase, which lead to alterations in the copy number and integrity of mtDNA associated with embryonic lethality and numerous heritable neuromuscular diseases8,9,10,11.
Twinkle belongs to the superfamily 4 DNA helicases, which assemble into ring-shaped oligomers that bind NTPs at each of the subunits interfaces and DNA in the central channel12,13,14. However, structural and biochemical data showed that Twinkle can also exist in other oligomeric states in solution, from broken rings to higher order ≥ 6-mer ring-like oligomers12,13,14,15,16. Each subunit of Twinkle could be divided into an N-terminal domain (NTD) and a C-terminal domain (CTD), joined by a flexible helical linker (Fig. 1a). The CTD of Twinkle, which shares high similarity with the CTD of the homologous bacteriophage T7 helicase17, contains the conserved helicase and ATPase motifs18. These motifs are located between two adjacent CTDs, so that the binding of the NTPs effectively connects the subunits and enables their coordinated movement after NTP hydrolysis. The terminal residues of the CTD form an unstructured tail (C-tail) with no sequence similarity with the C-terminal tail of T7 helicase, but well conserved among most mammals19. Deletion of the C-tail increased NTP hydrolysis12,19,20, suggesting that the C-tail of the full-length helicase provides negative regulatory function. Twinkle NTD exhibits DNA binding activity, providing stability and uniformity to Twinkle oligomers4,21,22. Although the NTD contains primase motifs, this function has been lost in human Twinkle due to its deficiency to bind Mg2+ in the ancestral catalytic site17. The N-terminal zinc-binding domain (ZBD) confers ssDNA binding activity, which in the homologous T7 helicase contributes to the priming process23. In humans, however, ZBD appears to lack three of the four conserved cysteine residues that coordinate Zn2+ in the T7 helicase15,17, and its function remains unknown.

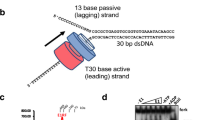
a (Top) Twinkle consists of an ancestral N-terminal primase domain and a C-terminal helicase domain, connected by a linker region. Residues (numbers) corresponding to the Zinc Binding Domain (ZBD, orange) and the carboxyl-terminal tail (C-Tail, magenta) were deleted in the ΔZBD and ΔC-tail variants, respectively. MTS-Mitochondrial Targeting Sequence. (Bottom) Representation of Twinkle hexamer, with ZBD in orange and the C-tail in magenta, prepared by superimposing AlfaFold270 predicted monomers with T7gp4 hexamer (PDB 1E0K). b (Left) Diagram of the optical tweezers-confocal assay to image eGFP-Twinkle (green) on individual DNA constructs containing a DNA fork attached between optically trapped beads (Methods). (Right) Schematics of the fork DNA construct showing the helicase loading site, (dT)30, the distance of the fluorophore to the junction (20 bp) and the lengths of the unwinding segment (446 bp) and dsDNA handles labeled with biotin (blue dots). c Example scans showing separately (left) the Atto 647N-labelled fork position (red) and (right) Twinkle diffraction-limited spots (green) on the DNA construct. d Distribution of initial positions of eGFP-Twinkle diffraction-limited spots on the DNA constructs showing preferential binding at the fork position (N = 50, PDF considers the error in the position of each oligomer as the standard deviation). e Representative kymographs of eGFP-Twinkle oligomers (green) binding directly (top) or diffusing before loading (bottom) at the fork position (in red). f Representative position vs. time plots of eGFP-Twinkle spots show that diffusion is halted upon finding the fork position (4 mM ATP). g Diffusion coefficients of individual Twinkle units (6 ± 2 monomers) on dsDNA (green, N = 39 independent Twinkle units), ssDNA (grey, N = 38, Supplementary Fig. 4) and DNA fork (red, N = 21) at different ATP concentrations or in the presence of ATPγS. Error bars represent standard error of the mean (s.e.). For this figure (f, g) source data are provided as a Source Data file.
Twinkle binds both single-stranded and double-stranded (ds)DNA8,24,25 and can self-load onto single-stranded (ss)DNA circles8,22,25. Recent AFM and HS-AFM studies on human and Lates calcarifer Twinkle (LcTwinkle) have shown that Twinkle ring-like oligomers can switch between close-to-open conformations, allowing them to load (and unload) onto dsDNA without the need for a dedicated helicase loader26,27. The dual ability of Twinkle to load onto both ds- and ss- DNA raises the question of how the helicase recognizes the DNA fork position within the ~16 kbp long mtDNA.
Upon loading at the DNA fork, Twinkle catalyzes the unwinding of duplex DNA in a 5′ to 3′ direction, driven by the hydrolysis of nucleoside triphosphate4,24,28. Recent biochemical and single-molecule studies suggest the existence of multiple ssDNA-binding sites on the helicase surface3,5,21, supporting a steric exclusion and wrapping model for DNA unwinding29. Interestingly, Twinkle exhibits a DNA unwinding activity limited to a few base pairs (20–40 bp)4,24,28, which is enhanced to kilo base pairs processivity by its partners at the replisome24,28; Polγ and the mitochondrial SSB protein (mtSSB). This behavior suggests strong regulatory mechanisms. However, the origin of regulation and the mechanism by which is relieved by other replication components remain unexplored.
In this study, we combine biochemical assays with single-molecule manipulation and visualization techniques to investigate the effects of ATP, helicase-DNA interactions and mtSSB on the loading and real-time DNA unwinding and rewinding kinetics of the human mitochondrial helicase Twinkle. To assess the putative regulatory roles of the N- and C-terminal domains, we compare the real-time kinetics of wild-type helicase (WT) with those of truncation variants (Fig. 1a): ΔZBD, which lacks the first 146 residues of the NTD (corresponding to the ZBD in the homologous T7 helicase2,23), and ΔC-tail, which lacks 66 residues of the unstructured C-terminal tail. The ΔZBD variant disrupts the stabilization of binding to the translocation strand without significantly affecting the ATPase activity22, while the ΔC-tail variant increases ATPase activity without altering DNA binding significantly9,12,19,20. Our results demonstrate that Twinkle rapidly diffuses on long dsDNA, scanning for and stably binding to the DNA fork. Functional loading of the helicase at the fork (i.e., loading followed by unwinding) is facilitated by interactions between Twinkle’s C-tail and mtSSB. During DNA unwinding, the real-time kinetics of Twinkle are strongly autoregulated by ZBD-DNA interactions and the control of ATPase activity by the CTD. Binding of mtSSB to ssDNA competes for ZBD-DNA interactions, alleviating the inhibitory effects of the ZBD. Furthermore, we show that the ZBD and C-tail play critical roles in controlling DNA rewinding events, a hallmark of Twinkle’s activity following DNA unwinding.
Results
Diffusion of Twinkle on dsDNA facilitates fork recognition
First, we investigated how Twinkle recognizes a single DNA fork within a long dsDNA compatible with the length of the mtDNA ( ~ 16 kbp). We used optical tweezers combined with confocal scanning microscopy and microfluidics30 to image the position of fluorescent Twinkle (eGFP-Twinkle) oligomers in real-time along individual 18 kbp-long dsDNA molecules containing a single DNA fork (446 bp), tethered under constant tension (3 or 10 picoNewtons, pN) between two optically trapped beads (Fig. 1b, Methods). The fork was labelled with Atto 647 N at the 3′-end to locate its position and contains a 30 nt long 5′-ssDNA gap to facilitate Twinkle DNA unwinding activity (Fig. 1b, c, Methods). Of note, the DNA unwinding activity of eGFP-Twinkle was indistinguishable from that of wild-type (WT) Twinkle on DNA forks with and without Atto 647 N as evidenced by both biochemical and single-molecule assays (Supplementary Figs. 1 and 2a).
Upon DNA attachment, eGFP-Twinkle (1 nM) was flown into the reaction chamber and detected as diffraction-limited spots on the DNA (Fig. 1c). In the presence of ATP (4 mM), all trapped DNA molecules contained one or several diffraction-limited fluorescent spots (Fig. 1c). We observed a wide distribution of initial binding positions, with a predominant peak at the DNA fork location, which constitutes only ~0.16% of the total length of the trapped DNA construct (Fig. 1d, number of molecules, N = 50). Fluorescence spots bound to the DNA fork remained apparently static (Fig. 1e). However, those bound to the dsDNA moved bidirectionally until they encountered the protein-free DNA fork, where movement ceased (Fig.1e, f). We confirmed that the fluorophore does not non-specifically halt Twinkle diffusion (Supplementary Fig. 2b, c). Additionally, in the absence of the DNA fork, eGFP-Twinkle did not show preferential binding, and its diffusion was not halted at specific positions on dsDNA (Supplementary Fig. 2d, e). These results show that loading of the helicase at the fork can be attained alternatively after scanning of the dsDNA.
The number of Twinkle monomers per diffraction-limited spot was estimated by normalizing their fluorescent intensity by that of a control eGFP-protein fusion (Methods and Supplementary Fig. 3). We considered oligomeric assemblies containing 6 ± 2 eGFPs as individual Twinkle units, whereas multiples of this value were considered either partial (broken rings) or higher oligomers (Supplementary Fig. 3).
Analysis of the motion of diffraction-limited spots compatible with one unit of Twinkle revealed that they presented an average diffusion coefficient on dsDNA (Dds) of 0.075 ± 0.006 μm2/s, which corresponds to 0.787 ± 0.065 kb2/s (4 mM ATP, N = 20). Dds was independent on ATP concentration and the presence of the non-hydrolysable ATP analog ATPγS (Fig. 1g, N = 39), showing that diffusive movement did not depend on ATP hydrolysis. Dds was also found to be independent on tension below 10 pN (Supplementary Fig. 3f). Because the inner diameter of Twinkle oligomers (~ 6-mer) is wide enough to accommodate dsDNA in the central channel13,14, we presume that the rapidly diffusive Twinkle is topologically linked to dsDNA. In fact, the measured Dds values are in line with those reported for the diffusion coefficients of other eukaryotic proteins known to encircle dsDNA, such as the Fanconi anemia D2-I complex31 and CMG helicase32,33. In sharp contrast, Twinkle remained static at the DNA fork at all ATP concentrations during the observation time, or eGFP-bleaching time (~ 120 s). The average diffusion coefficient at the fork (Dfork) was ~0.04 kb2/s (N = 21) at all ATP concentrations tested (Fig. 1g), which probably provides a lower limit on the measurable diffusion constant under our imaging conditions. In an independent set of control experiments, we measured that Twinkle units also diffuse on ssDNA with a diffusion coefficient (Dss) of 0.21 ± 0.02 kb2/s (N = 38, 4 mM ATP), Fig. 1g and Supplementary Fig. 4. This rate is 5-times faster than that of Twinkle on the DNA fork, indicating that the static binding at the fork position is not caused by interaction of Twinkle with the ssDNA portion of the fork. Instead, interactions with the two strands of the DNA fork are more likely required to stall diffusion of the helicase.
Modulation of real-time DNA unwinding kinetics by ATP
We used dual-beam counter-propagating optical tweezers34 to monitor the real-time kinetics of wild-type and two deletion variants of Twinkle on individual forked-like DNA constructs, Fig. 2a, b (Methods). DNA unwinding traces were measured under constant tension monitoring the increase in the end-to-end extension of the tether as each base pair of the DNA hairpin was converted into two single-stranded nucleotides, Fig. 2c (Methods). No such extension changes were observed when assaying a Twinkle variant (K421A) defective in ATP hydrolysis and DNA unwinding35 (Supplementary Fig. 1). In many cases, unwinding activities were followed by DNA rewinding, detected as a reduction in the end-to-end length of the tether, Fig. 2c. We analyzed unwinding and rewinding events independently and will discuss them here sequentially.

a Schematic of optical trapping assays. A DNA fork-like construct is tethered between two functionalized micron-sized beads. One bead is hold in the optical trap (red cone), while the other is immobilized on a micropipette. In the presence of ATP, DNA unwinding is recorded as a change in tether extension (Δx) under constant mechanical tension (F). The blue arrow indicates the 5′ to 3′ translocation of the helicase along the hairpin. b The DNA construct consists of a 559 bp DNA hairpin (sequence in Supplementary Methods) with a ∼2.6-kb long dsDNA handle labelled with digoxigenin (red dots) and a 5′-poly(dT)35 labeled with biotin (blue dot). The single stranded poly(dT)35 tail serves as a helicase loading site (Methods). c Representative traces of the WT Twinkle (blue), ΔZBD (orange) and ΔC-tail (magenta) variants (F = 6 pN). DNA unwinding (increase in bp) is often followed by rewinding (decrease in bp). Raw data (grey background) was smoothed using a low-pass 10 Hz filter. d–g ATP dependencies of the average unwinding processivity (d), unwinding rate (e), pause-free velocity (f), and pause occupancy (g) for the WT helicase (blue), and ΔC-tail (magenta) variant. The dotted line in (f) represents the Michaelis-Menten fit for the pause-free velocity of WT Twinkle as a function of ATP concentration (R2 = 0.89). Data in (d, g) were recorded at F = 6 pN, the minimal tension enabling consistent activity detection across all ATP concentrations (WT, N = 46 independent activities; Δ-Ctail, N = 36). h–k Tension dependencies of the average unwinding processivity (h), unwinding rate (i), pause-free velocity (j) and pause occupancy (k) for WT Twinkle (blue, N = 45) and the ΔZBD variant (orange, N = 32). For all panels, data points represent to the average of multiple independent measurements, and error bars indicate the s.e. For this figure (d–k) source data are provided as a Source Data file.
Similar to other helicases36,37,38,39,40, ATP turnover is expected to regulate the real-time kinetics of DNA unwinding by Twinkle. To investigate this, we first examined the effect of varying ATP concentrations (ranging from 0.2 mM to 10 mM) on the real-time kinetics of DNA unwinding of wild-type (WT) Twinkle (5 nM, tension F = 6 pN, N = 46, Fig. 2d–g). Analysis of individual DNA unwinding traces revealed an average unwinding processivity of ~40 bp, which was independent on ATP concentration (Fig. 2d and Table 1). Processivity was not limited by the length (559 bp) and/or specific sequence (i.e., GC clusters) of the DNA fork (Supplementary Methods) and was consistent with previous biochemical studies of this helicase4,24,28. The increase of ATP concentration stimulated the average DNA unwinding rate 5-fold, from ~0.8 bp/ s to ~4 bp/ s, Fig. 2e and Table 1. The average unwinding rate averages out active unwinding burst with pauses, or transient inactive states, which frequently interrupted the helicase advancement (Fig. 1c). To discern the effect of ATP concentration on the active and pause states, we calculated the pause-free unwinding rate41 and the pause occupancy (Methods). The latter was defined as the ratio between the average rate and the pause-free rate and estimates the probability of finding the helicase in a pause state. This analysis showed that while the pause-free rate presented the expected sigmoidal increase (Fig. 2f), the pause occupancy decreased almost twofold with increasing ATP concentrations (Fig. 2g and Table 1). These findings point to ATP binding as a strong regulator of the characteristic high pause occupancy of Twinkle, which in turns would modulate the unwinding kinetics of the helicase at near physiological ATP concentrations42,43.
In line with these findings, we observed that the Twinkle C-terminal tail deletion variant (ΔC-tail, 5 nM), which exhibits enhanced ATPase activity in vitro, showed similar average processivity but a 5-times faster average unwinding rate than the WT helicase at ≤4 mM ATP (N = 36, Fig. 2d, e and Table 1). The deletion of the C-tail did not affect significantly the pause-free velocity of the variant with respect to that of the WT (Fig. 2f), showing that its increased average unwinding rate is mainly due to a reduced pause occupancy (Fig. 2g and Table 1). At ATP concentrations ≤4 mM, pause occupancy of the ΔC-tail variant was about half that of the WT. Accordingly, we showed that this variant is two- to fivefold more effective than the WT helicase in DNA unwinding in vitro (Supplementary Fig. 5). Overall, these findings suggest that the C-terminal tail hinders ATP turnover, which in turn, would increase pause occupancy slowing down the DNA unwinding kinetics of Twinkle.
Modulation of real-time DNA unwinding kinetics by helicase-fork interactions
Helicase-DNA interactions are crucial for coupling ATP turnover with DNA unwinding, and mechanical tension applied to the opposite strands of the DNA fork influences these interactions44,45,46,47. Thus, we aimed to determine the role of Twinkle-fork interactions in DNA unwinding by measuring the helicase’s DNA unwinding kinetics in response to increasing mechanical tension. Measurements were performed using the experimental setup described above (Fig. 2a) over a range of constant tension below that required to unfold the hairpin mechanically (~ 12 pN) at 4 mM ATP.
Individual DNA unwinding traces of WT Twinkle were detected at a minimum tension of 5 pN, with an average processivity of ~40 bp (N = 45, Fig. 2h and Table 1). Interestingly, processivity was found to decrease slightly with increasing tensions, Fig. 2h. In contrast, tension stimulated by threefold the average unwinding rate from ~0.8 bp/s to ~2.5 bp/s (Fig. 2i and Table 1). Because, tension had no significant effect on the pause-free rate (~ 8 bp/s, Fig. 2j), stimulation of the average velocity could be attributed to the effect of tension on decreasing the pause occupancy of the helicase, from ~90% (5 pN) to ~70% (11 pN) (Fig. 2k and Table 1). This stimulatory effect of mechanical tension could be interpreted as if tension would disrupt helicase-fork interactions that slow down the average velocity. However, we cannot discard the possibility of tension favoring the ATPase activity of the helicase.
To distinguish between these two possibilities, we measured the activity of the Twinkle deletion variant ΔZBD, which exhibits similar ATPase activity but reduced DNA binding compared to the WT Twinkle16,22. Initially, we corroborated that under our experimental conditions the DNA unwinding kinetics of ΔZBD depend on ATP concentration in a manner similar to that of the WT helicase, as expected for this variant (Supplementary Fig. 6). Next, we measured the effect of mechanical tension on the real-time kinetics of the variant (N = 32). Individual DNA unwinding traces of ΔZBD were detected consistently at tension as low as 2 pN (3 pN lower than that required to detect WT activity) and with an enhanced average processivity of ~80 bp (Fig. 2h). These results indicate that elimination of ZBD favors functional loading (loading followed by initiation of DNA unwinding) at the fork and promotes processivity. As in the case of the WT, the average processivity of the ΔZBD variant decreased gradually with tension (Fig. 2h). In addition, the average unwinding rate of ΔZBD was 2–3 times faster, and its pause occupancy lower, than those of the WT Twinkle (< 8 pN) and they did not show any significant tension dependencies (Fig. 2i–k). These results go in line with in vitro biochemical assays showing an enhanced DNA unwinding activity of the ΔZBD variant (Supplementary Fig. 5) and overall, point to the ZBD as a source of pauses during DNA unwinding. The fact that tension did not promote the average rate of the ΔZBD variant up to values found at higher ATP concentrations, suggested that tension does not have an effect on the ATPase activity of the enzyme. Therefore, the observed dependency of the WT helicase average unwinding rate with tension could be attributed to the effect of tension on affecting helicase-fork interactions. A putative scenario compatible with our data is that tension would disrupt the interactions established by ZBD of the WT helicase and the DNA, which otherwise, would downregulate DNA unwinding rate by favoring pause occupancy at near physiological ATP concentrations.
Modulation of real-time DNA unwinding kinetics by mtSSB
mtSSB is known to stimulate the DNA unwinding activity of Twinkle in vitro20,24,26. However, the specific mechanism behind this stimulation remains unclear. To investigate this, we measured the effect of mtSSB (5 nM) on the tension-dependent real-time kinetics of WT Twinkle and the two variants used in this study (at 4 mM ATP, Fig. 3a, b and Table 1). In the case of the WT (N = 57), mtSSB favored the detection of DNA unwinding activities at tension 3 pN lower than that in the absence of mtSSB. This result indicates that mtSSB binding to the 5′ ssDNA tail favors the functional loading of the helicase at the fork. This in agreement with the stimulation of Twinkle DNA unwinding activity by mtSSB, as measured in our bulk biochemical assays (Supplementary Fig. 5). At the lowest tension (2 pN), mtSSB stimulated by ~3–4 times the average processivity and DNA unwinding rate of the WT helicase with respect to the values expected at this tension in the absence of mtSSB, Fig. 3c, d. The stimulus in the average velocity aligns well with the effects of mtSSB and RPA on Twinkle and CMG DNA unwinding rates measured previously in vitro, respectively26,33. Our results showed that the stimulation of average velocity was due to the effect of mtSSB on decreasing the pause occupancy rather than to increasing the pause-free velocity, which remained unaltered (Fig. 3e, f). These stimulatory effects on the real-time kinetics of DNA unwinding seemed specific to mtSSB because significant stimulation was not detected with identical concentrations of the homologous Escherichia coli SSB, consistent with previous bulk studies24 (Supplementary Fig. 7). These two SSB proteins share a high degree of sequence, structural, and functional homology, including similar binding modes, and affinities for ssDNA48,49,50,51,52. Interestingly, the stimulatory effect of mtSSB on the helicase kinetics ceased as tension increased; stimulation was no longer detected above ~6 pN (Fig. 3c, d, f). Tension reduces the binding mode or binding footprint of mtSSB to ssDNA51,52, suggesting that the mtSSB binding mode is relevant for stimulation of helicase activity.

a Schematic of optical trapping assays with mtSSB. The experimental setup is identical to that described in Fig. 2a, with the addition of mtSSB (5 nM, green shape) in the reaction buffer. Experiments were performed at 4 mM ATP. b Representative unwinding traces with mtSSB. Traces show the activity of WT Twinkle (blue, F = 2 pN), ΔZBD (orange, F = 2 pN) and ΔC-tail (magenta, F = 6 pN) variants in the presence of mtSSB. Under these conditions, only the ΔC-tail exhibited significant rewinding events upon unwinding. Raw data (grey background) was smoothed using a low-pass 10 Hz filter. c–f Tension dependencies of the average unwinding processivity (c), unwinding rate (d), pause-free velocity (e) and pause occupancy (f) for WT Twinkle in the absence (blue empty symbols, N = 45) and presence (solid blue symbols, N = 57) of mtSSB. The shaded box highlights the tension range where mtSSB’s stimulatory effects are most apparent. g–j panels show same metrics as above (c–f) for the ΔZBD variant in the absence (empty orange symbols, N = 32) and presence (full orange symbols, N = 29) of mtSSB. k–n panels show same metrics as above (c–f) for the ΔC-tail variant in the absence (empty magenta symbols, N = 27) and presence (solid magenta symbols, N = 18) of mtSSB. For all panels, data points represent the average of multiple independent measurements, and error bars indicate the s.e. For this figure (c–n) source data are provided as a Source Data file.
In contrast, the binding of mtSSB to DNA did not favor the detection of activities at lower tension, nor did it have a significant impact on the DNA unwinding kinetics of the ΔZBD and ΔC-tail Twinkle variants at any mechanical tension, compared to conditions when it was absent (Fig. 3g–n, ΔZBD N = 29, ΔC-tail N = 18). Biochemical analysis further confirmed that mtSSB does not stimulate the DNA unwinding activity of Twinkle variants (Supplementary Fig. 5d). These results highlight the relevance of the ZBD and C-tail on mediating functional and/or physical interactions with the mtSSB that would facilitate functional loading of the helicase onto the DNA fork and/or promote DNA unwinding kinetics.
ATP binding and ZBD-DNA interactions control rewinding events
Single-molecule experiments revealed that many individual unwinding traces were followed by rewinding events (Fig. 2c). We did not observe in any case rapid or instantaneous rewinding of the DNA fork itself, which would suggest fast helicase slippage or detachment. Interestingly, significant differences were observed in the rewinding probabilities and kinetics of the WT and helicase variants under study, depending on ATP concentration and mechanical tension (Fig. 4a–f).

a Bar plots shows the probability of rewinding events following unwinding for the WT helicase (blue bars, N = 46), ΔZBD (orange bars, N = 25), and ΔC-tail (magenta bars, N = 36) at varying ATP concentrations. b Average rewinding/unwinding ratios for the WT (blue symbols, N = 17), ΔZBD (orange symbol, N = 4) and ΔC-tail (magenta symbols, N = 29) as a function of ATP concentration. Ratios ~1 indicate that the number of rewound bp is similar to the number unwound bp. Data for the ΔZBD variant is shown only at 10 mM ATP due to insufficient rewinding events at lower ATP concentrations for meaningful analysis. c Average rewinding velocities for the WT (blue symbols, N = 20), ΔZBD (orange symbol, N = 4) and ΔC-tail (magenta symbols, N = 25) variants as a function of ATP concentration. d Probabilities of rewinding events following unwinding for the WT helicase (blue bars, N = 45), ΔZBD (orange bars, N = 32) and ΔC-tail (magenta bars, N = 27) variants at varying mechanical tension. e Average rewinding/unwinding ratios for the WT (blue symbols, N = 15), and ΔC-tail (magenta symbols, N = 24) variant as a function of tension. f Average rewinding velocities for the WT (blue symbols, N = 16) and ΔC-tail (magenta symbols, N = 23) variant under varying tension. ATP dependencies were measured at 6 pN and tension dependencies were measured at 4 mM ATP. Data points represent the average of multiple independent measurements, and error bars indicate the s.e. For this figure (b–f) source data are provided as a Source Data file.
In the case of the WT helicase, the probability of occurring of rewinding events increased from ~35 to ~80% with ATP concentration (Fig. 4a, F = 6 pN). These rewinding probabilities are in good agreement with those reported previously for WT Twinkle at comparable ATP concentrations by single-molecule FRET studies5. Remarkably, the number of rewound base pairs was generally smaller than that of unwinding. For WT Twinkle the average rewinding/unwinding ratio (i.e., average number of rewound base pairs divided by the number of unwound base pairs) was ~0.5 at <10 mM ATP, Fig. 4b. This ratio, along with the average velocities of rewinding, exhibited increasing trends with ATP concentration (Fig. 4b, c). These ATP dependencies indicate that ATP turnover favors the rewinding events. In line with this hypothesis is that the ΔC-tail variant (with augmented ATPase activity) presented higher rewinding probabilities (~ 80–100%), rewinding/unwinding ratios and rewinding rates than those of the WT at ATP concentrations below 10 mM, Fig. 4a–c. In sharp contrast, the rewinding probability of the ΔZBD variant was just ~5–10% at <10 mM ATP and only increased to 20% at 10 mM (Fig. 4a). These results indicate that the ZBD domain and/or ZBD interactions with the DNA are essential to promote rewinding events.
Interestingly, mechanical tension above 5–6 pN had no significant effect on the average unwinding/rewinding ratios and rates, but it did affect the rewinding probabilities of WT Twinkle and ΔC-tail variant differently (Fig. 4d–f). In the case of the WT, mechanical tension decreased the occurrence of rewinding events from 60% (5 pN) to 0% (11–12 pN), Fig. 4d. These results suggest that mechanical tension would disrupt WT helicase-DNA interactions relevant for rewinding. In line with this hypothesis, we measured that the presence of mtSSB (and E.coli SSB) inhibited the occurrence of the WT rewinding events at all tensions, suggesting mtSSB would outcompete the WT helicase for DNA interactions relevant for rewinding. In sharp contrast, mechanical tension and mtSSB did not affect the rewinding probability of the ΔC-tail variant (Fig. 4d and Supplementary Fig. 8). These results highlight again that elevated ATP turnover would favor rewinding events even if helicase-DNA interactions are compromised by application of mechanical tension to the DNA fork or outcompeted by mtSSB binding to the DNA (Supplementary Fig. 8). Altogether, our data indicates that helicase-DNA interactions, probably mediated by ZBD, together with ATP binding, control rewinding events.
Discussion
Here, we presented single-molecule studies complemented by biochemical assays to provide unique insights into Twinkle’s DNA fork recognition, real-time DNA unwinding kinetics, and the regulatory mechanisms of the ZBD and C-terminal tail in modulating helicase activity. Table 1 summarizes the measured ATP and tension dependencies, while Fig. 5 outlines our main findings. Briefly, we demonstrate that Twinkle diffuses on long dsDNA molecules, scanning for and stably binding to the DNA fork. Functional loading at the fork is facilitated by interactions between Twinkle’s C-terminal tail and mtSSB. We further reveal that Twinkle is a strongly autoregulated helicase: its real-time DNA unwinding kinetics are slowed down by high pause occupancy, which is governed by ZBD-DNA interactions and the regulation of ATPase activity by the C-terminal tail. The binding of mtSSB to ssDNA competes with ZBD-DNA interactions, partially alleviating the inhibitory effects of the ZBD. Additionally, our results indicate that the ZBD and C-terminal tail play critical roles in controlling DNA rewinding events, a characteristic of Twinkle’s activity following DNA unwinding.

Twinkle scans DNA to locate the fork junction through diffusion. In the absence of mtSSB, ZBD-DNA interactions restrict functional loading at the fork and promote pauses (and rewinding events) during DNA unwinding. During this reaction, the C-tail downregulates ATP turnover, leading to additional pauses and reduced DNA unwinding rate. In the presence of mtSSB, interactions between the mtSSB and the C-terminal tail facilitate functional loading of the helicase onto the fork. mtSSB binding to the translocation strand outcompetes the inhibitory ZBD-DNA interactions, enhancing functional loading, decreasing pause occupancy during DNA unwinding, and reducing the probability of rewinding events.
Loading of replicative helicases onto the DNA fork is often required for replisome assembly and therefore, a critical regulatory step in DNA replication53,54. Our data supports that Twinkle can diffuse on dsDNA for thousands of bp with a diffusion coefficient similar to that of other eukaryotic proteins known to encircle dsDNA31,33. This diffusion contributed to targeting efficiently the fork position, which represents ~0.16% of the total length of the scanned DNA. This ability would confer to the helicase an alternative mechanism for fork recognition within the ~16 kbp long mtDNA. Twinkle remained stably bound at the fork, likely due to its interactions with the two strands of fork structure, as our data shows that ssDNA does not stall the helicase diffusion. Whether diffusion on dsDNA facilitates ATP-independent Twinkle activities, such as annealing and strand exchange, remains to be elucidated.
In addition, we demonstrate that functional loading (i.e., loading followed by DNA unwinding initiation) is regulated by the ZBD and C- terminal tail of the helicase, in conjunction with the mtSSB. Without mtSSB, truncating the ZBD allowed the helicase variant to function at 3 pN lower tensions than the WT, suggesting that ZBD-DNA interactions hinder functional loading. However, in the presence of mtSSB, the WT helicase operated at significantly lower tensions, suggesting that mtSSB outcompetes the restrictive ZBD-DNA interactions, thereby facilitating functional loading. Notably, this stimulatory effect of mtSSB was absent in the ΔC-tail variant, which lacks the unstructured C-terminal tail. Given that the C-terminal tail of the homologous T7 helicase mediates physical protein-protein interactions55, our results suggest that physical interactions between Twinkle’s C-terminal tail and mtSSB may also play a role in promoting functional loading at the DNA fork. These findings align with recent suggestions that mtSSB influences the loading mechanism of Twinkle on DNA26.
Upon functional loading, Twinkle unwound DNA with average rates (0.7–4 bp/s at 1–10 mM ATP, respectively, F = 5 pN) in line with those estimated previously for this helicase in air-AFM studies26 and similar to those of other AAA+ helicases56,57. The average DNA unwinding rate of Twinkle was dominated by the high probability of occupancy of transient state(s) that do not support DNA unwinding (considered as pauses in our work). Similarly, frequent pause states have been shown to modulate the average DNA unwinding rates of other prokaryotic and the eukaryotic replicative helicases44,57. However, the origin of pauses during DNA unwinding is poorly understood.
Our results showed that truncation of the ZBD or C-terminal tail from Twinkle decreased significantly the pause occupancy of these variants compared to the WT (ATP ≤ 4 mM and F ≤ 6 pN), supporting the implication of these domains in regulating the DNA unwinding kinetics of the helicase. On the one hand, the differences in response to mechanical tension between the real-time kinetics of WT and ΔZBD helicases suggest that interactions between ZBD and ssDNA favor pause occupancy. One possible explanation is that the flexible and dynamic nature of the NTD of Twinkle13,27, where the ZBD is located, may facilitate regulatory interactions with the two single-stranded DNA strands at the replication fork. These interactions would be diminished by application of mechanical tension explaining the decrease of pause occupancy of WT with increasing tension.
On the other hand, the pause occupancy of WT and ΔZBD depend strongly on ATP concentration, whereas the ΔC-tail variant with enhanced ATPase activity shows no such dependency. These findings, together with the relatively high Km value of Twinkle for ATP (~ 1.4 mM)4,8, point to ATP binding or exchange as a limiting factor and the C-terminal tail of the helicase as a relevant regulatory region that modulates ATP turnover and influences pause occupancy and DNA unwinding kinetics. Notably, the alphafold3 prediction of human Twinkle in the presence of ATP, which matches the structure of LcTwinkle bound to DNA and ATP27, shows the C-tail of Twinkle covering the ATP binding pocket and stabilizing the nucleotide (Supplementary Fig. 9). The basic C-tail may function as an auto-inhibitory lid, modulating ATP binding and exchange. This in turn, would hinder the proper coordination between ATP turnover and DNA binding by ZBD needed to couple translocation with DNA unwinding at near physiological ATP concentrations. Similarly, auto inhibitory basic C-terminal domains have been reported to hamper the activity of repair helicases58.
Autoregulation of Twinkle’s DNA unwinding rate by its ZBD and C-tail domains would be critical to prevent the helicase from surpassing the DNA polymerase when the two enzymes occasionally decouple at the replication fork. However, this self-regulation must be modulated by other replisome partners during DNA replication. Our results showed that mtSSB stimulates DNA unwinding rate of the WT helicase up to threefold, primarily by reducing the pause occupancy during unwinding. Notably, the real-time kinetics of the WT helicase in the presence of mtSSB were identical to those of ΔZBD variant in the absence of mtSSB at low tension (2 pN). This suggests that mtSSB outcompetes the ZBD for DNA binding, effectively releasing the ZBD’s restrictive regulatory effect on unwinding kinetics. The stimulation was specific to mtSSB, ruling out stimulatory mechanisms based on ‘passive’ coverage of newly generated ssDNA by the SSB, which may otherwise facilitate fraying of the fork. Additionally, the stimulatory effect of mtSSB decreased rapidly as mechanical tension increases. Since tension below 12 pN is known to reduce the binding mode or binding footprint of mtSSB to ssDNA52, these findings imply that the mtSSB’s binding mode is critical for stimulating helicase activity.
The WT Twinkle stalled after unwinding ~40 bp of the DNA fork, regardless of the unwinding rate. Notably, truncating the ZBD increased the average number of unwound nucleotides by up to threefold, suggesting that ZBD-DNA interactions regulate helicase processivity. Together with previous studies indicating that Twinkle establishes multiple contacts with the displaced strand on its surface3,5,6,21, our findings support a model in which increasing interactions between the ZBD, the helicase surface and the two strands of the fork generate torsional stress or a mechanical barrier between the helicase and the fork during unwinding. This barrier would be reached after unwinding a specific number of bases (or establishing a specific number of helicase-DNA interactions), regardless of the unwinding rate, explaining the absence of correlation between unwinding rate and processivity. This mechanism likely acts as a ‘molecular brake’29, potentially decoupling ATPase activity and/or DNA binding from motor function, thereby limiting processivity and preventing excessive DNA unwinding in the absence of other replisome factors. Importantly, mtSSB binding to the DNA increased the processivity of the WT by up to 3 times but had no effect on the ΔZBD variant. These results strongly suggest that mtSSB competes with the ZBD for DNA interactions, alleviating mechanical stress and enhancing the helicase processivity at the low tension.
When Twinkle stalls, partial rewinding events were typically observed. On average the WT helicase rewound ~20 bp, which contrasts with its unwinding processivity of ~40 bp (Fig. 4b, e). This implies that rewinding stops 20 nucleotides before reaching the primer-template junction, a length that coincides with Twinkle DNA-binding size4. This suggest that during rewinding, the helicase tracks the displaced strand until it encounters the dsDNA primer-template junction. Our data supports a rewinding mechanism in which, upon stalling, helicase interactions with the two DNA strands and ATP turnover favor a helicase-DNA conformation prone to slide back toward the primer-template junction, pushed by the regression pressure of the fork. The ZBD and C-tail regions would play contrasting and likely complementary, roles in regulating rewinding. ZBD-DNA interactions promote rewinding, while downregulation of the ATPase activity by the C-tail hinders this process. Additionally, the presence of mtSSB would compete with the helicase for DNA interactions, inhibiting rewinding at near physiological ATP concentrations. While the requirements for rewinding resemble those of Twinkle’s ‘intermolecular’ DNA annealing activity4, the two processes may be mechanistically different.
In summary, our findings demonstrate that Twinkle’s activity is tightly autoregulated by its amino and carboxyl terminal domains. Interactions between the ZBD and DNA along with C-terminal tail’s control of ATP hydrolysis act as critical regulatory checkpoints, modulating the helicase’s functional loading, DNA unwinding kinetics, and rewinding activities at the fork. While mtSSB partially alleviates this autoregulation, it does not fully override the inhibitory effects of these domains. Our work paves the way for future in singulo studies to explore the real-time kinetics and regulation of Twinkle within the human mitochondrial replisome.
Methods
Recombinant proteins
Recombinant wild-type (WT, i.e., 43–684), N- (∆ZBD, i.e., Δ146) and C- terminal (∆C-tail, i.e., 43–633) truncation variants and, the ATP hydrolysis defective K421A mutant of the human mtDNA helicase Twinkle were prepared from Sf9 cells12. Briefly, cells were harvested at 72 h post-infection and lysed in 25 mM Tris-HCl pH 8.0, 1 mM PMSF, 10 mM sodium metabisulfite, 2 μg/ml leupeptin, 10 mM 2-mercaptoethanol, followed by addition of NaCl to the final concentration of 1 M. The homogenate was next centrifuged, and supernatant was diluted with an equal volume of 50 mM Tris-HCl pH 8.0, 0.6 M NaCl, 10% glycerol. Initial purification was performed over NiNTA resin and the target was eluted with 250 mM imidazole. Peak fractions were pooled, diluted to ionic equivalent of 150 mM NaCl, and purified further over Heparin Sepharose equilibrated with 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol, 0.5 mM EDTA, 1 mM PMSF, 10 mM sodium metabisulfite, 2 μg/ml leupeptin, 10 mM 2-mercaptoethanol. Bound protein was eluted with 0.6 and 1 M NaCl. Finally, pooled peak fractions were loaded onto 12–30% glycerol gradients in 35 mM Tris-HCl pH7.5, 330 mM NaCl, 1 mM EDTA, 1 mM PMSF, 10 mM sodium metabisulfite, 2 μg/ml leupeptin, 10 mM 2-mercaptoethanol, and centrifuged at 37,000 rpm for 30 h at 4 °C in a Beckman SW41 rotor. Peak fractions were pooled, frozen in liquid nitrogen, and stored in aliquots at −80 °C.
Cloning and purification of eGFP-Twinkle. The Twinkle DNA sequence was synthesized (gBlockTM, IDT) and cloned into a pRK5 vector for mammalian expression with a C-terminal eGFP-3xFLAG-6xHIS tag and a 3C-Prescission protease site using IVA cloning59. Expi293FTM (ThermoFisher Scientific) cultured in Expi293FTM Expression Medium (ThermoFisher Scientific) at 3 × 106 cells/mL density were transfected with 1 ng/ml of plasmid and 6 ng/ml PeiMax® (Linear Polyethylenimine Hydrochloride: Polysciences) as a transfection reagent after 30 min of incubation at RT. Cells were harvested 48 h after the transfection and frozen in liquid N2 for preservation and lysis. Frozen cells were resuspended in a ratio 1:3 (mass:volume) in Lysis buffer (25 mM Hepes pH7.5, 500 mM KCl, 10 mM MgCl2, 5% glycerol, 2 μg/ml Aprotinine, 5 μg/ml Leupeptine, 1 mM Benzamidine and 0.1 mg/ml AEBSF) and fully lysed by sonication in a Vibra-Cell 75042 sonicator (BioBlock Scientific) for 5 min, with pulses ON/OFF of 3 s ON-2 second OFF at 37% amplitude in an ice bath. Lysate was then cleared by centrifugation at 50.000 G for 1 h and filtered using a 0.22 μm syringe filter. ANTI-FLAG M2 Affinity beads (Sigma-Aldrich) were added to the cleared lysate and incubated for at least 3 h at 4° in rotation. After the binding step, the beads were washed three times using the lysis buffer and transferred to a SigmaPrepTM spin column (Sigma-Aldrich in which we incubated the beads with the washing buffer with 300 ng/μL of 3 × Flag peptide (ChinaPeptide). After 30 min, the elution were recovered by centrifugation and frozen in liquid N2.
Preparation of eGFP-Polγ holoenzyme is described in Supplementary Methods60.
Recombinant mtSSB was prepared from E. coli including gel filtration step as follows61. Fraction IIb was chromatographed on a Superdex 75 gel filtration column equilibrated with buffer containing 50 mM Tris-HCl, pH 7.5, 8% glycerol, 150 mM KCl, 2 mM EDTA at a flow rate of 1 ml/min at 4 °C. Fractions containing the mtSSBs were pooled (fraction III) and dialyzed against buffer containing 35 mM Tris-HCl, pH 7.5, 8% glycerol, 100 mM NaCl, 2 mM EDTA, 2 mM dithiothreitol. Recombinant E.coli SSB was purchased from Thermofisher.
Bulk biochemical assays
Biochemical assays are described in the Supplementary Methods.
DNA constructs for single molecule experiments
Four DNA constructs were synthetized to quantify the diffusion on dsDNA and fork recognition by eGFP-Twinkle. (1) Biotinylated dsDNA was generated from plasmid 89DIR.Poly-BbvCI (18709 bp)62. This plasmid was linearized with SalI-HF (NEB #R3138S) and the resulting ends ligated to the complementary ends of biotinylated DNA handles amplified by PCR62. (2) dsDNA containing a single forked-like structure was generated upon linearization of vector 89DIR.Poly-BbvCI with SalI-HF (NEB #R3138S). Then specific locations between positions 6389 and 6452 bp of one strand of the plasmid were nicked with endonuclease Nt.BbvCI (NEB #R0632S). The small DNA fragments between the nicked positions were thermally denatured by raising temperature to 80 °C rendering a 63 nt ssDNA gap. Then the two protruding regions ((dN)30) of a 446 bp long stem loop were hybridized and ligated overnight to the complementary ssDNA gap. Simultaneously, the plasmid ends were ligated to the complementary ends of biotinylated DNA fragments digested with XhoI-HF. Upon ligase denaturation, the DNA strand complementary to the section in which the hairpin was included (6389 and 6449 bp) was nicked at several positions with Nb.BbvCI (NEB #R0632S). In the optical tweezers, individual DNA constructs tethered between streptavidin beads (see below) to induce the mechanical denaturation of the nicked region. This procedure generates a forked-liked structure with two ssDNA regions flanking a DNA hairpin. (3) dsDNA labelled with Atto647N at position ~6400 was obtained following the protocol described for the forked-DNA (construct 2) but replacing the stem loop by a 60 nt long ssDNA oligonucleotide labelled with Atto647N at the 5′ end. In this case, no additional nicking reaction was run upon ligation of the oligo. (4) Single-stranded DNA molecules (17303 nt) were obtained in the optical tweezers upon mechanical denaturation of a linear dsDNA, Supplementary Fig. 4. All protocols avoid DNA purification steps and enzymes were thermally denatured.
The fork-like DNA construct for measuring the force and ATP-dependent unwinding kinetics of Twinkle was synthetize as follows52,63,64. Briefly, the 3′- and 5′- ends of a 559 bp stem loop were ligated to a 2686 bp DNA handle (pUC19 DNA vector, NEB #N3041S) labeled with digoxigenin and to a poly(dT)35 oligonucleotide (IDT) functionalized with biotin, respectively. The poly(dT)35 tail facilitates loading of the helicase at the fork.
Buffers
Single molecule studies were carried out in the reaction buffer containing 50 mM Tris pH 8.5, 30 mM KCl, 10 mM DTT, 4 or 10 mM MgCl2, 0.2 mg/ml BSA and ATP concentrations ranging from 1 mM to 10 mM.
Single-molecule fluorescence imaging and data analysis
Single-molecule imaging experiments of eGFP-Twinkle were performed on an instrument that combines three color confocal fluorescence microscopy (488, 532, 635 nm), with optical tweezers and microfluidics (C-trap® LUMICKS). Trap stiffness was adjusted to 0.32 pN/nm in both traps. DNA-molecules were trapped between two streptavidin-coated polystyrene beads (4.38 µm diameter, Spherothech). The tethering of single DNA molecules was confirmed by analyzing the force-extension curve. The DNA was then transferred to another channel (channel 4) containing a 20 bp DNA oligo (50 nM) complementary to the 3′-tail of the forked structure and labelled with Atto 647 N. Next, the DNA was transfer to channel 5 (previously passivated with BSA (2 mg/ ml)), the distance between both beads was fixed to achieve a tension of 3 or 10 pN, and then Twinkle-eGFP (1 nM as hexamer) was flown into the channel. We used the 488 nm excitation laser for visualization of eGFP-Twinkle (emission filter of 500–525 nm) and the 635 nm excitation laser for visualization of the fork position labeled with Atto 647 N (emission filter 650–750 nm). For confocal images (scans) the confocal pixel size was set to 100 nm and a scan velocity of 1 µm/s−1. Kymographs were generated by single line scans covering the entire DNA stretched between the two beads and the edge of both beads. Pixel size was set to 100 nm and the illumination time per pixel was 0.1 ms, resulting in a typical time per line of 28.1 ms.
Software and code
We used Python 3.8 with several libraries for image processing: numpy = = 1.26.2, lumicks.pylake = = 1.0.0, matplotilb = = 3.7.1, scipy = = 1.10.1, pandas = = 2.0.1.
Trace tracking
We used the greedy algorithm65,66,67 implemented in Pylake Python library (https://github.com/lumicks/pylake // https://doi.org/10.5281/zenodo.4280788 to track the position (µm) of individual fluorescent spots with time (seconds). Kymograms were aligned using the position of the beads as fiducial points. The distance in µm was converted to base pairs (bp) using the theoretical average extension per bp according to the Worm Like Chain model. Similarly, we used the Freely Jointed Chain model of polymer elasticity to convert the distance in µm to single-stranded nucleotides. We used the interp1 MATLAB function to interpolate data in time and in position.
Determination of number of fluorophores per diffraction-limited spot
The fluorescence background was quantified and subtracted from all scan images and kymograms. We used a eGFP-labelled version of the human mitochondrial polymerase holoenzyme (eGFP-Polγ) as a control to estimate the average number of eGFP-molecules per Twinkle diffraction–limited spot. Polγ holoenzyme is a heterotrimeric complex, consisting of the catalytic PolγA subunit and two PolγB accessory processivity factors. We labelled the PolγB subunit with eGFP. The reconstituted eGFP-Polγ holoenzyme, which binds stably to the primer-template position of the fork, provides an internal control to determine the intensity of two eGFP molecules. This intensity value was used to estimate the average number of eGFP-subunits per Twinkle diffraction–limited spots assuming a linear increase of the intensity with the number of eGFP molecules. First, we fit the first 50 maximum intensity data points of eGFP-Polγ diffraction–limited spots to a normal distribution to calculate the average intensity (IPolγ) and standard deviation (σPolγ) of two eGFPs 3.692 ± 0.788 intensity units (N = 19 independent molecules). Then, an identical procedure was used to determine the average intensity of eGFP-Twinkle (ITw) diffraction limited spots and its associated standard deviation (σTw). As the fluorophore intensity increases linearly with the number of eGPF per spot, upon determining the intensity of two eGFPs, we calculated the number of eGFP-labeled per Twinkle spot. The associated measurement error has the form: \({\sigma }_{{\#GFP}}=\sqrt{{\left(\frac{2}{{I}_{{Pol}\gamma }}{\sigma }_{{I}_{{Tw}}}\right)}^{2}+{\left(\frac{-2{I}_{{Tw}}}{{I}_{{Pol}\gamma }^{2}}{\sigma }_{{I}_{{Pol}\gamma }}\right)}^{2}}\)
Diffusion analysis
We used the DSigma2_Est MATLAB function (https://www.nist.gov/programs-projects/fluctuations-and-nanoscale-control-software-archived) to estimate the Mean Squared Displacement vs time lag (τ) for each trace as: \(\left(\tau \right)=2D\tau+2{\sigma }_{x}^{2}\), where D is the diffusion coefficient and \({\sigma }_{x}\) is the location measurement error. The appropriate range of delay times were calculated as previously published66,68.
Single-molecule force spectroscopy experiments and data analysis
A counter propagating dual-beam optical tweezers instrument34 was used to manipulate individual DNA constructs functionalized with biotin or digoxigenin at each end. The DNA was tethered between a streptavidin- and anti-digoxigenin-coated polystyrene beads (3 μm, Spherotech) held on top of a micropipette and in the optical trap, respectively. Twinkle (WT and variants) and SSB proteins were diluted to 5 nM in the reaction buffer and flown inside the flow cell. We note that mtSSB concentrations higher than 5 nM prevented detection of single-molecule DNA unwinding activities. This finding is in line with the deleterious effect of high mtSSB concentrations on the in vitro activity of Twinkle20,24. Data was monitored at 5100 Hz at 22 ± 1 °C using a feedback loop to maintain a constant mechanical tension on the DNA construct below 13 pN. The trap stiffness was k = 0.135 ± 0.0043 pN nm−1 (3.0 μm beads). Data was collected and processed using custom made C++ (http://tweezerslab.unipr.it/cgi-bin/software.pl/Search) and MatLab scripts (MatLab R2021a).
At constant mechanical tension below 12 pN (the mechanical opening tension of the DNA hairpin), the extension of the DNA tether increases (∆x in Fig. 2a) as the helicase unwinds the hairpin and generates new ssDNA between the beads. The number of unwound base pairs (processivity) was obtained by dividing the increase of the tether extension by the change in extension at a given tension accompanying during each catalytic step the generation of two new ssDNA nucleotides (or SSB-bound nucleotides). We used the average extensions per nucleotide of free- and SSB-bound ssDNA as a function of tension reported previously52.
The average DNA unwinding rate was determined by a line fit to the traces showing the number of replicated nucleotides versus time. The final average rate at each mechanical tension was obtained by averaging over all of the traces taken at that tension. The average replication rate without pauses (pause-free velocity) for each trace was obtained by analyzing each trajectory with an algorithm that computes instantaneous velocities, averaging the position over sliding time windows41,69.
The pause occupancy represents the probability of finding the helicase in a transient, not active or pause state. The pause occupancy at each tension (F) was calculated as \({Pocc}\left(F\right)=(1-{Vmean}(F)/V\max ( F))\), where \({Vmean}(F)\) and \(V\max (F)\) represent the average and pause-free replication rates at each tension, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Bulk and single-molecule data generated in this study have been deposited in the IMDEA Nanociencia Research Data repository and can be found at https://hdl.handle.net/20.500.12614/3946. The datasets are also available from the corresponding authors upon request. Source data are provided with this paper.
Code availability
The code used in the current study to analyze confocal-optical tweezers data is available at https://hdl.handle.net/20.500.12614/3946.
References
Spelbrink, J. N. et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 28, 223–231 (2001).
Peter, B. & Falkenberg, M. TWINKLE and other human mitochondrial DNA helicases: structure, function and disease. Genes 11, 408 (2020).
Sen, D., Patel, G. & Patel, S. S. Homologous DNA strand exchange activity of the human mitochondrial DNA helicase TWINKLE. Nucleic Acids Res. 44, 4200–4210 (2016).
Sen, D., Nandakumar, D., Tang, G.-Q. & Patel, S. S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 287, 14545–14556 (2012).
Khan, I. et al. Biochemical characterization of the human mitochondrial replicative twinkle helicase: SUBSTRATE SPECIFICITY, DNA BRANCH MIGRATION, AND ABILITY TO OVERCOME BLOCKADES TO DNA UNWINDING. J. Biol. Chem. 291, 14324–14339 (2016).
Singh, A., Patel, G. & Patel, S. S. Twinkle-catalyzed toehold-mediated DNA strand displacement reaction. J. Am. Chem. Soc. 145, 24522–24534 (2023).
Hensen, F. et al. Mitochondrial RNA granules are critically dependent on mtDNA replication factors Twinkle and mtSSB. Nucleic Acids Res. 47, 3680–3698 (2019).
Longley, M. J., Humble, M. M., Sharief, F. S. & Copeland, W. C. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J. Biol. Chem. 285, 29690–29702 (2010).
Matsushima, Y. & Kaguni, L. S. Differential phenotypes of active site and human autosomal dominant progressive external ophthalmoplegia mutations in Drosophila Mitochondrial DNA helicase expressed in Schneider cells. J. Biol. Chem. 282, 9436–9444 (2007).
Tyynismaa, H. et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 102, 17687–17692 (2005).
Tyynismaa, H. & Suomalainen, A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. 10, 137 (2009).
Ziebarth, T. D., Farr, C. L. & Kaguni, L. S. Modular architecture of the hexameric human mitochondrial DNA helicase. J. Mol. Biol. 367, 1382–1391 (2007).
Fernández-Millán, P. et al. The hexameric structure of the human mitochondrial replicative helicase Twinkle. Nucleic Acids Res. 43, 4284–4295 (2015).
Riccio, A. A. et al. Structural insight and characterization of human Twinkle helicase in mitochondrial disease. Proc. Natl. Acad. Sci. USA 119, e2207459119 (2022).
Kaguni, L. S. & Oliveira, M. T. Structure, function and evolution of the animal mitochondrial replicative DNA helicase. Crit. Rev. Biochem. Mol. Biol. 51, 53–64 (2016).
Ziebarth, T. D. et al. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J. Biol. Chem. 285, 14639–14647 (2010).
Shutt, T. E. & Gray, M. W. Twinkle, the mitochondrial replicative DNA helicase, is widespread in the eukaryotic radiation and may also be the mitochondrial DNA primase in most eukaryotes. J. Mol. Evol. 62, 588–599 (2006).
Singleton, M. R., Dillingham, M. S. & Wigley, D. B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 (2007).
Matsushima, Y., Farr, C. L., Fan, L. & Kaguni, L. S. Physiological and Biochemical Defects in Carboxyl-terminal Mutants of Mitochondrial DNA Helicase. J. Biol. Chem. 283, 23964–23971 (2008).
Rodrigues, A. P. C. & Oliveira, M. T. Stimulation of variant forms of the mitochondrial DNA helicaseDNA helicases twinkle by the mitochondrial single-stranded DNA-binding proteinmitochondrial single-stranded DNA-binding protein (mtSSB). In Single Stranded DNA Binding Proteins (ed. Oliveira, M. T.) 313–322. https://doi.org/10.1007/978-1-0716-1290-3_20 (Springer US, New York, NY, 2021).
Johnson, L. C., Singh, A. & Patel, S. S. The N-terminal domain of human mitochondrial helicase Twinkle has DNA-binding activity crucial for supporting processive DNA synthesis by polymerase γ. J. Biol. Chem. 299, 102797 (2023).
Farge, G. et al. The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res. 36, 393–403 (2008).
Lee, S.-J., Zhu, B., Akabayov, B. & Richardson, C. C. Zinc-binding domain of the bacteriophage T7 DNA primase modulates binding to the DNA template. J. Biol. Chem. 287, 39030–39040 (2012).
Korhonen, J. A., Gaspari, M. & Falkenberg, M. TWINKLE Has 5′ → 3′ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 278, 48627–48632 (2003).
Jemt, E. et al. The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res. 39, 9238–9249 (2011).
Kaur, P. et al. Single-molecule level structural dynamics of DNA unwinding by human mitochondrial Twinkle helicase. J. Biol. Chem. 295, 5564–5576 (2020).
Li, Z. et al. Structural and dynamic basis of DNA capture and translocation by mitochondrial Twinkle helicase. Nucleic Acids Res. 50, 11965–11978 (2022).
Korhonen, J. A., Pham, X. H., Pellegrini, M. & Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 23, 2423–2429 (2004).
Carney, S. M. & Trakselis, M. A. The excluded DNA strand is SEW important for hexameric helicase unwinding. Methods 108, 79–91 (2016).
Candelli, A., Wuite, G. J. L. & Peterman, E. J. G. Combining optical trapping, fluorescence microscopy and micro-fluidics for single molecule studies of DNA–protein interactions. Phys. Chem. Chem. Phys. 13, 7263–7272 (2011).
Alcón, P. et al. FANCD2–FANCI surveys DNA and recognizes double- to single-stranded junctions. Nature 632, 1165–1173 (2024).
Sánchez, H. et al. DNA replication origins retain mobile licensing proteins. Nat. Commun. 12, 1908 (2021).
Wasserman, M. R., Schauer, G. D., O’Donnell, M. E. & Liu, S. Replication fork activation is enabled by a single-stranded DNA gate in CMG helicase. Cell 178, 600–611.e16 (2019).
Smith, S. B., Cui, Y. & Bustamante, C. [7] Optical-trap force transducer that operates by direct measurement of light momentum. In Methods in Enzymology, Vol. 361 (eds. Marriott, G. & Parker, I.) 134–162 (Academic Press, 2003).
Wanrooij, S., Goffart, S., Pohjoismäki, J. L. O., Yasukawa, T. & Spelbrink, J. N. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 35, 3238–3251 (2007).
Sun, B. et al. ATP-induced helicase slippage reveals highly coordinated subunits. Nature 478, 132–135 (2011).
Yuan, Z. et al. DNA unwinding mechanism of a eukaryotic replicative CMG helicase. Nat. Commun. 11, 688 (2020).
Spinks, R. R., Spenkelink, L. M., Dixon, N. E. & van Oijen, A. M. Single-molecule insights into the dynamics of replicative helicases. Front. Mol. Biosci. 8, 741718 (2021).
Bocanegra, R., Plaza, G. I., Pulido, C. R. & Ibarra, B. DNA replication machinery: insights from in vitro single-molecule approaches. Comput. Struct. Biotechnol. J. 19, 2057 (2021).
Schlierf, M., Wang, G., Chen, X. S. & Ha, T. Hexameric helicase G40P unwinds DNA in single base pair steps. eLife 8, e42001 (2019).
Cerrón, F. et al. Replicative DNA polymerases promote active displacement of SSB proteins during lagging strand synthesis. Nucleic Acids Res. 47, 5723–5734 (2019).
Kennedy, H. J. et al. Glucose generates sub-plasma membrane ATP microdomains in single islet β-cells: POTENTIAL ROLE FOR STRATEGICALLY LOCATED MITOCHONDRIA. J. Biol. Chem. 274, 13281–13291 (1999).
Gajewski, C. D., Yang, L., Schon, E. A. & Manfredi, G. New insights into the bioenergetics of mitochondrial disorders using intracellular ATP reporters. Mol. Biol. Cell 14, 3628–3635 (2003).
Ribeck, N., Kaplan, D. L., Bruck, I. & Saleh, O. A. DnaB helicase activity is modulated by DNA geometry and force. Biophys. J. 99, 2170–2179 (2010).
Ribeck, N. & Saleh, O. A. DNA unwinding by ring-shaped T4 helicase gp41 is hindered by tension on the occluded strand. PLoS ONE 8, e79237 (2013).
Manosas, M., Spiering, M. M., Ding, F., Croquette, V. & Benkovic, S. J. Collaborative coupling between polymerase and helicase for leading-strand synthesis. Nucleic Acids Res. 40, 6187–6198 (2012).
Johnson, D. S., Bai, L., Smith, B. Y., Patel, S. S. & Wang, M. D. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell 129, 1299–1309 (2007).
Yang, C., Curth, U., Urbanke, C. & Kang, C. Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 Å resolution. Nat. Struct. Biol. 4, 153–157 (1997).
Qian, Y. & Johnson, K. A. The human mitochondrial single-stranded DNA-binding protein displays distinct kinetics and thermodynamics of DNA binding and exchange. J. Biol. Chem. 292, 13068–13084 (2017).
Curth, U. et al. Single-stranded-DNA-binding proteins from human mitochondria and Escherichia coli have analogous physicochemical properties. Eur. J. Biochem. 221, 435–443 (1994).
Suksombat, S., Khafizov, R., Kozlov, A. G., Lohman, T. M. & Chemla, Y. R. Structural dynamics of E. coli single-stranded DNA binding protein reveal DNA wrapping and unwrapping pathways. eLife 4, e08193 (2015).
Morin, J. A. et al. DNA synthesis determines the binding mode of the human mitochondrial single-stranded DNA-binding protein. Nucleic Acids Res. 45, 7237–7248 (2017).
O’Shea, V. L. & Berger, J. M. Loading strategies of ring-shaped nucleic acid translocases and helicases. Curr. Opin. Struct. Biol. 25, 16–24 (2014).
Bleichert, F., Botchan, M. R. & Berger, J. M. Mechanisms for initiating cellular DNA replication. Science 355, eaah6317 (2017).
Lee, S.-J. & Richardson, C. C. Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 15, 580–586 (2011).
Joo, S., Chung, B. H., Lee, M. & Ha, T. H. Ring-shaped replicative helicase encircles double-stranded DNA during unwinding. Nucleic Acids Res. 47, 11344–11354 (2019).
Burnham, D. R., Kose, H. B., Hoyle, R. B. & Yardimci, H. The mechanism of DNA unwinding by the eukaryotic replicative helicase. Nat. Commun. 10, 2159 (2019).
Richards, J. et al. Structure of the dna repair helicase hel308 reveals dna binding and autoinhibitory domains. J. Biol. Chem. 283, 5118–5126 (2008).
García-Nafría, J., Watson, J. F. & Greger, I. H. IVA cloning: a single-tube universal cloning system exploiting bacterial In Vivo Assembly. Sci. Rep. 6, 27459 (2016).
Fernández-Moreno, M. A., Farr, C. L., Kaguni, L. S. & Garesse, R. Drosophila melanogaster as a Model system to study mitochondrial biology. in Mitochondria: Practical Protocols (eds. Leister, D. & Herrmann, J. M.) 33–49 https://doi.org/10.1007/978-1-59745-365-3_3 (Humana Press, Totowa, NJ, 2007).
Ciesielski, G. L. et al. Mitochondrial single-stranded DNA-binding proteins stimulate the activity of DNA polymerase γ by organization of the template DNA. J. Biol. Chem. 290, 28697–28707 (2015).
Aicart-Ramos, C., Hormeno, S., Wilkinson, O. J., Dillingham, M. S. & Moreno-Herrero, F. Chapter Twelve - Long DNA constructs to study helicases and nucleic acid translocases using optical tweezers. In Methods in Enzymology (ed. Trakselis, M. A.) Vol. 673 311–358 (Academic Press, 2022).
Morin, J. A. et al. Active DNA unwinding dynamics during processive DNA replication. Proc. Natl. Acad. Sci. USA 109, 8115–8120 (2012).
Morin, J. A. et al. Manipulation of single polymerase-DNA complexes: a mechanical view of DNA unwinding during replication. Cell Cycle Georget. Tex 11, 2967–2968 (2012).
Berglund, A. J., McMahon, M. D., McClelland, J. J. & Liddle, J. A. Fast, bias-free algorithm for tracking single particles with variable size and shape. Opt. Express 16, 14064–14075 (2008).
Berglund, A. J. Statistics of camera-based single-particle tracking. Phys. Rev. E 82, 011917 (2010).
Sbalzarini, I. F. & Koumoutsakos, P. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 151, 182–195 (2005).
Michalet, X. & Berglund, A. J. Optimal diffusion coefficient estimation in single-particle tracking. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 85, 061916 (2012).
Plaza-G A, I. et al. Mechanism of strand displacement DNA synthesis by the coordinated activities of human mitochondrial DNA polymerase and SSB. Nucleic Acids Res. 51, 1750–1765 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Acknowledgements
We dedicate this work to Prof. Laurie S. Kaguni, whose friendship, mentorship and enduring passion for science have been a constant source of inspiration. We also thank her generous gift of expression constructs for Twinkle WT and ΔZBD and ΔC-tail variants. We thank Cameron Young for helping with preparation of the figures. We thank the members of our labs for useful discussions. This study was supported by MCIN/AEI/10.13039/501100011033 (grants PGC2018-099341-B-I00 and PID2021-126755NB-I00 to BI; BFU2017- 87316-P and PID2020-120258GB-I00 to R.F.L.; PID2020-112998GB-I00 and PID2023-146255NB-I00 to F.M.H.), ‘la Caixa’ Foundation under the grant agreement HR24-00604 to R.F.L. and B.I. The National Institute of General Medical Sciences of the National Institutes of Health under Award GM139104 to G.L.C.
Author information
Authors and Affiliations
Contributions
G.C. and B.I. conceived and supervised the study. I.P.G.A., M.O.R. and K.M.L. perfomed single-molecules studies and data analysis. S.P.B. and S.M.-A. perfomed biochemical studies. R.F.L. and F.M.H. provided materials and techniques for single-molecule confocal experiments. G.C. and B.I. wrote the manuscript with input from all the authors. All authors were involved in the discussion of the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sjoerd Wanrooij and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Plaza-G A, I., Ortiz-Rodríguez, M., Buchanan, S.P. et al. Autoregulation of the real-time kinetics of the human mitochondrial replicative helicase. Nat Commun 16, 5460 (2025). https://doi.org/10.1038/s41467-025-60289-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60289-0
This article is cited by
-
Diagnosis of mitochondrial defects: livestock diseases
Animal Diseases (2025)
