
Abstract
The growth of lithium (Li) dendrites and the accumulation of dead Li (i.e., Li metal regions which are electronically disconnected from the current collector) significantly undermine the safety and performance of Li metal batteries. This study employs kilogram–scale atomic layer deposition technology to construct zinc oxide with a preferential (002) crystal orientation, which homogeneously forms on commercial carbon nanotube papers. Our approach emphasizes the importance of achieving a moderate Li adsorption energy and low Li migration energy barriers to suppress Li dendrite growth. In this work, we introduce the concept of “catalytic” effect for dead Li reconversion, as validated through time–of–flight secondary ion mass spectrometry, leading to a Li plating/stripping efficiency of 99.89%. The Ah–level Li metal pouch cells with high–nickel positive electrodes achieve a specific energy of 380 Wh kg−1 (based on the mass of the whole pouch cell) and demonstrate stable cycling under demanding conditions. Analysis of the cycled pouch cells confirms the structural integrity and provides insights into the mechanism of the dead Li “catalytic” conversion.
Similar content being viewed by others


Introduction
Given the growing global demand for clean energy and the widespread utilization of smart devices, lithium (Li) secondary batteries have emerged as a favored choice for energy storage. Safety concerns and specific energy are paramount considerations for the practical utilization of Li–ion batteries. It is noteworthy that the current commercial graphite negative electrode has nearly reached its theoretical capacity limit (372 mAh g−1)1,2,3. Fortunately, Li metal electrode (LME) is considered one of the most promising options in current research due to its high specific energy (3860 mAh g−1) and the lowest reduction potential of −3.04 V (vs. standard hydrogen electrodes) among negative electrode materials4,5,6. As a result, it has garnered significant attention and become a prominent research topic in recent times. However, the practical implementation of LME in Li metal batteries (LMBs) faces two major challenges: (1) low Coulombic efficiency (CE) and (2) uncontrollable reaction sequence and serious heat release arising from dendrite growth7,8. The formation of Li dendrites is primarily attributed to the inhomogeneous Li nucleation sites and accumulation of dead Li (i.e., Li metal regions which are electronically disconnected from the current collector, simplified as LiD) on the negative electrode surface9,10,11. Furthermore, the buildup of LiD is a crucial factor contributing to the instability of the battery interface and rapid capacity deterioration, which also poses significant safety hazards in LMBs. Therefore, the eradication of Li dendrites or the implementation of effective strategies to enhance the utilization rate of LiD holds paramount significance in advancing the practicality of LMBs.
Currently, researchers are actively pursuing several mainstream strategies to mitigate the growth of Li dendrites in batteries. These strategies encompass the following: (1) designing functional electrolytes12,13,14,15, (2) creating artificial solid electrolyte interphase (SEI)16,17,18, (3) constructing specialized “lithiophilic” substrate, and others19,20,21. Among these strategies, the establishment of a “Li–friendly accommodation space” within the host material of LME is widely recognized as one of the most effective approaches for fabricating composite Li negative electrodes and inhibiting the growth of Li dendrites22,23. Carbon materials were initially favored as substrates for composite negative electrode materials in LMBs due to several advantages24. Firstly, carbon materials are abundantly available, providing a rich resource for their applications25. Secondly, carbon materials exhibit appropriate electrical conductivity and mechanical stability, which are crucial factors for their successful integration into LMBs26. Lastly, but not least, carbon materials possess a lightweight nature and can be tailored to achieve desirable porosity, enabling precise control over their properties27,28. More recently, Qian et al.29 verified LiZn/Li2O configurations can effectively decompose the Li clusters to prevent Li aggregation, which ensures efficient electron transfer throughout the configuration and enables stable LME. However, while LiZn/Li₂O plays a significant role in inhibiting Li dendrite growth, the role of zinc oxide (ZnO) in early-stage Li nucleation should not be overlooked. As the target material, ZnO is the key determinant in achieving dense Li deposition in later stages30,31. Furthermore, even with well–designed Li affinity sites and spaces, the accumulation of significant amounts of LiD on the surface remains unavoidable32. Consequently, enhancing the utilization of LiD remains an ongoing scientific problem and a challenge in addressing the safety concerns associated with LMBs33,34.
In this work, we utilized an autonomously designed and scalable atomic layer deposition (ALD) technology to construct a 3D thin carbon nanotubes (CNTs) paper (CP) to confront the challenges posed by Li dendrites and the accumulation of LiD. This design facilitates the implementation of dendrite–free LME. Through this design, we introduce the concept of the LiD “catalytic” effect, which enables the maximum recycling of Li resources. Furthermore, unlike well–known strategies such as electrodeposition, thermal diffusion, and mechanical rolling, which have the potential to inhibit the formation of Li dendrites while facilitating large–scale production, the advanced ALD technique we employ allows for material processing on a kilogram scale in a single step. This approach ensures precise control over the coating layer, providing industrial competitiveness in the production of thin Li interlayers/composite LME. As shown in Fig. 1, the CNT paper loaded with ZnO, exhibiting a preferential (002) crystal plane–denoted as CP@ZnO(002)–plays two critical roles. First, ZnO(002) provides abundant nucleation sites for early Li seed growth. The generated Li₂O and LiZn, with their favorable localized free electron distribution, facilitate the decomposition of Li clusters, preventing aggregation29,35. Second, CP@ZnO(002) captures collapsed LiD, which loses electrical contact with the current collector due to anisotropic Li stripping, while the interwoven CP@ZnO(002) framework restores electron conductivity, enabling LiD reutilization. Consistently, density functional theory (DFT) calculations also reveal that the specific crystal structure of ZnO(002) induces a dual “Li affinity–Li migration” effect, providing a crucial driving force for the initial nucleation and deposition of the Li layer. As a result, the 20 μm CP@ZnO(002) film, serving as an “accommodation space”, has enabled Li metal pouch cells using high–nickel–layered positive electrodes (Li[Ni0.90Co0.05Mn0.05]O2, simplified as NCM90) to achieve an Ah–level capacity. This cell system is denoted as Li/CP@ZnO(002) | | NCM90. Under demanding conditions, including a 30 μm Li foil, an E/C ratio of 2.45, and a high positive electrode areal capacity of 5.3 mAh cm−2, the Li/CP@ZnO(002) | | NCM90 pouch cells have achieved a breakthrough in the pack–level specific energy, reaching 380 Wh kg−1 (based on the mass of the whole pouch cell), and have demonstrated stable cycling for 50 cycles. These exciting findings and results indicate the significance of the mechanism and materials we proposed in advancing the development of Li metal composite negative electrode materials. It was also demonstrated that the matching of LME with high nickel or high–capacity positive electrodes is essential to harness the high specific energy advantages of LME.

Schematics of a CNTs after ozone treatment, b CNTs modified with ZnO by ALD technology, c Li deposition on CP@ZnO(002), and d ZnO (002) “catalyzing” the uniform spreading of Li metal on CNTs with Li2O and LiZn alloys. e–g Adsorption structures and energies of Li on different crystal planes of ZnO. h–j Top views of Li migration pathways on different surfaces of ZnO and LiZn. k, l Adsorption structures and energies of Li on different crystal planes of LiZn alloy. m A photo of a large–area CP product. n HRTEM and o–r TEM elemental mapping images of a single CNT@ZnO(002) nanowire.
Results
The thin Interlayer design and manufacture
As illustrated in Fig. 1a, commercial CNTs (Supplementary Fig. 1) were treated with ozone (O3) to introduce surface defects and oxygen–containing functional groups. The O3 treatment duration significantly influences the growth behavior of ALD–deposited ZnO (002). Our results show that a 30–minute treatment creates an optimal concentration of defects and oxygen–containing functional groups, enabling uniform deposition of ZnO (002) via ALD across the CNT surface (Supplementary Figs. 2, 3, and 4). The treated CNTs were then modified uniformly with ZnO dots using our spatially designed ALD technique (Fig. 1b). Remarkably, ZnO is one of the earliest materials discovered to possess preferred nucleation sites for Li deposition36. However, previous studies have shown that the deposition and subsequent growth of Li cannot solely rely on the material’s affinity to Li, but also on the Li migration capability37,38,39. These two features synergistically ensure a more homogenized formation of initial Li nucleation sites, resulting in dendrite-free LME. Modulating crystal plane orientations of the target material to balance Li adsorption and Li migration capability offers a different avenue in inducing the growth of dendrite–free LMBs. Therefore, we conducted first–principle calculations to study the adsorption energies and migration barriers of Li on three typical crystal planes of ZnO: (002), (202), and (020). The computational results indicate that Li exhibits different performances on the various crystal planes of ZnO (Fig. 1e–g, Supplementary Fig. 5). Specifically, the (002) plane of ZnO exhibits moderate adsorption energy (3.40 eV) and low Li migration barrier compared to the (020) (1.61 eV) and the (202) (4.32 eV) planes. Specifically, the ZnO (002) plane not only promotes the preferential adsorption of Li but also enhances the rapid migration of Li (Fig. 1h–j). This dual property is essential for achieving ideal initial Li nucleation sites, which is also crucial in achieving the ultimate dense Li deposition morphology (Fig. 1c, d). Moreover, the adsorption and nucleation of Li+ on ZnO result in the formation of Li2O40,41, exhibiting lithiophilicity, and LiZn42,43 alloy with appropriate ionic conductivity (showing the lowest Li mobility barrier of 0.017 eV), as shown in Fig. 1k–l and Supplementary Fig. 6.
To further elucidate the crystallographic orientation of ALD–deposited ZnO, the results of transmission electron microscopy (TEM) characterization indicate that ZnO deposited at ALD temperatures of 70 °C, 150 °C, and 200 °C (or below 70 °C) shows preferential orientations along the (200), (202), and (002) planes, respectively (Supplementary Fig. 7). Additionally, crystallographic properties were examined through selective area electron diffraction (SAED) analysis (Supplementary Fig. 8). Further detailed spectrogram analysis (Supplementary Fig. 9) indicated that the reflection of the (002) lattice plane was dominant at 50 °C, while the (200) and (202) peaks began to strengthen as the temperature increased to 70 °C and 150 °C, with the (002) peak regaining dominance at 200 °C. Moreover, a thorough examination of the atomic force microscopy (AFM) test results revealed significant morphological differences on the ALD–deposited ZnO surfaces at different temperatures (Supplementary Figs. 10, 11). At 50 °C, ZnO exhibits a rod–like morphology on the single–crystal silicon surface (Supplementary Fig. 10a), aligning with the (002) crystal facet. As the temperature rises to 70 °C, 150 °C, ZnO transitions to a needle–like and columnar–like growth pattern corresponding to the (200) and (202) crystal facets, respectively (Supplementary Figs. 10b, c), with features of the ZnO (002) crystal facet re–emerging at 200 °C (Supplementary Fig. 10d). Raman spectroscopy results indicated that we successfully obtained the desired ZnO(002) product, aligning consistently with prior studies44,45.
By employing a drawing technique with CNT fiber (Supplementary Fig. 12) and biofiber (Supplementary Fig. 13), we achieved a structurally stable large–area (0.5 meter*1 meter) CP with a stable morphology, high porosity, and a thickness of merely 20 μm. The scanning electron microscope (SEM) images (Supplementary Fig. 14) clearly illustrate the uniform dispersion and abundant porosity of the CNT@ZnO(002) structure formed through ALD. This special morphology is advantageous for facilitating efficient electron and ion conduction, while also offering ample space for Li deposition (Fig. 1c, d). The X–ray photoelectron spectroscopy (XPS) and X–ray diffraction (XRD) analysis results reveal a significant binding energy and 2 theta degree of the coated CNTs with ZnO(002) (Supplementary Figs. 15, 16), the spatial confinement of XPS technique further confirming the uniform distribution of ZnO(002) particles on the surface of CNTs. The illustration in Fig. 1m demonstrates the substantiation of the considerable potential for scalable production of CP@ZnO(002) and the thermogravimetry analysis (TGA) reveals that the mass fraction of ZnO(002) in CP@ZnO(002) film is as low as 12.41% (Supplementary Fig. 17), which not only demonstrates the precise of ALD technology but also avoids significant mass burdens on the battery. The TEM and SAED images show that the CNT@ZnO(002) nanowires are uniformly dispersed (Supplementary Fig. 18). The High–resolution TEM (HRTEM) characterization (Fig. 1n) demonstrates that the deposition of ZnO on the surface of CNTs is discontinuous and sparse, with an average particle size of ZnO ranging between 5 and 10 nm. These gaps promote preferential Li deposition and facilitate radial diffusion of Li along the CNTs, enabling a more uniform Li deposition. This aspect is crucial in the choice of ZnO(002) deposition, which exhibits moderate adsorption energy and low Li diffusion barrier. The well–grown ZnO(002) in the form of particles on CNTs not only guarantees the presence of homogeneous Li nucleation sites (Fig. 1o–r) but also enables moderate adsorption and swift migration of Li on the ZnO (002) planes. The Li ions will react with ZnO(002) to produce desired ionic conductive Li2O and LiZn alloy, facilitating lateral Li migration and preventing the concentration of Li nucleation. Furthermore, the electron conductive CNTs (Supplementary Fig. 19) provide a strong electric field driving force for rapid Li/Li+ migration and diffusion, making the CP@ZnO(002) capable for achieving efficient Li utilization and the LiD “catalytic” effect.
The deposition observation of Li with SEM and in situ optical microscopy
To visually demonstrate the moderate “lithiophilic” ability of CP@ZnO(002) and the corresponding “catalytic” effect of LiD, we assembled CP@ZnO(002) film with Li foil to create a half–cell for investigating the morphology evolution of Li deposition. Figure 2a–c, Supplementary Fig. 20a–c, and Fig. 2d–f show physical images of bare CP, O3–treated CP (O3–CP), and CP@ZnO(002) with Li deposition capacities of 1, 3, and 5 mAh cm−2 at 1 mA cm−2, respectively. It’s obvious that the CP and O3–CP gradually became enveloped by Li metal, resulting in the appearance of a large amount of silver–white Li metal on the surface as the Li deposition capacity increased. The corresponding SEM images (Fig. 2g–i and Supplementary Fig. 20d–e) further shows at low Li deposition capacities ( ≤ 3 mAh cm−2), O3–CP can enhance the deposition morphology compared to CP, attributed to the ability of carbon’s defective surface to adsorb Li ions, forming uniform Li nucleation sites. Simultaneously, the high porosity of the CP framework enables it to fully accommodate the deposited Li below 3 mAh cm−2. However, when the deposition capacity of Li reaches practical levels (5 mAh cm−2), a substantial amount of silver–white Li metal appears on the surface of CP and O3–CP, accompanied by numerous Li dendrites. This occurrence stems from the fact that while carbon defects can induce Li nucleation, strong defects hinder Li adsorption, resulting in insufficient Li diffusion, leading to Li aggregation during deposition and ineffective suppression of Li dendrite growth. In contrast, after continuous Li deposition on CP@ZnO(002), optical photographs show no significant precipitation of Li metal. Correspondingly the SEM images (Fig. 2j–l) confirm the uniform deposition of Li metal within the CP@ZnO(002) film framework, thereby achieving a dendrite–free LME with desirable Li accommodation ability. On the path towards the practical application of LMBs, the volume expansion effect and Li dendrite growth in LME pose significant challenges that cannot be ignored. To address this, we characterized the thickness of the deposited CP and CP@ZnO(002) at various capacities. Cross–sectional SEM images reveal the thickness of CP increased from an initial value of 19.8 μm to 80.6 μm (5 mAh cm−2) as the Li deposition capacity increased, resulting in a volume expansion rate of 300% (Supplementary Fig. 21a–d). This is mainly attributed to the lack of “lithiophilic” properties in CPs, which leads to preferential Li aggregation and dendritic growth. The expansion of large Li metal particles further amplifies the spacing between CP frameworks, ultimately causing significant volume changes in the Li composite negative electrode. In contrast, CP@ZnO(002) exhibits a continuous increase in Li deposition thickness, ranging from 20.1 μm (1 mAh cm−2) to 43.8 μm (5 mAh cm−2), with reasonable changes (volume expansion rate 100%) (Supplementary Fig. 21e–h). This observation suggests that the CP@ZnO(002) possesses a robust Li accommodation capability, which, in conjunction with its appropriate electronic conductivity network, plays a pivotal role in enabling “catalytic” conversion and re–utilization of LiD.

Optical photographs and SEM images of CP with a, g 1, b, h 3, and c, i 5 mAh cm−2 of deposited Li at 1 mA cm−2 current density. Optical photographs and SEM images of CP@ZnO(002) with d, j 1, e, k 3, and (f, l) 5 mAh cm−2 of deposited Li. In situ optical microscopy images depicting electrochemical deposition behavior of Li on (m–r) CP and s–x CP@ZnO(002) at the conditions of a current density of 18.75 mA cm−2 and a capacity of 7.8 mAh cm−2. Schematics of Li deposition mechanism on y CP and z CP@ZnO(002).
Furthermore, we conducted real–time observations of the morphological evolution of Li deposition on CP and CP@ZnO(002) using in situ optical microscopy imaging technique under simulated electrochemical conditions. The current of Li deposition is 1.5 mA, and the deposition window size is 0.4*0.2 cm2, so the corresponding specific areal current density of the Li deposition reaches 18.75 mA cm−2. Figure 2m–r illustrates the 25–minute evolution of Li deposition morphology on CP. It is evident that after 10 min of Li deposition, growth of Li dendrite began to occur on the CP surface (Supplementary Movie 1). On the contrary, during the initial 15 min of Li deposition on CP@ZnO(002) (Fig. 2s–x), there was almost no precipitation of metallic Li. As the Li deposition time extended, a large number of Li dendrites emerge haphazardly on the CP surface, posing a critical safety concern for the battery’s performance (Fig. 2y), CP@ZnO(002) demonstrating the adequate Li accommodation capability of CP@ZnO(002) (Supplementary Movie 2). This can be attributed to several factors: (1) the moderate lithiophilicity of ZnO (002), which induces uniform Li nucleation; (2) the low Li migration barrier of ZnO (002) and LiZn alloy, which avoids local Li accumulation; and (3) the electron conductive CNTs that establish an efficient electron network for CP@ZnO(002). Consequently, Li not only deposits on the surface of CNTs but also effectively utilize the available free volume within the CP framework for Li confinement, thus achieving a safe dendrite–free LME (Fig. 2z). However, we also observed that after 25 min of deposition, a small quantity of Li dendrites began to emerge on the surface of CP@ZnO(002), indicating the full occupation of interstitial space within CP@ZnO(002). This emphasizes that matching the capacity of the positive electrode is another important factor in designing safe LMBs.
The CE calculation with Aurbach method
To access the application potential of CP@ZnO(002) in LMBs, the most important indices, CE, are tested for LME armed with CP@ZnO(002). We initially utilized the Aurbach method46 to determine the CE of Li deposition/stripping on both CP, O3–CP and CP@ZnO(002) (Fig. 3a and Supplementary Fig. 22). It is worth noting that the initial deposition exhibits a limited efficiency, which is close to the average cycling efficiency. Therefore, the average cycling efficiency can be calculated by the following equation: where X is the CE; N is the number of cycles; qa, ql, and qr are the cycling capacity, pre–deposited Li capacity (3 mAh cm−2), and finally stripped Li capacity, respectively.

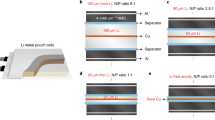
a The CE of CP and CP@ZnO(002) calculated using Aurbach method. The CE and corresponding potential profiles at different current densities: (b, d) 1, and (c, e) 5 mA cm−2. f Hysteresis voltage of Li/CP | | CP/Li and Li/CP@ZnO(002) | | CP@ZnO(002)/Li symmetrical cells. g Cycling performance of Li/CP | | NCM811 and Li/CP@ZnO(002) | | NCM811 full cells. h Schematic diagram of Li/CP@ZnO(002) | | NCM90 pouch cell. i Cycling performance of Li/CP@ZnO(002) | | NCM90 pouch cells. j Performance comparison of Li/CP@ZnO(002) | | NCM90 pouch cell with related cutting–edge works.
Upon pre–depositing 3 mAh cm−2, followed by cycles at 1 mA cm−2 and 1 mAh cm−2, the CP and O3–CP exhibit average CEs of 91.62% and 90.66%, respectively, while CP@ZnO(002) achieved an impressive 99.89%. This signifies the ideal Li adsorption capability of ZnO(002) and the low ion diffusion barrier that promotes uniform Li nucleation and growth, reducing the occurrence of interface side reactions. However, CP accumulates LiD due to its non–affinity for Li, leading to low CE, whereas O3–CP experiences low CE owing to its excessive adsorption affinity towards Li, it hinders Li diffusion. To further elucidate the mechanistic role of ZnO in the early-stage Li nucleation process, we conducted Li deposition/stripping tests under the same conditions using ZnO as the electrode (Supplementary Fig. 23). In the voltage profile, the prominent signal at ~0.5 V corresponds to the electrode reaction of ZnO (ZnO + 2Li++ 2e− → Zn + Li2O)40,41. A peak at 0.2 V signifies the electrolyte decomposition, SEI layer growth, and formation of Li–Zn alloy (Zn + Li++ e− → LiZn). This peak also represents a significant factor contributing to the capacity when the Li deposition voltage reaches 0 V in the Li | |CP@ZnO(002) cell. Furthermore, we also conducted a CE assessment for CP@ZnO(020), CP@ZnO(202), and CP@ZnO (fabricated at ALD 100 °C, Supplementary Fig. 24) as illustrated in Supplementary Fig. 25 to validate the proposed mechanism that highlights moderate adsorption energy and low ion migration barrier as optimal features for achieving efficient Li deposition/stripping performance. The results reveal that the average CEs for CP@ZnO(020), CP@ZnO(202), and CP@ZnO are 95.53%, 96.48%, and 96.46%, respectively, all of which are lower than that of CP@ZnO(002). This indirect evidence serves to underscore that strong adsorption and higher Li migration barriers impede the full utilization of Li, thereby providing selection criteria for the future design of more suitable Li deposition substrates.
Comprehensive electrochemical test
Based on the in situ optical microscopic test results, we further employed CP and CP@ZnO(002) as the working electrodes and Li foil as the counter electrode to study the Li deposition/stripping efficiency in Li | |CP and Li | |CP@ZnO(002) half–cells. Figure 3b and Supplementary Fig. 26 present a comparative analysis of the long–term CE between CP and CP@ZnO(002), and a more detailed presentation is shown in Supplementary Fig. 27. It is evident that CP exhibits significant CE fluctuations after 40 cycles at 1 mA cm−2. Previous work9 has demonstrated that the forms of Li loss in LMBs can be assessed based on CE performance: when the CE is low and exhibits substantial fluctuations, the accumulation of LiD becomes the primary driver of Li resource loss, whereas when the CE is high and remains stable, the loss of Li predominantly arises from the formation of the SEI Li+. Therefore, the test results of CE indicate that CP fails to achieve efficient Li deposition and stripping. The accumulation and activation of LiD on the surface of CP result in erratic fluctuations in CE, which seriously jeopardizes the safe operation of batteries. Additionally, through XPS tests on the Li | |CP and Li | |CP@ZnO(002) half–cells subjected to multiple Li deposition/stripping cycles, we discovered the CP@ZnO(002) effectively promotes a higher abundance of LiF at the Li deposition interface during the stripping process. This observation indicates that, in comparison to the CP, the CP@ZnO(002) possesses more favorable composition and distribution of SEI components. Consequently, it facilitates the achievement of uniform and densely packed Li deposition (Supplementary Fig. 28). In contrast, the corresponding SEM images show a notable accumulation of LiD on the CP surfaces (Supplementary Fig. 29a), while CP@ZnO(002) realizes dendrite–free morphology with a smooth surface (Supplementary Fig. 29b). The synergistic effect of ZnO (002) in CP@ZnO(002) offers uniform Li nucleation sites and promotes rapid Li migration, thereby enabling the deposition of planar Li and stabilizing the negative electrode interface layer. As a result, the CP@ZnO(002) achieves stable Li deposition/stripping for up to 2000 and 180 cycles at 0.5 mA cm−2 / 0.5 mAh cm−2 and 1 mA cm−2 / 1 mAh cm−2 conditions, respectively, and increases the CE lifespan by nearly 6–fold.
The post–cycling CP and CP@ZnO(002) underwent additional microscopic tests for further comparison. The cross–section SEM images show a significant accumulation of LiD on CP surfaces after 200 cycles at a low current density of 0.5 mA cm−2, with a thickness of up to 75.9 μm following Li stripping (Supplementary Fig. 30a). This indicates that, due to CP’s non–lithiophilic character, a large amount of metallic Li deposits on the CP surfaces during the short–term process of Li deposition and stripping, and then strip to cause the accumulation of LiD. On the contrary, CP@ZnO(002) exhibits an intact three–dimensional structure and a clean surface even after 200 cycles, with a thickness around 50.7 μm following Li stripping (Supplementary Fig. 30b). These observations demonstrate that CP@ZnO(002) has sufficient sites and ample space to allow the homogeneous growth of Li during long–term deposition and stripping, which further promotes the activation and recycling of LiD. In particular, when the current density increases to 5 mA cm−2, the CP experiences a sharp decline in CE after approximately 10 cycles. In contrast, CP@ZnO(002) demonstrates a stable Li deposition/stripping behavior and achieves nearly 10 times the CE of CP (Fig. 3c, Supplementary Fig. 31). This remarkable improvement can primarily be attributed to the combination of lower Li nucleation barriers and faster Li migration rates in CP@ZnO(002). Additionally, the electron conductivity of CNTs further enhances the kinetics of Li oxidation/reduction. The corresponding first–cycle potential profiles, conducted at the capacity of 1 mAh cm−2 and different current densities of 1 mA cm−2 and 5 mA cm−2 (Fig. 3d, e), reveal that CP@ZnO(002) possesses lower Li deposition/stripping barriers compared to CP. This finding further demonstrates that moderate adsorption and rapid Li diffusion ability are necessary for achieving efficient Li deposition/stripping. Another important factor influencing the practicality of LME is their cycle life and overpotential at different current densities and deposition capacities. Here, we introduce an thin (20 μm) CP@ZnO(002) interlayer between the Li metal and the separator, constructing Li | |Li symmetric cells for testing. We found that, regardless of high (3 mA cm−2 / 3 mAh cm−2) or low (1 mA cm−2 / 1 mAh cm−2) current densities, the cells with CP@ZnO(002) exhibited lower overpotential ( ~ 10 mV) and significantly extended cycle life compared to those with CP (Fig. 3f, Supplementary Fig. 32), which further proves the promising application prospects of CP@ZnO(002) in improving the electrochemical performance of LME.
Due to the inherent Li content in the LME itself, it is possible to pair LME with almost all positive electrode materials, and LMBs have been regarded as the next–generation energy storage devices capable of achieving breakthrough specific energy. Over the past five years, the specific energy milestones in LMBs have been mostly limited to the Li | |NCM systems4,47,48,49,50,51. This preference arises not only from the higher theoretical capacities of NCM positive electrode materials compared to LiFePO4 (LFP) materials but also due to the more promising commercial outlook of NCM than higher–capacity sulfur positive electrodes. To investigate the practical potential of CP@ZnO(002) as a Li accommodation interlayer, we initially used CP@ZnO(002) as an intermediate accommodating layer for Li deposition and paired it with a high–nickel ternary positive electrode (Li[Ni0.8Co0.1Mn0.1]O2, simplified as NCM811) with a critical areal loading of 4.5 mAh cm−2. As shown in Fig. 3g, the Li/CP@ZnO(002) | | NCM811 full cell demonstrates a stable cycling performance and with a capacity retention rate of 92.6% after 100 cycles. Throughout these cycles, the Li/CP | | NCM811 full cell, although devoid of capacity fade tendency, exhibits the presence of abundant LiD after 30 cycles (Supplementary Fig. 33), while CP@ZnO(002) continues to exhibit stable and uniform Li deposition/stripping, as well as a clear CNTs framework after 100 cycles (Supplementary Fig. 34). This occurrence is mainly due to the accumulation of Li dendrites and LiD at the negative electrode interphase, leading to a severe soft short circuit within the high–nickel positive electrode–equipped LMBs. Consequently, despite its high capacity retention rate, the Li/CP | | NCM811 suffers unexpected capacity drops and failures. Therefore, Li/CP@ZnO(002), as both a Li accommodation space and an activation network for LiD, perfectly addresses this safety hazard and improves the capacity degradation prediction for LMBs. Furthermore, through electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) tests (Supplementary Fig. 35), we found that the NCM811 full cell with CP@ZnO(002) exhibited a more stable interface impedance and efficient electrochemical reaction kinetics. This finding confirms that CP@ZnO(002) promotes the sustainable deposition and stripping of Li, leading to a stable SEI membrane. Additionally, CP@ZnO(002) enhances the migration and uniform deposition of Li ions on the negative electrode side due to the electron conductive CNTs networks and the adequate Li accommodation space, which also constitute essential mechanisms for improving battery cycle life. Remarkably, CP@ZnO(002) has demonstrated a notable enhancement in cycling performance in high–voltage (4.6 V) Li/CP@ZnO(002) | | LCO (LiCoO2) batteries, compared to Li/CP in Li/CP | | LCO batteries (Supplementary Fig. 36). The efficacy of CP@ZnO(002) in optimizing interface and kinetics, as well as enabling dendrite–free LME, is further verified and substantiated (Supplementary Figs. 37, 38). However, for higher specific–energy anode–free LMBs systems, constrained by the active Li consumption brought about by the side reactions between conventional organic electrolytes with Li, and ZnO with Li, utilizing CP@ZnO(002) as the anode–free electrode of LMBs will lead to a 30% capacity decay after 5 cycles (based on CE from Figs. 3b and 3c). This underscores the need for subsequent integration of other optimization strategies for LMBs (such as electrolyte engineering) to collectively tackle this challenge.
Li metal pouch cell
In order to fully exploit the capacity of LME and overcome the specific energy limitations of LMBs, the continuous advancement of NCM positive electrode technology has led to the emergence of higher–nickel–content (Ni ≥ 90%) NCM materials with enhanced capacity. In this context, we have synergistically combined CP@ZnO(002) with a high–nickel ternary positive electrode (NCM90) featuring an high areal loading (5.3 mAh cm−2), alongside a 30 μm–thick Li metal negative electrode. Notably, with limited electrolyte usage (2.45 g Ah−1) (Fig. 3h), we successfully assembled practical Li/CP@ZnO(002) | | NCM90 pouch cells, demonstrating an average specific energy of 380 Wh kg−1 (based on the mass of the whole pouch cell) and remarkable stability over 50 cycles at 0.33 C, along with an impressive average capacity retention rate of 94.3% (Fig. 3i). This achievement signifies a promising future trend for LME technology to match higher–nickel–content NCM positive electrodes (Fig. 3j). Moreover, after cycling in the Li/CP@ZnO(002) | | NCM90 pouch cells for 50 cycles, the electrodes maintain intact physical structures with a smooth and the microscopic image of CP@ZnO(002) show no structural degeneration and no Li cluster (Supplementary Fig. 39). This observation provides compelling evidence for the remarkable capability of CP@ZnO(002) in enduring Li deposition restoration and accommodation, thereby promoting the reactivation and utilization of LiD. These findings also align well with the mechanisms for dendrite–free LME and LiD “catalytic” effect observed in Li/CP@ZnO(002) | | NCM811 and Li/CP@ZnO(002) | | LCO full cells. Consequently, these results fully demonstrate the promising potential of CP@ZnO(002) with its unique structure and chemical properties in practical application for advancing LMBs.
Analysis and discussion of LiD
The quantification of LiD content after cell cycling serves as a crucial indicator for validating the utilization efficiency of active Li and plays a significant role in ensuring the safety of LMBs. Therefore, we employed the time–of–flight secondary ion mass spectrometry (TOF–SIMS) characterization technique to identify the surface configuration and concentration of Li in cycled LMBs (1 mA cm−2 current densities and in the discharged state). The test outcomes exhibit remarkable universality and persuasiveness due to the expansive testing area as 100*100 μm2. In the negative ion mode, there is a significant presence of anions on the surface of the CP after Li stripping. The predominant anions observed include F− (m/z = 19), OH− (m/z = 17), while organic species such as C2 − (m/z = 24), C2H − (m/z = 25), and CN− (m/z = 26) are also found in substantial quantities on the CP surfaces (Fig. 4a, Supplementary Figs. 40, 41). This finding indicates that the decomposition of the electrolyte solvent on the CP surface cannot be overlooked and we speculate this phenomenon is primarily due to the ineffective suppression of Li dendrite growth by the CP, leading to severe deformation of the CP interface during continuous Li deposition and stripping process. Consequently, the SEI film becomes vulnerable, causing the dynamic reaction of fresh Li with the electrolyte and thus the formation of various Li–compounds adhering to the CP surface. Additionally, during Li stripping process, the collapsed Li dendrites lose electronic conductivity due to their encapsulation by the SEI film, resulting in the depletion of active Li and the accumulation of fragile SEI fragments. In contrast, the residual anion species on the CP@ZnO(002) surface are significantly decreased, and the content of organic components is greatly suppressed (Fig. 4b, Supplementary Figs. 42, 43). From the depth elemental distribution results, it can be inferred that compared to the CP, CP@ZnO(002) exhibits a more uniform elemental distribution and a smaller amount of Li salt decomposition/reduction products (F− (m/z = 19)/LiNO3 − (m/z = 69)/LiSNOF− (m/z = 88)). This result suggests that during the initial stages of battery operation, a dense and desired ion–conductive SEI film is completely established on the CP@ZnO(002) surface due to the uniform and dense Li deposition. Consequently, the decomposition of Li salt and solvent is effectively inhibited. In this way, a higher ionic transference number in the electrolyte is ensured, while the formation of organics–dominated and fragile SEI films is avoided. Moreover, in the cation mode, the elemental spectra and mappings show that the CP (Fig. 4c, Supplementary Figs. 44, 45) exhibits a more complex pattern of cationic complexes compared to CP@ZnO(002) (Fig. 4d, Supplementary Figs. 46, 47). Moreover, the presence of characteristic information representing metallic Li (Li+ (m/z = 7), Li2 + (m/z = 14))52 provides robust evidence for the LiD “catalytic” conversion within CP@ZnO(002), which is instrumental in achieving stable cycling of LMBs under limited Li resources.

Mapping distributions of the corresponding Li stripped negative electrodes in negative mode of a CP and b CP@ZnO(002). Mapping distributions of the corresponding Li stripped negative electrodes in positive mode of c CP and d CP@ZnO(002). Schematics of e Li foil dendrite growth, and f LiD “catalytic” effect of CP@ZnO(002) at Li deposition/stripping.
Discussion
In this work, we conducted first–principles calculations to investigate the Li affinity and migration energy barriers of ZnO with different crystal orientations, revealing an inherent trade–off between these two properties in the target material. By utilizing kilogram–scale ALD technology with specific design, we successfully obtained precise ZnO(002)–modified carbon nanotubes with moderate Li adsorption and low Li migration energy barriers. In situ optical microscopy observations provide compelling evidence that CP@ZnO(002) exhibits sufficient and persistent Li accommodation capability. Electrochemical tests demonstrate that CP@ZnO(002) achieves an Li deposition/stripping efficiency of 99.89% and exhibits a cycle life that is 10 times longer than that of CP. Furthermore, by conducting practical Li metal pouch cell validation, CP@ZnO(002) achieves Ah–level Li metal pouch cells with high–nickel positive electrodes (Li/CP@ZnO(002) | | NCM90). The pouch cells exhibit an impressive specific energy of 380 Wh kg−1 (based on the mass of the whole pouch cell) and stable cycling for 50 cycles at 0.33 C under critical conditions (positive electrode areal capacity: 5.3 mAh cm−2, E/C ratio: 2.45, N/P ratio: 1.13). It is worth noting that the specific energy is up to ~424 Wh kg−1, excluding the mass of packing foil, which could be a valuable index for evaluating the specific energy of larger–size batteries. This achievement demonstrates that the matching of LME with high nickel or higher capacity positive electrodes is essential to harness the high specific energy advantages of LME. Finally, TOF–SIMS characterization results reveal that the CP@ZnO(002) surface of the LMBs exhibits less electrolyte decomposition product and Li metal on after cycling, which not only confirms the stability of the negative electrode interface and the high Li utilization rate but also sheds light on the underlying mechanism of the LiD “catalytic” conversion. We believe that this work offers valuable insights into the selection suitable Li–hosting materials and demonstrates the immense potential of our proposed CP@ZnO(002) material in advancing the commercialization of LMBs.
Methods
Materials
Carbon nanotubes (CNTs) were acquired from M–Grade MWNT’s, and the number is NTL–12112. Biofibers was obtained from the corporation of Hainan Guang Yu Biotechnology. Anhydrous ethanol (AR, ≥ 99.8%) was purchased from Xin Shen Shi corporation (Wuhan). Atomic layer deposition (ALD) equipment was from Yun Mao technology corporation (Xiamen), and ALD technique was provided by BattFlex corporation (Wuhan). Li foil and 30 um Li/Cu/Li foil were purchased from China Energy Lithium Co., Ltd. Separator used was Celgard 2400 monolayer PP film. ZnO powder (Purity = 99.9%), Polyvinylidene difluoride (PVDF) (average Mw ~880,000), N–Methyl pyrrolidone (NMP) (AR, ≥ 99%), the solvent, 1,2–dimethoxyethane (DME) (AR, ≥ 99.5%), dioxolane (DOL) (Purity ≥ 99%), ethylene carbonate (EC) (Purity > 99%), dimethyl carbonate (DMC) (Purity ≥ 99%), vinylene carbonate (VC) (Purity ≥ 99.5%), diethyl carbonate (DEC) (Purity ≥ 99%), ethyl methyl carbonate (EMC) (Purity ≥ 98%), fluoroethylene carbonate (FEC) (Purity ≥ 98%), lithium bis(fluoromethyl)imide (LiFSI) (Purity = 99.9%), lithium hexafluorophosphate (LiPF6) (Purity = 99.5%), lithium bis(trifluoromethanesulphonyl)imide (LiTFSI) (Purity ≥ 99.9%), and lithium nitrate (LiNO3) (Purity = 99.99%) were purchased from Sigma–Aldrich without further purification. Carboxyl methyl Cellulose (CMC) (Purity: column chromatography), Styrene, 1,3–butadiene polymer (SBR) (average Mw ~140,000) was purchased from Sinopharm Chemical Reagent Co., Ltd. Super P was purchased from AkzoNobel corporation. NCM90 (Li[Ni0.90Co0.05Mn0.05]O2), NCM811 (Li[Ni0.8Co0.1Mn0.1]O2), and LCO (LiCoO2) positive electrode materials were obtained from Rong Bai technology corporation. Aluminum (Al), copper (Cu) and nickel (Ni) foil purchased from Aladdin Scientific Corp., specifications for battery grade, where the thickness of Al and Cu foil are 10, 6 μm respectively, and the areal weight are 2.7 mg cm−2 and 5.88 mg cm−2 respectively.
DFT computational method
The adsorption and migration simulations of Li on ZnO and LiZn alloy surfaces are conducted by Vienna Ab–initio Simulation Package53 based on density functional theory. The ZnO (002), (020), (202) surfaces and LiZn (220), (112) surfaces are considered according to the experimental XRD tests and the structural regularity. The surface models were created from their geometry optimized bulk structures, and the vacuum thickness between periodic mirror images were set to 20 Å. The atom numbers and the dimensions of optimized surfaces are listed in Supplementary Table 1, the calculation parameters and configurations of Li and ZnO (002), ZnO (020), ZnO (202), LiZn (220), LiZn (112) in Supplementary Data 1–5. In adsorption simulations of Zn (002) and (202) surfaces, two cases distinguished by the category of the top atoms (either Zn or O) were involved. For geometry optimizations, Perdew–Burke–Ernzerhof functional54 was applied to deal with the exchange–correlation interactions, and projector augmented–wave (PAW) method55 with energy cutoff of 520 eV was involved. The k–mesh of Brillouin zone is sampled using Gamma scheme with a density of 0.04*2π/Å. The energy threshold of electronic self–convergence iteration is 10−5 eV, and the force threshold for geometry optimization is 0.02 eV/Å. In all the surface simulations, the positions of bottom two layers of atoms were fixed to approximate the solid parts. The van der Waals interaction was considered using Grimme–D3 method with Becke–Johnson damping function56. Climbing Image Nudged Elastic Band (NEB) method57 was employed for simulating the transport of Li on different surfaces, the calculation parameters for NEB and the corresponding initial and final configurations of Li atoms on ZnO (002), ZnO (020) and LiZn (220) in Supplementary Data 6. The binding energy of Li is calculated by summing the energies of a surface and a Li atom, and then subtracting the energy of a surface–Li adsorption structure.
Materials synthesis
After a 12 h drying process, the CNTs were coated with a 15 nm thick ZnO(002) layer using ALD technology, resulting in the formation of CNT@ZnO(002). The specifications of CNTs used in this work are shown in Supplementary Table 2, including the radius and length of CNTs. In addition, the four–probe method was used to test the compaction density, resistivity and conductivity with pressure of CNTs, the pressure set was 2.000–30.000 MPa. As pressure escalated, the compaction density rose, leading to a decrease in the resistivity of the CNTs and a subsequent enhancement in conductivity, the specific test specifications and parameters are shown in Supplementary Table 3. Before the deposition process, there are several experimental procedures we had to do with the reaction chamber: it was evacuated and filled with argon for cleaning at first, and then ozone was introduced under vacuum conditions to create a large number of defects on CNTs surfaces, followed by the re–evacuation and re–fill of argon for cleaning again. During the deposition process, diethylzinc (DEZ) was introduced to attach to the CNTs surfaces, and the unattached DEZ was completely drained out. Subsequently, the CNTs was put into deionized water (H2O), which reacted with DEZ to produce the target product ZnO(002). After the reaction, argon was applied again to completely remove the gaseous H2O, to elaborate, the included details such as pulse time, gas flow, carrier gas, among others, with specifics on pulse time and carrier gas conditions presented in Supplementary Fig. 48. The attainment of gas pressure within the process chamber upon the introduction of DZE and H2O into the source is illustrated in Supplementary Fig. 49, delineating three interim pressure points preceding a conclusive pressure point. This arrangement aims to enhance the adherence of DEZ to defects on the surface of CNTs and promote a more comprehensive subsequent reaction. Repeated above ALD steps to obtain the ideal ZnO(002) deposition, when the above cavity is fed, it is divided into three small reach pressures of 6–7 Torr, and the reaction is finally carried out under the condition of 20 Torr, all above reaction the temperatures are 200 °C. According to areal density 1 mg cm−2, CNT@ZnO(002) were mixed with biofibers in anhydrous ethanol solvent with the mass ratio of 9:1. To ensure complete dispersion of the CNT@ZnO(002), the mixture was subjected to ultrasonic vibration for 30 min. Subsequently, the uniformly dispersed mixture was filtered on a 500–mesh cloth to obtain the CP@ZnO(002) paper with dimensions of 200 cm × 80 cm. After drying at 80 °C, the CP@ZnO(002) paper was stripped from the carbon cloth. Last but not least, the CP@ZnO(002) was further dried at 100 °C in a vacuum oven for 12 h, which was cut using a 16 mm diameter slicer, followed by carbonization at 600 °C for 5 h in a tube furnace with flowing argon gas. During the carbonization, the heating rate was 2 °C per minute. The CP film was obtained through the same process, while skipping the ALD deposition process.
Assembly of cells
The electrochemical performance of the CP@ZnO(002) was tested in stainless–steel cells assembled in an argon gas–filled glovebox (MIKROUNA). All the Li | |ZnO, Li | |CP, Li | |CP@ZnO(002) half–cells, Li/CP | | CP/Li, Li/CP@ZnO(002) | | CP@ZnO(002)/Li symmetric cells, Li/CP | | NCM811, Li/CP@ZnO(002)/NCM811, Li/CP | | LCO, and Li/CP@ZnO(002)/LCO full cells were assembled in CR2032 coin cells. For Li | |ZnO, Li | |CP, Li | |CP@ZnO(002) Li | |CP@ZnO(020), Li | |CP@ZnO(202), Li | |CP@ZnO half–cells, the diameter of ZnO, CP CP@ZnO(002), CP@ZnO(020) and CP@ZnO(202) negative electrode slices is 12 mm. For Li/CP | | CP/Li, Li/CP@ZnO(002) | | CP@ZnO(002)/Li symmetric cells, the diameter of CP and CP@ZnO(002) negative electrodes slices are 16 mm. For Li/CP | | NCM811, Li/CP@ZnO(002)/NCM811, Li/CP | | LCO, and Li/CP@ZnO(002)/LCO full cells, the diameters of NCM811 and LCO positive electrodes slices are 12 mm, and those of CP and CP@ZnO(002) slices are 12 mm. In all coin cells, the Li electrode slices have a diameter of 16 mm and a thickness of 100 μm, while the separator has a diameter of 16.5 mm, a thickness of 25 μm, and an average porosity of 41%. 50 μl of DME/DOL (1:1, v/v) electrolyte containing 1 M LiTFSI and 2 wt% LiNO3 was added to Li | |CP, Li | |CP@ZnO(002), Li | |ZnO half–cells and Li/CP | | CP/Li, Li/CP@ZnO(002) | | CP@ZnO(002)/Li symmetric cells. For Li/CP | | NCM811, Li/CP@ZnO(002)/NCM811, Li/CP | | LCO, and Li/CP@ZnO(002)/LCO full cells, 50 μl of electrolyte composed of a mixed solvent EC/DMC (1:1, v/v), 1 M LiPF6, and 2.0 wt% VC was added for each cell. The assembly and tests of the half-cells, symmetric cells and full cells were carried out in a constant temperature room at 25 ± 0.5 °C.
Preparation of Li metal deposition CP, CP@ZnO(002) and after cycled CP, CP@ZnO(002)
Li | |CP, Li | |CP@ZnO(002) half–cells were put to rest for 24 h for electrolyte to ensure complete electrolyte infiltration and adaptation to constant temperature conditions, and the Li plating was carried out at 1 mA cm−2 current density. For each cell, 50 μl of electrolyte composed of DME/DOL (1:1, v/v) electrolyte containing 1 M LiTFSI and 2 wt% LiNO3 was added to both Li | |CP and Li | |CP@ZnO(002) half–cells. Half-cells and Li/CP | | NCM811, Li/CP@ZnO(002) | | NCM811 full cells with varied Li deposition capacities (1, 3, 5 mAh cm⁻²) were disassembled in an argon-filled glovebox (H₂O/O₂ <0.1 ppm). Electrodes were sequentially processed through In situ optical microscopy analysis, solvent cleaning (DME/DOL, 1:1 v/v) to remove residual Li salts, complete solvent evaporation under glovebox atmosphere, and vacuum transfer to SEM for surface/cross-sectional imaging. For chemical characterization, solvent-cleaned electrodes underwent TOF-SIMS analysis, and XPS measurements, both conducted under ultrahigh vacuum. All cycling experiments were performed in a temperature-controlled environmental chamber (25.0 ± 0.5 °C) using a LANDMon V7 system. Cell disassembly for post-test analysis was strictly conducted under identical thermal conditions to prevent temperature-induced morphological changes.
In situ optical microscopy
The dimensions of the Li, CP, and CP@ZnO(002) electrodes in the Li | |CP and Li | |CP@ZnO(002) half–cells in situ pool were all 0.8 * 0.8 cm2 and the deposition window size is 0.4 * 0.2 cm2. Commercial Li discs with a diameter of 16 mm and a thickness of 100 μm were used, the separator with a window size of 0.4 * 0.2 cm2 and a thickness of 0.9 mm. A total of 4 ml of DME/DOL (1:1, v/v) electrolyte containing 1 M LiTFSI and 2 wt% LiNO3 was added to both Li | |CP and Li | |CP@ZnO(002) half–cells. For the half–cells, in situ optical microscopy imaging technique was employed under the conditions of a current density of 18.75 mA cm−2 and a capacity of 7.8 mAh cm−2. All electrochemical energy storage performance evaluations were conducted in a temperature-controlled environmental chamber with rigorously maintained ambient conditions at 25.0 ± 0.5 °C throughout the cell cycling experiments.
Preparation of NCM811, LCO positive electrodes and ZnO electrode
The positive electrode mixture slurry comprised NCM811 (96.5 wt%), super P (1.0 wt%), PVDF (2.0 wt%) solution (PVDF 5.0 wt%, the solvent is NMP), and multi–wall carbon nanotubes (MWCNTs, 0.5 wt%). The LCO positive electrode slurry was formulated with 80 wt% active material, 10 wt% Super P conductive agent, and 10 wt% PVDF (5.0 wt% in NMP). For the ZnO electrode, 92 wt% ZnO, 2.0 wt% Super P, and 5.0 wt% CMC (1.5 wt% aqueous solution) were homogenized for 4 h, followed by addition of 1.0 wt% SBR (SBR, 50 wt% aqueous dispersion) with 30 min further mixing. Slurries were blade-coated onto Al foil using gap heights of 150 μm (NCM811), 20 μm (LCO) and 15 μm (ZnO), dried at 60 °C for 12 h under vacuum, and punched into 12 mm discs. The NCM811, LCO and ZnO active material loading was calibrated to 20 mg cm⁻², 2.5 mg cm⁻² and 0.8 mg cm−2, respectively. All electrode preparations were conducted in a climate-controlled environment (25.0 ± 0.5 °C, humidity <30% RH) to ensure thermal stability during processing.
Pouch cell
The fabrication components of Li/CP@ZnO(002) | | NCM90 pouch cells are provided in Supplementary Table 4. Commercial NCM90 (Rong Bai Tech) electrodes were precision-cut using a die-cutting system to dimensions of 4.5 × 6.2 cm² (positive electrode) and 4.7 × 6.5 cm² (negative electrode/CP@ZnO(002)). Stacking configuration followed negative-to-separator-to-positive sequence (6 negative electrode/5 positive electrode layers) prior to ultrasonic tab welding (0.3 mm-thick Al/Ni tabs for positive electrode/negative electrode respectively). Pouch cells were vacuum-sealed after electrolyte injection and 24 h aging at 25.0 ± 0.5 °C. The negative electrode–to–positive electrode capacity (N/P) ratio of Li metal versus NCM90 was controlled at 1.13, enabling multi-layered pouch cells to achieve 424 Wh kg⁻¹ core-specific energy. Electrode manufacturing was executed in a − 55 °C dew-point controlled dry room, implementing strict lithium metal handling protocols to mitigate oxidation risks and ensure electrochemical stability. All cycling experiments were performed in a temperature-controlled environmental chamber (25.0 ± 0.5 °C) using a Neware BTS Client 8.0 system. Cell disassembly for post-test analysis was strictly conducted under identical thermal conditions to prevent temperature-induced morphological changes.
Electrochemical tests
The Aurbach CE test follows the protocol outlined below: (1) an initial formation cycle involves Li deposition of 3 mAh cm−2 at 0.3 mA cm−2, followed by stripping to 1 V; (2) Li deposition of 3 mAh cm−2 occurs at 1 mA cm−2; (3) ten cycles of stripping and deposition of Li repeat at 1 mAh cm−2 and 1 mA cm−2; (4) finally, all Li is stripped to 1 V. For the Coulombic efficiency (CE) tests of Li | |CP, Li | |CP@ZnO(002) half–cells, three cycles of pre–cycling were applied under 0.2 mA cm−2 and 1 mAh cm−2 for SEI construction. To determine the CE, 1 mAh cm−2 of Li was deposited onto CP and CP@ZnO(002) film at 1, 5 mA cm−2, respectively and subsequently stripped to 1 V. To determine the the electrochemical reaction mechanism of Li | |ZnO half–cell, 1 mAh cm−2 of Li was deposited onto ZnO film at 1 mA cm−2, and subsequently stripped to 1 V. Li/CP | | CP/Li and Li/CP@ZnO(002) | | CP@ZnO(002)/Li symmetric cells were tested under 1, 3 mAh cm−2 areal capacities and 1, 3 mA cm−2 current densities, respectively. The cycle performance of Li | |NCM811 full–cells was evaluated within a voltage range of 2.8–4.3 V and a charge/discharge rate of 0.5 C. Similarly, the cycle performance of Li | |LCO full–cells was assessed within a voltage range of 3.0–4.6 V and a charge/discharge rate of 1 C. Li/CP@ZnO(002) | | NCM90 pouch cells underwent an initial two cycles of formation process at a rate of 0.1 C / 0.1 C (1 C = 1400 mA) (Supplementary Fig. 50), followed by the cycling performance measurement at a charge/discharge rate of 0.33 C, within a voltage range of 2.75–4.25 V and an external pressure of 0.34 MPa. Cyclic voltammetry (CV) measurements were conducted on Li/CP | | NCM811 and Li/CP@ZnO(002) | | NCM811 coin cells using an Autolab PGSTAT 302 N instrument. The scanning rates were set at 0.1 mV s−1, and a 12–hour rest period was allowed for electrolyte infiltration and interface stabilization. The electrochemical characterization voltage window for these cells was defined as 2.8–4.3 V. Similarly, CV measurements were performed on Li/CP | | LCO and Li/CP@ZnO(002) | | LCO coin cells under identical conditions, with the voltage window set at 3.0–4.6 V. CV measurements were performed on Li | |ZnO coin cells under identical conditions, with the voltage window set at 0–1.0 V. Electrochemical impedance spectroscopy (EIS) measurements of Li/CP | | NCM811 and Li/CP@ZnO(002) | | NCM811 coin cells were performed after one activation cycle and discharging to 2.8 V, the same process but discharging to 3.0 V for Li/CP | | LCO and Li/CP@ZnO(002) | | LCO for EIS test in potentiostatic mode, a frequency range from 4 MHz to 0.01 Hz, sinus amplitude is 10 mV, data points are acquired by measuring at per decade of frequency, 0 V quasi-stationary potential applied before carrying out the EIS measurement. All electrochemical energy storage performance evaluations were conducted in a temperature-controlled environmental chamber with rigorously maintained ambient conditions at 25.0 ± 0.5 °C throughout the cell cycling experiments.
Structural/Physicochemical characterization
X–ray diffraction (XRD) characterization was performed using a non–monochromated Cu Kα X–ray source on an X–ray diffractometer (Miniflex600). Time–of–flight secondary ion mass spectrometry (TOF–SIMS) was conducted using the ToF.SIMS 5–100 instrument (IONTOF GmbH) in a vacuum chamber to prevent exposure to ambient air. A primary beam of Bi3 + + (30 kV) was utilized for sample detection. Scanning electron microscopy (SEM) images were acquired using the JEOL JSM–7100 F and Hitachi SU–8010 electron microscopes. Transmission electron microscopy (TEM), selective area electron diffraction (SAED) and high–resolution TEM (HRTEM) images were obtained using the Titan G2 60–300 instrument equipped with an energy dispersive spectroscopy (EDS) image corrector. Thermogravimetric (TG) curves were generated using a thermal analyzer (Netzsch STA 449 C), while X–ray photoelectron spectroscopy (XPS) measurements were conducted using the VG MultiLab 2000 instrument. In situ microimaging was performed using an in situ characterization device provided by Beijing Zhongyan Huanke Science & Technology Corporation. Raman spectral data of the carbon paper were collected using a Witec–Raman Spectrometer Alpha 300 device with an excitation wavelength of 532 nm. Oxygen elemental analysis was performed using an Eltra ONH2000 analyzer (Eltra GmbH, Germany).
Data availability
All data supporting the findings described in this manuscript are available within the paper and its Supplementary Information, and the source data is available on the public data repository Figshare (https://doi.org/10.6084/m9.figshare.28682144). Source data are provided with this paper.
References
Yu, S. et al. Composite Lithium Metal Structure to Mitigate Pulverization and Enable Long–Life Batteries. Adv. Energy Mater. 13, 2302400 (2023).
Zhao, L. et al. Revisiting the Roles of Natural Graphite in Ongoing Lithium–Ion Batteries. Adv. Mater. 34, 2106704 (2022).
Jiao, X. et al. Multi–Physical Field Simulation: A Powerful Tool for Accelerating Exploration of High–Energy–Density Rechargeable Lithium Batteries. Adv. Energy Mater. 13, 2301708 (2023).
He, Q. et al. Ultra–Uniform and Functionalized Nano–Ion Divider for Regulating Ion Distribution toward Dendrite–Free Lithium–Metal Batteries. Adv. Mater. 35, 2302418 (2023).
Zhang, S. et al. Spatially Hierarchical Carbon Enables Superior Long–term Cycling of Ultrahigh Areal Capacity Lithium Metal Anodes. Matter 5, 1263–1276 (2022).
Kim, E. et al. Functionality of 1–Butyl–2, 3–Dimethylimidazolium Bromide (BMI–Br) as a Solid Plasticizer in PEO–Based Polymer Electrolyte for Highly Reliable Lithium Metal Batteries. Adv. Energy Mater. 13, 2301674 (2023).
Xu, X. Q. et al. Dendrite–accelerated thermal runaway mechanisms of lithium metal pouch batteries. SusMat 2, 435–444 (2022).
Qian, S. et al. CuCl2–Modified Lithium Metal Anode via Dynamic Protection Mechanisms for Dendrite–Free Long–Life Charging/Discharge Processes. Adv. Energy Mater. 12, 2103480 (2022).
Fang, C. et al. Quantifying Inactive Lithium in Lithium Metal Batteries. Nature 572, 511–515 (2019).
Li, S. et al. A Dynamically Stable Mixed Conducting Interphase for All–Solid–State Lithium Metal Batteries. Adv. Mater. 36, 2307768 (2023).
Cheng, X. B. et al. Electrochemically and Thermally Stable Inorganics–Rich Solid Electrolyte Interphase for Robust Lithium Metal Batteries. Adv. Mater. 36, 2307370 (2023).
Ma, Q. et al. Designing Bidirectionally Functional Polymer Electrolytes for Stable Solid Lithium Metal Batteries. Adv. Energy Mater. 13, 2203892 (2023).
Kim, S. C. et al. High–entropy Electrolytes for Practical Lithium Metal Batteries. Nat. Energy 8, 814–826 (2023).
Jiang, F. N. et al. Thermoresponsive Electrolytes for Safe Lithium-Metal Batteries. Adv. Mater. 35, 2209114 (2023).
Xu, J. et al. Revealing the Anion–Solvent Interaction for Ultralow Temperature Lithium Metal Batteries. Adv. Mater. 36, 2306462 (2023).
Yu, Z. et al. A Solution–Processable High–Modulus Crystalline Artificial Solid Electrolyte Interphase for Practical Lithium Metal Batteries. Adv. Energy Mater. 12, 2201025 (2022).
Fan, L. et al. A Dual–Protective Artificial Interface for Stable Lithium Metal Anodes. Adv. Energy Mater. 11, 2102242 (2021).
Naren, T. et al. Reactive Polymer as Artificial Solid Electrolyte Interface for Stable Lithium Metal Batteries. Angew. Chem. Int. Ed. 62, e202305287 (2023).
Shang, J. et al. Metallic Glass–Fiber Fabrics: A New Type of Flexible, Super–Lightweight, and 3D Current Collector for Lithium Batteries. Adv. Mater. 35, 2211748 (2023).
Qing, P. et al. Highly Reversible Lithium Metal Anode Enabled by 3D Lithiophilic–Lithiophobic Dual–Skeletons. Adv. Mater. 35, 2211203 (2023).
Sun, J. et al. Robust Transport: An Artificial Solid Electrolyte Interphase Design for Anode–Free Lithium–Metal Batteries. Adv. Mater. 35, 2209404 (2023).
Li, Y. et al. Artificial Graphite Paper as a Corrosion–Resistant Current Collector for Long–Life Lithium Metal Batteries. Adv. Funct. Mater. 33, 2214523 (2023).
Huang, A. et al. Lithiophilic Mo2C Clusters–Embedded Carbon Nanofibers for High Energy Density Lithium Metal Batteries. Adv. Funct. Mater. 33, 2303111 (2023).
Guan, W. et al. Impact of Morphological Dimensions in Carbon–Based Interlayers on Lithium Metal Anode Stabilization. Adv. Energy Mater. 13, 2302565 (2023).
Chen, C. et al. Lotus–Root–Like Carbon Fibers Embedded with Ni–Co Nanoparticles for Dendrite–Free Lithium Metal Anodes. Adv. Mater. 33, 2100608 (2021).
Wang, C. et al. A Large–area Lithium Metal–carbon Nanotube Film for Precise Contact Prelithiation in Lithium–ion Batteries. Energy Environ. Sci. 16, 4660–4669 (2023).
Pathak, R. et al. Advanced Strategies for the Development of Porous Carbon as a Li Host/Current Collector for Lithium Metal Batteries. Energy Storage Mater. 41, 448–465 (2021).
Wang, Z. Y. et al. A Dendrite–Free Lithium/Carbon Nanotube Hybrid for Lithium–Metal Batteries. Adv. Mater. 33, 2006702 (2020).
Qian, H. et al. LiZn/Li2O Induced Chemical Confinement Enabling Dendrite–Free Li–Metal Anode. Adv. Funct. Mater. 34, 2310143 (2024).
Wu, Z. et al. Growing single-crystalline seeds on lithiophobic substrates to enable fast-charging lithium-metal batteries. Nat. Energy 8, 340–350 (2023).
Li, N. et al. Normalized Lithium Growth from the Nucleation Stage for Dendrite–Free Lithium Metal Anodes. Angew. Chem. Int. Ed. 58, 18246–18251 (2019).
Xiao, Y. et al. The Regulation of Lithium Plating Behavior by State of Stripping in Working Lithium Metal Anode. Adv. Energy Mater. 13, 2300959 (2023).
Wang, C., Yang, C., Du, Y., Guo, Z. & Ye, H. Spherical Lithium Deposition Enables High Li–Utilization Rate, Low Negative/Positive Ratio, and High Energy Density in Lithium Metal Batteries. Adv. Funct. Mater. 33, 2303427 (2023).
Jiao, S. et al. Behavior of Lithium Metal Anodes under Various Capacity Utilization and High Current Density in Lithium Metal Batteries. Joule 2, 110–124 (2018).
He, D. et al. Electronic Localization Derived Excellent Stability of Li Metal Anode with Ultrathin Alloy. Adv. Sci. 9, 2105656 (2022).
Liu, Y. et al. Lithium-coated polymeric matrix as a minimum volume-change and dendrite-free lithium metal anode. Nat. Commun. 7, 10992 (2016).
Wang, X. et al. Glassy Li Metal Anode for High–Performance Rechargeable Li Batteries. Nat. Mater. 19, 1339–1345 (2020).
Li, Y. et al. Lithiophilicity: The Key to Efficient Lithium Metal Anodes for Lithium Batteries. J. Energy Chem. 77, 123–136 (2023).
Yang, T., Li, L., Wu, F. & Chen, R. A Soft Lithiophilic Graphene Aerogel for Stable Lithium Metal Anode. Adv. Funct. Mater. 30, 2002013 (2020).
Kim, M. S. et al. Suspension Electrolyte with Modified Li+ Solvation Environment for Lithium Metal Batteries. Nat. Mater. 21, 445–454 (2022).
Wood, K. N. et al. Operando X–ray Photoelectron Spectroscopy of Solid Electrolyte Interphase Formation and Evolution in Li2S–P2S5 Solid–state Electrolytes. Nat. Commun. 9, 2490 (2018).
Yu, J. et al. Smart Construction of Multifunctional Li1.5Al0.5Ge1.5(PO4)3|Li Intermediate Interfaces for Solid–state Batteries. Energy Storage Mater. 46, 68–75 (2022).
Ouyang, Y. et al. In Situ Formed LiZn Alloy Skeleton for Stable Lithium Anodes. ACS Appl. Mater. Interfaces 12, 25818–25825 (2020).
Malm, J., Sahramo, E., Perälä, J., Sajavaara, T. & Karppinen, M. Low–temperature Atomic Layer Deposition of ZnO Thin Films: Control of Crystallinity and Orientation. Thin Solid Films 519, 5319–5322 (2011).
Cai, J. et al. A revisit to atomic layer deposition of zinc oxide using diethylzinc and water as precursors. J. Mater. Sci. 54, 5236–5248 (2019).
Aurbach, D., Gofer, Y. & Langzam, J. The Correlation Between Surface Chemistry, Surface Morphology, and Cycling Efficiency of Lithium Electrodes in a Few Polar Aprotic Systems. J. Electrochem. Soc. 136, 3198–3205 (2019).
Xu, Q. et al. High Energy Density Lithium Metal Batteries Enabled by a Porous Graphene/MgF2 Framework. Energy Storage Mater. 26, 73–82 (2020).
Zhao, Q. et al. Upgrading Carbonate Electrolytes for Ultra–stable Practical Lithium Metal Batteries. Angew. Chem. Int. Ed. 61, e202116214 (2022).
Gao, Y. et al. Effect of the Supergravity on the Formation and Cycle life of Non–aqueous Lithium Metal Batteries. Nat. Commun. 13, 5 (2022).
Zhang, L. et al. Practical 4.4 V Li||NCM811 Batteries Enabled by a Thermal Stable and HF Free Carbonate–based Electrolyte. Nano Energy 96, 107122 (2022).
He, B. et al. Scalable Fabrication of a Large–area Lithium/Graphene Anode towards a Long–Life 350 Wh kg−1 Lithium Metal Pouch Cell. J. Mater. Chem. A 9, 25558–25566 (2021).
Nanda, S. & Manthiram, A. Lithium Degradation in Lithium–sulfur Batteries: Insights into Inventory Depletion and Interphasial Evolution with Cycling. Energy Environ. Sci. 13, 2501–2514 (2020).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total–energy calculations using a plane–wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented–wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT–D) for the 94 elements H–Pu. J. Chem. Phys. 132, 154104 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
This work was financially supported by the Major Research Plan of the National Natural Science Foundation of China (92372207,J. Lu), the National Natural Science Foundation of China (52302258, Z. Li), Hubei Provincial Natural Science Foundation of China (2024AFB1003, Z. Li), Wuhan Knowledge Innovation Special project of China (2022010801010369, Y. Li), University–Level Research Project Funding Program of JHUN (2022XKZX01, Y. Li) and Excellent Discipline Cultivation Project of JHUN (2023XKZ011, M. Xie). Jiantao Li and Khalil Amine gratefully acknowledge support from the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy, Vehicle Technologies Office.
Author information
Authors and Affiliations
Contributions
Z.H. Li, J. T. Li, Y. Y. Li, M.Xie and J.Lu conceived and directed the project. H.S. Shen, Z.H. Li and J.T. Li lead the syntheses and characterizations. Q. He and Y. Zhao carried out the density functional theory calculations. K. Amine, M. J. Li, Y.N. Tian and G.N. Wu helped analyze the data. X.X. Zhang and J.J. Xiao guided the welding of CCs with metal tabs. All authors participated in the discussion of the results, commented on the implications, and fully approved the content of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jong Hun Kang and the other, anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, H., Li, Z., Li, J. et al. Industrializable interlayer with catalytic conversion of dead lithium for Ah–level Nickel–rich lithium metal batteries. Nat Commun 16, 5462 (2025). https://doi.org/10.1038/s41467-025-60609-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60609-4
