
Abstract
Asthma is a prevalent respiratory condition with over 100 genetic loci identified through genome-wide association studies (GWAS). However, the genetic basis of asthma in East Asians remains underexplored. To address this, we performed a comprehensive analysis of shared genetic mechanisms between asthma and white blood cell (WBC) traits in East Asians, aiming to identify potential pleiotropic loci. Using linkage disequilibrium score regression (LDSC), we identified a significant genetic correlation between asthma and eosinophil count, further supported by Mendelian randomization (MR) analysis. A multi-trait analysis of GWAS (MTAG) uncovered 52 genome-wide significant loci, including 31 previously unreported loci specific to East Asians. Notably, we discovered a missense variant (rs75326924) in the CD36 gene that exhibits increased expression in lymphocytes and type 2 innate lymphoid cell (ILC2)-enriched cells in asthma patients, confirmed by flow cytometry. Proteomic profiling demonstrated downregulation of immune-related proteins such as Interleukin-7, Oncostatin M, and VEGFA in carriers of rs75326924, a variant previously associated with CD36 deficiency. Our findings provide insights into genetic loci and candidate genes underlying asthma in East Asians, offering potential targets for therapeutic interventions tailored to this population.
Similar content being viewed by others

Introduction
Asthma, a prevalent allergic respiratory disorder globally, is characterized by airway mucosal inflammation, wheezing, and shortness of breath. Its pathogenesis involves complex interactions between environmental triggers and genetic susceptibilities1. Recently, the Global Biobank Meta-analysis Initiative (GBMI) conducted the largest genome-wide association study (GWAS) on asthma2. This study collected data from asthma cohorts worldwide, encompassing over 150,000 patients with asthma and more than 1.6 million healthy controls. In total, 179 genetic loci associated with asthma susceptibility were identified, indicating a polygenic model of inheritance. However, the vast majority of genetic data in this study originated from European populations. Consequently, although there are several genetic loci shared between European and Asian populations, genetic underpins of asthma in East Asian populations remain to be unearthed. East Asians comprise approximately 20% of the global population and are genetically diverse, including Chinese, Japanese, Korean, and Mongolian populations. In the GBMI study, East Asian data are primarily derived from the China Kadoorie Biobank (CKB), Taiwan Biobank (TWB), and BioBank Japan (BBJ). This reflects the broader trend in genomic research, where Chinese and Japanese populations are commonly used as representative East Asian groups, as seen in major reference datasets such as the 1000 Genomes Project and HapMap3.
Recent advancements in statistical methodologies for GWAS analyses have facilitated joint analyses of traits with shared genetic architecture3,4,5. Multiple studies have focused on identifying shared genetic risk factors and pathways between asthma and coexisting conditions, including other allergic diseases and obesity6. However, only a few studies have examined if shared genetic mechanism exist between asthma and traits associated with white blood cells (WBC), which act as important indicators of immune function and play a critical role in allergic reactions. For instance, eosinophilic asthma is marked by a significant elevation of eosinophils7. Eosinophils are a type of WBC tending to accumulate at sites of allergic inflammation, which are involved in the development of asthma exacerbation8. Intriguingly, a recent study highlighted a shared genetic architecture between blood eosinophil counts and asthma risk in individuals of European ancestry, implicating a genetic link to the STAT6 signaling pathway in these two traits9.
In this study, we first confirmed strong positive genetic correlations between asthma and WBC traits in East Asians. To further elucidate the genetic architecture of asthma in East Asians, we jointly analyzed the GWAS results of asthma and eosinophils using multi-trait analysis of GWAS (MTAG) and identified multiple pleiotropic loci shared by asthma and WBC traits in East Asians, specifically. Finally, we validated our findings in a Chinese cohort through comprehensive analysis of multi-omics data, affirming the importance of newly discovered loci and their associated genes in asthma susceptibility.
Results
Study design
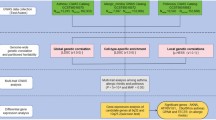
An overview of the study design is shown in Fig. 1. We performed a genome-wide cross-trait analysis to quantify genetic correlation, identify pleiotropic loci, detect expression–trait associations, and infer causal relationships. The analysis included genetic correlation analysis, Mendelian randomization, and multi-trait GWAS, leading to the identification of significant loci, including a missense variant in the CD36 gene. These findings were validated through gene expression, flow cytometry, protein expression analysis, and further confirmed in a separate Chinese population GWAS.

The figure illustrates the multi-trait analysis pipeline used to identify genetic loci associated with asthma and eosinophil traits in East Asians. Key steps include genetic correlation analysis, Mendelian randomization, and validation of findings through multi-omics data and replication cohorts.
Shared heritability between asthma and hematological traits
SNP-based heritability estimates were calculated by Linkage Disequilibrium score regression (LDSC)10 to assess the proportion of phenotypic variance explained by the tested variants. Heritability estimates on the observed scale using GWAS summary statistics of East Asians were 2.30%, 9.30%, 11.37%, 11.51%, 8.83%, 9.19%, 12.55% and 16.55% for asthma, basophils, eosinophils, neutrophils, lymphocytes, monocytes, erythrocytes and platelets, respectively (Supplementary Tables S1, 2). We next investigated the genetic correlations of asthma and hematological traits using LDSC10. Significant genetic correlations were detected between asthma and certain WBC traits including eosinophils (Rg = 0.50, P = \(2.38\times {10}^{-15}\)), basophils (Rg = 0.27, P = \(7.93\times {10}^{-5}\)) and neutrophils (Rg = 0.19, P = 0.0006) in East Asians (Fig. 2A and Supplementary Table S3). We further investigated the pattern of SNP-heritability between asthma and various leukocyte traits across chromatin marks and nine cell types. Enrichment patterns further support the overlap in genetic influences on asthma and leukocyte counts, particularly in immune-related cell types. This was most prominently observed in annotations related to active regulatory elements, such as DNase I hypersensitive sites and histone modifications (H3K27ac and H3K4me3, Supplementary Fig. S1). These findings suggest a potential common genetic architecture underlying asthma susceptibility and leukocyte regulation, emphasizing the role of immune cell-specific genetic variation in the pathogenesis of asthma.

A Global genetic correlations between asthma and hematological traits estimated using LD score regression based on GWAS summary statistics. Squares represent the point estimates of genetic correlation (rg), and horizontal error bars represent 95% confidence intervals (CI). Error bars indicate 95% CI, and the center corresponds to the estimated rg value. Sample sizes: Asthma (GBMI): 341,204 (18,549 cases, 322,655 controls); Basophil count: 81,042; Eosinophil count: 86,890; Lymphocyte count: 89,266; Monocyte count: 88,929; Neutrophil count: 78,744; Leukocyte count: 151,807; Platelet count: 145,648; Erythrocyte count: 150,708. All GWAS were conducted in East Asian populations. B Bi-directional causal relationships between asthma and hematological traits, assessed using two-sample Mendelian randomization (MR). The left panel shows the causal effect of asthma (exposure) on hematological traits (outcomes); the right panel shows the reverse. Each point represents the odds ratio (OR), and horizontal error bars represent the 95% CI. Error bars indicate 95% CI, and the center corresponds to the estimated OR value. No. SNPs: number of instrumental variants used in the MR analysis. Genetic correlation analyses in panel (A) were conducted using LD score regression with two-sided P-values. Bi-directional Mendelian randomization in panel (B) was performed using the inverse-variance weighted (IVW) method, also using two-sided tests. No multiple testing correction was applied to P-values shown, as results are interpreted in the context of a hypothesis-driven analysis of selected traits. Source data are provided as a Source Data file.
We further performed bi-directional Mendelian randomization (MR) analysis to investigate potential causal relationships between asthma and correlated white blood cell traits (eosinophils, basophils, and neutrophils) in East Asians. Our results indicate a strong and significant causal effect of eosinophil count on asthma (OR = 1.75, P = 1.87 × 10−13, Fig. 2B), as well as a significant causal effect of neutrophil count on asthma (OR = 1.53, P = 2.68 × 10−6, Fig. 2B). These findings support the role of eosinophilic and neutrophilic inflammation in asthma pathogenesis. Meanwhile, our reverse MR analysis showed that asthma has a significant causal effect on eosinophil count (OR = 1.23, P = 3.26 × 10−21, Fig. 2B) and neutrophil count (OR = 1.08, P = 1.17 × 10−7, Fig. 2B), suggesting bidirectional relationships. However, basophil count showed a modest association with asthma when used as an exposure (OR = 1.05, P = 0.02, Fig. 2B) but did not demonstrate a significant causal effect in the reverse direction (OR = 1.04, P = 0.29, Fig. 2B).
Local genetic correlations between asthma and hematological traits
We next scanned the entire genome to identify distinct genomic loci associated with shared heritability among genetically correlated trait pairs. After accounting for multiple testing, six significantly correlated regions were pinpointed between asthma and eosinophils in East Asians (Supplementary Fig. S2 and Supplementary Table S4). Conversely, only the leukocyte antigen (HLA) region exhibited correlation between asthma and neutrophils or asthma and monocytes in East Asians. Notably, no significant correlated region was observed between asthma and basophils.
Multi-trait GWAS analysis between asthma and eosinophil counts
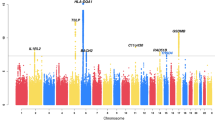
Considering the strongest genetic correlation observed between asthma and eosinophils, we conducted MTAG analysis utilizing the East Asian asthma data from GBMI and eosinophils data from GWAS catalog for MTAG analysis. To maximize the discovery of potential loci, we also performed MTAG analysis using asthma data from BBJ and eosinophils data. Loci with MTAG P-values less than 5 × 10−8 in either analysis were considered significant pleiotropic loci. With increased statistical power, our analysis identified 52 genome-wide significant loci, including 31 loci not previously reported in the original asthma GWAS in East Asians (Fig. 3 and Supplementary Table S5). Among these, nine newly identified loci have been previously documented in Europeans, suggesting a shared regulatory mechanism between Europeans and East Asians. Additionally, we observed a few signals in East Asians overlapping with those in Europeans within the same genomic regions, such as 8q24.21 (rs16902875) near MYC and 17q21.33 (rs2671655) near gene ZNF652, indicating potential shared genetic susceptibility to asthma between the two populations. In contrast, nine loci were found to be specific to East Asian populations. Among them, the 18q21.1 locus (rs57631119) is located near the SMAD2 gene, which has been associated with airway remodeling in asthma11. Interestingly, multiple genes in the SMAD gene family, including SMAD3, SMAD4, and SMAD7, have been significantly associated with asthma in GWASs of Europeans. Moreover, in vitro experiments have demonstrated that inhibiting the TGF-β/Smad signaling pathway can alleviate inflammation and allergic reactions, contributing to the amelioration of asthma symptoms12,13. Notably, we detected a signal at the 7p21.11 locus. The lead variant of the 7p21.11 locus (rs75326924) is a loss-of-function missense variant of the CD36 gene, found exclusively in East Asians. This variant is associated with a reduced risk of asthma, indicating a protective effect against the disease. According to the ClinVar database, rs75326924 is classified as pathogenic due to its role in causing platelet glycoprotein IV deficiency, thereby reinforcing the variant’s functional significance and establishing CD36 as a causal gene. In further support of this, a prior study using a mouse model demonstrated that CD36 plays a crucial role in mediating asthma induced by house dust mites14. Collectively, these findings suggest that CD36 play an important role in asthma pathogenesis and may serve as a promising therapeutic target, particularly in East Asian populations.

Circular plot illustrating pleiotropic loci shared between asthma and eosinophil traits across the genome. The outermost ring represents human chromosomes (chr1 to chr22), while the subsequent inner rings display significant associations from MTAG-GBMI, MTAG-BBJ, Asthma-GBMI, Asthma-BBJ, and eosinophils. Newly identified loci are highlighted in red, and previously known loci are shown in black. MTAG-GBMI: Multi-trait analysis of GWAS applied to asthma data from the Global Biobank Meta-analysis Initiative (GBMI). This combines asthma and eosinophil GWAS data to identify shared loci. MTAG-BBJ: MTAG analysis using asthma data from BioBank Japan (BBJ) alongside eosinophil GWAS data. Asthma-GBMI: Single-trait GWAS results for asthma from GBMI. Asthma-BBJ: Single-trait GWAS results for asthma from BBJ. Eosinophils: Single-trait GWAS results for eosinophil counts. Source data are provided as a Source Data file.
Replication of pleiotropic loci in the Chinese population
We next conducted a GWA study in a Chinese cohort consisting of 1040 asthmatic patients and 2506 healthy controls. Our analysis successfully replicated six previously established loci and identified five previously undetected pleiotropic loci using the MTAG approach in East Asian populations (Supplementary Data 1; P < 0.05). Notably, this included the missense variant rs75326924 in CD36, which exhibited a consistent effect direction with the MTAG findings (BETA = −1.08, P = 0.038). To further validate this association, we assessed rs75326924 in an independent GWAS of a Korean cohort [19], observing a similar effect direction (BETA = −0.54) with a P-value approaching significance (P = 0.086). This suggests that an increased sample size could potentially lead to a statistically significant result.
Furthermore, we stratified our asthma cases into eosinophilic and non-eosinophilic subtypes to assess subtype-specific genetic associations. Our findings revealed that the loci 5q22.1 (TSLP, rs1837253), 6p22.1 (PPP1R11, rs886997) and 17q21.1 (GSDMB, rs56750287) were significantly associated with eosinophilic asthma (Supplementary Data 2; P < 0.05), but did not replicate in the non-eosinophilic asthma group, implying potential genetic differences between eosinophilic and non-eosinophilic asthma subtypes in East Asian populations.
Differential gene expression and flow cytometry analysis of CD36 in asthma
We analyzed RNA sequencing data of primary bronchial epithelial cells from 88 asthmatic patients and 42 healthy controls15, which were downloaded from the GEO database. Differential expression analysis reveals a significant up-regulation of CD36 gene expression in the asthmatic patient group compared to the healthy control group (Supplementary Fig. S3).
We next conducted flow cytometric analysis on peripheral blood immune cells obtained from 22 asthmatic patients and 23 healthy non-allergic volunteers (Fig. 4). Comparative analysis between asthmatic patients and healthy cohorts revealed no significant difference in the proportions of granulocytes or monocytes. However, there was a significant increase in the proportion of Type 2 Innate Lymphoid Cells (ILC2)-enriched cell populations (CD4−, CRTH2+) within the asthmatic population, aligning with prior research indicating the involvement of ILC2 in asthma progression (Fig. 4). Further examination of CD36 expression within each cell subset showed no significant difference in CD36 expression proportions among granulocyte or monocyte clusters between asthmatic patients and controls. However, an elevated proportion of CD36 expression was observed in lymphocytes and ILC2-enriched cell populations (Fig. 4). These findings corroborate our genomic and transcriptomic analyses, providing additional evidence that CD36 may serve as a potential therapeutic target in asthma treatment.

A Flow cytometry was used to detect the proportion of granulocytes, monocytes, and lymphocytes, as well as the expression of CD36 and CRTH2 in peripheral blood samples from asthmatic and healthy individuals. B Quantification of the proportion of granulocytes (P = 0.4076), monocytes (P = 0.1499), and ILC2-enriched (CD4- CRTH2+) cells (P = 0.0415) in asthma patients and controls. C Quantification of CD36+ cell percentages in granulocytes (P = 0.1114), monocytes (P = 0.1230), and lymphocytes (P = 0.0063), as well as the mean fluorescence intensity (MFI) of CD36 in CD4- CRTH2+ cells (P = 0.0147). Flow cytometric analysis was performed on peripheral blood immune cells from 22 asthmatic patients and 23 healthy non-allergic volunteers. Data are presented as mean ± standard deviation (SD). Error bars indicate SD. Statistical significance was assessed using a two-sided unpaired Student’s t test without correction for multiple comparisons (**P < 0.01; *P < 0.05; ns not significant). Source data are provided as a Source Data file.
Differential proteomic analysis of CD36 missense variant (rs75326924)
To investigate potential downstream targets regulated by CD36, we analyzed the proteomic data from 1056 Chinese individuals who underwent Olink Explore inflammation proteomic profiling. Among them, five individuals carried the rs75326924 mutation at the T locus. Following Wilcoxon rank-sum test, we found significant expression alterations in 43 proteins (P < 0.05) between carriers and non-carriers. Notably, all of the top eight proteins (P < 0.01, Supplementary Fig. S4 and Supplementary Data 3) were down-regulated. These proteins encompassed key players implicated in immune response and allergic reaction, including Interleukin-7 (IL7), Oncostatin M (OSM), and Vascular Endothelial Growth Factor A (VEGFA).
Polygenic risk prediction
We developed a polygenic risk score (PRS-32) for the Chinese population based on the count of risk alleles carried at 32 genome-wide significant loci identified in East Asians (Supplementary Data 4). Each PRS was weighted by the overall effect sizes of the included alleles derived from the association analysis conducted in East Asians. PRS-32 exhibited a trend towards higher OR values in the second top quintiles (Fig. 5). In comparison, we constructed another PRS (PRS-63), which included 31 additional pleiotropic loci identified by MTAG for asthma and eosinophils. (Supplementary Data 5). PRS-63 showed a steady rise in OR across the quintiles, with the highest risk observed in the top quintile group (Fig. 5). Consistent with this, the mean variance explained by the PRS (Liability R²) was 2.6% for PRS-32 and 2.9% for PRS-63, indicating the better predictive capability of PRS-63.

Two different polygenic risk score (PRS) models, PRS-32 and PRS-63, are presented. The x-axis represents PRS quintiles, and the y-axis shows the odds ratios (ORs) for asthma, with 95% confidence intervals (CIs) compared to the lowest PRS group (reference). Error bars represent 95% CI, and the center indicates the estimated OR. The analysis was based on 1100 asthmatic patients and 2506 controls of East Asian ancestry. Source data are provided as a Source Data file.
Disscussion
In this study, we performed a genome-wide multi-trait analysis that systematically investigated the shared genetic architecture between asthma and WBC traits in East Asians. Leveraging the well-established genetic correlation between asthma and eosinophils, our analysis identified multiple East Asian-specific loci which have not been detected previously. Subsequent analysis using multi-omics approaches further validated these findings within the Chinese population.
Our genetic correlation and MR analyses consistently identify eosinophils as the pivotal WBC, indicating the intricate relationship between immune cell profiles and asthma susceptibility in East Asians. This observation aligns with previous analyses conducted in European populations, reinforcing its robustness across different ethnic groups. Moreover, it is supported in epidemiological studies and in line with our current understanding of asthma pathogenesis16,17.
The combined analysis of asthma and eosinophils has significantly enhanced our statistical power in detecting potential pleiotropic loci in East Asians. Our analysis has identified 31 significant loci, which were not reported in East Asians previously, including nine loci previously reported in Europeans. In addition to this, three signals in East Asians overlap with those observed in Europeans within the same genomic regions, indicating shared genetic mechanisms between the two populations. Several loci, including GLB1, IL13, and HBS1L, showed strong associations with eosinophil counts but only marginal associations with asthma, suggesting that these variants primarily influence eosinophil biology rather than directly contributing to asthma risk. This highlights the complexity of asthma genetics, where not all eosinophil-associated loci necessarily translate into strong asthma risk loci. One possible explanation is that these variants regulate eosinophil levels, which may indirectly affect asthma susceptibility, but their direct effect on asthma may be weak or context-dependent. Pleiotropic effects could also be at play, where these genes influence multiple biological pathways, with a primary role in eosinophil regulation but only a minor contribution to asthma. Additionally, differences in statistical power between eosinophil and asthma GWAS may explain the discrepancies in significance levels. Our Mendelian randomization (MR) analysis supports a causal relationship between eosinophil count and asthma, reinforcing the importance of eosinophilic inflammation in asthma pathogenesis while suggesting that not every eosinophil-associated variant is a direct asthma risk locus.
Moreover, we have identified nine East Asian-specific loci, which exhibit a high frequency in East Asian populations compared to other ethnic groups. Intriguingly, we observed a significant association between rs75326924 and reduced risk of asthma. This missense variant results in a loss-of-function mutation in CD36 and is exclusively present in East Asians. Unlike most GWAS signals that are found in non-coding regions, making it challenging to pinpoint the causal variant, our findings strongly suggestes rs75326924 as a protective variant in asthma. Additionally, studies have indicated that rs75326924 leads to decreased CD36 protein expression in platelets18,19, which in turn causes platelet glycoprotein IV deficiency disorder20. Despite limited research on CD36 and asthma, a previous study has shown that mice lacking CD36 exhibit impaired house dust mite uptake, reduced epithelial production of TSLP and IL-33, decreased frequencies of lung ILC2 populations, and suppressed allergic disease development14. In our pursuit of direct evidence linking CD36 to protection of asthma in humans, we conducted flow cytometric analysis on peripheral blood immune cells, comparing samples from asthmatic patients to those from healthy controls. Notably, our findings revealed a significant increase in CD36 expression within lymphocytes and ILC2-enriched cell populations among asthmatic individuals. This compelling observation suggests a significant role of CD36 in the pathogenesis of asthma. Through the analysis of inflammation-related proteomic data from 1,056 Chinese individuals, we observed significant downregulation of inflammation-related proteins in samples carrying the rs75326924 variant in CD36. Notably, many of these proteins are implicated in immune hypersensitivity, indicating potential pathways and targets influenced by CD36 dysregulation. For instance, IL7 promotes T and B cell proliferation and activation, while OSM influences immune cell activation and cytokine production. Dysregulation of IL7 and OSM can contribute to allergies and asthma by exacerbating immune responses21,22.
Considering that differences in living environments between East Asian and European populations may lead to environmental influences outweighing genetic factors, potentially affecting the reliability of our identified loci, we have calculated the heritability of asthma in East Asian populations (2.3%) and found it to be highly comparable to that observed in European populations (2.6%). Moreover, our analysis identified a substantial number of loci shared between European and East Asian populations. We further estimated the genetic correlation for asthma between East Asian and European populations to be 0.64 (P = \(8.27\times {10}^{-6}\)), indicating a strong genetic overlap23. For eosinophil counts, the heritability was 11.4% in East Asians and 19.6% in Europeans, with a genetic correlation of 0.26 (P = 0.028). This consistency in heritability estimates, the high genetic correlation, and the presence of shared genetic loci reinforce the genetic basis of asthma in East Asians and support the reliability of our identified loci, independent of environmental differences.
We developed two distinct sets of PRSs for our Chinese cohort. These scores were constructed by utilizing results from MTAG outcomes and single-trait GWAS results. Through comparative analysis, we found that integrating GWAS data from asthma and eosinophils using MTAG improved the accuracy of predicting asthma susceptibility. This strategy offers a promising direction for asthma research, particularly valuable in non-European populations with limited sample sizes.
Our study has limitations. While PRS-63 performed better than PRS-32, the unexpected lower risk in the fourth quintile four compared to the third quintile highlights the need for better SNP selection. These inconsistencies may reflect limitations in the genetic dataset. Future studies incorporating newly discovered risk-associated SNPs may improve the accuracy of PRS models for asthma. In addition, our study primarily focuses on genetic factors, and we acknowledge that environmental influences such as air pollution, smoking, and lifestyle factors, which were not accounted for in our analysis, may contribute to asthma susceptibility and PRS performance. The lack of environmental data in the available datasets limits our ability to fully capture gene-environment interactions. Future studies incorporating comprehensive environmental exposure data alongside genetic information will be crucial to provide a more holistic understanding of asthma risk and improve the predictive power of PRS models.
Taken together, by elucidating the intricate genetic interplay between asthma and WBC traits in East Asians, our study not only enhances our understanding of the underlying mechanisms driving asthma susceptibility but also provides valuable insights into potential therapeutic targets and personalized treatment strategies tailored to this population.
Methods
GWAS data of asthma and hematological traits
GWAS summary statistics of asthma in East Asians were downloaded from GBMI study2, and summary statistics Japanese were obtained from studies of BioBank Japan through GWAS Catalog. The asthma GWAS in GBMI contains 18,549 cases and 322,655 controls, while the asthma GWAS in BioBank Japan includes 13,015 cases and 162,933 controls. Summary statistics of the largest GWAS for hematological traits24 were obtained from GWAS Catalog25. Blood cell counts of basophils, eosinophils, neutrophils, lymphocytes, monocytes, erythrocytes and platelets were analyzed in this study. Details of each dataset were summarized in Supplementary Table S1.
Estimation of genetic correlation and SNP-based heritability
Genetic correlation rg between asthma and hematological traits was estimated by LDSC using GWAS summary statistics overlap with HapMap3 variants as recommended10. SNP-based heritability of analyzed traits was also estimated by LDSC v1.0.110. Pre-computed linkage disequilibrium scores for HapMap3 SNPs calculated based on East-Asian-ancestry or European-ancestry individuals from the 1000 Genomes Project were used in the analysis, and SNP markers with an imputation INFO score <0.9 were excluded. Detailed methodological information on genetic correlation calculations is available in the supplementary materials of the publication by Bulik-Sullivan et al.10 We corrected multiple testing for LDSC P-values by the Bonferroni method and a P-value of 0.00625 (0.05/8) was considered as the significance level for LDSC analysis.
Cell-type-specific enrichment of SNP heritability
Cell-type-specific SNP heritability enrichment was evaluated using stratified linkage disequilibrium score regression (S-LDSC) to identify functional categories or cell types that substantially contribute to the heritability of the traits investigated26. Annotation data from the Roadmap Epigenomics project, encompassing six chromatin marks (DHS, H3K27ac, H3K36me3, H3K4me1, H3K4me3, and H3K9ac) across 88 diverse cell types and tissues, were used to partition the SNP heritability of each trait. These cell-type annotations were organized into seven categories: central nervous system (CNS), digestive system, cardiovascular, musculoskeletal and connective tissue, immune and blood, pancreas, and others. Enrichment values for each annotation were scaled and visualized using hierarchical clustering, providing a comprehensive overview of cell-type-specific contributions to trait heritability.
Mendelian randomization
Instrumental variables (IVs) were selected from exposure GWAS data through LD clumping (r2 threshold: 0.01, P-value threshold: \(5\times {10}^{-8}\), window size: 10 mb). Data corresponding to these IVs were then extracted from both exposure and outcome datasets and harmonized. Bidirectional Mendelian randomization analysis was conducted using the Inverse Variance Weighted (IVW) method implemented in the R package TwoSampleMR v0.6.427,28, which combines effects across multiple SNPs to estimate causal effects.
Local genetic correlation analysis
Given that genetic correlation, as estimated by LDSC, integrates data from all genetic variants across the genome, we proceeded to assess the pairwise local genetic correlation using ρ-HESS v0.5.3 (heritability estimation from summary statistics)29. ρ-HESS is designed to quantify the local genetic correlation between pairs of traits within each of the 1703 pre-specified LD-independent segments, with an average length of 1.6 Mb. We considered statistical significance with a Bonferroni correction, setting the threshold at P < 0.05/1703.
Multi-trait GWAS analysis
Multi-trait GWAS meta-analysis for asthma and different hematological traits was performed by MTAG. MTAG method can increase the power to detect loci from correlated traits by analyzing GWAS summary statistics jointly. The first step of MTAG is to filter variants by removing non common SNPs, duplicated SNPs, or SNPs with strand ambiguity. MTAG then estimates the pairwise genetic correlation between asthma and hematological traits using LDSC10 and uses these estimates to calibrate the variance-covariance matrix of the random-effect component. MTAG next performs a random-effect meta-analysis to generate the SNP-level summary statistics. Loci are considered as significant with the trait of interest if the P-value is less than \(5\times {10}^{-8}\) in the MTAG analysis and the P-value is less than 0.01 in the original GWAS.
GWAS analysis of Chinese individuals
Replication analysis was performed using a Chinese cohort recruited through community-based health screenings and hospital outpatient services in Shandong Province. Asthma cases were clinically diagnosed, while controls were selected based on questionnaire responses indicating no history of allergic or respiratory conditions. As participation was voluntary, some self-selection bias may exist. Additionally, the cases and controls were recruited from different sources, which could introduce recruitment-related bias. However, these samples were used only for replication, and all individuals were of East Asian ancestry. Standardized quality control procedures were applied to minimize systematic bias.
A total of 1100 asthmatic patients were recruited from The First Affiliated Hospital of Shandong First Medical University. Patients diagnosed with asthma were identified based on the criteria outlined in the 2023 GINA Report (Global Strategy for Asthma Management and Prevention)30. Additionally, asthmatic patients from the CAS cohort who are currently undergoing asthma treatment were included based on healthcare and lifestyle questionnaires. The cohort consisted of 50.5% males and 49.5% females, with a mean age of 28.4 years. A total of 2506 control subjects were selected from the CAS cohort based on questionnaire information indicating no history of allergies or respiratory diseases. Among these participants, 48.9% were male and 51.1% were female, with a mean age of 39.1 years. Sex were included as covariates in the statistical analyses to account for potential confounding effects. Age was not included as a covariate in the association analyses due to substantial differences in age distribution between cases and controls, which could introduce bias when adjusting. Therefore, analyses were conducted without age adjustment. The CAS cohort is a prospective multi-omics cohort comprising 3197 employees (49.0%) from various institutes or offices of the Chinese Academy of Sciences in Beijing, China31,32,33,34. This study was approved by the Institutional Review Boards of The First Affiliated Hospital of Shandong First Medical University, Beijing Institute of Genomics (Chinese Academy of Sciences) and Beijing Zhongguancun Hospital.
Genotyping was conducted using the Infinium Asian Screening Array. Individuals with low genotype call rate (<95%, n = 31), gender mismatch (n = 0), possible contamination (n = 9) or departure from Chinese Han population (n = 13) were removed before association test. SNPs were excluded if they were not on autosomal chromosomes, had a missing call rate ≥5%, had a minor allele frequency ≤1%, or had a Hardy–Weinberg equilibrium P value ≤ \(1\times {10}^{-5}\). Principal component analysis (PCA) was performed, and the PC1 vs. PC2 scatter plot was used to assess potential population stratification, ensuring genetic homogeneity within the study cohort before association testing (Supplementary Fig. S5). After quality control, a total of 3546 samples is left for further analyses. Imputation was done by Minimac3 using 1000 Genomes Project Phase 3 version 5 genotype data as reference. Multivariable logistic regression was employed to examine the association between genetic variants and the status of asthma diagnosis, utilizing PLINK 1.935. The covariates included in the regression model were sex (predicted using PLINK) and the first five principal components (PCs).
Differential expression analysis
Differential expression analysis was conducted using RNA sequencing data from the GEO dataset GSE201955, which includes primary bronchial epithelial cells from 88 asthmatic patients and 42 healthy controls15. RNA was extracted using the QIAGEN AllPrep DNA/RNA/miRNA Universal Kit, and quality was assessed with the Agilent 2100 Bioanalyzer. cDNA libraries were prepared with the Illumina TruSeq RNA Library Prep Kit v2 and sequenced on Illumina HiSeq 2500 or 4000 platforms. Quality control was performed using FastQC and VerifyBamID, with no contamination detected and sample swaps corrected. Reads were aligned to the reference genome using STAR, and genes with low counts or those on the X, Y, and mitochondrial chromosomes were excluded. Raw counts were normalized using the trimmed mean of M-values (TMM) method. Differential expression of CD36 was evaluated using the two-sided Wilcoxon rank-sum test.
Flow cytometry analysis
Peripheral blood (2 ml) was collected in anticoagulant tubes and diluted 1:1 with phosphate-buffered saline (PBS). The diluted blood was carefully layered over Ficoll-Paque™ PLUS (Cytiva, 17144002) in a 15 ml centrifuge tube. Following density gradient centrifugation, the peripheral blood mononuclear cell (PBMC) layer was collected. PBMCs were freshly isolated from heparinized blood, washed, and resuspended in PBS.
For flow cytometry staining, cells were incubated with fluorochrome-conjugated monoclonal antibodies: anti-CD4-FITC (BioLegend, clone: OKT4, 317407), diluted 1:400; anti-CD36-APC (BioLegend, clone: 5-271, 336207), diluted 1:200; and anti-CRTH2-DAPI (BioLegend, clone: BM16, 350129), diluted 1:100. All antibodies were diluted in PBS. For each sample, 100 μl of diluted antibody mixture was added and incubated at 4 °C for 30 min in the dark. After incubation, 150 μl of PBS was added to stop the staining reaction.
Cells were analyzed on a BD-FACSAria™ Fusion cytometer. Data acquisition was performed using BD FACSDiva software, and flow cytometry data were analyzed using FlowJo (version 10, FlowJo LLC). Cell populations, including monocytes, granulocytes, and lymphocytes, were identified based on size, granularity, and scatter properties. ILC2-enriched cells were further identified within the lymphocyte population by their CD4- CRTH2+ phenotype.
Olink proteomics analysis
In order to identify inflammatory signatures associated with the CD36 missense variant (rs75326924), plasma proteins were measured in the CAS cohort 1 K multi-omics subgroup (n = 1056) using the Olink Explore 384 Inflammation panel. The concentrations of proteins were quantified as Normalized Protein Expression (NPX), which represents Olink’s normalized relative unit on a log2 scale. Three protein assays (BCL2L11, BID, and MGLL) were excluded due to Olink QC warnings, leaving a total of 365 proteins for subsequent analysis. Differential protein expression was assessed using a two-sided t-test between CC and CT groups based on the rs75326924 genotype.
Polygenic risk analysis
Two sets of PRS for the Chinese cohort were constructed using the PLINK software. The first set, PRS-32, utilized a total of 32 GWAS significant loci previously identified in East Asians to construct the PRS. The second set, PRS-63, utilized a total of 63 significant pleiotropic loci identified in this study for PRS construction. For each PRS set, the PLINK score command was employed to calculate the PRS. This command integrates the weighted sum of risk alleles across the specified loci for each individual in the study cohort. Subsequently, the PRS values were standardized based on the mean and standard deviation of the PRS distribution in the population to facilitate comparison and interpretation. In the evaluation phase, Nagelkerke’s R2 was computed to assess the predictive power of the model. This was accomplished by comparing the full model incorporating PRS and five PCs against a null model devoid of PRS. Subsequently, Nagelkerke’s R2 was transformed from the observed scale to the liability scale, with consideration of a population prevalence of 5%.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets analyzed in this study are obtained from publicly available sources. Summary statistics for the BioBank Japan (BBJ) dataset were accessed via the GWAS Catalog (https://www.ebi.ac.uk/gwas/summary-statistics) under their data usage policies. Data from the Global Biobank Meta-analysis Initiative (GBMI) were used in accordance with their access guidelines (https://www.globalbiobankmeta.org/resources). Additional genotype and phenotype data from the Chinese replication cohort are not publicly available due to participant privacy restrictions. Access can be requested from the corresponding author (changxiao@sdfmu.edu.cn) and requires ethical approval and a data use agreement. Summary statistics for genome-wide significant loci generated in this study are provided in the Supplementary Data. Source data for figures are provided with this paper. Source data are provided with this paper.
Code availability
We utilized a range of publicly available software tools to conduct the analyses in this study. The computational environment included Python v2.7.18, Perl v5.16.3, and R v4.2.3. We employed LDSC v1.0.1 to estimate genetic correlations between asthma and hematological traits, and also to perform stratified LDSC (S-LDSC) for evaluating the enrichment of cell-type-specific SNP heritability. Local genetic correlation analysis was carried out using ρ-HESS v0.5.3. Bidirectional Mendelian randomization was conducted with the R package TwoSampleMR v0.6.4. For multi-trait GWAS meta-analysis of asthma and hematological traits, we used MTAG, and GWAS in the Chinese population was performed using PLINK v1.9. The differential expression of CD36 was assessed in R using the wilcox.test function, applying a two-sided Wilcoxon rank-sum test with continuity correction. The custom scripts used in this study have been deposited in Zenodo and are available at: https://zenodo.org/records/15258788. The repository includes scripts used for data processing, statistical analyses, and figure generation.
References
Zhu, Z. et al. A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat. Genet. 50, 857–864 (2018).
Tsuo, K. et al. Multi-ancestry meta-analysis of asthma identifies novel associations and highlights the value of increased power and diversity. Cell Genom. 2, 100212 (2022).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Zhu, Z., Anttila, V., Smoller, J. W. & Lee, P. H. Statistical power and utility of meta-analysis methods for cross-phenotype genome-wide association studies. PLoS One 13, e0193256 (2018).
Song, Y. et al. Multitrait genetic analysis identifies novel pleiotropic loci for depression and schizophrenia in East Asians. Schizophr. Bull. https://doi.org/10.1093/schbul/sbae145 (2024).
Zhu, Z. et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J. Allergy Clin. Immunol. 145, 537–549 (2020).
Berry, M. et al. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax 62, 1043–1049 (2007).
Bel, E. H. et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N. Engl. J. Med. 371, 1189–1197 (2014).
Li, B. et al. Shared genetic architecture of blood eosinophil counts and asthma in UK Biobank. ERJ Open Res. 9, https://doi.org/10.1183/23120541.00291-2023 (2023).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Sagara, H. et al. Activation of TGF-beta/Smad2 signaling is associated with airway remodeling in asthma. J. Allergy Clin. Immunol. 110, 249–254 (2002).
Li, M. et al. Scutellarin alleviates ovalbumin-induced airway remodeling in mice and TGF-beta-induced pro-fibrotic phenotype in human bronchial epithelial cells via MAPK and Smad2/3 signaling pathways. Inflammation, https://doi.org/10.1007/s10753-023-01947-7 (2024).
Xuan, A., Yang, M., Xia, Q. & Sun, Q. Downregulation of NOX4 improves airway remodeling and inflammation by the TGF-beta1-Smad2/3 pathway in asthma. Cell Mol. Biol. 69, 201–206 (2023).
Patel, P. S. & Kearney, J. F. CD36 and platelet-activating factor receptor promote house dust mite allergy development. J. Immunol. 199, 1184–1195 (2017).
Magnaye, K. M. et al. DNA methylation signatures in airway cells from adult children of asthmatic mothers reflect subtypes of severe asthma. Proc. Natl Acad. Sci. USA 119, e2116467119 (2022).
Mallah, N., Rodriguez-Segade, S., Gonzalez-Barcala, F. J. & Takkouche, B. Blood eosinophil count as predictor of asthma exacerbation. A meta-analysis. Pediatr. Allergy Immunol. 32, 465–478 (2021).
Kerkhof, M. et al. Association between blood eosinophil count and risk of readmission for patients with asthma: historical cohort study. PLoS One 13, e0201143 (2018).
Xu, X. et al. Variants of CD36 gene and their association with CD36 protein expression in platelets. Blood Transfus. 12, 557–564 (2014).
Masuda, Y. et al. Diverse CD36 expression among Japanese population: defective CD36 mutations cause platelet and monocyte CD36 reductions in not only deficient but also normal phenotype subjects. Thromb. Res. 135, 951–957 (2015).
Kashiwagi, H. et al. Analyses of genetic abnormalities in type I CD36 deficiency in Japan: identification and cell biological characterization of two novel mutations that cause CD36 deficiency in man. Hum. Genet. 108, 459–466 (2001).
Kelly, E. A. et al. Potential contribution of IL-7 to allergen-induced eosinophilic airway inflammation in asthma. J. Immunol. 182, 1404–1410 (2009).
Pothoven, K. L. et al. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. J. Allergy Clin. Immunol. 136, 737–746.e734 (2015).
Brown, B. C., Asian Genetic Epidemiology Network Type 2 Diabetes, C., Ye, C. J., Price, A. L. & Zaitlen, N. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet 99, 76–88 (2016).
Chen, M. H. et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182, 1198–1213 e1114 (2020).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, https://doi.org/10.7554/eLife.34408 (2018).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081 (2017).
Shi, H., Mancuso, N., Spendlove, S. & Pasaniuc, B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am. J. Hum. Genet. 101, 737–751 (2017).
Venkatesan, P. 2023 GINA report for asthma. Lancet Respir. Med. 11, 589 (2023).
Zheng, Z. et al. DNA methylation clocks for estimating biological age in Chinese cohorts. Protein Cell, https://doi.org/10.1093/procel/pwae011 (2024).
Zhang, Q. X. et al. Searching across-cohort relatives in 54,092 GWAS samples via encrypted genotype regression. PLoS Genet. 20, e1011037 (2024).
Peng, Q. et al. Analysis of blood methylation quantitative trait loci in East Asians reveals ancestry-specific impacts on complex traits. Nat. Genet. https://doi.org/10.1038/s41588-023-01494-9 (2024).
Du, Z. et al. Whole genome analyses of Chinese population and de novo assembly of a Northern Han Genome. Genomics Proteom. Bioinforma. 17, 229–247 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Acknowledgements
The authors would like to express their sincere gratitude for the generous financial support provided by various funding sources. This study was supported by the Special Funds of Taishan Scholar Project, China (tsqn202211224, X.C.), the Excellent Youth Science Fund Project (Overseas) of Shandong, China (2023HWYQ-082, X.C.), and the National Natural Science Foundation of China (32270661, X.C.). Additional support was provided by the National Natural Science Foundation of China (82371830, HT, 81971552, H.T.), the Major Research Plan of the National Natural Science Foundation of China (92042306, H.T.), the National Basic Research Program of China (2015CB943203, H.T.), and the Academic Promotion Program of Shandong First Medical University (2019QL007, H.T.). We are also deeply grateful to all the study participants, including asthma patients and healthy controls, for their willingness to participate and their valuable contributions to this study. Their participation has been crucial in advancing our understanding of asthma genetics.
Author information
Authors and Affiliations
Contributions
Conceptualization: X.C., H.H., H.T. Methodology: L.Z., Q.Z., Y.J., L.Y., L.L., Y.S., B.P., C.Z., H.J., R.L., F.M., J.G., P.J. Investigation: L.Z., Q.Z., Y.J., L.Y., L.L., Y.S., J.G., F.M., R.L. Visualization: Y.J., L.L., Y.S. Formal analysis: Q.Z., Y.J., L.L., B.P. Data curation: B.P., C.Z., H.J. Funding acquisition: H.T., X.C., H.H. Project administration: X.C., H.H., H.T. Supervision: X.C., H.H., H.T. Writing—original draft: L.Z., Q.Z., Y.J. Writing—review & editing: X.C., H.H., H.T., P.J. L.Z., Q.Z., and Y.J. contributed equally to this work. X.C., H.H., and H.T. jointly supervised this study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks William Cookson, Amaury Vaysse and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhi, L., Zheng, Q., Jiang, Y. et al. Multi-trait genetic analysis of asthma and eosinophils uncovers pleiotropic loci in East Asians. Nat Commun 16, 5081 (2025). https://doi.org/10.1038/s41467-025-60405-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60405-0
