
Abstract
Asymmetric hydrophosphorylation of unsaturated compounds is a straightforward and atom-economic approach to obtain chiral organophosphorus compounds. Herein, we describe a ligand-controlled regiodivergent and enantioselective hydrophosphorylation of styrenes facilitated by Pd catalysis and a PC-Phos ligand. This methodology enables the facile synthesis of both Markovnikov and anti-Markovnikov alkyl phosphorus compounds with high to excellent regio- and enantioselectivity, achieving up to >95:5 rr and 92% ee. Deuterium-labeling experiment and computational mechanistic studies unveil that the migratory insertion is the enatio-determining step, in which the O···H hydrogen bonding between the H-phosphonate and the ligand is identified as a crucial factor. Furthermore, our investigations demonstrate that both migratory insertion and reductive elimination contribute to achieving high regioselectivity and enantioselectivity. These findings not only advance the field of asymmetric hydrophosphorylation of simple styrenes, but also deepen our understanding of noncovalent interactions in ligand design.
Similar content being viewed by others

Introduction
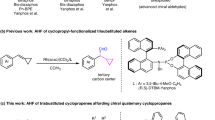
Enantioenriched organophosphorus compounds have garnered substantial attention in recent decades owing to their frequent occurrence in bioactive molecules and functional materials1,2,3,4,5. Among the numerous chiral organophosphorus compounds, α-chiral alkyl phosphorus compounds have received significant attention owing to their biological activities6,7,8,9,10,11 and application as chiral ligands and catalysts in asymmetric catalysis (Fig. 1a)12,13,14,15. Conventional methods to access these compounds always involve various chiral auxiliaries and multiple steps16,17,18,19. Therefore, the pursuit of novel methods for synthesizing these enantioenriched phosphorus-containing compounds has been a subject of considerable interest. The predominate routine to access these compounds is asymmetric hydrogenation of α-substituted vinylphosphonates with Rh20,21,22, Ir23,24 and Ru25,26,27. Notably, Zhang and coworkers recently reported an elegant nickel-catalyzed asymmetric hydrogenation of α-substituted vinylphosphonates28. Besides the asymmetric hydrogenation, C-P bond coupling reactions also provide a convenient routine for α-chiral alkyl phosphorus compounds. For instance, Liu and coworkers presented an enantioconvergent radical Michaelis-Becker-type C-P bond coupling between H-phosphonates with alkyl halides via Cu catalysis (Fig. 1b)29,30.

a Representative example of α-chiral alkyl phosphorus compounds. b Current strategies of catalytic access α-chiral alkyl phosphorus. c Previous work: catalytic asymmetric addition of phosphorus nucleophiles to unsaturated bonds. d Our design: enantioselective hydrophosphorylation of styrenes by Pd catalysis.
Apart from the hydrogenation and C-P coupling, the past few decades also witnessed great development on asymmetric addition of phosphorus nucleophiles to unsaturated bonds, including alkynes31,32,33,34,35,36,37,38, dienes39,40, allenes41,42, and alkenes43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59. It is regarded as one of the most economical and straightforward methods to access enantioenriched organophosphorus compounds. In this regards, Duan and Leung pioneerly developed the asymmetric asymmetric Michael addition of diarylphosphines to enones. Later, Dong and coworkers reported a seminal work of enantioselective coupling of terminal 1,3-dienes and phosphine oxides to afford allylic phosphine oxides by palladium catalysis. Meanwhile, this protocol was also applied to bicyclic alkenes and other active alkene substrate. Despite these advances, the alkene partner involved in the asymmetric addition is still confined to electro-biased Michael acceptors43,44,45,46,47,48,49,50,51,52,53, or other activated alkenes (Fig. 1c)54,55,56,57,58,59. Since the initial report of hydrophosphorylation of alkenes by Han and Tanaka60, sporadic efforts have been devoted to the enantioselective hydrophosphorylation of alkenes, particularly styrenes61,62, which represent one of the most straightforward ways to access α-chiral alkyl phosphorus compounds. However, these attempts have been plagued by poor regioselectivity or enantioselectivity, which is known to be challenging to control. The primary difficulty arises from the lack of coordination site and low reactivity of styrenes compared with Michael acceptors and other activated alkenes. Moreover, the potential regioselectivity issue further complicates the situation63. Additionally, the facile tautomerism of the hydrogen phosphoryl compounds between the P(V) and P(III) forms, allows them to act as ligands to transition metal, potentially deactivating the catalyst64,65,66,67,68. Nevertheless, we believe that the particular ligand is pivotal in overcoming these challenges.
Recently, our groups have become increasingly intrigued by the formation of P-C bonds through the Pd-catalyzed P-C cross coupling reactions69,70 and the atom-economical hydrophosphinylation of alkynes33. Building on our continuous interest in the synthesis of P-containing enantioenriched compounds, herein we present our work on the Pd-catalyzed enantioselective hydrophosphorylation of styrenes. Notably, it enables facile access to different α and β-arylphosphonic acid derivatives by simply switching the ligands (Fig. 1d).
Results
Optimization of the reaction conditions
At the outset, the asymmetric hydrophosphorylation reaction was carried out using 4-phenylstyrene (1a) with pinacol H-phosphonate (4,4,5,5-tetramethyl-1,3,2-dioxaphospholane 2-oxide, 2a)60 as the model reaction to verify the feasibility. As shown in Table 1, a range of commercially available chiral phosphine ligands, including bidentate P, P-ligands (L1-L3, L5-L6), N, P-ligands L7, and monodentate ligand (L4), were initially examined. Unfortunately, most of the commercial ligands did not yield good results. Although the bisphosphine ligands (L1, L2, L5) are promising in facilitating the reaction, it predominantly produced the anti-Markovnikov product. In contrast, when monophosphine ligands (L4) or N, P-ligands (L6, L7) were used, the reaction was dominated by Markovnikov products, but the reactivity and enantioselectivity of the reaction were suboptimal. These observations underscore the critical role of the ligand in influencing the reactivity, regioselectivity and enantioselectivity. Our recently developed Sadphos, known for their excellent performance in hydrophosphinylation of alkynes33, prompted further investigation of their efficacy in the hydrophosphorylation reaction. Xu-Phos, Xiang-Phos and TY-Phos, like the other commercially available ligands, exhibit unsatisfactory catalytic performance. However, our exploration revealed intriguing insight: Wei-Phos ligands demonstrated good reactivity, albeit suffering from poor enantioselectivity, as observed with Segphos (L1). The use of Xiao-Phos improved reactivity, but shared similar regioselectivity control as Ming-Phos and PC-Phos. Notably, PC1, in contrast to Ming1, exhibited a slight improvement in enantioselectivity (Table 1, Entry 1). Therefore, we selected PC-Phos as the ligand backbone for the reaction modification. Initial modifications involved replacing the aryl group with the alkyl group, such as tBu (PC2) and Ad (PC3), resulting in significantly increased reactivity, but a minor decrease in the enantioselectivity control (Table 1, Entries 3–4). Subsequent modifications fine-tuned the substituents on aryl group of the PC-Phos (Table 1, Entries 4–10). Remarkably, the enantioselectivity (44%–78% ee) was progressively improved with increasing aryl-neighboring site hindrance (Me, iPr, tBu) on PC-Phos (Table 1, Entries 8–10). Upon identifying PC10 as the optimal ligand, we investigated the catalyst precursor for the reaction. Using [Pd(allyl)Cl]2 as a catalyst further improved the reaction yield and enantioselectivity without compromising regioselectivity (Table 1, Entry 11). We also fine-tuned other parameters, such as solvents, additives, and catalyst loading, to enhance yield and enantioselectivity (see pages S6-S8 of the Supplementary information). Notably, the reaction could not proceed without the addition of (PhO)2P(O)OH (Table 1, Entry 15). Other H-P reagents, such as H-phoshpine oxide, H-phosphinates and other acylic H-phosphonates showed relatively low reactivity than 2a (See Supplementary Fig. 1 for details). Ultimately, under the optimal reaction conditions, product 3aa from the hydrophosphorylation reaction was obtained in high yield (87%) with high enantioselectivity (90% ee) and excellent regioselectivity (>95:5 rr) (Table 1, Entry 16).
Reaction scope study
Subsequently, the scope of substrates for vinylarenes 1 was examined under optimal conditions. As shown in Fig. 2, vinylarenes with electron-donating and electron-withdrawing substituents at the para-, meta-, or ortho-positions furnished the desired products (3aa-3qa) in moderate to good yields with excellent regioselectivities (up to >95:5 rr) and good enantioselectivities (up to 92% ee). The reaction displayed good tolerance of functional groups, including fluoro (3ha, 3na, 3qa), chloro (3oa), ester (3ia), cyano (3ja), alkyl (3ca, 3da), alkoxy (3ea, 3ma, 3pa) and aryl (3aa) or alkenyl (3la) groups. It highlighted that substrates containing thioethers (1f) and boron esters (1g), which were typically incompatible in metal-catalyzed coupling reactions, delivered the desired products (3fa, 3ga) in good yields with excellent regioselectivities and enantioselectivities. Similarly, polysubstituted aryl (3ta) and naphthyl groups (3ra, 3sa) exhibited favourable results in asymmetric hydrophosphorylation reactions. Additionally, heteroaryl (1u) and ferrocene-substituted substrates (1v) proved suitable for this reaction system, though the enantioselectivity of the ferrocene-substituted product (3va) was slightly lower. The absolute configuration of 3aa was confirmed by X-ray crystallography analysis, and those of the others were assigned analogously.

aReaction condition A: [a] 1 (0.22 mmol), 2a (0.2 mmol), (PhO)2P(O)OH (25 mol%), [Pd(allyl)Cl]2 (3 mol%) and PC10 (15 mol%) in DCM (1 ml) at 100 °C for 24 h. [b] 15 mol% TsOH·H2O was used instead of (PhO)2P(O)OH. Reaction condition B: 1 (0.22 mmol), 2a (0.2 mmol), (PhO)2P(O)OH (30 mol%), Pd(OAc)2 (6 mol%) and rac-BINAP (15 mol%) in dioxane (0.6 ml) at 80 °C for 24 h.
During the screening of the conditions, it came to our attention that enantioselective synthesis of ent-3aa could be achieved by employing diastereoisomers of Ming-Phos and PC-Phos-like ligands (see pages S4-S5 of the Supplementary information). Subsequently, we initiate trials using the diastereoisomer of the best ligand (RS,SC)-PC10. The result indicated that the reaction performance was comparable when (RS,RC)-PC10 was used instead of (RS,SC)-PC10. Therefore, (RS,RC)-PC10 was used as ligand for the synthesis of ent-3 and the reactions consistently delivered the corresponding enantiomer products ent-3 in good yields (up to 80%), regioselectivities (up to >95:5 rr), and enantioselectivities (up to 92% ee). This result highlights a novel approach for the rapid synthesis of product enantiomers, especially considering that the two diastereomers of the ligand were easily accessible by varying the organometallic reagent RLi or RMgX71.
Likewise, in our ligand screening, we observed that bisphosphine ligands had the capacity to modulate the regioselectivity of the hydrophosphorylation reaction, favoring the anti-Markovnikov product. To delve deeper into this phenomenon, common bisphosphine ligands were subsequently screened. It was ultimately discovered that using rac-BINAP enabled the reaction to favor the anti-Markovnikov product 4aa in high yield with excellent regioselectivity (see pages S8-S9 of the Supplementary information). As illustrated in Fig. 2, we systematically investigated the substrate scope of the reaction, which demonstrated good compatibility of functional groups and furnished the corresponding target products 4 in good yields (up to 84%) with excellent regioselectivities (up to >95:5 rr). Alkyl substituted alkenes were also examined and proved to be not compatible in current system, resulting in poor reactivity. Extensive exploration of ligands was conducted using allylarene 1w-1aa as the substrate. It is pleasing to detect the chain-walking hydrophosphorylation products 3wa-3aaa, though the regioselectivity is relatively poor.
To investigate the mechanism, a series of control experiments were conducted as outlined in Fig. 3. Initially, deuterium-labeled pinacol H-phosphonate d−2a was subjected to the standard reaction conditions (Fig. 3c). In this experiment, deuterium incorporation was observed at both the α (0.10 D) and β (0.58 D) positions of d−3aa. Similarly, deuterium incorporation was also observed at both the α (0.34 D) and β (0.34 D) positions of d−4aa under standard condition B. An attempt to extend the current asymmetric hydrophosphinylation to the alkyl substituted alkene proved to be unsuccessful. As shown in Fig. 3d, the reaction with allylarene 1wa afforded the chain-walking product 3wa in poor yield and regioselectivitivity, albeit with good enantioselectivity. Additionally, a slight negative linear relationship between the ee values of the PC10 and product 3aa was observed, suggesting that minor oligomeric aggregates of catalyst may play a role in influencing the stereochemistry of the asymmetric hydrophosphorylation reaction (Fig. 4e)72,73,74. Based on literature precedence37 and our observations, we propose a plausible mechanism depicted in Fig. 3f. The Pd(0) precatalyst I undergoes oxidative addition to diphenylphosphoric acid ((PhO)2P(O)OH) to form PdH species II. Notably, a significant loss of yield was observed in the absence of the acid additive ((PhO)2P(O)OH). A related oxidative addition has been implicated as a key step in the hydrophosphinylation of terminal alkynes35,36. Following this, the ligand exchange and two distinct modes of migratory insertion lead to the formation of species V and IV. Subsequent reductive elimination furnishes the product and regenerates the catalyst.

a Gram scale reaction. b Synthetic transformations. c Deuterium-labeling experiments. d Reaction with allylarene. e nonlinear effect study. f proposed mechanism.

a Calculated reaction pathway. b Distortion/interaction and independent gradient model based on Hirshfeld partition (IGMH) analysis.
To probe the origin of the enantioselectivity and regioselectivity control, we performed DFT calculations on the reaction pathway (Fig. 4a). Following the oxidative addition of diphenylphosphinic acid and subsequent ligand exchange, the migratory insertion of Int3 with styrene occurs, leading to the major benzyl-Pd intermediate Int4 with an energy barrier of 4.8 kcal/mol and the minor benzyl-Pd intermediate ent-Int4 with an energy barrier of 7.1 kcal/mol. While, the energy barrier to generate the linear alkyl-Pd intermediate Int5 is 6.3 kcal/mol. As the energy of the following reductive elimination from TS4 and ent-TS4 is relatively lower than the corresponding TS2 and ent-TS2. Therefore, we speculate the migratory insertion is the enantio-determining step.c The high regioselectivity is attributed to the elevated energy barrier of reductive elimination (TS5: 33.7 kcal/mol) to afford the anti-Markovnikov product (The possibility of the migratory insertion occurring before the ligand exchange cannot be completely excluded). The energy of the following reductive elimination from TS4 and ent-TS4 is relatively lower than the corresponding TS2 and ent-TS2. According to our DFT calculations, migratory insertion is considered to be the enantio-determining transition state. The comparison of TS2 and ent-TS2 is shown in Fig. 4b. The ΔΔG≠(2.3 kcal/mol) was well consistent with the experimental enantioselectivity. Steric environment analysis of the transition state indicated that TS2 possessed a stronger hydrogen bonding interaction between the phosphonate P = O and the NH, as well as the tert-butyl CH on the ligand. This weak hydrogen bonding interaction may contribute to the energy difference of migratory insertion. Control experiment showed a significantly decrease in enantioselectivity and reactivity when the reaction was conducted in high polarity solvent, such as DMF and EtOH, or when the NH is absent (see Supplementary Fig. 3 for details). Furthermore, distortion/interaction model analysis75,76,77 suggested that the barrier differences between TS2 and ent-TS2 originate primarily from the much greater interaction between the substrate and catalyst (Fig. 4b). An independent gradient model based on Hirshfeld partition (IGMH)77,78,79,80,81 indicated a significant interaction between the tert-butyl group of the ligand and the aromatic ring of styrene (the green region implied attractive interactions).
Subsequently, the origin of the regioselectivity control to obtain the anti-Markovnikov product while using rac-BINAP is also probed by DFT calculation. Similarly, the calculations revealed that migrataroy is the regio-determining step. Again, the hydrogen bonding interaction between the phosphonate P = O and the aryl CH on the ligand is observed to be stronger in the anti-Markovnikov transition state (see Supplementary Fig. 4 for details).
Discussion
We presented a ligand-controlled palladium-catalyzed asymmetric hydrophosphorylation of simple styrenes, enabling regio- and stereoselective access to both branched and linear alkyl phosphorus compounds by simply switching the ligands. Mechanistic studies and DFT calculation have provided valuable insights into the reaction process, revealing the reversibility of the migratory insertion and underscoring the importance of noncovalent interactions (O···H hydrogen bonding, ect) between the phosphonate P = O and the ligand. These findings not only advance the field of asymmetric hydrophosphorylation but also deepen our understanding of the noncovalent interactions in ligand design. Given the high importance and broad synthetic application of enantioenriched organophosphorus compounds, we believe that this protocol will pave the way for future developments of hydrophosphorylation.
Methods
Typical procedure for ligand-controlled regiodivergent and enantioselective hydrophosphorylation of styrenes by palladium
Under a nitrogen atmosphere, vinylarenes 1 (0.22 mmol), 2a (0.2 mmol), [Pd(allyl)Cl]2 (3 mol%), PC10 (15 mol%) and (PhO)2P(O)OH (25 mol%) were added to a dry 10 mL sealed tube. Then, DCM (1.0 mL) was added to the reaction tube under a nitrogen atmosphere. The resulting mixture was stirred with 1000 rpm at 100 °C in an oil bath. After the reaction was complete as monitored by TLC, the reaction mixture was filtered to remove insoluble and further purified by flash column chromatography on silica gel (Petroleum ether: Ethyl acetate) to afford the desired product 3 or ent-3.
Under a nitrogen atmosphere, vinylarenes 1 (0.22 mmol), 2a (0.2 mmol), Pd(OAc)2 (6 mol%), rac-BINAP (15 mol%) and (PhO)2P(O)OH (30 mol%) were added to a dry 10 mL sealed tube. Then, dioxane (0.6 mL) was added to the reaction tube under a nitrogen atmosphere. The resulting mixture was stirred with 1000 rpm at 80 °C in an oil bath. After the reaction was complete as monitored by TLC, the reaction mixture was filtered to remove insoluble and further purified by flash column chromatography on silica gel (Petroleum ether: Ethyl acetate) to afford the desired product 4.
Data availability
We declare that all the data supporting this study, including the experimental details, data analysis, and spectra for all unknown compounds, are available within the article and its Supplementary Information Files. Source Data are provided with this paper. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2248216 ((R)-3aa)). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Source data are provided with this paper.
References
Börner, A. Phosphorus ligands in asymmetric catalysis: synthesis and applications (Wiley-VCH, Weinheim) (2008).
Joly, D. et al. White organic light-emitting diodes based on quench-resistant fluorescent organophosphorus dopants. Adv. Funct. Mater. 22, 567–576 (2012).
Carroll, M. P. & Guiry, P. J. P,N ligands in asymmetric catalysis. Chem. Soc. Rev. 43, 819–833 (2014).
Guo, H., Fan, Y. C., Sun, Z., Wu, Y. & Kwon, O. Phosphine organocatalysis. Chem. Rev. 118, 10049–10293 (2018).
Xie, C., Smaligo, A. J., Song, X.-R. & Kwon, O. Phosphorus-based catalysis. ACS Cent. Sci. 7, 536–558 (2021).
Andaloussi, M. et al. Design, synthesis, and x-ray crystallographic studies of α-aryl substituted fosmidomycin analogues as inhibitors of mycobacterium tuberculosis 1-deoxy-d-xylulose 5-phosphate reductoisomerase. J. Med. Chem. 54, 4964–4976 (2011).
Bellucci, C. et al. Negative inotropic and calcium antagonistic activity of alkyl and arylalkyl phosphonates. Farmaco 44, 1167–1191 (1989).
Bondarenko, N. A. et al. Biological activity of 1-arylethylphosphonic acids. Pharm. Chem. J. 37, 226–228 (2003).
Charandabi, M. R. M. D., Ettel, M. L., Kaushik, M. P., Huffman, J. H. & Morse, K. W. Characterization and properties of some dialkyl 1-(N,N-dialkylamino)alkylphosphonates and their hydrochloride salts. Phosphorus, Sulfur, and Silicon and the Related Elements, 223-234 (1989).
Kyung Woon, J., Janda, K. D., Sanfilippo, P. J. & Wachter, M. Syntheses and biological evaluation of two new naproxen analogs. Bioorg. Med. Chem. Lett. 6, 2281–2282 (1996).
Seto, H. & Kuzuyama, T. Bioactive natural products with carbon–phosphorus bonds and their biosynthesis. Nat. Prod. Rep. 16, 589–596 (1999).
Tang, W. & Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 103, 3029–3070 (2003).
Zhang, W., Chi, Y. & Zhang, X. Developing chiral ligands for asymmetric hydrogenation. Acc. Chem. Res. 40, 1278–1290 (2007).
Gillespie, J. A., Zuidema, E., van Leeuwen, P. W. N. M. & Kamer, P. C. J. Phosphorus ligand effects in homogeneous catalysis and rational catalyst design. in phosphorus(iii) ligands in homogeneous catalysis: design and synthesis. (2012).
Fernández-Pérez, H., Etayo, P., Panossian, A. & Vidal-Ferran, A. Phosphine-phosphinite and phosphine-phosphite ligands: preparation and applications in asymmetric catalysis. Chem. Rev. 111, 2119–2176 (2011).
Omelanzcuk, J. et al. Photo-Arbuzov rearrangements of benzyl phosphites. J. Am. Chem. Soc. 110, 6908–6909 (1988).
Cairns, S. M. & Bentrude, W. G. Photorearrangements of benzyl phosphites. Stereochemistry at phosphorus. Tetrahedron Lett. 30, 1025–1028 (1989).
Bennani, Y. L. & Hanessian, S. The asymmetric synthesis of α-substituted α-methyl and α-phenyl phosphonic acids: design, carbanion geometry, reactivity and preparative aspects of chiral alkyl bicyclic phosphonamides. Tetrahedron 52, 13837–13866 (1996).
Bhanthumnavin, W., Arif, A. & Bentrude, W. G. Photo-Arbuzov rearrangements of benzylic phosphites. stereochemistry at migratory carbon. J. Org. Chem. 63, 7753–7758 (1998).
Burk, M. J., Stammers, T. A. & Straub, J. A. Enantioselective synthesis of α-hydroxy and α-amino phosphonates via catalytic asymmetric hydrogenation. Org. Lett. 1, 387–390 (1999).
Wang, D.-Y. et al. Enantioselective synthesis of chiral α-aryl or α-alkyl substituted ethylphosphonates via Rh-catalyzed asymmetric hydrogenation with a P-stereogenic bophoz-type ligand. J. Org. Chem. 74, 4408–4410 (2009).
Fernández-Pérez, H. et al. Access to α-aminophosphonic acid derivatives and phosphonopeptides by [Rh(P–OP)]-catalyzed stereoselective hydrogenation. J. Org. Chem. 85, 14779–14784 (2020).
Cheruku, P., Paptchikhine, A., Church, T. L. & Andersson, P. G. Iridium-N,P-ligand-catalyzed enantioselective hydrogenation of diphenylvinylphosphine oxides and vinylphosphonates. J. Am. Chem. Soc. 131, 8285–8289 (2009).
Liu, X., Han, Z., Wang, Z. & Ding, K. Synthesis of chiral 1,3-bis(1-(diarylphosphoryl)ethyl)-benzenes via Ir-catalyzed double asymmetric hydrogenation of bis(diarylvinylphosphine oxides). Sci. China Chem. 57, 1073–1078 (2014).
Henry, J. C. et al. Asymmetric hydrogenation of vinylphosphonic acids and esters with chiral Ru(II) catalysts. Tetrahedron Lett. 39, 3473–3476 (1998).
Beghetto, V., Matteoli, U. & Scrivanti, A. Synthesis of Chiraphos via asymmetric hydrogenation of 2,3-bis(diphenylphosphinoyl)buta-1,3-diene. Chem. Commun. 155-156 (2000).
Goulioukina, N. S. et al. A practical synthetic approach to chiral α-aryl substituted ethylphosphonates. Tetrahedron Asymmetry 12, 319–327 (2001).
Wei, H., Chen, H., Chen, J., Gridnev, I. D. & Zhang, W. Nickel-catalyzed asymmetric hydrogenation of α-substituted vinylphosphonates and diarylvinylphosphine oxides. Angew. Chem. Int. Ed. 62, e202214990 (2023).
Wang, L.-L. et al. A general copper-catalysed enantioconvergent radical Michaelis–Becker-type C(sp3)–P cross-coupling. Nat. Synth. 2, 430–438 (2023).
Zhou, H. et al. Copper-catalyzed chemo- and enantioselective radical 1,2-carbophosphonylation of styrenes. Angew. Chem. Int. Ed. 62, e202218523 (2023).
Cai, B. et al. Asymmetric hydrophosphinylation of alkynes: facile access to axially chiral styrene-phosphines. Angew. Chem. Int. Ed. 62, e202215820 (2023).
Chen, T., Zhao, C. Q. & Han, L. B. Hydrophosphorylation of alkynes catalyzed by palladium: generality and mechanism. J. Am. Chem. Soc. 140, 3139–3155 (2018).
Dai, Q., Liu, L., Qian, Y., Li, W. & Zhang, J. Construction of P-chiral alkenylphosphine oxides through highly chemo-, regio-, and enantioselective hydrophosphinylation of alkynes. Angew. Chem. Int. Ed. 59, 20645–20650 (2020).
Ji, D. et al. Palladium-catalyzed asymmetric hydrophosphination of internal alkynes: atroposelective access to phosphine-functionalized olefins. Chem 8, 3346–3362 (2022).
Liu, X. T. et al. Ni-catalyzed asymmetric hydrophosphination of unactivated alkynes. J. Am. Chem. Soc. 143, 11309–11316 (2021).
Yang, Z., Gu, X., Han, L. B. & Wang, J. Palladium-catalyzed asymmetric hydrophosphorylation of alkynes: facile access to: P-stereogenic phosphinates. Chem. Sci. 11, 7451–7455 (2020).
Zhang, Y.-Q. et al. Ni-catalyzed asymmetric hydrophosphinylation of conjugated enynes and mechanistic studies. Chem. Sci. 13, 4095–4102 (2022).
Jiang, Y., Cheng, K. W., Yang, Z. & Wang, J. Pd-catalyzed enantioselective and regioselective asymmetric hydrophosphorylation and hydrophosphinylation of enynes. Chin. Chem. Lett. 36, 110231–110237 (2025).
Nie, S. Z., Davison, R. T. & Dong, V. M. Enantioselective coupling of dienes and phosphine oxides. J. Am. Chem. Soc. 140, 16450–16454 (2018).
Long, J., Li, Y., Zhao, W. & Yin, G. Nickel/Brønsted acid dual-catalyzed regio- and enantioselective hydrophosphinylation of 1,3-dienes: access to chiral allylic phosphine oxides. Chem. Sci. 13, 1390–1397 (2022).
Yang, Z. & Wang, J. Enantioselective palladium-catalyzed hydrophosphinylation of allenes with phosphine oxides: access to chiral allylic phosphine oxides. Angew. Chem. Int. Ed. 60, 27288–27292 (2021).
Tang, M.-Q., Yang, Z.-J., Han, A.-J. & He, Z.-T. Diastereoselective and enantioselective hydrophosphinylations of conjugated enynes, allenes and dienes via synergistic Pd/Co catalysis. Angew. Chem. Int. Ed. 64, e202413428 (2025).
Sadeer, A. et al. Desymmetrization of achiral heterobicyclic alkenes through catalytic asymmetric hydrophosphination. Chem. Asian J. 13, 2829–2833 (2018).
Li, Y.-B., Tian, H. & Yin, L. Copper(I)-catalyzed asymmetric 1,4-conjugate hydrophosphination of α,β-unsaturated amides. J. Am. Chem. Soc. 142, 20098–20106 (2020).
Teo, R. H. X., Chen, H. J., Li, Y., Pullarkat, S. A. & Leung, P. H. Asymmetric catalytic 1,2-dihydrophosphination of secondary 1,2-diphosphines – Direct access to free P*- and P*,C*-diphosphines. Adv. Synth. Catal. 362, 2373–2378 (2020).
Yue, W.-J., Xiao, J.-Z., Zhang, S. & Yin, L. Rapid synthesis of chiral 1,2-bisphosphine derivatives through copper(i)-catalyzed asymmetric conjugate hydrophosphination. Angew. Chem. Int. Ed. 59, 7057–7062 (2020).
Das, S., Hu, Q., Kondoh, A. & Terada, M. Enantioselective protonation: hydrophosphinylation of 1,1-vinyl azaheterocycle N-oxides catalyzed by chiral bis(guanidino)iminophosphorane organosuperbase. Angew. Chem. Int. Ed. 60, 1417–1422 (2021).
Pérez, J. M. et al. Manganese(I)-catalyzed H–P bond activation via metal–ligand cooperation. J. Am. Chem. Soc. 143, 20071–20076 (2021).
Wang, C., Huang, K., Ye, J. & Duan, W.-L. Asymmetric synthesis of P-stereogenic secondary phosphine-boranes by an unsymmetric bisphosphine pincer-nickel complex. J. Am. Chem. Soc. 143, 5685–5690 (2021).
Wu, Z.-H. et al. Cobalt-catalysed asymmetric addition and alkylation of secondary phosphine oxides for the synthesis of P-stereogenic compounds. Angew. Chem. Int. Ed. 60, 27241–27246 (2021).
Ge, L. & Harutyunyan, S. R. Manganese(i)-catalyzed access to 1,2-bisphosphine ligands. Chem. Sci. 13, 1307–1312 (2022).
Yang, Q., Zhou, J. & Wang, J. Enantioselective copper-catalyzed hydrophosphination of alkenyl isoquinolines. Chem. Sci. 14, 4413–4417 (2023).
Sinnema, E. G., Ramspoth, T.-F., Bouma, R. H., Ge, L. & Harutyunyan, S. R. Enantioselective hydrophosphination of terminal alkenyl aza-heteroarenes. Angew. Chem. Int. Ed. 63, e202316785 (2024).
Zhang, Y., Jiang, Y., Li, M., Huang, Z. & Wang, J. Palladium-catalyzed diastereo- and enantioselective desymmetric hydrophosphination of cyclopropenes. Chem. Catal. 2, 3163–3173 (2022).
Zhang, S., Jiang, N., Xiao, J.-Z., Lin, G.-Q. & Yin, L. Copper(I)-catalyzed asymmetric hydrophosphination of 3,3-disubstituted cyclopropenes. Angew. Chem. Int. Ed. 62, e202218798 (2023).
Zhou, J., Meng, L., Lin, S., Cai, B. & Wang, J. Palladium-catalyzed enantio- and regioselective ring-opening hydrophosphinylation of methylenecyclopropanes. Angew. Chem. Int. Ed. 62, e202303727 (2023).
Lin, X. et al. Diastereo- and enantioselective hydrophosphination of cyclopropenes under lanthanocene catalysis. Angew. Chem. Int. Ed. 62, e202308488 (2023).
Lu, Z. et al. Asymmetric hydrophosphination of heterobicyclic alkenes: facile access to phosphine ligands for asymmetric catalysis. ACS Catal. 9, 1457–1463 (2019).
Xu, Q. & Han, L. B. Palladium-catalyzed asymmetric hydrophosphorylation of norbornenes. Org. Lett. 8, 2099–2101 (2006).
Han, L. B., Mirzaei, F., Zhao, C. Q. & Tanaka, M. High reactivity of a five-membered cyclic hydrogen phosphonate leading to development of facile palladium-catalyzed hydrophosphorylation of alkenes. J. Am. Chem. Soc. 122, 5407–5408 (2000).
Barta, K., Franciò, G., Leitner, W., Lloyd-Jones, G. C. & Shepperson, I. R. A new class of 3′-sulfonyl binaphos ligands: Modulation of activity and selectivity in asymmetric palladium-catalysed hydrophosphorylation of styrene. Adv. Synth. Catal. 350, 2013–2023 (2008).
Ananikov, V. P., Khemchyan, L. L., Beletskaya, I. P. & Starikova, Z. A. Acid-free nickel catalyst for stereo- and regioselective hydrophosphorylation of alkynes: synthetic procedure and combined experimental and theoretical mechanistic study. Adv. Synth. Catal. 352, 2979–2992 (2010).
Dondoni, A. & Marra, A. Validating the alkene and alkyne hydrophosphonylation as an entry to organophosphonates. Org. Biomol. Chem. 13, 2212–2215 (2015).
Duncan, J. A. S., Hedden, D., Roundhill, D. M., Stephenson, T. A. & Walkinshaw, M. D. Synthesis of novel iridium and rhodium complexes containing diphenylphosphinito and dimethylphosphito ligands. Angew. Chem. Int. Ed. Engl. 21, 452–453 (1982).
Janesko, B. G., Fisher, H. C., Bridle, M. J. & Montchamp, J.-L. P(═O)H to P–OH tautomerism: a theoretical and experimental study. J. Org. Chem. 80, 10025–10032 (2015).
Li, G. Y. The first phosphine oxide ligand precursors for transition metal catalyzed cross-coupling reactions: C-C, C-N, and C-S bond formation on unactivated aryl chlorides. Angew. Chem. Int. Ed. 40, 1513–1516 (2001).
Shaikh, T. M., Weng, C.-M. & Hong, F.-E. Secondary phosphine oxides: Versatile ligands in transition metal-catalyzed cross-coupling reactions. Coord. Chem. Rev. 256, 771–803 (2012).
Wang, X.-B., Goto, M. & Han, L.-B. Efficient asymmetric hydrogenation of α-acetamidocinnamates through a simple, readily available monodentate chiral H-phosphinate. Chem. Eur. J. 20, 3631–3635 (2014).
Dai, Q., Liu, L. & Zhang, J. Palladium/Xiao-Phos-catalyzed kinetic resolution of sec-phosphine oxides by P-benzylation. Angew. Chem. Int. Ed. 60, 27247–27252 (2021).
Dai, Q., Li, W., Li, Z. & Zhang, J. P-chiral phosphines enabled by palladium/Xiao-Phos-catalyzed asymmetric P-C cross-coupling of secondary phosphine oxides and aryl bromides. J. Am. Chem. Soc. 141, 20556–20564 (2019).
Zhang, Z.-M. et al. A new type of chiral sulfinamide monophosphine ligands: Stereodivergent synthesis and application in enantioselective gold(I)-catalyzed cycloaddition reactions. Angew. Chem. Int. Ed. 53, 4350–4354 (2014).
Blackmond, D. G. Kinetic aspects of nonlinear effects in asymmetric catalysis. Acc. Chem. Res. 33, 402–411 (2000).
Satyanarayana, T., Abraham, S. & Kagan, H. B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 48, 456–494 (2009).
Wang, Z. et al. Highly enantioselective michael addition of pyrazolin-5-ones catalyzed by chiral metal/N,N′-dioxide complexes: metal-directed switch in enantioselectivity. Angew. Chem. Int. Ed. 50, 4928–4932 (2011).
Liu, F., Paton, R. S., Kim, S., Liang, Y. & Houk, K. N. Diels-Alder reactivities of strained and unstrained cycloalkenes with normal and inverse-electron-demand dienes: activation barriers and distortion/interaction analysis. J. Am. Chem. Soc. 135, 15642–15649 (2013).
Wang, J. W. et al. Nickel-catalyzed remote asymmetric hydroalkylation of alkenyl ethers to access ethers of chiral dialkyl carbinols. J. Am. Chem. Soc. 145, 10411–10421 (2023).
Li, B., Xu, H., Dang, Y. & Houk, K. N. Dispersion and steric effects on enantio-/diastereoselectivities in synergistic dual transition-metal catalysis. J. Am. Chem. Soc. 144, 1971–1985 (2022).
Huo, J. et al. Palladium-catalyzed enantioselective carbene insertion into carbon-silicon bonds of silacyclobutanes. J. Am. Chem. Soc. 143, 12968–12973 (2021).
Johnson, E. R. et al. Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Lu, T. & Chen, Q. Independent gradient model based on Hirshfeld partition: a new method for visual study of interactions in chemical systems. J. Comput. Chem. 43, 539–555 (2022).
Acknowledgements
We gratefully acknowledge the funding support of the National Key R&D Program of China (2021YFF0701600), the National Natural Science Foundation of China (22271053, 22031004, and 22471040), Shanghai Municipal Commission of Science and Technology (21ZR1445900) and Shanghai Municipal Education Commission (20212308).
Author information
Authors and Affiliations
Contributions
J.Y. and J.Z. conceived the project, analyzed the data and wrote the manuscript. C.M. performed the most of experiments. X.W. and T.S. participated in some of the experiments. J.Y. did the DFT calculations. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, C., Wang, X., Soós, T. et al. Ligand-controlled regiodivergent and enantioselective hydrophosphorylation of styrenes by palladium. Nat Commun 16, 5436 (2025). https://doi.org/10.1038/s41467-025-60621-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60621-8
