
Abstract
Combining the chemo- and biocatalytic sites within an integrated catalyst to orchestrate complex, multi-step reactions is highly desirable yet remains a significant challenge. Here, we introduce an ionic organic cage platform for such a chemoenzymatic catalyst, achieved by electrostatic complexation of cationic molecular cage-encapsulated Pd clusters (Pd@C-Cage+) and anionic Candida antarctica lipase B. The spatial compartmentalization provided by the cage scaffold averts undesirable coordination interactions between metal and enzyme, while also facilitating substrate channelling between dual active sites in the one-pot tandem dynamic kinetic resolution of amines, resulting in 2.1-2.7 folds enhancement in product yield within the same reaction time, compared to the physical mixture of individual analogues, and even one order of magnitude higher than the mixture of commercial immobilized lipase Novozym 435 and Pd/C. Additionally, the well-defined pore aperture and charged cage skeleton enables precise microenvironment engineering of confined metal sites, providing stringent site/shape selectivity towards substrates featuring different substituents and sizes. This platform is further demonstrated by integrating other metal clusters (e.g., Ru) and enzymes (e.g., Candida antarctica lipase A, Thermomyces lanuginosus lipase, and Glucose oxidase) for a variety of chemoenzymatic reactions, with 2.1–5.3 folds enhancement compared to the physical mixture of individual analogues.
Similar content being viewed by others


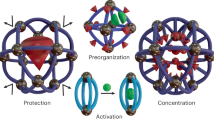
Introduction
Chemoenzymatic catalysts hold great promise in merging the reactivity of chemical catalysts with the selectivity of biological catalysts, offering significant potential for industrial development and organic synthesis1,2. By combining chemical and biological catalysis in a cascade manner, these catalysts hold the ability to substantially reduce reaction time, cost, and waste generation, while overcoming challenges associated with unstable intermediates and ultimately enhancing overall selectivity and yield3,4. Nevertheless, the mismatched reaction parameters (solvent, temperature, etc.) often lead to reduced efficiency or deactivation of one catalyst during the cascade process, thus limiting the full potential of synergistic catalysis. The distinct structural matrix in terms of size, composition, and surface physical chemistry, as well as active-site reactivity, poses challenges in the rationally integrating both catalyst types into single hybrids without compromising their reactivity and stability. In recent years, various strategies have been developed to integrate natural enzymes with synthetic catalysts using porous supports such as mesoporous silica, metal-organic frameworks (MOFs), and porous polymeric matrices5,6,7,8,9. However, challenges such as poor support-enzyme compatibility, limited exposure of active sites, low reactivity/selectivity, and inefficient mass transport still need to be addressed to advance chemoenzymatic catalysis and fully unlock its potential in diverse applications. Of particular concern is the precise control of location of multiple catalytic sites, as the diffusion of chemical catalysts onto the protein surface may result in undesired coordination binding with amino acid residues, thereby compromising catalyst’s activity10,11,12,13,14.
Organic molecular cages, as a class of porous materials with discrete pore architectures, have garnered increasing attention in recent years15,16,17,18,19,20,21,22,23,24. Their precisely tailored molecular structures and inherent cavities bestow them with a wide range of functionalities and applications, including molecular recognition25, guest absorption and separation, etc26,27,28. In particular, the permanent cavities can accommodate chemical catalysts, such as metal clusters (MCs), without obstructing accessibility of substrates to the active sites, thereby enhancing catalytic performance29,30,31,32,33,34,35,36,37,38,39,40. In addition, their easily modifiable architecture allows for functionalization of the cage polyhedrons to create advanced composite catalysts in combination with the hosted MCs, which work synergistically to achieve high-performance cascade reactions41,42,43.
Herein, we report the synthesis of an integrated chemoenzymatic catalyst through a dual-solvent assisted ion-exchange approach. Specifically, by electrostatically coupling a Pd cluster-loaded cationic cage (Pd@C-Cage+) with the anionic Candida antarctica lipase B (CALB−), we successfully fabricate a Pd@C-Cage-CALB catalyst with spatially organized active sites that are compartmentalized by the cage skeleton (Figs. 1 and 2). This unique arrangement effectively prevents undesired mutual quenching or deactivation resulting from coordinated complexation between the catalyst components. The proximity of two active centers facilitates the channelling of the substrates during the catalysis, leading to exceptional activity and recyclability in the one-pot tandem reaction of dynamic kinetic resolution (DKR) of amines, outperforming both the physical mixture of individual analogues and a combination of commercial immobilized lipase Novozym 435 (Supplementary Fig. 1) and Pd/C catalysts under mild conditions. Impressively, the C-Cage+ host can serve as a versatile platform for modulating the microenvironment surrounding the encapsulated Pd cluster based on the following two aspects. 1) The nanosized pore window of molecular cage imparts shape-selective catalytic activity for the amine substrates of different sizes in the initial step of cascade reaction; 2) the ionic electron-deficient nature of quaternary ammonium moiety on the C-Cage+ scaffold allows for precise regulation of the surface electronic state of the encapsulated Pd cluster. This modulation, in turn, exerts a significant influence on the catalytic activity and site selectivity observed in the model catalysis of DKR using the amine-bearing substrates with different substituents. Moreover, the current ionic organic cage can integrate other MCs (Ru) and enzymes (Candida antarctica lipase A (CALA), Thermomyces lanuginosus lipase (TLL), and Glucose oxidase (GOx)) for a broad of chemoenzymatic reactions with enhanced activity compared to the physical mixture of individual analogues. This work exemplifies the potential of customizable molecular cages in tailoring optimized chemoenzymatic catalysts, showcasing the integration of enzymatic and heterogeneous catalysis for enhanced efficiency and selectivity.

Electrostatic complexation between Pd@C-Cage+ and CALB−, along with the engineered substrate channel, facilitates efficient intermediate transfer and enhances the tandem catalytic process.

Intense agitation of an aqueous CALB-TBA solution and a DCM Pd@C-Cage-TFSI solution leads to a rapid combination of hydrophobic TBA+ and TFSI− species in the oil phase. Simultaneously, the cationic Pd@C-Cage+ component dissociates from the oil phase, merging with CALB− in the aqueous phase to maintain the charge balance. This results in the formation of the integrated chemoenzymatic catalyst (Pd@C-Cage-CALB) through electrostatic complexation.
Results
Pd@C-Cage-CALB construction and structural characterization
First, a cationic cage (C-Cage+) was selected as the host material to accommodate Pd clusters, acting as a chemical catalyst. The C-Cage+ was synthesized through the HCl-acidification of its neutral amine cage precursor (A-Cage) (Supplementary Figs. 2–5). It exhibits excellent chemical stability, and is characterized by its easily attainable synthesis method, with readily accessible Cl− serving as the counteranion. Zeta potential analysis revealed that the aqueous solution of C-Cage+ possesses a value of +32.6 mV (Fig. 3a). The encapsulation of Pd cluster followed our previous method42. Transmission electron microscopy (TEM) and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) confirmed the uniform dispersion of Pd clusters with an average diameter of 0.7 ± 0.1 nm, closely matching the cavity size of C-Cage+ (diameter: 0.72 nm) (Fig. 3b). To eliminate the possibility of Pd clusters being stabilized by multiple cages, two-dimensional diffusion-ordered spectroscopy (2D DOSY) NMR experiments were conducted on both C-Cage+ and Pd@C-Cage+ under identical conditions. The results reveal very similar diffusion coefficients of 1.81 × 10−10 m2 s−1 for C-Cage+ and 1.86 × 10−10 m² s−1 for Pd@C-Cage+. These findings indicate that the encapsulation process does not significantly alter the size or shape of the cage (see detailed analysis in Supplementary Figs. 6 and 7).

a Zeta potential histograms of C-Cage+ (red) and CALB− (blue). The error bar states the standard error based on three experimental trials. b HAADF-STEM images of Pd@C-Cage-CALB and the size distribution histograms (each experiment was repeated independently three times, yielding similar results. Data were calculated from 30 counts). c FT-IR spectra of CALB (blue), Pd@C-Cage+ (green), and Pd@C-Cage-CALB (red). d XPS spectrum of Pd@C-Cage-CALB confirming the presence of Pd from Pd@C-Cage+ and the O from CALB−. e SEM images and corresponding elemental mapping of Pd@C-Cage-CALB obtained by freeze-drying. f Molecular dynamics (MD) simulation illustrating the intimate interaction between Pd@C-Cage+ and CALB− after 8 ns (each experiment was repeated independently three times, yielding similar results).
The CALB protein was used as the enzymatic catalyst in current work. It is worth noting that CALB carries a negative charge when the pH exceeds its isoelectric point (pI = 6). Conversely, when the pH falls below the isoelectric point, the protein acquires a positive charge44,45,46. Consequently, electrostatic complexation will occur when the environmental pH surpasses the isoelectric point of CALB, facilitating the interaction with the positively charged Pd@C-Cage+. However, it should be emphasized that direct mixing of Pd@C-Cage+ with CALB− in aqueous solution does not guarantee quantitative complexation between the Pd@C-Cage+ and CALB−. To enable the effective combination of the two catalysts, a dual-solvent assisted ion-exchange approach was developed, as illustrated in Fig. 2. Initially, the Pd@C-Cage-Cl was dissolved in water and subjected to an exchange reaction with 1.2 equivalents of bis(trifluoromethane)sulfonimide lithium (LiTFSI). This process resulted in the formation of a white precipitate (Pd@C-Cage-TFSI), which could be readily dissolved in the oil phase (dichloromethane, DCM). In the case of the lipase, the pH of the system was adjusted to a value higher than the isoelectric point by the addition of tetrabutylammonium bromide (TBAB). As a result, a negatively charged CALB− species equipped with TBA+ counterions was formed. Zeta potential analysis confirmed that the CALB− solution possesses a charge of −16.3 mV (Fig. 3a). Next, the DCM solution of Pd@C-Cage-TFSI was mixed with the aqueous solution of CALB-TBA. Vigorous agitation promoted the association of the hydrophobic groups TFSI− and TBA+ within the organic phase, resulting in the formation of a white precipitate referred to as TBA-TFSI. Concurrently the cationic Pd@C-Cage+ component dissociated from the oil phase and entered the aqueous phase, thereby maintaining charge balance. Consequently, the integrated chemoenzymatic catalyst (Pd@C-Cage-CALB) was generated through the electrostatic complexation.
The obtained Pd@C-Cage-CALB sample was further isolated from aqueous solution by a freeze-drying process for characterizations. Fourier-transform infrared (FT-IR) spectra revealed the typical spectrum of CALB proteins with absorption bands associated with amide groups47,48, featuring characteristic peaks of amide I and amide II at 1641 and 1540 cm−1 in the spectrum of the Pd@C-Cage-CALB. Furthermore, the characteristic bands of C-Cage+ at 1454 cm–1 attributed to the vibrations of benzene rings were present in the Pd@C-Cage-CALB. Additionally, the vibrations of N-H for both C-Cage (3347 cm–1) and CALB (3280 cm–1) merged together within Pd@C-Cage-CALB. These observations confirmed the successful formation of the electrostatic complex of Pd@C-Cage-CALB (Fig. 3c). This finding is consistent with the corresponding elements observed in the X-ray photoelectron spectroscopy (XPS) test (Fig. 3d). Scanning electron microscopy (SEM) revealed that the freeze-dried composite consists of multiple lamellar structures. Elemental mapping analysis further showed the uniform distribution of all elements from both components throughout the Pd@C-Cage-CALB (Fig. 3e). Inductively coupled plasma (ICP) analysis revealed a Pd content of 0.49%. Additionally, UV-Vis spectroscopy was employed for quantitative analysis of the components of cationic cage (Pd@C-Cage+) and anionic protein (CALB−) in the chemoenzymatic catalyst, yielding a corresponding molar ratio of ~ 4:1 (see detailed analysis in Supplementary Fig. 8).
Molecular dynamics (MD) simulations were employed to further demonstrate the electrostatic interaction between Pd@C-Cage+ and CALB− (Fig. 3f). The results revealed the formation of a negative electrostatic field surrounding the protein surface, originating from its negative charge (Supplementary Fig. 9). As the cationic Pd@C-Cage+ approached, it adsorbed onto the protein surface driven by the electrostatic attraction (Movie S1 and Supplementary Figs. 10 and 11). In contrast, when the protein carries positive charges, the two positively charged entities experienced repulsion and moved apart, resulting in larger average distance between their catalytic centers (Movie S2 and Supplementary Fig. 12)49.
Catalytic activity of Pd cluster in Pd@C-Cage-CALB catalyst
After the successful assembly of the integrated catalyst, the catalytic properties of Pd cluster in Pd@C-Cage-CALB were evaluated in the racemization of (S)−1-phenylethylamine ((S)−1-PEA). For comparison, a commercially available Pd/C catalyst was used as a reference (Supplementary Fig. 13). Notably, Pd@C-Cage-CALB exhibited significantly enhanced catalytic activity compared to Pd/C over a wide temperature range. In particular, the conversion within 0.5 h of reaction increased rapidly and reached a maximum value with Pd@C-Cage-CALB at 60 °C (the optimum temperature for lipase, see below). This value is more than 95 times higher than that achieved with commercial Pd/C (Supplementary Fig. 14).
To comprehend the intrinsic high activity of the Pd clusters encapsulated within the C-Cage+, we conducted Density Functional Theory (DFT) calculations to unveil the Bader charge, a metric that reveals the surface electronic state of the Pd clusters influenced by interfacial interaction with cage host50. Based on the optimized geometries of typical Pd13 model within the C-Cage+, it was observed that 2.26 electrons were transferred from the Pd cluster to the host cage (Fig. 4a). The electron density distribution (EDD) profile revealed that the electron transfers mainly occurred at the region between Pd clusters and linkers. Specifically, there was an accumulation of electron density on the cage host, while a reduction of electron density was observed at the Pd clusters. As a result, the Pd exhibited a positive charge, which was consistent with the Bader charge analysis (Fig. 4b). For comparison, DFT calculations were also performed on Pd13 within the neutral A-Cage, which featured a fixed cage skeleton but different functional amine groups on the skeleton (Fig. 4c). It should be noted that the Pd@A-Cage can be easily prepared by neutralized C-Cage+ component to the A-Cage via adding a stoichiometric amount of NaOH (12 equiv to C-Cage+), as confirmed by 1H NMR spectroscopy (Supplementary Fig. 15). In this case, 1.80 electrons were transferred from the Pd cluster to the host cage (Fig. 4a). This distinct behavior was directly related to the electron-donating effect of functional amine/ammonium groups on the cage skeleton to the inner Pd clusters. The degree of electron donation followed the order of secondary amine > quaternary ammonium, which provided a compelling explanation (charge compensation from the functional groups to Pd clusters) for the observed reversed order of electron transfer numbers in the calculations51,52. Moreover, the catalytic activity towards the racemization of (S)−1-PEA over Pd@A-Cage catalyst was lower than that of Pd@C-Cage+ (Supplementary Fig. 16). These results indicated that the microenvironment discrepancy around Pd cluster played a significant role in their discriminated activities. Moreover, this modification of electronic state of Pd resulted in a difference in the binding energy of Pd 3 d peaks in the XPS spectra (Fig. 4d), with an apparent 3d5/2 peak shifting from 335.8 eV (Pd@A-Cage) to 336.1 eV (Pd@C-Cage+) (both are higher than the zero-valent pure Pd NPs in the metallic state at 335.6 eV53, (Fig. 4d). Indeed, the CO adsorption spectra collected on attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) provided additional insights. A noticeable redshift to 2048 cm−1 for Pd@C-Cage+ in comparison to Pd@A-Cage with a peak at 2037 cm−1 was observed. This shift was associated with the reinforcement of the C-O bond and concurrent weakening of the Pd-CO bond, attributed to reduced electron donation from occupied d-states on the Pd cluster (Supplementary Fig. 17)54.

a Quantitative analysis of electron transfers determined using Electron Density Difference (EDD). b Diagram illustration (top) and EDD analysis (down) confirming the substantial charge transfer from Pd clusters to the C-Cage+ host. c Diagram illustration (top) and EDD analysis (down) confirming the relatively smaller charge transfer from Pd cluster to A-Cage host (the blue color indicates the reduction of electrons, and the red color indicates the accumulation of electrons; the image provides a frontal view of the cage matrix, with part of the Pd cluster surface covered by transferred electrons and thus not shown.) d The Pd 3 d XPS spectra of neutral Pd NPs (the synthetic procedure is detailed in Section S1.15), Pd@C-Cage+ and Pd@A-Cage exhibit the peak shifts, indicating changes in the surface electronic structure. e Theoretical models revealing the high activity of Pd in Pd@C-Cage+. A comparison of free-energy profiles for the (S)−1-PEA racemization on distinct Pd surfaces.
Previous studies have established the involvement of an imine intermediate in the Pd-catalyzed racemization of (S)−1-PEA55. Building upon this knowledge, a reaction pathway was investigated, wherein (S)−1-PEA is initially adsorbed on the positively charged Pd surface, followed by transformation into the imine intermediate, and eventually recovery to (R)- or (S)−1-PEA (Fig. 4e). The calculated free-energy profiles revealed that both the positively charged Pd surface exhibited higher catalytic activity compared to that of neutral Pd. In particular, the Pd@C-Cage+ catalyst showed a lower adsorption energy (PEA → PEA*) with a reduction of 3.2 eV for Pd@C-Cage+, and a lower dehydrogenation/hydrogenation reaction energy (PEA*→intermediate→PEA*) with a decrease of by 1.38 eV for Pd@C-Cage+ was achieved, compared to the neutral Pd surface. In summary, the microenvironment engineering of the cage skeleton, which leads to the positive charge on the Pd surface, plays a crucial role in promoting catalytic efficiency of (S)−1-PEA racemization at mild reaction conditions.
Catalytic activity of CALB enzyme in Pd@C-Cage-CALB catalyst
After successfully elucidating the catalytic activity of Pd cluster within the Pd@C-Cage-CALB catalyst, we conducted further investigations to assess its influence on the catalytic activity of CALB. Importantly, the incorporation of the Pd cluster within C-Cage+ cavity did not induce any noticeable changes in the secondary and tertiary structure of CALB, as confirmed by FT-IR and fluorescence spectroscopy (see details in Supplementary Figs. 18 and 19)56,57,58. Moreover, the relative activities of lipase in Pd@C-Cage-CALB in aqueous solution, determined using the standard method with p-nitrophenyl butyrate (p-NPB) as the substrate at 25 °C, were comparable to the activity of native CALB at the same protein amount (Supplementary Fig. 20). In the kinetic resolution of (±)-1-PEA in toluene at 60 °C (the optimum temperature of lipase) (Supplementary Fig. 21), Pd@C-Cage-CALB displayed an approximately 11.5 and 2.1-fold higher apparent activity compared to free CALB and commercial immobilized lipase Novozym 435, respectively, with the same amount of CALB as in Pd@C-Cage-CALB. The notable enhancement in CALB activity within Pd@C-Cage-CALB in toluene can be reasonably attributed to the highly dispersed form of catalyst, facilitated by the presence of C-Cage+, as free CALB tends to aggregate in organic solvents (Supplementary Fig. 22). The Novozym 435 is a well-established immobilized lipase known for its high activity and stability in organic solvents. However, at temperatures ranging from 30 to 70 °C, the highly dispersed Pd@C-Cage-CALB exhibited approximately 1.5-2.1 folds higher activity than Novozym 435 (Supplementary Fig. 21).
One-pot tandem DKR reactions catalyzed by Pd@C-Cage-CALB
At the optimum temperature of 60 °C, as shown in Table 1 entries 1-4, the Pd@C-Cage-CALB catalyst exhibited exceptional catalytic efficiency in the coupled reaction of Pd-catalyzed racemization of (S)−1-PEA and lipase-catalyzed acylation of (R)−1-PEA to produce 2-methoxy-N-[(1 R)−1-phenylethyl]acetamide. The performance of Pd@C-Cage-CALB was compared to the combination of Novozym 435 and Pd/C at the same temperature. Within 2.5 hours, the Pd@C-Cage-CALB catalyst achieved nearly 100% conversion and an enantiomeric excess (e.e.) value of over 99% at a substrate concentration of 0.067 M (Table 1 entry 4). In contrast, the combination of Novozym 435 and Pd/C only achieved a conversion rate of ~9.02% under the same conditions, with the specific activity of CALB being much lower (Table 1 entry 5). In previous investigations of the DKR of amines, obtaining over 90% conversion required more than 4 hours at temperatures above 60 °C under a H2 atmosphere59,60,61. To reach 99% conversion, it usually took more than 12 h (Table S1). However, due to the high activity of Pd clusters, combined with the spatially separated metal and protein sites within the integrated Pd@C-Cage-CALB, the conversion rate exceeded 99% within just 2.5 h at a temperature of 60 °C. The calculated the total turnover number (TTN) of CALB enzyme within in the Pd@C-Cage-CALB catalyst reached a high value of 5033 mol mol−1, surpassing those of reported Pd-CALB chemoenzymatic catalysts in the literature (Table S1).
The impressive enhancement in catalytic activity observed in the assembled nanocomposite catalyst brings to mind the concept of substrate channelling commonly observed in natural enzyme-catalyzed sequential reactions62. In such reactions, an intermediate generated by one enzyme can be directly transferred to a neighboring enzyme without diffusing through the bulk environment. This substrate channelling effect accelerates substrate transportation and minimizes side reactions, thereby promoting catalytic efficiency and enhancing product yields in multistep reactions. In the present catalyst, the dual catalytic sites (Pd cluster and CALB) are compartmentalized by the organic cage, which would facilitate intermediate diffusion and improve reaction efficiencies in the cascade catalysis process. To assess this effect, a control experiment was conducted using a physical mixture of Pd@C-Cage+ and free CALB catalysts, referred to as Pd@C-Cage+ + CALB (the counteranion for Pd@C-Cage+ is Cl−), for the same reaction (Note: the two catalytic active sites are randomly dispersed in the solution due to the lack of electrostatic attraction, see Movie S3 and Supplementary Fig. 23). Clearly, a significantly reduced catalytic efficiency with only approximately 37.69% conversion was achieved under the same conditions (Table 1 entry 6). In addition, to avoid the low dispersibility of free CALB in toluene from influencing mass diffusion, we further performed another control experiment using C-Cage-CALB instead to physically mix with Pd@C-Cage+, referred to as Pd@C-Cage+ + C-Cage-CALB, for the above reaction (Note: the amounts of Pd and CALB are the same as those in the integrated Pd@C-Cage-CALB catalyst). In this case, only approximately 48.49% conversion was achieved under the same conditions (Table 1 entry 7).
To further highlight the advantage of the current integrated system in tandem reactions, the cascade was then challenged by adding an inhibitor to the second enzyme. Previous studies have established that the impact of such challenges is minimal in cases of high substrate channelling. Conversely, in the absence of channelling, the cascade activity experiences a significant reduction, given that the inhibitor hinders a crucial reaction step63,64. Here, we introduced an enzyme inhibitor (methanol, acting on CALB) initially65, deactivating CALB and thereby disrupting the second step of the cascade reaction (Fig. 5a). As shown in Fig. 5b, when the cascade reaction was catalyzed by a physical mixture of Pd@C-Cage+ and CALB catalysts or a physical mixture of Pd@C-Cage+ + C-Cage-CALB catalysts in solution, a notably lower residual activity, below 1%, was observed for both catalysts at a high inhibitor content (60 μL of methanol). However, the channelling phenomenon in the integrated cascade reaction facilitated by the Pd@C-Cage-CALB catalyst may mitigate the inhibitory effect of inhibitors on the catalysts. This is achieved by reducing the diffusion distance of intermediates to the second active sites, resulting in an approximate 72% maintenance of residual activity at the same inhibitor content. Indeed, the shorter diffusion path in the integrated Pd@C-Cage-CALB catalyst is conducive to the efficient conversion of intermediates into the final products, as also demonstrated by a comparison of reaction kinetics with a physical mixture of Pd@C-Cage+ and CALB catalysts and Pd@C-Cage+ + C-Cage-CALB catalysts under the same conditions (Fig. 5c, d and Supplementary Figs. 24 and 25). Therefore, the enhanced activity observed within the current system can be attributed to channelling the substrate to proximal dual catalysts compartmentalized by C-Cage+. This drastically enhances the overall activity in comparison to their freely dispersed analogues in the solution.

a Diagram of a one-pot tandem DKR reaction showing the impact of an enzyme inhibitor on the key steps of the catalytic reaction. b Relative residual activity for the one-pot tandem DKR cascade reaction with different methanol contents by the integrated Pd@C-Cage-CALB catalyst, the physical mixture of Pd@C-Cage+ + CALB catalysts and Pd@C-Cage+ + C-Cage-CALB catalysts in the solution. The methanol concentration ranges from 0 to 3.23% v/v (Vmethanol/Vtotal solvents). c Schematic illustration of the substrate channel within the integrated Pd@C-Cage-CALB catalyst, facilitating enhanced mass transfer in cascade reaction. d Kinetic curve depicting the contents of starting reagent (blue), intermediate product (yellow) and final product (red) over reaction time in the presence of the Pd@C-Cage-CALB catalyst. The error bar states the standard error based on three experimental trials. Reaction condition: 0.2 mmol of (S)−1-PEA, 3 mL of toluene, and 1 atm H2 at 60 °C, using 3 mg of Pd@C-Cage-CALB (1.31 mg of CALB).
To further investigate the location of Pd nanoparticles on the cage support and their impact on catalytic performance, we prepared different chemoenzymatic catalysts with Pd nanoparticle sizes larger than the cage cavity (i.e. Pd nanoparticles immobilized on the surface of cage support), referred to as Pd1/C-Cage-CALB (average size: 11.8 ± 1 nm), and Pd2/C-Cage-CALB (average size: 254.3 ± 20 nm) (Supplementary Figs. 26 and 27). The catalytic performance of these catalysts in the DKR reaction was evaluated, and the results were summarized in Table 1 (entries 8 and 9). As the size of the metal nanoparticles increased, the overall catalytic yield gradually decreased. Moreover, in control experiments where Pd nanoparticles were directly immobilized onto the enzyme surface (denoted as Pd/CALB, average size: 6.6 ± 1 nm, Supplementary Fig. 28), complete enzyme deactivation occurred (Table 1 entry 10). These findings suggest that without the compartmentalization of the cage skeleton, the intimate contact between the metal nanoparticles and the enzyme surface likely disturbs the secondary/tertiary structure of CALB, leading to reduced or even quenched catalytic activity. This hypothesis was further compellingly evidenced by FT-IR and FL spectra, which provided insights into the structural changes induced by the close contact of Pd nanoparticles with CALB (Supplementary Figs. 18 and 19).
Furthermore, the Pd@C-Cage-CALB catalyst demonstrated excellent recyclability. After each reaction cycle, the catalyst could be easily recovered by centrifugation and subsequent freeze-dried for further use. Even after six cycles of reuse, the Pd@C-Cage-CALB catalyst maintained over 95% of its initial activity and retained a high enantiomeric excess (e.e.) value exceeding 99% (Supplementary Fig. 29). The Pd clusters embedded within the catalyst remained stable without detachment or aggregation (Supplementary Fig. 30), demonstrating the robustness of the catalyst during the recycling process.
In addition to the development of a highly active integrated catalyst for chiral amine synthesis, our catalyst has demonstrated high selectivity towards substrates of different molecular sizes. Besides (S)-1-PEA (size: 5.18 Å) (the sizes of reactants are summarized in Supplementary Table S2), other amine precursors with sizes ranging from 5.28 to 5.45 Å have shown complete conversion into the corresponding chiral products, achieving high yields and enantiomeric excess (e.e.) values within a short reaction time of 2.5 hours (Table 2 entries 2-6). However, with larger substrates such as (S)-1,2,3,4-tetrahydro-1-naphthalenamine ((S)-1-TNA, size: 6.91 Å) and (S)-1-naphthalen-2-yl-ethylamine ((S)-1-NEA, size: 6.92 Å), no conversion was observed even after an extended reaction time of 6 h (Table 2 entries 7 and 8). This intriguing observation can be attributed to the size selectivity of the cage aperture (size: 5.8 Å), which restricts the diffusion of certain molecules to reach the inner active sites of catalyst66,67,68. MD simulations and guest capture experiments further support this finding, revealing that the cage window does not permit the passage of larger (S)-1-TNA and (S)-1-NEA molecules (Movie S4-S6, Supplementary Figs. 31–39 and Fig. 6a). Interestingly, the selectivity of the current system extends beyond substrate size and encompasses the nature of substituents attached to the substrate. An intriguing observation was made regarding the catalyst’s unresponsiveness towards substrates with electron-withdrawing substituents, such as (S)-1-phenylethylamine with a fluorine substituent ((S)-1-FPA) (Table 2 entry 9). Surprisingly, in this scenario, the molecule size (5.18 Å) was not the determining factor for the lack of reactivity, suggesting that the substituents may impact the adsorption mode of the substrate on the catalyst surface (Fig. 6b)69. To gain further insights, we conducted time-dependent competitive adsorption experiments over ATR-FTIR spectrometer using a mixture of (S)-1-PEA and fluorobenzene (FB) (molar ratio 1:1). An overwhelming adsorption peaks attributed to FB over (S)-1-PEA were observed (Supplementary Fig. 40). Additionally, DFT calculation revealed that the fluorine substituent (-F) carries a larger negative charge (-0.248 e) compared to the amino group (-NH2) −0.064 e) (Supplementary Fig. 41a). According to Coulomb’s law, the larger negative charge of the -F group implies a stronger attraction to the positively charged Pd surface in Pd@C-Cage-CALB with a relatively lower adsorption energy (Supplementary Fig. 41b). This stronger interaction may hinder the progress of the reaction, leading to the observed inertness of substrates ((S)-1-FPA) bearing electron-withdrawing substituents. A parallel low conversion was also observed in the Pd cluster catalyst encapsulated within A-Cage under the same conditions (Supplementary Fig. 42).

a Size selectivity towards the different substrates. The aperture of cage’s window restricts larger substrates ((S)−1-NEA and (S)−1-TNA) from accessing the active site. b Site selectivity toward substrates with different substituents. The presence of -F substituents alters the adsorption mode of the substrate on the catalyst surface. Specifically, the more negatively charged -F group implies a stronger attraction to the positively charged Pd surface, thus hindering the reaction progress.
Versatility of ionic organic cages in enhanced chemoenzymatic catalysis
To further illustrate the versatility of current ionic organic cages as platforms for integrating chemical and enzymatic sites, we prepared three additional chemoenzymatic catalysts: Pd@C-Cage-CALA (CALA = Candida antarctica lipase A), Pd@C-Cage-TLL (TLL = Thermomyces lanuginosus lipase), and Ru@C-Cage-GOx (GOx = Glucose oxidase) (Fig. 7a, b the detailed synthetic procedure can refer to Supplementary Methods). It is noteworthy that all the enzymes can be negatively charged when the pH exceeds their isoelectric point (Fig. 7a and Supplementary Fig. 43), facilitating electrostatically-driven assemblies similar to Pd@C-Cage-CALB (Supplementary Fig. 44). The successful encapsulation of the Ru clusters within cage was characterized by HAADF-STEM, showing an average size of 0.68 ± 0.1 nm (Supplementary Figs. 45–46). The formation of electrostatically complexed composites was confirmed by FT-IR spectra (Supplementary Fig. 47). The quantitative analysis of the components of cationic cages and anionic proteins in the chemoenzymatic catalysts was conducted using the UV-Vis spectroscopy (Supplementary Fig. 44). Importantly, the secondary and tertiary structure of enzymes were maintained in these catalysts, as confirmed by FT-IR and FL spectroscopy (Supplementary Figs. 48–49). The catalysts were then examined in the following reactions, involving 1) Pd-catalyzed racemization of (S)−1-PEOH ((S)−1-phenylethanol) and CALA-catalyzed acylation of (R)−1-PEOH to produce acetic acid (R)-1-phenylethyl ester, 2) Pd-catalyzed benzaldehyde reduction and TLL-catalyzed benzyl alcohol acylation to produce benzyl hexanoate, and 3) GOx -catalyzed the conversion of glucose to produce hydrogen peroxide, which Ru clusters then decomposed to generate ·OH radicals, followed by the oxidization of ABTS (2,2´-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) to ABTS˙+ (detailed experimental results are listed in Supplementary Figs. 50–53). In general, the integrated catalysts exhibited higher activity, with enhancements of 2.1- to 5.3-fold compared to the physical mixture of individual analogues (Fig. 7c). The reaction kinetics for each chemoenzymatic reaction further revealed that the integrated catalysts, with shorter diffusion path, are favorable for the efficient conversion of intermediates into the final products, as compared to the physical mixture of individual analogues (Supplementary Figs. 50–53)70. These results provide solid proof for the adaptability of our strategy of using the ionic organic cage platform in chemoenzymatic tandem catalysis. To verify the substrate channeling effect, we evaluated the tandem reaction over the representative Ru@C-Cage-GOx catalyst and its physical mixture, introducing thiourea as a competitive reagent. In this case, the hydrogen peroxide, serving as the intermediate product, can be consumed either in the oxidation of ABTS or by reacting with thiourea, thereby establishing a competitive reaction system (Supplementary Fig. 54). As shown in Supplementary Figs. 55 and 56, the integrated Ru@C-Cage-GOx catalyst exhibited much higher residual activity compared to the physically mixed catalyst at the same competing agent concentration, further highlighting the channeling effect within the system. To further assess the robustness of the electrostatically complexed catalyst under varying ionic strength conditions (using a NaCl aqueous solution), we conducted catalytic activity assays using the representative Ru@C-Cage-GOx catalyst (Supplementary Fig. 57). Notably, a sharp decline in catalytic conversion efficiency was observed when the NaCl concentration reached approximately 0.3 M. This result indicates that the electrostatically integrated catalyst can maintain its activity under moderate ionic strength conditions.

a The isoelectric point information of the proteins used in this work. b A schematic diagram of the tandem reactions using three different integrated chemoenzymatic catalysts. c The relative catalytic activity of the integrated MCs@C-Cage-enzyme compared to the physical mixture catalyst MCs@C-Cage+ + C-Cage-Enzyme \((R{elative\; Catalytic\; Activity}=\frac{{T}_{\left({physcially}\; {mixed}\; {catalyst}\right)}}{{T}_{\left({integrated\; catalyst}\right)}}\times 1.0)\) T: The time required for the reaction to complete.
Discussion
In conclusion, we have successfully engineered a chemoenzymatic catalyst by electrostatically coupling a molecular cage-encapsulated Pd cluster with Candida antarctica lipase B. The cage scaffold spatially separates the catalytic sites while facilitating substrate transfer between them. This unique approach results in enhanced efficiency for one-pot tandem DKR of amines under mild conditions. Furthermore, the cationic cage, with its well-defined pore aperture and electronically deficient skeleton, provides a versatile platform for precise modulation of microenvironment of the Pd cluster, imparting strict site and shape selectivity toward substrates with different substituents and molecular sizes. The versatility of current strategy is further demonstrated with three additional chemoenzymatic catalysts for enhanced chemoenzymatic tandem catalysis. Given the design adaptability of molecular cages with tailored inner cavities and modifiable skeleton functionality, the current system is expected to expand to integrate a library of metal clusters and enzymes. Overall, this study will inspire further research on precise design and synthesis of advanced chemoenzymatic catalysts for highly efficient and selective substrate conversion via a supramolecular approach.
Methods
Materials
All chemicals were commercially available and can be used without further purification. Methanol, dichloromethane, trichloromethane, toluene, L−1-phenylethylamine, ethyl methoxyacetate, bis(trifluoromethane)sulfonimide lithium salt (TFSI), tetrabutylammonium bromide (TBAB), sodium borohydride, potassium hexachloropalladate, and Glucose oxidase (GOx) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd., Amplex Red (10-Acetyl-3,7-dihydroxyphenoxazine, ADHP, fluorescent Red dye) purchased from Beyotime Biotechnology Technology Co., Ltd., (R,R)−1,2-diaminocyclohexane was purchased from Sigma-Aldrich Co. LLC., 1,3,5-triformylbenzene was purchased from Beijing Kaida Technology Development Co., Ltd. Candida antarctica lipase A (CALA, NovoCor AD L), Candida antarctica lipase B (CALB), and Thermomyces lanuginosus lipase (TLL) were purchased from Novozymes (China) Biotechnology Co., Ltd.
Nuclear magnetic resonance (NMR) spectra
The proton nuclear magnetic resonance (1H NMR) measurements were performed using a Bruker Ascend III HD 400 MHz spectrometer at 298 K. For 2D diffusion-ordered spectroscopy (2D DOSY) 1H NMR experiments (in D2O at 298 K), a Bruker Avance III HD 700 MHz spectrometer was employed.
Zeta potential
The zeta potential of the samples was measured using a Zetasizer Nano ZS (Malvern Instruments, UK) equipped with a laser Doppler electrophoresis system. Prior to the measurement, the samples were dispersed in solutions with the desired pH using ultrasonic treatment to achieve a uniform dispersion. The resulting suspension was then transferred into a folded capillary cell for measurement. All measurements were conducted at room temperature (25 ± 1 °C), and each sample was measured at least three times to ensure reproducibility. The average zeta potential values and standard deviations were reported.
Ultraviolet–visible spectra (UV-Vis)
UV-Vis spectra were recorded using a UV-6100 spectrophotometer in the wavelength range of 190−1100 nm. Prior to measurement, the samples were diluted to an appropriate concentration using the corresponding solvent to ensure that the absorbance values fell within the linear range of the instrument. The measurements were conducted using quartz cuvettes with a 1.0 cm optical path length. A baseline correction was performed using the corresponding pure solvent as a reference. Each spectrum was collected with a scanning speed of 200 nm/min and a spectral resolution of 1 nm. All measurements were carried out at room temperature (25 ± 1 °C).
Fluorescence spectra
A F97 Pro fluorescence spectrophotometer was used with an excitation wavelength of 280 nm. The solution samples were measured in quartz cuvettes with a light path of 1 cm. The testing voltage was 650 V, the width of the excitation and emission slits was 5 nm, the scanning speed was 300 nm/min.
Fourier-transform infrared (FT-IR) spectra
FT-IR spectra were collected on a Thermo Scientific Nicolet iS 50 spectrometer in the wavenumber range of 4000–400 cm−1. The CO and substrate adsorption spectra were recorded on a Bruker ALPHA II attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectrometer. The whole set of devices was built up in our laboratory including a gas control device, an adsorption chamber and the main FTIR analysis apparatus.
X-ray photoelectron spectra (XPS)
XPS analysis was performed using an ESCALAB 250Xi spectrometer (Thermo Fisher Scientific) equipped with a monochromatized Al Kα X-ray source (hv = 1486.6 eV). The measurements were conducted under an ultra-high vacuum (UHV) environment with a base pressure below 1 × 10−9 mbar. Survey spectra were recorded to identify the elemental composition, followed by high-resolution scans of relevant core-level regions. The pass energy was set to 100 eV for survey spectra and 20 eV for high-resolution spectra. Charge compensation was applied using a low-energy electron flood gun. All binding energies were calibrated against the C 1 s peak of neutral carbon at 284.6 eV. The obtained data were analyzed using Thermo Avantage software with Shirley background subtraction and Gaussian-Lorentzian peak fitting.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)
Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were obtained using an FEI Talos F200X microscope (Thermo Fisher Scientific) operated at an accelerating voltage of 200 kV. The samples were dispersed in ethanol and ultrasonicated to achieve a uniform suspension, followed by drop-casting onto carbon-coated copper grids and drying under ambient conditions before analysis. HAADF-STEM imaging was conducted in STEM mode using a high-angle annular dark-field detector to provide Z-contrast imaging.
Field-emission scanning electron microscopy (FE-SEM)
FE-SEM images were collected on FEI Nova NanoSEM with an accelerating voltage of 5 kV.
High performance liquid chromatography (HPLC) and gas chromatography (GC)
The catalytic process was analyzed using an Agilent high-performance liquid chromatography system (LC1260infinity II) equipped with an IA-3 chiral column and the Agilent 8860 gas chromatography (GC) with Agilgent CP-Chirasil Dex CB (df = 0.25 μm, 0.32 mm i.d. × 25 m) chiral column and a J&W HP-5 column. The mobile phase consisted of n-hexane and isopropyl alcohol in an 80:20 ratio, with a flow rate of 1 mL/min, and detection at a wavelength of 254 nm. GC conditions: Carrier gas, N2 (flow 30 mL/min); injection temp, 250 °C; initial column temperature 80 °C, then progress rate, 3 °C/min; final column temperature, 180 °C for 30 min; The Glucose samples were centrifuged at 12840 g for 3 min, and the supernatant was filtered through a 0.22 μm filter for detection. Glucose Concentrations were analyzed using HPLC with an Aminex HPX-87H column (300 × 7.8 mm; Bio-Rad, Hercules) equipped with RID systems. The mobile phase was 5 mmol/L H2SO4, and the elution was carried out at 50 °C with a flow rate of 0.5 mL/min.
Inductively coupled plasma optical emission spectrometer (ICP-OES)
The Pd contents in the catalysts were quantified using an Agilent 5800 ICP-OES instrument.
Density functional theory (DFT) computation
DFT calculations were conducted using the Vienna Ab-initio Simulation Package (VASP), with full computational details provided in the Supplementary Information.
Molecular dynamic (MD) simulation
MD simulations were performed using GROMACS, with the RESP charges for Pd clusters, C-Cage+, and A-Cage derived from Gaussian-based calculations; full simulation protocols and parameterization details are provided in the Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the article, Supplementary Information and from corresponding author(s) upon request. The 1H NMR spectra and chromatography of catalytic products are listed in Figs. S58–S80. The main data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Hönig, M., Sondermann, P., Turner, N. J. & Carreira, E. M. Enantioselective chemo‐and biocatalysis: partners in retrosynthesis. Angew. Chem. Int. Ed. 56, 8942–8973 (2017).
Rudroff, F. et al. Opportunities and challenges for combining chemo-and biocatalysis. Nat. Catal. 1, 12–22 (2018).
González‐Granda, S., Escot, L., Lavandera, I. & Gotor‐Fernández, V. Chemoenzymatic cascades combining biocatalysis and transition metal catalysis for asymmetric synthesis. Angew. Chem. Int. Ed. 62, e202217713 (2023).
Gonzalez-Granda, S., Albarran-Velo, J., Lavandera, I. & Gotor-Fernández, V. Expanding the synthetic toolbox through metal–enzyme cascade reactions. Chem. Rev. 123, 5297–5346 (2023).
Dutta, S. et al. Highly mesoporous metal‐organic frameworks as synergistic multimodal catalytic platforms for divergent cascade reactions. Angew. Chem. Int. Ed. 132, 3444–3450 (2020).
Wang, K.-Y. et al. Bioinspired framework catalysts: from enzyme immobilization to biomimetic catalysis. Chem. Rev. 123, 5347–5420 (2023).
Huang, S., Chen, G. & Ouyang, G. Confining enzymes in porous organic frameworks: from synthetic strategy and characterization to healthcare applications. Chem. Soc. Rev. 51, 6824–6863 (2022).
Man, T. et al. Hierarchically encapsulating enzymes with multi-shelled metal-organic frameworks for tandem biocatalytic reactions. Nat. Commun. 13, 305 (2022).
Wang, X., Lan, P. C. & Ma, S. Metal–organic frameworks for enzyme immobilization: beyond host matrix materials. ACS Cent. Sci. 6, 1497–1506 (2020).
Shi, J. et al. Bioinspired construction of multi-enzyme catalytic systems. Chem. Soc. Rev. 47, 4295–4313 (2018).
Li, X., Cao, X., Xiong, J. & Ge, J. Enzyme–metal hybrid catalysts for chemoenzymatic reactions. Small 16, 1902751 (2020).
Pamies, O., Dieguez, M. & Bäckvall, J.-E. Artificial metalloenzymes in asymmetric catalysis: key developments and future directions. Adv. Synth. Catal. 357, 1567–1586 (2015).
Nicolaou, K. C. & Chen, J. S. The art of total synthesis through cascade reactions. Chem. Soc. Rev. 38, 2993–3009 (2009).
Denard, C. A. et al. Cooperative tandem catalysis by an organometallic complex and a metalloenzyme. Angew. Chem. Int. Ed. 53, 465–469 (2014).
Swamy, S. I. et al. A metal− organic framework with a covalently prefabricated porous organic linker. J. Am. Chem. Soc. 132, 12773–12775 (2010).
Mukhopadhyay, R. D., Kim, Y., Koo, J. & Kim, K. Porphyrin boxes. Acc. Chem. Res. 51, 2730–2738 (2018).
Zhang, G., Presly, O., White, F., Oppel, I. M. & Mastalerz, M. A permanent mesoporous organic cage with an exceptionally high surface area. Angew. Chem. Int. Ed. 53, 1516–1520 (2014).
Lavendomme, R., Ronson, T. K. & Nitschke, J. R. Metal and organic templates together control the size of covalent macrocycles and cages. J. Am. Chem. Soc. 141, 12147–12158 (2019).
Beuerle, F. & Gole, B. Covalent organic frameworks and cage compounds: design and applications of polymeric and discrete organic scaffolds. Angew. Chem. Int. Ed. 57, 4850–4878 (2018).
Yang, X., Ullah, Z., Stoddart, J. F. & Yavuz, C. T. Porous organic cages. Chem. Rev. 123, 4602–4634 (2023).
Wang, H. et al. Constraining homo-and heteroanion dimers in ultraclose proximity within a self-assembled hexacationic cage. J. Am. Chem. Soc. 142, 20182–20190 (2020).
He, C. et al. A porous metal-organic cage liquid for sustainable CO2 conversion reactions. Nat. Commun. 14, 3317 (2023).
Cao, W. et al. Ionic organic cage-encapsulated metal clusters for switchable catalysis. Cell Rep. Phys. Sci. 2, 9 (2021).
Yang, J., Hu, S.-J., Cai, L.-X., Zhou, L.-P. & Sun, Q.-F. Counteranion-mediated efficient iodine capture in a hexacationic imidazolium organic cage enabled by multiple non-covalent interactions. Nat. Commun. 14, 6082 (2023).
Wang, Z. et al. Networked cages for enhanced CO2 capture and sensing. Adv. Sci. 5, 1800141 (2018).
He, A. et al. A smart and responsive crystalline porous organic cage membrane with switchable pore apertures for graded molecular sieving. Nat. Mater. 21, 463–470 (2022).
Li, X. et al. Polycage membranes for precise molecular separation and catalysis. Nat. Commun. 14, 3112 (2023).
Liu, S.-H. et al. Sub-8 nm networked cage nanofilm with tunable nanofluidic channels for adaptive sieving. Nat. Commun. 15, 2478 (2024).
Sun, J.-K., Zhan, W.-W., Akita, T. & Xu, Q. Toward homogenization of heterogeneous metal nanoparticle catalysts with enhanced catalytic performance: soluble porous organic cage as a stabilizer and homogenizer. J. Am. Chem. Soc. 137, 7063–7066 (2015).
Sun, N. et al. Multifunctional Tubular Organic Cage‐Supported Ultrafine Palladium Nanoparticles for Sequential Catalysis. Angew. Chem. Int. Ed. 58, 18011–18016 (2019).
Yang, X., Sun, J.-K., Kitta, M., Pang, H. & Xu, Q. Encapsulating highly catalytically active metal nanoclusters inside porous organic cages. Nat. Catal. 1, 214–220 (2018).
Gou, X.-X., Liu, T., Wang, Y.-Y. & Han, Y.-F. Ultrastable and Highly Catalytically Active N‐Heterocyclic‐Carbene‐Stabilized Gold Nanoparticles in Confined Spaces. Angew. Chem. Int. Ed. 59, 16683–16689 (2020).
Zhang, S.-Y. et al. Ionic organic cage-encapsulating phase-transferable metal clusters. Chem. Sci. 10, 1450–1456 (2019).
Hua, M. et al. Hierarchically porous organic cages. Angew. Chem. Int. Ed. 133, 12598–12605 (2021).
Koo, J. et al. Gigantic porphyrinic cages. Chem 6, 3374–3384 (2020).
Xu, N., Su, K., El-Sayed, E.-S. M., Ju, Z. & Yuan, D. Chiral proline-substituted porous organic cages in asymmetric organocatalysis. Chem. Sci. 13, 3582–3588 (2022).
Luo, N., Ao, Y.-F., Wang, D.-X. & Wang, Q.-Q. Exploiting anion–π interactions for efficient and selective catalysis with chiral molecular cages. Angew. Chem. Int. Ed. 60, 20650–20655 (2021).
Zhang, S.-Y. et al. Accelerating crystallization of open organic materials by poly (ionic liquid) s. Angew. Chem. Int. Ed. 59, 22109–22116 (2020).
Lu, J.-N. et al. Synergistic Metal‐Nonmetal Active Sites in a Metal‐Organic Cage for Efficient Photocatalytic. Synthesis of Hydrogen Peroxide in Pure Water. Angew. Chem. Int. Ed 62, e202308505 (2023).
Yan, D.-N. et al. An Organo‐Palladium Host Built from a Dynamic Macrocyclic Ligand: Adaptive Self‐Assembly. Induced‐Fit Guest Binding, and Catalysis. Angew. Chem. Int. Ed 61, e202209879 (2022).
Li, J.-Y., Yang, X.-D., Chen, F.-X. & Sun, J.-K. The marriage of porous cages and metal clusters for advanced catalysis. Mater. Chem. Front. 7, 5355–5376 (2023).
Tan, L., Zhou, J.-H., Sun, J.-K. & Yuan, J. Electrostatically cooperative host-in-host of metal cluster⊂ ionic organic cages in nanopores for enhanced catalysis. Nat. Commun. 13, 1471 (2022).
Yang, X.-D., Zhang, Y.-J., Zhou, J.-H., Liu, L. & Sun, J.-K. Air‐Stable Radical Organic Cages as Cascade Nanozymes for Enhanced Catalysis. Small 19, 2206127 (2023).
Tanasković, S. J. et al. Immobilization of Candida antarctica lipase B on kaolin and its application in synthesis of lipophilic antioxidants. Appl. Clay Sci. 135, 103–111 (2017).
Schreiber, S. et al. New application of depth filters for the immobilization of Candida antarctica lipase B. Appl. Microbiol. Biotechnol. 101, 599–607 (2017).
Du, P., Xu, S., Xu, Z.-K. & Wang, Z.-G. Bioinspired Self‐Assembling Materials for Modulating Enzyme Functions. Adv. Funct. Mater. 31, 2104819 (2021).
Foresti, M. L., Valle, G., Bonetto, R., Ferreira, M. L. & Briand, L. E. FTIR, SEM and fractal dimension characterization of lipase B from Candida antarctica immobilized onto titania at selected conditions. Appl. Surf. Sci. 256, 1624–1635 (2010).
Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta, Bioenerg. 1767, 1073–1101 (2007).
Rindfleisch, S. et al. Ground-state destabilization by electrostatic repulsion is not a driving force in orotidine-5′-monophosphate decarboxylase catalysis. Nat. Catal. 5, 332–341 (2022).
Chen, D. et al. Boosting catalysis of Pd nanoparticles in MOFs by pore wall engineering: the roles of electron transfer and adsorption energy. Adv. Mater. 32, 2000041 (2020).
Esnal, I. et al. Reaction of amines with 8‐MethylthioBODIPY: Dramatic optical and laser response to amine substitution. Chem. - Asian J 8, 2691–2700 (2013).
Liu, Q. et al. Electron-donating amine-interlayer induced n-type doping of polymer: nonfullerene blends for efficient narrowband near-infrared photo-detection. Nat. Commun. 13, 5194 (2022).
Kibis, L. S., Stadnichenko, A. I., Koscheev, S. V., Zaikovskii, V. I. & Boronin, A. I. Highly oxidized palladium nanoparticles comprising Pd4+ species: spectroscopic and structural aspects, thermal stability, and reactivity. J. Phys. Chem. C 116, 19342–19348 (2012).
Zeinalipour-Yazdi, C. D., Willock, D. J., Thomas, L., Wilson, K. & Lee, A. F. CO adsorption over Pd nanoparticles: A general framework for IR simulations on nanoparticles. Surf. Sci. 646, 210–220 (2016).
Zhu, J. et al. High-efficient dynamic kinetic resolution of amines with a core-shell hollow mesoporous MIL−101@ Pd@ ZIF-8 nanocatalyst and lipase. Microporous Mesoporous Mater 329, 111490 (2022).
Gao, S. et al. Incorporation of metals and enzymes with porous imine molecule cages for highly efficient semiheterogeneous chemoenzymatic catalysis. ACS Catal 11, 5544–5553 (2021).
Wang, H., Zhang, Y., Yue, W., Liang, J. & Su, W. Application of magnetic field (MF) as an effective method to improve the activity of immobilized Candida antarctica lipase B (CALB). Catal. Sci. Technol. 12, 5315–5324 (2022).
Liang, W. et al. Enhanced Bioactivity of Enzyme/MOF Biocomposite via Host Framework Engineering. J. Am. Chem. Soc. 145, 20365–20374 (2023).
Wang, Q. et al. Functionalized poly (ionic liquid) s bridging Pd NPs and lipase for highly efficient dynamic kinetic resolution of chiral amine. Appl. Catal. Gen. 666, 119426 (2023).
Gao, L. et al. Co-immobilization of metal and enzyme into hydrophobic nanopores for highly improved chemoenzymatic asymmetric synthesis. Chem. Commun. 56, 13547–13550 (2020).
Jin, Q., Jia, G., Zhang, Y. & Li, C. Modification of supported Pd catalysts by alkalic salts in the selective racemization and dynamic kinetic resolution of primary amines. Catal. Sci. Technol. 4, 464–471 (2014).
Wheeldon, I. et al. Substrate channelling as an approach to cascade reactions. Nat. Chem. 8, 299–309 (2016).
Sweetlove, L. J. & Fernie, A. R. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 9, 2136 (2018).
Trujillo, M., Donald, R. G., Roos, D. S., Greene, P. J. & Santi, D. V. Heterologous Expression and Characterization of the Bifunctional Dihydrofolate Reductase− Thymidylate Synthase Enzyme of Toxoplasma gondii. Biochemistry 35, 6366–6374 (1996).
Fang, X., Zhan, Y., Yang, J. & Yu, D. A concentration-dependent effect of methanol on Candida antarctica lipase B in aqueous phase. J. Mol. Catal. B: Enzym. 104, 1–7 (2014).
Assen, A. H. et al. Ultra‐tuning of the rare‐earth fcu‐MOF aperture size for selective molecular exclusion of branched paraffins. Angew. Chem. Int. Ed. 54, 14353–14358 (2015).
Li, P.-Z. et al. A triazole-containing metal–organic framework as a highly effective and substrate size-dependent catalyst for CO2 conversion. J. Am. Chem. Soc. 138, 2142–2145 (2016).
Jiang, Z. et al. A highly permeable porous organic cage composite membrane for gas separation. J. Mater. Chem. A 11, 6831–6841 (2023).
Li, L. et al. Integration of Pd nanoparticles with engineered pore walls in MOFs for enhanced catalysis. Chem 7, 686–698 (2021).
Tian, D. et al. Multi-compartmental MOF microreactors derived from Pickering double emulsions for chemo-enzymatic cascade catalysis. Nat. Commun. 14, 3226 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22471018, 22071008, and 52003029), the High-level Overseas Talents Program of China, the Excellent Young Scholars Research Fund from the Beijing Institute of Technology, and the Central University Basic Research Fund of China (2021CX01024). The technical support from Analysis & Testing Center of Beijing Institute of Technology is also appreciated.
Author information
Authors and Affiliations
Contributions
K.Z. (Methodology: Lead; Writing – review & editing: Supporting). L.-X.T. (Software: Supporting; Writing – review & editing: Supporting). N.G. (Writing – review & editing: Supporting). J.-K.S. (Conceptualization: Lead; Data curation: Lead; Formal analysis: Lead; Project administration: Lead; Validation: Lead; Writing – original draft: Lead; Writing –review & editing: Lead).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Taeyoung Yoon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, K., Tan, LX., Gao, N. et al. Ionic organic cage as a versatile platform for immobilizing chemical and enzymatic sites for chemoenzymatic catalysis. Nat Commun 16, 5698 (2025). https://doi.org/10.1038/s41467-025-60292-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60292-5
