
Abstract
Steroid hormones, crucial for development and physiology, were traditionally believed to diffuse passively through membranes. However, recent evidence shows insect steroid ecdysone being secreted via regulated exocytosis, but the mechanisms ensuring successful hormone release into circulation remain unclear. Our study identifies specialized membrane protrusions, signaling filopodia, in the Drosophila prothoracic gland as essential for vesicle-mediated steroid release. Confocal imaging reveals that these actin- and tubulin-rich structures form a membrane-intertwined basal domain critical for secretion. Disrupting filopodia by interfering with basement membrane interactions—Perlecan or β-integrin—or filopodia-specific protein expression—α-actinin—significantly reduces ecdysone signaling by impairing its release, despite proper production in the gland. Additionally, filopodia dynamics, such as length and density, align with secretion timing and hormone circulating levels, suggesting their role in synchronizing release with physiological needs. The systematic presence of membrane protrusions in steroid-secreting glands across species prompts a comprehensive re-evaluation of steroid release mechanisms.
Similar content being viewed by others


Introduction
Steroid hormones are essential bioactive molecules that regulate a wide range of physiological processes in both animals and plants, including immune response, salt and water balance, glucose metabolism, and developmental sexual maturation1,2. Rapid fluctuations in circulating steroid hormone levels are essential for exerting their effects on distant target tissues. Despite their critical roles, the mechanisms driving steroid hormone release from endocrine tissues remain poorly understood. Endocrine gland cells are typically organized in cords or clumps, attached laterally to neighboring cells, and often lack classical apical-basal polarity. This unique organization raises an important question: How do endocrine glands efficiently direct rapid surges of hormone secretion outward?
While steroid hormones were traditionally thought to diffuse across lipid bilayers, recent studies have revealed a more complex and regulated process. For instance, in Drosophila, the release of the steroid hormone ecdysone involves a regulated vesicular trafficking mechanism that depends on calcium signaling, Rab3, Synaptotagmin 1 (Syt1), and an ABC transporter (Atet), which together facilitate ecdysone transport across the lipid bilayer3. In humans, steroid hormones are detected in blood and urine within exovesicles4. These findings suggest that steroid hormone secretion is regulated, potentially involving multiple pathways depending on the tissue or signaling context.
Regulated exocytosis and multivesicular body (MVB) recycling pathways require a fusion event between the plasma membrane and the membrane of the vesicle or MVB, which poses a significant energetic challenge for cells5. The mass secretion of steroids during maturation exacerbates this issue, highlighting the need for efficient mechanisms to reduce energy demands. Cells can mitigate these demands by altering their lipid composition6,7, membrane tension8, or curvature9. The energy required for stalk and pore formation during membrane fusion is lower at membranes with higher curvature9,10. Therefore, by optimizing these critical membrane properties, cells might enhance membrane fusion efficiency, facilitating vesicle secretion.
Interestingly, electron microscopy studies from the 1960s on the crab endocrine Y-organ revealed cell surface irregularities with steroid-containing vesicles11, suggesting a direct link between these membrane structures and steroid hormone secretion. Similar observations have been made in the prothoracic gland of lepidopterans12 and dipterans13, as well as in vertebrate steroid-secretory glands like the adrenal cortex14 and luteal cells15,16,17. These irregularities, described as blebs, microvilli, or filopodia depending on the tissue and fixation technique (note that membrane projection properties are affected by fixation methods18), appear to be common features related to steroid secretion across different species. This crucial aspect has largely been overlooked.
Filopodia, cytonemes, microvilli, and blebs are dynamic membrane protrusions that play essential roles in cellular processes, including migration, cell adhesion, cell-to-cell communication, signaling, and wound healing19,20. Primarily composed of tightly bundled actin filaments, these slender, finger-like structures can also incorporate microtubules. The interplay between actin and microtubules is vital for maintaining their stability21,22. Notably, microtubules act as scaffolds for vesicle transport within protrusions, raising the question of whether filopodia containing microtubules not only facilitate the outward transport of steroid vesicles but also locally modify plasma membrane curvature23, a key factor for membrane fusion and subsequent vesicle secretion.
In this study, we explore the role of membrane projections in mediating the secretion of ecdysone in the Drosophila prothoracic gland (PG). We provide a detailed description of the PG’s structure, highlighting a bilayer organization surrounded by a basement membrane (BM) lacking classical apical-basal polarity. Through clonal analyses and ex vivo experiments, we identified filopodia projections concentrated in the sub-BM region. These filopodia contain actin, β-integrin, and microtubules, forming an asymmetric membrane network. Disruption of the endocytic/MVB pathway via Rab11 downregulation altered PG cell morphology, decreased filopodia abundance, and delayed development. Furthermore, the asymmetric distribution of filopodia depends on BM components like Perlecan/Trol and β-integrin. We also found that filopodia contain ecdysone secretory machinery, including Syt1 and Atet. Silencing filopodia-specific components resulted in developmental delays and reduced ecdysone secretion, confirming their crucial role in secretion. Filopodia dynamic significantly changes during the late larval stages in terms of their size and density, correlating with ecdysone secretion peaks. In conclusion, our findings demonstrate that a specialized asymmetric membrane domain, formed by filopodia projections, is required for steroid hormone secretion in the insect prothoracic endocrine glands. This membrane network structure is essential for the efficient release of ecdysone.
Results
The PG lacks classical apical-basal polarity but exhibits polarized secretion properties
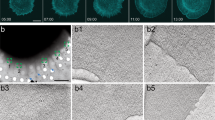
To assess whether the PG shares traits with other endocrine glands in vertebrates and invertebrates, we analyzed the arrangement of PG cells in the ring gland using confocal microscopy. Z-stacks and orthogonal XZ/YZ sections revealed a well-organized tissue with two layers: a dorsal layer containing a greater number of smaller cells and a ventral one (Fig. 1a; Supplementary Fig. 1a). Only the ventral layer is in direct contact with tracheal cells (Fig. 1a). A 3D reconstruction is shown in Fig. 1a’. As expected, no lumen was observed between the layers (Supplementary Fig. 1b, c). PTTH neurons were restricted to the interface of the two layers (Fig. 1b, b”, see arrowheads), and the PTTH receptor, Torso (Tor), was nicely concentrated at postsynaptic regions of the middle cell membrane (Fig. 1c). Using the terribly reduced optic lobes (trol)-GFP knock-in reporter (a homolog of vertebrate Perlecan), we observed a BM surrounding the gland (Fig. 1d, d’), also containing Viking (Vkg), Nidogen (Ndg), and Laminin-B1 (LanB1) confirmed by knock-in GFP fusion protein reporters (Supplementary Fig. 1d-d”’). Unexpectedly, we found BM protein dots within the tissue along the lateral and middle PG cell membranes (Fig. 1d, d’, arrowheads; Supplementary Fig. 1d-d”’). These dots correspond to Collagen IV intercellular concentrations (CIVIC) structures, as they are composed of Collagen IV (a heterotrimer formed by two α1-like chains encoded by Collagen at 25C (Cg25C) and one α2-like chain encoded by viking (vkg)), Trol, Ndg, and LanB1 as shown by the colocalization of Cg25C with vkg-GFP, trol-GFP, Ndg-GFP, and LanB1-GFP. (Supplementary Fig. 1d-d”’).

a, a’ The PG is organized into a dorsal and ventral layer. Dotted lines in the ventral layer image outline the trachea (a). 3D visualization of the ring gland (a’) composed of the PG (prothoracic gland), the corpora allata (CA), and the corpora cardiaca (CC). The aorta and trachea are indicated. b-b” Maximal projection of prothoracicotropic hormone (PTTH) staining (b) and two Z-sections revealing that PTTH neurons project their axons at the midline of the PG (arrowheads in b’ and b”). n ≥ 10 independent experiments (c) Synaptic PTTH boutons reveal PTTH in the presynapse and Torso in the postsynapse. nexperiment ≥ 3. d, d’ Expression of trol-GFP in the BM plane and in a lateral plane (d). The XZ section (d’) facilitates the observation of the trol-GFP dots. These dots (indicated by arrowheads) are CIVICs. nexperiment ≥ 10. e Z-sections reveal that the adherens junction proteins Arm and ECad, the septate junction markers Cora, Dlg1, as well as βPS, are uniformly distributed along the PG cell membranes. nexperiment ≥ 10. f Heatmap of Syt1-GFP in an XZ section of a PG cell. Flip-out Syt1 clones were generated by crossing the heat shock Flipase (hsFLP); Act > CD2>Gal4 line with UAS-Syt1-GFP. Following heat shock, CD2 is excised in random cells, enabling Syt1-GFP expression under Act-Gal4 control, here abbreviated as hsFLP; Act»Syt1-GFP. The quantification, shown below, is made across ROI boxes appearing in dotted lines, nclones = 7 (see Methods). Data are presented as mean ± 95% CI and were subjected to the Friedman test followed by Dunn’s multiple comparison tests (ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Source data are provided as a Source Data file. White scale bars: 20 μm; yellow scale bar: 5 μm. g Schematic representation of a PG dorsal layer and XZ cut.
Despite its epithelial origin24, PG cells exhibit CIVIC structures, which are typically associated with unpolarized mesodermal fat body cells25. Next, we examined whether PG cells display apical-basal polarity by staining them for epithelial markers. Apical markers Bazooka (Baz)-GFP and Crumbs (Crb)-YFP were absent from the membranes (Supplementary Fig. 1e, f). Adherens junction proteins Armadillo (Arm), E-cadherin (Ecad), the septate junction markers Coracle (Cora) and Disc large 1 (Dlg1), as well as the βPS integrin subunit encoded by myospheroid (mys) were uniformly distributed along the membrane (Fig. 1e). The N-cadherin (Ncad) was only expressed in the corpora allata (Supplementary Fig. 1g). These findings strongly suggest that Drosophila PG lacks typical apical-basal polarity and preserves mesodermal properties acquired at the late embryonic stages24, a characteristic also seen in other endocrine glands like the adrenal gland26.
Interestingly, a Z-stack analysis of individual PG cells overexpressing Syt1-GFP, a secretory marker, achieved through the generation of flip-out clones27, revealed that Syt1-GFP is concentrated twice as much in the sub-BM region compared to the lateral or midline membranes (Fig. 1f), indicating a polarized secretion. Therefore, three distinctive cell membrane domains are identified in the PG: a sub-BM membrane domain right below the BM enriched in Syt1, a lateral membrane one only in contact laterally with the neighboring cells of the same layer, and the middle membrane in contact with the PTTH neuron projections and cells of the other layer (schematic in Fig. 1g).
PG cells exhibit polarized features highlighted by an uneven distribution of filopodia in the sub-BM domain
We induced random fluorescent clones within the PG using the flip-out technique and visualized individual cell shapes. Confocal images revealed that PG cells exhibit yet unidentified membrane extensions of several µm resembling filopodia (Fig. 2a, arrowheads). These fluorescent extensions are visible only when in contact with a neighboring black cell that does not express the fluorescent signal. These projections were predominantly observed in the sub-BM region, where PG cells face the BM, and were largely absent from the lateral and middle membrane domains. This was evidenced by the distinct distribution observed using two independent membrane markers such as myristoylated (myr)-GFP (Fig. 2b, arrowheads) and the PH domain of Phospholipase C (PLC)-GFP (Fig. 2c, arrowheads). Overexpression of myr-GFP and PLC-GFP in the fat body, another highly secretory larval tissue composed of polyploid cells like the PG, resulted in smooth edges without the presence of filopodia-like projections (Fig. 2d), indicating that these membrane projections are specific to PG cells.

a Flip-out clones randomly expressing the fluorescent marker RFP in PG cells extend membrane protrusions (see arrowheads in the inset). The image corresponds to a maximum projection of a Z-stack. b Flip-out clone expressing the membrane marker myr-GFP. Long filopodia are observed on the sub-BM side (arrowheads), while fewer and shorter filopodia are observed in a lateral plane. The YZ section reveals a long filopodium present in the sub-BM region (arrowhead). c Flip-out clone expressing the membrane marker PLC-GFP shows filopodia in the sub-BM region (arrowheads) but not in the lateral plane. d myr-GFP (left) or PLC-GFP (right) flip-out clones induced in the fat body. No filopodia are observed. e Two neighboring cells (RFP positive due to CoinFLP) express complementary GFP fragments (spGFP1–10 or spGFP11) using the GRASP technique. Reconstructed GFP signal is detected on the sub-BM side, revealing filopodia contact between the two neighboring cells (arrowheads). A GFP signal is also observed in the lateral plane, appearing as a thin stripe. f Flip-out clones expressing an RNAi against mys (RFP positive) in a PG stained for βPS (green). In the sub-BM plane, βPS-decorated endogenous filopodia appear as punctate signals resembling projections in two WT cells (RFP negative, arrowheads in the zoomed image). No filopodia are detected in the lateral plane. g Flip-out PLC-GFP clone expressing cofilin RNAi. In the sub-BM plane, long filopodia containing F-actin are observed (arrowheads). In contrast, no actin-based filopodia are detected in the lateral plane despite F-actin stabilization. h Flip-out clone marked by PLC-GFP and the actin reporter Lifeact-Ruby, which label filopodia (arrowheads). i Flip-out clone expressing RFP in a PG stained against αTub85E. Microtubules are present in the filopodia (arrowheads in the zoomed images). j ex vivo experiment showing filopodia tracking through myr-GFP expression. Blue, white, and red arrowheads indicate different filopodia over time. Pictures are representative (n = 276 filopodia across 15 PG). White scale bars: 20 μm; yellow scale bars: 5 μm. k Schematic representation of filopodia distribution in PG cells, showing the presence of F-actin, tubulin, and βPS.
To further investigate the organization of these membrane projections, we used the CoinFlp combined with GRASP (GFP Reconstitution Across Synaptic Partner) systems28,29, allowing us to observe interactions between neighboring PG cells. When complementary parts of GFP (spGFP1–10 and sp-GFP11) are expressed in adjacent cells, GFP reconstitution revealed intertwined membrane projections primarily located in the sub-BM region (Fig. 2e, arrowheads). In contrast, in lateral sections, a distinct stripe of GFP signal was evident (Fig. 2e), reflecting the reduced number of these projections in this plane, confirming a clear asymmetry in the projection distribution between cells.
These membrane projections resemble filopodia, thin, finger-like structures formed by bundles of actin filaments that interact with the extracellular matrix (ECM) through the transmembrane protein βPS (the β subunit of the integrin dimer). To determine whether the membrane projections are indeed filopodia, we aimed to visualize the localization of βPS. To do so, we induced random clones silencing mys, the gene encoding for βPS, in PG cells to employ the black background clonal method to visualize filopodia. Under this mosaic expression pattern for βPS, we observed the presence of endogenous irregular punctuated projections in the sub-BM region consistent with filopodia decorated by βPS (Fig. 2f, arrowheads in inset).
Next, we analyzed the presence of F-actin using phalloidin staining. Unexpectedly, distinct actin filaments were not visible in the sub-BM region when staining with phalloidin; instead, we observed a diffuse punctate pattern, even though actin was clearly present in the cell cortex (Supplementary Fig. 2a, a’). We suspected that a balance between F-actin polymerization and depolymerization might have hindered its detection. To test this, we reduced actin dynamics by creating random PG cell clones with silenced cofilin (twinstar), which codes for a protein responsible for F-actin depolymerization30. Under these conditions, we observed a significant accumulation of F-actin in the sub-BM area (Fig. 2g, arrowheads); no filopodia could be seen in the lateral part despite F-actin stabilization (Fig. 2g). We next used the Lifeact-Ruby reporter, which revealed robust actin localization in filopodia, highlighted by the PLC-GFP membrane marker (Fig. 2h, arrowheads). This finding is further supported by GMA-GFP and Utrophin (UTRN)-GFP reporters, two actin-binding proteins (Supplementary Fig. 2b). Together, these data reinforce the idea that the PG membrane projections are defined by a specific enrichment of actin at the cell cortex.
The presence of microtubules in secretory filopodia is not widely observed31, although some exceptions have been reported18,32. However, microtubules are well-established as critical players in the dynamics of other filopodia types, such as those in growth cones and migratory lamellipodia33. Given the high expression of the αTub85E subunit in active secretory PGs34, we stained the gland to assess whether this α-tubulin subunit is present in PG filopodia. Surprisingly, we observed microtubules within the filopodia structures (Fig. 2i, arrowheads). Moreover, a lateral section revealed the presence of αTub85E in the cytoplasm in a uniform, non-polymerized manner, suggesting the absence of αTub85E microtubules within the cytoplasm (Supplementary Fig. 2a, a’).
Since filopodia are delicate and often compromised during fixation, we performed ex vivo experiments to capture their true structures and dynamics. In other systems, filopodia display diverse dynamics—for instance, extending and retracting over approximately 5 min in Drosophila abdominal histoblasts35 or completing extension and retraction within 30–50 s in zebrafish primordial germ cells36. Therefore, we conducted 15-min ex vivo time-lapse imaging on several independent PGs to examine PG filopodia dynamics (see Methods). Surprisingly, we observed neither the formation of new filopodia nor the complete retraction of any of the 276 filopodia analyzed, as illustrated in Fig. 2j (see arrowheads) and Supplementary Movie 1. These results suggest that PG filopodia exhibit lower dynamics than filopodia from other systems. This aligns with previous findings showing that the association between F-actin and microtubules provides mechanical stability21,22.
Our findings demonstrate that PG cells possess filopodia extensions composed of F-actin, integrins, and microtubules, which exhibit a polarized distribution predominantly beneath the BM. This organization indicates that PG cells have polarized properties based on the asymmetric distribution of cytoskeleton components (Fig. 2k).
The MVB/recycling pathway is required for the asymmetric membrane identity of PG cells and for timing development
Previous electron microscopy studies have revealed an uneven distribution of multivesicular bodies (MVBs) primarily near the cell cortex on the BM-facing side of PG cells13. Since MVBs are visualized only in highly secretory PG cells13 we evaluated the function of the PG MVB/recycling pathway on developmental timing, which is defined by the ecdysone surges. Our experiments revealed that downregulation of the small GTPase Rab11 leads to a 190-h delay in development (Fig. 3a). Additional experiments silencing other MVB pathway components, including Vps25, Vps36, and Shrb, confirmed a developmental delay ranging from 65 to 80 h (Fig. 3b). These findings underscore the critical role of the MVB pathway in the PG functionality; therefore, we investigated potential mechanisms behind these effects. First, we evaluated the direct secretion of ecdysone into exovesicles, by altering the MVB/recycling pathway. Indeed, human steroid hormones have been detected in exosomes, whose secretion depends on the MVB pathway4. Therefore, we expressed Rab11-GFP in PG cells and purified circulating exosomes from the hemolymph using an ultracentrifugation protocol37. Even though we detected the presence of Rab11-GFP PG containing exosomes in our exosome purified fraction by Western blot, our ecdysone measurement showed only negligible amounts of ecdysone in this fraction (Supplementary Fig. 3a), indicating that the MVB/recycling pathway does not directly secrete ecdysone. This aligns with previously reported Rab3-dependent ecdysone secretory pathway3.

a Silencing Rab11 results in a 190-h delay in metamorphosis onset compared to the control. Number of sample tubes: nphm>xw1118 = 8, nRNAi-Rab11xw1118 = 18, nphm>RNAi-Rab11 = 10. b Silencing Vps25, Vps36, or shrb in PG cells resulted in a several-hour delay compared to the control. Number of sample tubes: nphm> xw1118 = 8, nRNAi-Vps25xw1118 = 10, nRNAi-Vps36xw1118 = 6, nRNAi-shrbxw1118 = 4, nphm>RNAi-Vps25 = 8, nphm>RNAi-Vps36 = 2, nphm>RNAi-shrb = 2. phm > RNAi-Rab11 PG lacks microtubule-based (c) and βPS-based (d) filopodia in the sub-BM region. Filopodia are visible with the PLC-GFP membrane marker in ex vivo control PG cells (e, arrowheads) but disappear upon Rab11 downregulation (f). Comparison of control w1118 PG (g) and phm > RNAi-Rab11 PG (h) showing F-actin (single section) and PTTH staining (maximum projection of a Z-stack) processed with a depth color code from a Fiji plugin. XZ cuts reveal PTTH axons positioned between the PG bilayer in the control (g, arrowheads), whereas upon recycling pathway disruption, PTTH axons are also mislocalized just below the BM (h, arrowheads). i Axon from a phm > RNAi-Rab11 PG at the sub-BM region. The PTTH staining overlaps with its receptor Torso, suggesting that the mislocalized synaptic buttons are functional. j In phm > RNAi-Rab11 PG at the sub-BM region, mislocalized PTTH-containing synaptic buttons exclude βPS expression. Insets 1 and 2 highlight the signal exclusion between PTTH and βPS expression. k Z sections of a trolJO271#49 mutant PG reveal the loss of a bilayer organization. l αTub85E staining in trolJO271#49 mutant PG shows short microtubules in the sub-BM region, while in a lateral plane, αTub85E staining appears normal. m Down-regulation of mys impedes microtubule extension. n Developmental timing of trolJO271#49 mutant vs w1118 individuals. The trolJO271#49 mutant enters pupariation 24 h after the control line. nw1118 = 10, ntrolJO271#49 = 6. White scale bars: 20 μm; yellow scale bars: 5 μm. Data are presented as means ± SEM in (a, b, n). Source data are provided as a Source Data file.
Interestingly, the MVB/recycling pathway is also involved in the polarized distribution of adhesion proteins (reviewed in refs. 38,39) and filopodia formation40. Therefore, we investigated whether this pathway impacts filopodia’s asymmetric and polarized distribution in Drosophila PG cells. We manipulated the expression of Rab11 in the PG cells using the PG-specific expression driver, phm-Gal441. We observed the formation of rounded cells lacking αTub85E staining between them (Fig. 3c), suggesting a marked disruption of the filopodia network within the sub-BM region. Furthermore, reduced Rab11 levels in the PG resulted in well-defined cell borders with βPS accumulation at the cell edges (Fig. 3d), suggesting a substantial reduction of filopodia extensions. To confirm these findings and knowing that filopodia could be disrupted by the fixation mechanism18, we performed ex vivo analyses of PG cells, showing that, unlike the control group (Fig. 3e, arrowheads), clone cells with Rab11 silencing for only 24 h failed to extend filopodia as reported with the PLC-GFP-marked (Fig. 3f).
Consistent with the loss of identity of the PG cells membrane facing the BM, we also observed alterations at the PG midline when silencing Rab11. In a control situation, PTTH neurons project their axons at the midline (Fig. 3g, arrowheads). However, down-regulation of Rab11 expression in the PG cells causes aberrant PTTH axon organization, often leading to mis-localized axons situated below the BM (Fig. 3h, arrowheads). Remarkably, despite the mis-localization, PTTH staining, found in the pre-synapse, co-localizes with its receptor Torso in the post-synapse, suggesting the potential functionality of these mislocalized synaptic buttons (Fig. 3i). It is interesting to note that when the PTTH neuron’s synaptic buttons are below the BM, we observed the absence of βPS expression at these membrane sites (Fig. 3j, arrowheads in insets), suggesting that filopodia extensions and PTTH neuron’s synaptic buttons are mutually exclusive structures. These findings underscore the critical role of the MVB pathway in the PG functionality, including its 3D organization, such as the uneven distribution of filopodia in the BM membrane domain and the restricted interaction of the middle membrane domain with PTTH neurons.
However, given the pleiotropic functions of Rab11, we cannot directly attribute the absence of filopodia to the strong developmental timing phenotype observed. This hypothesis will be further explored in subsequent experiments.
Basement membrane Perlecan/Trol and integrins shape the PG 3D structure
Given Rab11’s role in regulating βPS composition in the plasma membrane via the MVB/recycling pathway42, and considering that β-integrin interacts with BM components at filopodia20, we investigated whether this interaction influences the asymmetric distribution of filopodia in PG cells. To develop a better methodological approach to address this question, we first determined the source of Drosophila Perlecan, named Trol, in the BM and CIVIC structures.
We conducted trol-RNAi clone expression analyses in the PG, marked with RFP, and observed that the presence of Trol in the internal dots of CIVICs disappeared, which are present in the unique wild-type (WT) non-RFP clone. However, in the same trol-RNAi clone analyses, we noticed that the levels of Trol at the BM were slightly higher autonomously in this WT cell than in the neighboring trol-RNAi clone cells (Supplementary Fig. 3b). This contribution of the PG on Trol in the BM was further confirmed when silencing trol in all the PG using the RNAi technique. In this case, we observed a strong reduction of Trol in the CIVIC structures, whereas the one present in the BM is still evident but slightly reduced when compared with the control (Supplementary Fig. 3c). Please note that Trol staining of control and trol-RNAi loss of function PGs was performed in the same tube for better comparison. Conversely, the trol JO27#49 hypomorphic mutant shows a strong reduction in Trol expression at the BM, maintaining some signal at the internal CIVIC dots (Supplementary Fig. 3c). These results indicate that the Trol protein present in the CIVICs is produced exclusively by the PG cells. In contrast, Trol in the BM comes partially from the hemolymph (as previously suggested in ref. 43) with a non-depreciable autonomous PG contribution.
Therefore, to address whether the BM plays a role in the asymmetric distribution of filopodia in the PG cells, we analyzed the PG in the hypomorphic allele trol JO27#49, which shows the absence of Trol in the BM (Supplementary Fig. 3c). These PGs are relatively round compared to WT glands and show aberration in the Z plane. Indeed, three layers of cells replace the PG two-layer organization (Fig. 3k). Examination of αTub85E staining in the sub-BM plane revealed punctuated staining of microtubules instead of the elongated signal observed in the controls (Fig. 2i). In contrast, in the cytoplasm, the αTub85E pattern is normal (Fig. 3l), suggesting a specific effect of Trol at the BM in αTub85E polymerization at the sub-BM plane. Accordingly, silencing trol specifically in the PG, affecting mainly the presence of Trol in the CIVICs, does not affect PG cell layers or αTub85E staining at the sub-BM (Supplementary Fig. 3d and e respectively). Moreover, direct downregulation of βPS expression in the PG cells causes a similar change in αTub85E staining in the sub-BM, shown by the reduced αTub85E signal at the PG cells boundaries (Fig. 3m), suggesting the absence of αTub85E polymerization at the filopodia. Moreover, trol JO27#49 mutants, as well as larvae that have βPS silenced exclusively in the PG cells, enter metamorphosis with an approximately 24-h delay compared to control (Fig. 3n; Supplementary Fig. 3f), suggesting that alteration in the 3D organization in these glands is relevant for the establishment of the large ecdysone peak inducing the onset of metamorphosis.
PG filopodia contain the machinery for ecdysone secretion
To assess whether filopodia participate in ecdysone secretion, we first examined if they harbor secretory vesicles. We stained for Cysteine String Protein (CSP), a key membrane component of regulated secretory organelles44,45,46, which is known to localize to neuronal synaptic vesicles along with the calcium-dependent secretory protein Syt1 previously linked to ecdysone secretion3. Using clonal analyses, we expressed PLC-GFP to visualize filopodia and performed immunostaining against CSP. We observed CSP-positive puncta within filopodia (Fig. 4a, arrowheads), suggesting their involvement in vesicular transport. Additionally, we detected punctate signals for Syntaxin1A (Syx1A)—a crucial SNARE complex component that mediates vesicle fusion with the plasma membrane47 —along the filopodia (Fig. 4b, arrowheads).

Zoomed-in images of a PLC-GFP-expressing clone showing that CSP (a) and Syx1A (b) localize in filopodia in the sub-BM area. Arrowheads indicate CSP and Syx1A puncta within the filopodia. c A Ypet-Atet-expressing clone showing Ypet-Atet at the membrane and in filopodia (arrowheads) at the sub-BM region. Zoomed-in images of Ypet-Atet-expressing clones showing CSP (d, arrowheads) and Syx1A (e, arrowheads) dots in filopodia. f A clone expressing Syt1-GFP where Syt1-GFP localized at the membrane and in filopodia (arrowheads). g ex vivo tracking of a Syt1-GFP vesicle (arrowhead) in a PG filopodium over time. Note that Ypet-Atet and Syt1-GFP were overexpressed for a short period of time (7 h) to achieve low protein expression levels. White scale bars: 20 μm; yellow scale bars: 5 μm. For each result shown here, n ≥ 3.
To determine if ecdysone-containing vesicles are present in filopodia, we analyzed the localization of Atet, a specific ABC transporter responsible for actively pumping ecdysone into vesicles3. Under low expression conditions, fluorescently tagged Atet (Ypet-Atet) localized to discrete puncta along filopodia (Fig. 4c, arrowheads). Note that to minimize artifacts such as mislocalization or clumping, we analyzed its distribution after a short overexpression period (7 h following the heat shock-induced clone, low expression level). Notably, Atet colocalized with CSP vesicles in filopodia (Fig. 4d, arrowheads). Atet-YFP vesicles are consistently surrounded by CSP-positive puncta (Supplementary Fig. 4a), which aligns with their respective topologies, Atet-YFP faces the vesicle interior3, while CSP associates with the vesicle surface44. Similarly, Atet colocalized with Syx1A in filopodia (Fig. 4e, arrowheads), but only when the reporter was near the plasma membrane or within filopodia (Supplementary Fig. 4b).
Although extending the overexpression period to 24 h (high expression level) doubled Atet’s signal intensity (Supplementary Fig. 4c, c’), its localization remained unchanged (Supplementary Fig. 4d). Together, these findings reinforce the idea that filopodia house secretory vesicles carrying ecdysone. A similar localization pattern was observed with Syt1-GFP that exhibited punctate fluorescence within filopodia under both low-expression (Fig. 4f, arrowheads) and high-expression conditions (Supplementary Fig. 4e). Finally, ex vivo live imaging of Syt1-GFP confirmed active vesicle movement within the filopodia (Fig. 4g, arrowheads).
Together, these findings strongly suggest that PG filopodia contain the essential molecular machinery for vesicular transport and ecdysone secretion. Actually, the localization of endogenous secretory proteins and ecdysone vesicles-related reporters—CSP, Syx1A, Atet, and Syt1—within filopodia strongly supports their role as specialized secretory structures that facilitate ecdysone release into the hemolymph.
Filopodia mediate ecdysone secretion
To confirm the direct role of filopodia in ecdysone secretion, we first disrupted filopodia in the PG and analyzed the effects on developmental timing, which depends directly on secreted ecdysone signaling. Using the PG-specific phm-Gal4 driver, we conducted a targeted RNAi screen focusing on genes involved in filopodia structure. First, we manipulated the expression of α- and β-tubulin subunits that function as heterodimers. Misexpression of αTub85E, βTub85D, and βTub56D resulted in a larval arrest at the third larval stage or significant developmental delays of 40 and 60 h, respectively (Fig. 5a), indicating disrupted ecdysone signaling. Interestingly, αTub85E is the α subunit shown in Fig. 2i as specifically enriched in the PG cell cortex.

a Screening for α- and β-tubulin subunits involved in timing development by expressing RNAi under the control of phm-Gal4. b Screening for general actin-associated proteins involved in developmental timing. c Down-regulation of filopodia-associated proteins diaphanous, enabled, or α-actinin leads to a significant developmental delay or arrest. d-d” ex vivo PG confocal images of control, dia PG-downregulation, and Actn silencing showing its effect on filopodia (d, arrowheads). Quantifying filopodia length and density using PLC-GFP clonal analysis confirms that dia and Actn down-regulation significantly impair both filopodia length and density (d”). nw1118 = 9 clones, nRNAi-dia33424 = 9, nRNAi-Actn7762 = 9 (d’). Compared to a control PG, dia or Actn PG silencing does not noticeably affect overall PG shape (d”). e 20E feeding rescues developmental delay caused by silencing dia or Actn expression in the PG. Data in (c) for RNAi-Actn7762 and RNAi-dia33424 are reused in (e) as controls without ecdysone feeding. The same phm>Dcr-2 x w1118 control has been used in (a–c, e). f-f” Actn down-regulation affects ecdysone secretion, not production. For the total ecdysone measurement, the number of independent samples is the following: nphm>Dcr-2xw1118 = 3, nRNAi-Actnxw1118 = 3, nphm>Dcr-2xRNAi-Actn = 2. For the % of circulating ecdysone, the number of independent samples is: nphm>Dcr-2xw1118 = 9, nRNAi-Actnxw1118 = 9, nphm>Dcr-2xRNAi-Actn = 6. Dib expression is unaffected upon Actn silencing, as immunostaining shows (f’). Similarly, nvd and phm RNA levels remain unchanged in RNAi-Actn conditions, as assessed by qPCR (f”). Number of independent samples: nphm>Dcr-2xw1118 = 3, nRNAi-Actnxw1118 = 3, nphm>Dcr-2xRNAi-Actn = 3. White scale bars: 20 μm; yellow scale bars: 5 μm. Data are presented as follows: means of the differences ± SEM (a–c, e), median ± IQR (d’), mean ± SEM (f, f”). Except for (e), data were subjected to the Kruskal-Wallis test, followed by Dunn’s multiple comparison tests. In (e), data were subjected to the two-tailed Mann–Whitney test. (ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Source data are provided as a Source Data file.
Next, we destabilized the actin cytoskeleton (Fig. 5b). Silencing Actin (Act5C) with two independent RNAi constructs led to either an early L3 stage arrest or a 50-h developmental delay. Disrupting actin polymerization or depolymerization by expressing chickadee (chic)48 or cofilin30 caused delays of 30 and 95 h, respectively. Knocking down Arpc3B, involved in actin branching29, and explicitly enriched in ring gland34, resulted in L3 arrest. We further quantified the total number of PG cells under each condition and found no significant differences, except for Act5C knockdown, which led to an approximately 30% reduction in cell number (Supplementary Fig. 5a). This reduction is consistent with previous reports of mitosis occurring in first instar larvae49. The downregulation of Act5C, cofilin, or chic in the PG led to an altered αTub85E immunostaining pattern in the cell cortex, previously linked to the presence of filopodia (Supplementary Fig. 5b). However, except for αTub85E, which shows specific localization at the cell cortex, altering the expression of these cytoskeletal proteins may have broader effects beyond filopodia, as they are essential for fundamental cellular functions22.
Therefore, we further altered the expression of cytoskeletal components already described as specific to filopodia (Fig. 5c). Targeting the formin diaphanous (dia) and enabled (ena), both involved in unbranched actin filament polymerization at filopodia tips20,50, resulted in either L3 stage arrest or a 12-h developmental delay. Downregulation of α-actinin (Actn), an actin cross-linking protein at the base of filopodia50, also caused significant developmental timing disruptions, showing either L3 arrest or around a 70-h delay. Moreover, silencing dia or Actn results in an altered αTub85E immunostaining pattern in the cell cortex with visible cell edges (Supplementary Fig. 5b). Their effect on the filopodia network was confirmed qualitatively by ex vivo experiments (Fig. 5d, arrowheads) and quantitatively in fixed tissues (Fig. 5d’), which showed that when dia expression was downregulated, there were shorter and less dense filopodia compared to the control group. A more significant effect on filopodia abrogation was observed when Actn was silenced in the PG. Notably, the PG retained its overall shape and morphology (Fig. 5d”).
Interestingly, feeding larvae with ecdysone, in which dia or Actn expression was downregulated in the PG, our most significant filopodia-specific candidates, significantly rescued the previously observed developmental timing defects (Fig. 5e). This indicates that the observed developmental defects were due to abnormal ecdysone signaling. Furthermore, to determine whether this defect in ecdysone signaling is due to a direct secretion defect, we determined the ecdysone secretory index as the percentage of ecdysone present in the circulating hemolymph relative to total larval ecdysone in the wandering stage. Under control conditions, around 20% of total ecdysone circulates during the wandering stage (Fig. 5f), as previously reported51. When Actn expression was downregulated in the PG, total ecdysone levels remained normal, indicating that PG cells with reduced Actn levels still produced ecdysone efficiently. However, the ecdysone secretory index dropped to just 3% of the total larval ecdysone (Fig. 5f).
To confirm that Actn loss does not affect PG cell functionality, we analyzed the expression of key ecdysone biosynthetic enzymes using two complementary approaches. First, immunostaining for Disembodied (Dib) showed comparable protein levels in WT PG cells and Actn-silenced clones (Fig. 5f’). Second, RT-qPCR analysis of neverland (nvd) and phantom (phm)—two transcriptionally regulated biosynthetic enzymes—revealed no significant differences between WT larvae and those with Actn silencing in the PG (Fig. 5f”). This confirms that Actn silencing in PG cells does not interfere with general transcription or cholesterol-based ecdysone production; however, it disrupts filopodia structures and ecdysone secretion. Consistent with our hypothesis, these findings suggest that the membrane-intertwined sub-BM filopodia network is essential for ecdysone secretion.
Filopodia developmental dynamics correlate with the establishment of the large metamorphosis-inducing ecdysone peak
Given the previous observation that several morphological changes occur in PG cells during late larval development13, which coincides with a peak of ecdysone production and secretion, we investigated whether filopodia exhibit developmental changes. We measured various filopodia parameters at early and late third instar larvae stages (eL3, lL3; Fig. 6a, arrowheads) following the analytical method shown in Supplementary Fig. 6a.

a Representative images of an eL3 and lL3 PG cell expressing Syt1-GFP. Filopodia are pointed out with arrowheads. b–e Filopodia length increases significantly from eL3 to lL3 (b), while their diameter remains stable (c). The density of filopodia also increases significantly during the L3 stage (d), resulting in a 70% increase in the exchange surface of the sub-BM area (e). Number of clones analyzed: neL3 = 11, nlL3 = 8. Scale bars: 20 μm. Data are presented as median of the means ± IQR in (b, c) and as median ± IQR in (d, e). Data were subjected to the two-tailed Mann-Whitney test (ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Source data are provided as a Source Data file.
We observed a significant increase in average filopodia length from approximately 1.8 μm in eL3 to 2.8 μm in lL3 (Fig. 6b). Note that no difference in filopodia length is observed between the cells of the dorsal layer and the ventral layer at both time points (Supplementary Fig. 6b, c). Also, filopodia diameter showed no significant difference between the two stages, ranging between 0.2 and 0.3 μm (Fig. 6c). Although the filopodia area increased between the early and late stages due to lengthening, the relative exchange surface of each filopodium, normalized to cell size, remained similar between the two time points (Supplementary Fig. 6d).
Notably, we observed a substantial increase in filopodia density, from 0.07 filopodia per μm² in eL3 to 0.33 filopodia per μm² in lL3 (Fig. 6d). Taken together, filopodia at the sub-BM surface increased the exchange surface by approximately 10% in eL3, whereas in lL3, the higher filopodia density led to a 70% increase in cell exchange surface (Fig. 6e). Our results indicate that filopodia are necessary not only for secretion but also to increase the secretory surface in the sub-BM area of the PG just before a significant surge of ecdysone is secreted into the hemolymph to induce pupariation.
Discussion
Filopodia structures, key players in ecdysone secretion
This work found that filopodia membrane projections mediate ecdysone secretion in the Drosophila PG. ex vivo experiments reveal that these structures harbor vesicles marked not only by ecdysone-specific transporters such as Atet (Fig. 4c) but also by classical secretory proteins like Syt1 (Fig. 4f). Moreover, we show that filopodia contain additional vesicle-associated components, including CSP (Fig. 4d) and Syx1A (Fig. 4e). Furthermore, the significant drop in circulating ecdysone levels following filopodia destabilization, despite intact ecdysone biosynthesis (Fig. 5f-f”), confirms that these structures act as specialized secretory sites, forming a stable membrane network that facilitates vesicle-mediated steroid release.
But why would filopodia be necessary for secretion? One possibility is that their curved nature introduces localized membrane curvature, optimizing the energetic efficiency of vesicle-membrane fusion and thus enhancing vesicle secretion. Another possibility is that filopodia facilitate the uptake of hemolymph components necessary for calcium signaling. Since filopodia act as antennae with signal receptors distributed along their length52,53, PG filopodia may enhance interactions between the predicted GPCR and its unknown ligand3 —a key step in triggering the calcium signaling required for vesicle secretion.
Polarized filopodia distribution in directed secretion
Our study reveals that the polarized distribution of filopodia serves as a mechanism for directed ecdysone secretion outward the gland. PG cells lack distinct apical and basolateral domains, showing an almost uniform distribution of basolateral factors (Fig. 1e). This absence of apico-basal polarity is also seen in other steroid-secreting glands, such as the zona glomerulosa rosettes of the adrenal cortex, which secrete aldosterone26. The lack of cell polarization in these highly secretory tissues raises an important question: How do these cells efficiently direct steroid hormones toward the circulatory system to elevate hormone levels rapidly?
Our results suggest that the asymmetric distribution of filopodia on PG cells is essential for efficient hormone secretion. In PG cells, these filopodia are concentrated near the BM (Fig. 2b, c). The movement of vesicles inside the filopodia (Fig. 4g) suggests a directional secretion process. Indeed, a distinguishing feature of PG filopodia structures is their remarkable stability (Supplementary Movie 1), which can be attributed to their tubulin composition (Fig. 2i) and their interactions with the BM (Fig. 3l). This stability forms a robust secretory platform facing the hemolymph. The identification of microtubules within PG filopodia suggests an essential role in polarized ecdysone secretion. Supporting this, research in Manduca sexta has shown PTTH signaling increases β-tubulin synthesis in PG cells54 and suggests that microtubules should be important for steroid precursor uptake55. These findings imply that the asymmetric distribution of cytoskeletal components in PG cells could be vital not only for ecdysone secretion but also for integrating local and systemic signals to control ecdysteroid biosynthesis, which requires further investigation.
Evolutionary significance of filopodia projections
The results presented here raise the question about the evolutionary significance of these findings. When delving into the older literature, we found that the filopodia observed in our study resemble the membrane invaginations previously reported in the PG cells of Drosophila melanogaster56, as well as similar structures in other insects like Manduca sexta57 and Lymantria dispar58. Electron microscopy studies showed these membrane invaginations as a network of membranes facing the BM, suggested to be involved in ecdysone secretion56. However, earlier research struggled to define the membrane network accurately, proposing various interpretations like invaginations or channels possible due to the non-preservation of filopodia projections in the former studies compared with current protocols59.
Our ex vivo and fixed PG protocols (see Methods) enabled the identification of filopodia. When considering all filopodia projections from PG cells, they collectively form a membrane network resembling the one observed in electron microscopy images13. Moreover, these membrane structures close to the BM in Lymantria dispar carry secretory vesicles and are essential for ecdysone secretion58, further supporting filopodia’s evolutionary conserved functional involvement in Insect steroid secretion.
Are filopodia key to hormone secretion beyond insects?
This idea is supported by similar mechanisms observed in other glands: cholecystokinin (CCK) is secreted by neuroendocrine cells through pseudopod-like processes known as neuropods60, and L cells in the gastrointestinal tract extend neuropods to release peptide PYY61. In insects, Inka cells, which secrete ecdysis-triggering hormones (ETH), harbor cytoplasmic extension62. In pancreatic delta cells, microtubule-containing filopodia facilitate somatostatin secretion18, and crustaceans have microvilli in the Y-organ that are suggested to be involved in the secretion of ecdysone63. The parallels suggest that membrane extensions are universally used for efficient, targeted hormone secretion, highlighting their potential evolutionary significance. Our study enriches this understanding by emphasizing the role of filopodia in endocrine signaling within the Drosophila PG, providing insights into the mechanisms of steroid hormone release.
Methods
Drosophila husbandry and strains
Flies were reared and crossed at 25 °C on standard cornmeal food containing, per liter, 15 g inactivated yeast powder, 80 g corn flour, 8 g agar (Sigma-Aldrich, A7002) and 3.5 g Nipagin (Sigma-Aldrich, W271004). The following fly strains were used: w1118 (BDSC#3605), hsFLP (BDSC#6), w1118;UAS-Dcr-2;phm22-Gal4 (gift of N. Yamanaka), w1118;phm-Gal441, P0206-Gal464, hsFLP;UAS-PLC-GFP;Act-CD2-Gal4 (gift of L. Boulan), Act-CD2-Gal4;UAS-RFP (BDSC#30558), CoinFLP-LexGAD/Gal4,LexAop-CD2-RFP;UAS-CD4-spGFP1–10,LexAop-CD4-spGFP11 (BDSC#58755), trol-GFP65, vkg-GFP (DGRC#110692), Ndg-GFP (VDRC#318629), LanB1-GFP (VDRC#318180), baz-GFP (BDSC#51572), crb-GFP (BDSC#99495), Sgs3-GFP (BDSC#5884), trolJO271#4966, UAS-CD8-RFP (BDSC#27391), UAS-myr-RFP (BDSC#7119), UAS-myr-GFP (BDSC#32197), UAS-PLC-GFP (BDSC#39693), UAS-Lifeact-Ruby (BDSC#35545), UAS-UTRN-GFP (BDSC#600555), UAS-GMA-GFP (BDSC#31775), UAS-Syt1-GFP (BDSC#6925), UAS-Ypet-Atet3, UAS-sec-GFP67, UAS-Rab11-GFP (BDSC#8506), UAS-RNAi-trol (BDSC#38298), UAS-RNAi-Rab11 (BDSC#27730), UAS-RNAi-Vps25 (VDRC#108105), UAS-RNAi-Vps36 (VDRC#107417), UAS-RNAi-shrb (VDRC#106823), UAS-RNAi-mys (BDSC#33642), UAS-RNAi-Act5C (BDSC#42651 and VDRC#101438), UAS-RNAi-cofilin (BDSC#65055), UAS-RNAi-chic (BDSC#34523), UAS-RNAi-Arpc3B (BDSC#27582), UAS-RNAi-dia (VDRC#103914 and VDRC#33424), UAS-RNAi-ena (VDRC#106484 and BDSC#43056), UAS-RNAi-Actn (VDRC#7762 and VDRC#110719), UAS-RNAi-αTub85E (VDRC#103202), UAS-RNAi-αTub84D (BDSC#64904), UAS-RNAi-αTub67C (BDSC#32502), UAS-RNAi-βTub97EF (BDSC#64858), UAS-RNAi-βTub85D (BDSC#65163), UAS-RNAi-βTub65B (BDSC#64875), UAS-RNAi-βTub60D (BDSC#64856), UAS-RNAi-βTub56D (BDSC#65028).
Developmental timing experiments
All developmental timing experiments were conducted at 25 °C. Embryos were collected after 4 h of egg laying on agar (1.5% w/v) dishes containing 3% (w/v) sucrose and were immediately transferred to tubes containing standard cornmeal food at a rate of 50 individuals per tube. The percentage of pupation as a function of time was determined for each tube by counting pupae number two times a day.
For rescue experiments using 20-HydroxyEcdysone (20E) feeding, 50 µL of a freshly prepared solution at 0.25 mg/mL in absolute ethanol (stock solution at 5 mg/mL, Sigma-Aldrich, H5142) were directly added on top of the food twice a day, starting at late second instar stage (before 72 h AEL).
Fluorescent immunostaining
Third instar larvae were dissected in PBS, fixed in 4% formaldehyde (16% stock solution, PolySciences, 18814-20) in PBS for 25 min at room temperature (RT), permeabilized three times with 0.1% Triton X-100 in PBS and blocked for 40 min in 1X ROTI®-Block (10X stock solution, Carl Roth, A151.1) in 0.1% Triton X-100 in PBS. Dissected larvae were incubated overnight at 4 °C in blocking solution containing primary antibodies, washed three times in blocking solution and incubated with fluorescent secondary antibodies diluted in blocking solution for 2 h at RT. Samples were washed three times in PBS and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, H-1200).
Primary antibodies were diluted as follows: rabbit α-Tor (1:200, this study), guinea pig α-Dib (1:400, this study), mouse α-Arm (1:50, DSHB N2 7A1), rat α-Ecad (1:50, DSHB DCAD2), mouse α-Cora (1:50, DSHB C615.16), mouse α-Dlg1 (1:50, DSHB 4F3), mouse α-βPS (1:50, DSHB CF.6G11), rat α-Ncad (1:50, DSHB DN-Ex #8), mouse α-αTub85E (1:50, DSHB 12G10), mouse α-CSP (1:100, DSHB ab49), mouse α-Syx1A (DSHB 8C3), guinea pig α-PTTH (1:400)68, rabbit α-Trol (1:3500)69, rabbit α-Cg25C (1:1000)70. Polyclonal antibodies against Tor were raised using the following peptide for rabbit immunization (Eurogentec): -WVQHSRGTEPAPNAT- and -PKRKLKPQPKKDSKQ-. Polyclonal antibodies against Dib were raised using the following peptide for guinea pig immunization (Eurogentec): -CKTLLINKPDAPVLIDLRLRRE-. Fluorescent secondary antibodies (Alexa Fluor or Fluor plus 488-, 546- or 555-, or 647-conjugated antibodies from Thermo Fisher Scientific) and TRITC-conjugated phalloidin (Sigma-Aldrich, P1951) were diluted at 1:200. To compare specimens of different genotypes, samples were stained in parallel using the same antibody solutions or within the same tube when tissue morphology allowed distinction, and imaged under identical acquisition settings. Confocal images were acquired on a Zeiss LSM 880 with Airyscan using the Fast mode of the Airyscan detector. XY pictures are either single optical sections or maximum intensity projections obtained from a Z-stack acquisition. XZ and YZ images are orthogonal sections extracted from a Z-stack acquisition. All images were processed and analyzed with Fiji software. 3D visualization of the ring gland was obtained with Vaa3D software71.
PG cells number
PG cell numbers were determined by manually counting nuclei stained with DAPI. The assignment of cells to the dorsal or ventral layer of the PG was based on Z-stack acquisition.
ex vivo imaging, live imaging and filopodia stability
The anterior part of late third instar larvae was dissected and directly mounted between two coverslips of different sizes in HL-3 saline solution (70 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 20 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose and 5 mM HEPES, pH 7.4). Confocal images were acquired using a Zeiss LSM 880 with Airyscan in Fast mode. Live imaging was performed using a time series scan with a 10s interval between frames. Filopodia stability was assessed by tracking the appearance and disappearance of 276 filopodia from 15 independent PGs over a 15-min period.
Ecdysone titration
Isolation of exosomes from hemolymph
This protocol was adapted from ref. 37. Briefly, 20 µL of hemolymph were collected from late third instar larvae, transferred into 500 µL of PBS + PTU (phenylthiourea 0.1 µg/mL, Merck, P7629), and centrifuged at 750 g for 15 min at 4 °C. The supernatant was transferred into a new tube and centrifuged again at 1500 × g for 10 min at 4 °C. The remaining supernatant was then centrifuged a last time at 100,000 × g for 2 h at 4 °C. The final pellet was suspended in 300 µL of methanol for ecdysone titration. Methanol samples were centrifuged at 12,000 × g for 5 min at 4 °C. First, the supernatant was kept, and the pellet was resuspended again in 300 µL of methanol and centrifuged in the same conditions. The second supernatant was pooled with the first and stored at −80 °C.
Ecdysone extraction from hemolymph and total larva
For hemolymph ecdysone, 5–7 µL of hemolymph were collected from late third instar larvae and resuspended in 90 µL of PBS + PTU. For total larva ecdysone, four late third instar larvae were homogenized in 100 µL of PBS + PTU with a Tissue Lyser (Qiagen) at 50 Hz for 1 min and centrifuged at 2000 × g for 10 min at 4 °C, and the supernatant was kept.
300 µL of methanol were added to both samples and centrifuged at 12,000 × g for 5 min at 4 °C. The first supernatants were kept, and pellets were resuspended in 300 µL of methanol and centrifuged in the same conditions. The second supernatants were pooled with the first and stored at −80 °C.
Ecdysone titration assay
Methanol from isolated exosomes, hemolymph, or total larva samples was removed using a Speed Vacuum for 2.5 h. Dried pellets were resuspended in 120 µL of EIA buffer from the 20-HydroxyEcdysone ELISA kit (Bertin Pharma, A05120). Ecdysone titration was then carried out according to the manufacturer’s protocol, using three independent replicates for each condition.
Filopodia measurements
Synchronized larvae of the indicated genotype (hsFLP; Act»Syt1-GFP or hsFLP; Act»PLC-PH-EGFP) were heat-shocked at 37 °C for 10 min and dissected 24 h later. Early or late third instar larvae were dissected in PBS and fixed in 4% formaldehyde for 10 min at RT. After three washes in PBS, PG were mounted in Vectashield mounting media with DAPI and assessed for the presence of isolated Syt1-GFP or PLC-GFP expressing clones, for proper visualization of the filopodia. Confocal images of isolated PG clones were acquired on a Zeiss LSM 880 with Airyscan using the Fast mode of the Airyscan detector. Maximum intensity projections of the slices corresponding to the sub-BM region were manually analyzed on ImageJ software. Sub-BM cell perimeters and areas without filopodia were quantified and, thanks to the contrast with the surrounding black background, filopodia located at the edge of the clone were counted and measured in their length (L) and diameter (d).
The number of filopodia was normalized to the perimeter on which they were counted to assess filopodia density per μm, and this value was used to estimate a filopodia density per μm2. Assuming the distribution of filopodia was homogenous throughout the entire sub-BM surface, the number of filopodia for a given clone cell was calculated by multiplying filopodia density (per μm2) by cell sub-BM area.
The exchange surface of each counted filopodia was calculated using the formula for the total surface area of a cylinder from which area of one circular base have been subtracted:
Filopodia exchange surface = \(\left[\Pi \times d\times (l+d/2)\right]-\,\left[\Pi \times {\left(d/2\right)}^{2}\right]\).
This filopodia exchange surface was also normalized to the cell sub-BM area and this parameter is referred to as “Filopodia exchange surface (relative to cell size)”.
For a given clone cell, the Δ exchange surface enrichment was calculated using the following formula:
Δ exchange surface enrichment (in %) = (Exchange surface with filopodia − Exchange surface without filopodia) × 100,
in which:
Exchange surface with filopodia = (number of filopodia × average exchange surface of 1 filopodia) + cell sub-BM area.
Exchange surface without filopodia = cell sub-BM area.
Fluorescence intensity measurement
Confocal images of Syt1-GFP clone cells were obtained using the same protocol as described above (see Filopodia measurements section). At least three distinct orthogonal sections (in the XZ or YZ planes) per clone were used to determine Syt1-GFP intensity at three different cell regions (referred to as sub-BM, lateral, and midline). For each of these cell regions, two to three separate measurements were taken on each orthogonal section using a region of interest (ROI) box of the same size (represented in Fig. 1f). These measurements were averaged to account for fluorescence variability. The mean of the three orthogonal sections were then averaged to obtain the Syt1-GFP intensity for a given region in each clone.
Confocal images of Ypet-Atet clone cells were obtained using the same protocol as described above, except that larvae were dissected 6 h or 24 h after heat-shock. Ypet-Atet intensity was measured at the sub-BM region of three distinct clones for each condition. For each clone, measurements were taken in two to three ROI boxes of the same size.
Statistical analysis
All statistics and plots were obtained with the GraphPad Prism software. For Fig. 1f, datasets are represented in a paired aligned dot plot displaying the mean ± 95% CI. Each dot represents fluorescence intensity for one sample, and the pairing line between dots represents the belonging to the same sample. In Fig. 3a, b, n and Supplementary Fig. 3f, developmental timings of the different genotypes were displayed in an XY plot, where the means ± SEM of the proportion (% pupariation) of all the tubes are plotted as a function of time (in hours) after egglaying (abbreviated hAEL). In Fig. 5a–c, e the differences in time at 50% pupariation (abbreviated t50%) between the different experimental conditions (phm>Dcr-2,RNAi-X) and the control (phm>Dcr-2 x w1118) were compared using a bar plot displaying the mean of the differences ± SEM. Each dot represents the value for one tube. For Fig. 5f, f” and Supplementary Figs. 1a, 3a, 4c’, 5a, datasets are also represented by bar plots showing the mean ± SEM. In Supplementary Fig. 1a, each PG is color-coded, and each colored dot represents the cell number for one given PG. In Fig. 5f” and Supplementary Fig. 3a, each dot represents the value for one sample. In Supplementary Fig. 4c’, each dot represents an independent measure. In Supplementary Fig. S5a, each dot represents the cell number for one PG. For Figs. 5d’ and 6d, e, datasets are represented by scatter dot plots indicating the median and the interquartile range (IQR). Each sample is color-coded, and each color-coded square represents the average for a given clone cell. For Fig. 6b, c, and Supplementary Fig. 6b, c and d datasets are represented by superplots in which each clone cell is color-coded. The colored dot represents individual filopodia for a given clone cell, and the corresponding colored-square represents different clone cells. The median of the means and IQR are displayed in these plots. For Fig. 1f, data were subjected to the Friedman test followed by Dunn’s multiple comparison tests. For Supplementary Fig. 1a, data were subjected to the two-tailed matched-pairs Wilcoxon test. For Fig. 5a, b, c, d’, f, f” and Supplementary Fig. 5a, data were subjected to the Kruskal–Wallis test, followed by Dunn’s multiple comparison tests. For Figs. 5e, 6b, c, d, e and Supplementary Fig. 6b–d data were subjected to the two-tailed Mann–Whitney test. The significance of statistical tests is reported in plots as follows: ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p-values are directly mentioned in figures when possible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Sisk, C. L. & Foster, D. L. The neural basis of puberty and adolescence. Nat. Neurosci. 7, 1040–1047 (2004).
Sapolsky, R. M., Romero, L. M. & Munck, A. U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 21, 55–89 (2000).
Yamanaka, N., Marqués, G. & O’Connor, M. B. Vesicle-mediated steroid hormone secretion in Drosophila melanogaster. Cell 163, 907–919 (2015).
Chu, L. et al. Sex steroid hormones in urinary exosomes as biomarkers for the prediction of prostate cancer. Clin. Chim. Acta 531, 389–398 (2022).
Schotten, S. et al. Additive effects on the energy barrier for synaptic vesicle fusion cause supralinear effects on the vesicle fusion rate. Elife 4, e05531 (2015).
Chasserot-Golaz, S., Coorssen, J. R., Meunier, F. A. & Vitale, N. Lipid dynamics in exocytosis. Cell. Mol. Neurobiol. 30, 1335–1342 (2010).
Ali Moussa, H. Y. et al. Requirement of cholesterol for calcium-dependent vesicle fusion by strengthening Synaptotagmin-1-induced membrane bending. Adv. Sci.10, e2206823 (2023).
Wei, L., Wang, X. & Wu, L.-G. How micron-sized exocrine vesicles release content: A comparison with sub-micron endocrine vesicles. J. Cell Biol. 222, e202310047 (2023).
McMahon, H. T., Kozlov, M. M. & Martens, S. Membrane curvature in synaptic vesicle fusion and beyond. Cell 140, 601–605 (2010).
Smirnova, Y. G. & Müller, M. How does curvature affect the free-energy barrier of stalk formation? Small vesicles vs apposing, planar membranes. Eur. Biophys. J. 50, 253–264 (2021).
Herman, W. S. The ecdysial glands of arthropods. Int. Rev. Cytol. 22, 269–347 (1967).
Sedlak, B. J., Marchione, L., Devorkin, B. & Davino, R. Correlations between endocrine gland ultrastructure and hormone titers in the fifth larval instar of Manduca sexta. Gen. Comp. Endocrinol. 52, 291–310 (1983).
Dai, J., Henrich, V. C. & Gilbert, L. I. An ultrastructural analysis of the ecdysoneless (l(3)ecd1ts) ring gland during the third larval instar of Drosophila melanogaster. Cell Tissue Res. 265, 435–445 (1991).
Pudney, J., Sweet, P. R., Vinson, G. P. & Whitehouse, B. J. Morphological correlates of hormone secretion in the rat adrenal cortex and the role of filopodia. Anat. Rec. 201, 537–551 (1981).
Chegini, N., Ramani, N. & Rao, C. V. Morphological and biochemical characterization of small and large bovine luteal cells during pregnancy. Mol. Cell. Endocrinol. 37, 89–102 (1984).
Adams, E. C. & Hertig, A. T. Studies on the human corpus luteum. I. Observations on the ultrastructure of development and regression of the luteal cells during the menstrual cycle. J. Cell Biol. 41, 696–715 (1969).
Gregoraszczuk, E. & Krzysztofowicz, E. The corpus luteum of the pig. Scanning electron microscopic study of surface features at different times of incubation. Acta Biol. Hung. 40, 145–156 (1989).
Arrojo e Drigo, R. et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nat. Comm. 10, 4218 (2019).
Wood, W. & Martin, P. Structures in focus—filopodia. Int. J. Biochem. Cell Biol. 34, 726–730 (2002).
Blake, T. C. A. & Gallop, J. L. Filopodia in vitro and in vivo. Annu. Rev. Cell Dev. Biol. 39, 307–329 (2023).
Kučera, O., Gaillard, J., Guérin, C., Théry, M. & Blanchoin, L. Actin–microtubule dynamic composite forms responsive active matter with memory. Proc. Natl Acad. Sci. 119, e2209522119 (2022).
Dogterom, M. & Koenderink, G. H. Actin–microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 20, 38–54 (2019).
McMahon, H. T. & Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 128, 1065–1070 (2015).
Sánchez-Higueras, C., Sotillos, S. & Castelli-Gair Hombría, J. Common origin of insect trachea and endocrine organs from a segmentally repeated precursor. Curr. Biol. 24, 76–81 (2014).
Dai, J., Ma, M., Feng, Z. & Pastor-Pareja, J. C. Inter-adipocyte adhesion and signaling by collagen IV intercellular concentrations in Drosophila. Curr. Biol. 27, 2729–2740 (2017).
Leng, S. et al. β-Catenin and FGFR2 regulate postnatal rosette-based adrenocortical morphogenesis. Nat. Comm. 11, 1680 (2020).
Pignoni, F. & Zipursky, S. L. Induction of Drosophila eye development by Decapentaplegic. Development 124, 271–278 (1997).
Pfeiffer, B. D. et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl Acad. Sci. 105, 9715–9720 (2008).
Bosch, J. A., Tran, N. H. & Hariharan, I. K. Coinflp: a system for efficient mosaic screening and for visualizing clonal boundaries in Drosophila. Development 142, 597–606 (2015).
Elam, W. A., Kang, H. & De La Cruz, E. M. Biophysics of actin filament severing by cofilin. FEBS Lett. 587, 1215–1219 (2013).
Hu, H. T. et al. The cellular protrusions for inter-cellular material transfer: similarities between filopodia, cytonemes, tunneling nanotubes, viruses, and extracellular vesicles. Front. Cell Dev. Biol. 12, 1422227 (2024).
Junyent, S. et al. Specialized cytonemes induce self-organization of stem cells. Proc. Natl Acad. Sci. 117, 7236–7244 (2020).
Schober, J. M., Komarova, Y. A., Chaga, O. Y., Akhmanova, A. & Borisy, G. G. Microtubule-targeting-dependent reorganization of filodopia. J. Cell Sci. 120, 1235–1244 (2007).
Christesen, D. et al. Transcriptome analysis of Drosophila melanogaster third instar larval ring glands points to novel functions and uncovers a cytochrome p450 required for development. G3 Bethesda 7, 467–479 (2017).
Simon, E. et al. Glypicans define unique roles for the Hedgehog co-receptors Boi and Ihog in cytoneme-mediated gradient formation. Elife 10, e64581 (2021).
Meyen, D. et al. Dynamic filopodia are required for chemokine-dependent intracellular polarization during guided cell migration in vivo. Elife 4, e05279 (2015).
Linnemannstöns, K. et al. Microscopic and biochemical monitoring of endosomal trafficking and extracellular vesicle secretion in an endogenous in vivo model. J. Extracell. Vesicles 11, e12263 (2022).
Kowalczyk, A. P. & Nanes, B. A. Adherens junction turnover: Regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell. Biochem. 60, 197–222 (2012).
De Franceschi, N., Hamidi, H., Alanko, J., Sahgal, P. & Ivaska, J. Integrin traffic – the update. J. Cell Sci. 128, 839–852 (2015).
McAtee, C. et al. Secreted exosomes induce filopodia formation. Preprint at https://doi.org/10.1101/2024.07.20.604139 (2024).
Guittard, E. et al. CYP18A1, a key enzyme of Drosophila steroid hormone inactivation, is essential for metamorphosis. Dev. Biol. 349, 35–45 (2011).
Eva, R. et al. Rab11 and its effector Rab coupling protein contribute to the trafficking of β1 Integrins during axon growth in adult dorsal root ganglion neurons and PC12 cells. J. Neurosci. 30, 11654–11669 (2010).
Pastor-Pareja, J. C. & Xu, T. Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and Perlecan. Dev. Cell 21, 245–256 (2011).
Zinsmaier, K. E., Eberle, K. K., Buchner, E., Walter, N. & Benzer, S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science 263, 977–980 (1994).
Brown, H. et al. Cysteine string protein (CSP) is an insulin secretory granule-associated protein regulating β-cell exocytosis. EMBO J. 17, 5048–5058 (1998).
Beckett, K. et al. Drosophila S2 cells secrete wingless on exosome-like vesicles but the wingless gradient forms independently of exosomes. Traffic 14, 82–96 (2013).
Schulze, K. L., Broadie, K., Perin, M. S. & Bellen, H. J. Genetic and electrophysiological studies of drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell 80, 311–320 (1995).
Scherl, A. et al. Functional proteomic analysis of human nucleolus. Mol. Biol. Cell 13, 4100–4109 (2002).
Ohhara, Y., Kobayashi, S. & Yamanaka, N. Nutrient-dependent endocycling in steroidogenic tissue dictates timing of metamorphosis in Drosophila melanogaster. PLoS Genet. 13, e1006583 (2017).
Li, R. & Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat. Rev. Mol. Cell Biol. 9, 860–873 (2008).
de la Riva-Carrasco, R. et al. The immunophilin Zonda controls regulated exocytosis in endocrine and exocrine tissues. Traffic 22, 111–122 (2021).
Heckman, C. A. & Plummer, H. K. Filopodia as sensors. Cell. Signal. 25, 2298–2311 (2013).
Davenport, R. W., Dou, P., Rehder, V. & Kater, S. B. A sensory role for neuronal growth cone filopodia. Nature 361, 721–724 (1993).
Rybczynski, R. & Gilbert, L. I. Prothoracicotropic hormone elicits a rapid, developmentally specific synthesis of β tubulin in an insect endocrine gland. Dev. Biol. 169, 15–28 (1995).
Watson, R. D., Ackerman-Morris, S., Smith, W. A., Watson, C. J. & Bollenbacher, W. E. Involvement of microtubules in prothoracicotropic hormone-stimulated ecdysteroidogenesis by insect (Manduca sexta) prothoracic glands. J. Exp. Zool. 276, 63–69 (1996).
King, R. C., Aggarwal, S. K. & Bodenstein, D. The comparative submicroscopic morphology of the ring gland of Drosophila melanogaster during the second and third larval instars. Z. Zellforsch. Mikrosk. Anat. 73, 272–285 (1966).
Hanton, W. K., Watson, R. D. & Bollenbacher, W. E. Ultrastructure of prothoracic glands during larval-pupal development of the tobacco hornworm, Manduca sexta: a reappraisal. J. Morphol. 216, 95–112 (1993).
Park, E. J., Yin, C.-M. & Burand, J. P. Baculovirus replication alters hormone-regulated host development. J. Gen. Virol. 77, 547–554 (1996).
Wood, B. M. et al. Cytonemes with complex geometries and composition extend into invaginations of target cells. J. Cell Biol. 220, e202101116 (2021).
Chandra, R., Samsa, L. A., Vigna, S. R. & Liddle, R. A. Pseudopod-like basal cell processes in intestinal cholecystokinin cells. Cell Tissue Res. 341, 289–297 (2010).
Bohórquez, D. V., Chandra, R., Samsa, L. A., Vigna, S. R. & Liddle, R. A. Characterization of basal pseudopod-like processes in ileal and colonic PYY cells. J. Mol. Hist. 42, 3–13 (2011).
Zitnan, D. & Adams, M. E. Neuroendocrine regulation of ecdysis. In Insect endocrinology (ed. L. Gilbert) 253–309 (Elsevier, 2012).
Bohórquez, D. V. et al. An enteroendocrine cell - Enteric glia connection revealed by 3D electron microscopy. PLoS One 9, e89881 (2014).
Colombani, J. et al. Antagonistic actions of ecdysone and insulins determine final size in Drosophila. Science 310, 667–670 (2005).
Besse, F. et al. The Ig cell adhesion molecule Basigin controls compartmentalization and vesicle release at Drosophila melanogaster synapses. J. Cell Biol. 177, 843–855 (2007).
Voigt, A., Pflanz, R., Schäfer, U. & Jäckle, H. Perlecan participates in proliferation activation of quiescent Drosophila neuroblasts. Dev. Dynam. 224, 403–412 (2002).
Entchev, E. V., Schwabedissen, A. & González-Gaitán, M. Gradient formation of the TGF-β homolog Dpp. Cell 103, 981–992 (2000).
Yamanaka, N. et al. Neuroendocrine control of Drosophila larval light preference. Science 341, 1113–1116 (2013).
Friedrich, M. V. K., Schneider, M., Timpl, R. & Baumgartner, S. Perlecan domain V of Drosophila melanogaster. Eur. J. Biochem. 267, 3149–3159 (2000).
Zang, Y. et al. Plasma membrane overgrowth causes fibrotic collagen accumulation and immune activation in Drosophila adipocytes. Elife 4, e07187 (2015).
Peng, H., Ruan, Z., Long, F., Simpson, J. H. & Myers, E. W. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat. Biotechnol. 28, 348–353 (2010).
Acknowledgements
We thank the plantBios ISA microscopy facility for technical support. We thank G. de Soussa, G. D’Angelo, S. Pizette, and all BES laboratory members for insightful discussions and comments on the scientific work. We thank the Vienna Drosophila RNAi Center, the Drosophila Genetics Resource Center, and the Bloomington Stock Center for providing Drosophila lines. We thank P. Léopold, F. Martin, and T. Kornberg for the critical reading of the manuscript. We thank N.Yamanaka for providing us the UAS-Ypet-Atet and the UAS-Dcr-2;phm22-Gal4 lines. We thank J.-C. Pastor-Pareja for providing us with the Cg25C antibody. We thank T. Perry for providing us with the phm-Gal4 line. This work was supported by the ATIP_AVENIR young group leader program (INSERM grant 2020/5 to N.M.R), the Marie Curie fellowship (EU MSCA to E.S), the ANRS MIE fellowship (ECTZ212526 to R.B), the ANRS MIE grant (ECTZ212478 to N.M.R), and the Labex Signalife program (grant NR-11-LABX-0028-01 and IDEX UCA Jedi ANR-15-IDEX-01 to N.M.R.).
Author information
Authors and Affiliations
Contributions
E.S., R.B., Y.M., and M.-P.N.-E. conducted the experiments. E.S., R.B., and N.M.R. designed and interpreted them. E.S. wrote the original manuscript, and E.S., R.B., and N.M.R. wrote, edited, and reviewed it. N.M.R supervised the work and led the conceptualization. All the authors were involved in the scientific discussions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Simon, E., Bonche, R., Maarouf, Y. et al. Filopodia are essential for steroid release. Nat Commun 16, 5501 (2025). https://doi.org/10.1038/s41467-025-60579-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60579-7
