
Abstract
Electroreduction of nitrate (NO3‒) to ammonia (NH3) is a promising approach for addressing energy challenges. However, the activity is limited by NO3‒ mass transfer, particularly at reduction potential, where an abundance of electrons on the cathode surface repels NO3‒ from the inner Helmholtz plane (IHP). This constraint becomes pronounced as NO3‒ concentration decreases, impeding practical applications in the conversion of NO3‒-to-NH3. Herein, we propose a generic strategy of catalyst bandstructure engineering for the enrichment of negatively charged ions through solid-liquid (S-L) junction-mediated charge rearrangement within IHP. Specifically, during NO3‒ reduction, the formation of S-L junction induces hole transfer from Ag-doped MoS2 (Ag-MoS2) to electrode/electrolyte interface, triggering abundant positive charges on the IHP to attract NO3‒. Thus, Ag-MoS2 exhibits a ~ 28.6-fold NO3‒ concentration in the IHP than the counterpart without junction, and achieves near-100% NH3 Faradaic efficiency with an NH3 yield rate of ~20 mg h‒1 cm‒2 under ultralow NO3‒ concentrations.
Similar content being viewed by others

Introduction
Ammonia (NH3), one of the most common industrial chemicals, is crucial for the production of agricultural fertilizers and holds immense potential as a green hydrogen-rich fuel1,2,3. The current global ammonia demand exceeds 180 million tons annually, primarily fulfilled through industrial-scale production of energy-intensive Haber-Bosch (H-B) process1,4. Within this process, steam-reformed hydrogen (H2) undergoes reaction with nitrogen (N2) under elevated temperature (~ 500 °C) and pressure (> 100 atm)5,6, which not only accounts for approximately 1.4% of global carbon dioxide (CO2) emissions, but also necessitates the consumption of 2% of the world’s total energy supply7,8. Recently, electrocatalytic methodologies have surfaced as a viable clean energy pathway for decentralized ammonia synthesis at room temperature, accommodating a range of infrastructure scales and potentially powered by locally sourced renewable energy sources9,10. Despite the substantial global demand that may sustain the traditional ammonia production route in the near future, the electrochemical ammonia synthesis can be promising complementary process to the Haber–Bosch technology for contributing to decarbonizing ammonia production11,12.
Recently, diverse electrochemical approaches have been explored to address the varied demands for NH3 production in the future energy landscape, encompassing the electrochemical N2 reduction reaction (NRR)13,14,15, lithium-mediated NRR16,17,18,19 and nitrate reduction reaction (NO3RR)20,21,22. Among them, the present NH3 yield via NO3RR surpasses those of NRR by two to three orders of magnitude, mainly due to the relatively lower dissociation energy of the N=O bond (204 kJ mol‒1) of nitrate anion (NO3‒) compared to the N≡N bond (941 kJ mol‒1)23,24. Besides, NO3‒ exhibits a ubiquitous presence within contaminated groundwater and industrial effluent25,26, with their availability further augmented through the oxidation process of atmospheric N2 as the source of nitrogen9,27,28. Consequently, the NO3RR pathway has emerged as one of the most potential in renewable NH3 production.
While continuous progress has been made in NO3RR under high NO3‒ concentrations (> 100 mM)11,29,30,31, achieving large throughput and effective NO3‒ reduction in groundwater with concentrations below 10 mM remains a challenge, presenting a limited NH3 yield rate below 5 mg h‒1 cm‒2 and a Faradaic efficiency (FE) below 85%22,32,33,34. On the other hand, a considerable portion of the groundwater, stemming from both agricultural and industrial sources35,36,37,38,39,40, has a diluted concentration, necessitating a platform suitable for operation at low concentration. More importantly, the removal process of NO3‒ continuously reduces its concentration, inevitably reaching the low concentration regime with reduced performance.
To mitigate the degradation in low concentration, significant efforts have been devoted to concentrating NO3‒ locally around electrodes, employing various strategies such as porous carbon framework encapsulation32, built-in electric field regulation41, and nitrogen-vacancy engineering42. However, these endeavors primarily focus on manipulating catalyst properties for NO3‒ enrichment, with limited exploration of interfacial NO3‒ transfer under operational conditions. Particularly, at the reduction potential, an excess of electrons accumulates on the cathode surface, leading to a repulsion of negatively charged NO3‒ ions from the inner Helmholtz plane (IHP), where the catalytic reactions occur27. This ubiquitous effect of charge repulsion hinders the mass transfer and sets up an intrinsic bottleneck for the catalytic process.
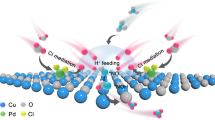
Here, we overcome the fundamental limitation through utilizing a solid-liquid (S-L) junction43,44 to manipulate the charge distribution within the IHP region. By employing proper bandstructure engineering of the catalyst (p-type Ag-doped MoS2, Ag-MoS2), the enhanced interfacial charge transfer endows reinforced S-L junction to facilitate the NO3‒ enrichment in IHP (Fig. 1), showcasing a ~ 27.6-fold increase in NO3‒ concentration compared to the counterpart without junction. As a proof of concept, the Ag-MoS2 platform demonstrates impressive performance, including a record-high NH3 yield rate of ~ 20 mg h‒1 cm‒2 and NH3 FE of ~ 100%, outperforming previous results obtained under low NO3‒ levels22,32,34,36. Furthermore, the high NO3‒-to-NH3 conversion efficiency (99.3%) enables the removal of nitrate from 100 mM to low levels of 0.54 mM within 3 h, in sharp contrast with the reference (still relatively high level under 48 h operation). Notably, we successfully convert NO3‒ into high-purity NH4Cl products with near-unity efficiency by coupling the NO3RR with an air stripping process, demonstrating the potential of large throughput ammonia production in a sustainable way.

Schematic diagram of S-L junction-mediated NO3‒ enrichment within the IHP region over Ag-MoS2 at the reduction potential. Eredox represents the theoretical redox potential of NO3‒/NH3. CBM and VBM express the conduction band minimum and valence band maximum of Ag-MoS2, respectively. Note: IHP contains surface holes, solvate molecules, and NO3‒ anions and cations. Electrons accumulate on the electrode surface under negative polarization. Ionized acceptors exist in the S-L junction (see detailed discussions in the supplementary discussions).
Results
Based on the merits of appropriate band gap (1.2–1.9 eV), excellent conductivity, adjustable Fermi level position, and two-dimensional characteristics of large specific surface area, MoS2 was selected as the platform for the electrocatalysis, with intentionally doped Ag as the enabler for the desired S-L junction formation. The Ag-doped MoS2 (Ag-MoS2) catalyst was grown in situ on a carbon cloth using a one-step hydrothermal method (Supplementary Fig. 1, see “Methods”). MoS2 as a reference was synthesized through a similar route, excluding the addition of a silver source. Scanning electron microscopy (SEM), transmission electron microscopy (TEM), and high-resolution TEM (HRTEM) revealed a nanoflower morphology of Ag-MoS2 with (100) crystal plane orientation (Fig. 2a–c and Supplementary Fig. 2), maintaining a morphology similar to that of MoS2 (Supplementary Figs. 3, 4). Energy-dispersive X-ray (EDX) elemental mapping illustrated the uniform distribution of Ag species on MoS2 nanoflowers (Fig. 2d). The X-ray diffraction (XRD) patterns of Ag-MoS2 and MoS2 were similar, indicating no formation of secondary phases after Ag doping (Fig. 2e). Inductively coupled plasma optical emission spectrometry (ICP-OES) determined the optimal Ag content in Ag-MoS2 to be 3.49 wt% (Supplementary Table 1).

a SEM, (b) TEM, (c) HRTEM, and (d) EDX mapping of Ag-MoS2. e XRD patterns of Ag-MoS2 and MoS2. f Fourier transformed k2-weighted EXAFS spectra of Ag-MoS2 and reference samples.
To analyze the surface chemical states of the catalysts, high-resolution X-ray photoelectron spectroscopy (XPS) was performed (Supplementary Fig. 5). The Mo 3 d and S 2p XPS spectra indicated the presence of Mo4+ and S2− in both Ag-MoS2 and MoS245,46. In the Ag 3 d spectra, characteristic peaks at 368.16 and 374.13 eV were assigned to the Ag-S bond47,48. To further elucidate the coordination structure of Ag species, X-ray absorption fine structure (XAFS) was investigated. The Mo K-edge X-ray absorption near edge structure (XANES) spectra exhibited a higher pre-edge absorption energy of Ag-MoS2 than those of Mo foil and MoS2 (Supplementary Fig. 6a), implying that the doping Ag species induced decreased electron density on the Mo site. From the Ag K-edge XANES spectra, the absorption energy of Ag-MoS2 located between those of Ag foil and Ag2S references (Supplementary Fig. 6b), indicating the valence state of the doped Ag within 0 to 1. The Fourier-transformed extended XAFS (EXAFS) of Ag-MoS2 exhibited characteristic peaks of the Ag-S bond at 1.9 Å, confirming the coordination of Ag with S atoms (Fig. 2f). These comprehensive results provide clear evidence of the synthesis of Ag-doped MoS2 catalysts.
To investigate the construction and regulation of the solid-liquid (S-L) junction, we conducted ultraviolet photoelectron spectra (UPS) and Mott-Schottky (M-S) measurements. The UPS results (Supplementary Fig. 7) demonstrated a significant increase in the work function of Ag-MoS2 (5.47 eV) compared to MoS2 (4.98 eV), indicating a downward-shifted Fermi level after Ag doping. In addition to the UPS method, KPFM measurements were carried out to collaboratively demonstrative the increase of work function by Ag doping (Supplementary Figs. 8, 9)49. The surface potential difference between Ag-MoS2 and FTO substrate was ~ 230 mV, while 20 mV for MoS2. Correspondingly, the work functions of Ag-MoS2 and MoS2 were 5.13 and 4.92 eV, respectively. The variation trend was in consistent with UPS results, indicating a relatively high work function of Ag-MoS2. M-S plots further revealed the Fermi level potential (EF) of Ag-MoS2 (1.07 V versus RHE) and MoS2 (0.53 V versus RHE), respectively (Fig. 3a), surpassing the theoretical redox potential of NO3–/NH3 (Eredox, 0.27 V versus RHE under neutral conditions (pH 6.71))50,51,52,53. The calculated carrier concentration of Ag-MoS2 is 1.14 × 1019 cm−3, 1.8 times higher than that of MoS2 (6.3 × 1018 cm−3, Supplementary Table 2). Thus, the Ag dopant as the electron acceptor to increase the carrier concentration and work function of MoS2 was verified.

a Mott-Schottky plots of Ag-MoS2, MoS2, and W-MoS2 catalysts. b The non-Faradaic capacitance-potential curves for the diverse catalysts. The potential of zero charge (PZC) describes the condition when the capacitance on a surface is minimal. c The fitted surface capacitance of the three catalysts. d The peak intensity of absorbed NO3– from in situ Raman spectra of catalysts at various potentials. e Bode plots of catalysts at the reduction potential of − 0.6 V versus RHE in 10 mM NO3– electrolyte. f The ECSA normalized NO3‒ adsorption capacity at different applied potentials on catalysts.
Upon contact with the NO3–-containing electrolyte, the potential difference between the EF and the Eredox (EF-Eredox) triggered charge transfer between the catalyst surface and the inner Helmholtz plane (IHP), forming positively charged IHP on the electrolyte side (Supplementary Fig. 10). Specifically, p-type MoS2 catalysts exhibited the characteristic of hole conduction. Under the drive of EF-Eredox, the holes (majority carrier) in the valance band would transfer from the semiconductor surface to the electrolyte, inducing charge rearrangement within the IHP region (Supplementary Fig. 10a)54. Meanwhile, the high carrier concentration (1.14 × 1019 cm−3) and thin space charge layer width (5.88 nm) of Ag-MoS2 can contribute to the increase of probability for hole tunneling effect (Supplementary Fig. 10b and Supplementary Tables 2, 3), which benefited positive charges distribution within IHP. Compared to MoS2 with a limited EF-Eredox (0.23 V), the larger EF-Eredox value of Ag-MoS2 (0.76 V) provoked 0.53 V more downward band bending, intensifying the built-in electric fields at S-L junctions (see detailed quantitative methods and discussion in Supplementary Figs. 11–13).
To explore the influence of S-L junction on the charge distribution within the IHP region, alternating current voltammetry (ACV) measurements were employed (Fig. 3b)55,56. ACV was a non-destructive technique which had been widely utilized to record the adsorption behavior change in the IHP range57,58. Compared to MoS2, Ag-MoS2 exhibited a negatively shifted potential of zero charges (PZC, 0.57 V versus RHE). PZC was adopted as an indicator for the IHP structure evolution, and the negative PZC indicated the adsorption of anion on the IHP region of Ag-MoS2 (Fig. 4 and Supplementary Figs. 14, 15)59,60. To investigate the charge distribution under reduction potential, surface capacitance (charge distribution within the solid-liquid interface) of the catalysts was obtained from Nyquist plots at − 0.6 V versus RHE (Supplementary Fig. 16). The capacitance of Ag-MoS2 (9.5 μC cm−2) was lower than that of MoS2 (32.4 μC cm−2), indicating significant positive charges distribution of Ag-MoS2 (Fig. 3c). High valence metals had been reported to serve as electron donors to regulate the EF away from VBM, for enhancing the n-type characteristics of semiconductors61. Thus, we also prepared the reference samples with tungsten (W) doping, to demonstrate the effect of the S-L junction. In contrast, a n-type W-doped MoS2 (W-MoS2) counterpart with no obvious S-L junction (Fig. 3a and Supplementary Figs. 7 and 17–21) had higher PZC value (0.67 V versus RHE, Fig. 3b) and capacitance (50.9 μC cm−2, Fig. 3c), suggesting extensive anions packed on its IHP. These results confirmed that Ag doping can effectively enhance the built-in electric field at the S-L junction, which consequently induced abundant positive charges distributed on the IHP of Ag-MoS2 at the reduction potential.

a W-MoS2 without S-L junction, (b) MoS2 with weak S-L junction, and (c) Ag-MoS2 with strong S-L junction at the reduction potential.
To investigate the S-L junction-mediated NO3‒ enrichment effect, in situ Raman spectra of the three catalysts were carried out (Supplementary Fig. 22). Apart from the observed H2O peak (at 1605 cm−1)62,63,64, the NO3‒ peak (at 1046 cm−1)65,66 was detected at the potentials from open circuit potential (OCP) to − 0.6 V versus RHE, indicating NO3‒ adsorption near the catalyst surface. Notably, Ag-MoS2 with a stronger NO3‒ peak than those of MoS2 and W-MoS2 at each reduction potential (Fig. 3d) demonstrated a pronounced NO3‒ enrichment effect. In Bode phase plots (Fig. 3e), compared with W-MoS2 (0.05 Hz) and MoS2 (0.08 Hz), the higher frequency of Ag-MoS2 (0.4 Hz) implied a facilitated interfacial charge transfer process, which can be ascribed to its intensified NO3‒ accumulation within the IHP region67,68. Elemental mapping displayed obvious N and O signals on Ag-MoS2 nanoflowers during the electrolysis of −0.6 V versus RHE in NO3‒-contained electrolyte, whereas they were relatively shallow or inconspicuous on MoS2 and W-MoS2 (Supplementary Figs. 23–25), disclosing a large number of NO3‒ migrated to the surface.
To quantify the NO3‒ concentration, the NO3‒ enrichment capacity of the three catalysts was measured by ion chromatography (IC) at applied potentials (Fig. 3f and Supplementary Fig. 26, see “Methods”69). By normalizing the NO3‒ adsorption capacity with respect to the electrochemical active surface area (ECSA), the concentration of NO3– adsorbed on Ag-MoS2 presented a normal distribution trend as the potential ranging from − 0.2 to − 1.0 V versus RHE (Fig. 3f and Supplementary Fig. 27), and reached the peak at − 0.6 V versus RHE (2.0 μg mL‒1 cm‒2ECSA). Due to the different Fermi level position, the adsorbed NO3– concentration of MoS2 and W-MoS2 peaked at more negative potentials of − 0.8 and − 1.0 V versus RHE, respectively. As the Ag dopant acted as the electron acceptor to increase the carrier concentration (Supplementary Table 2), it induced an intensified surface band bending when the Ag-MoS2 was contacted with the NO3– contained electrolyte. Thus, the maximum adsorbed NO3– concentration of Ag-MoS2 surpassed MoS2 and W-MoS2 by factors of 8.8 and 27.6, respectively, at their optimal adsorption potentials. The NO3‒ adsorption concentration was also normalized by specific surface area (SSA), this trend aligned with the ECSA normalization results (Supplementary Fig. 28), highlighting superior intrinsic NO3‒ adsorption capability of Ag-MoS2. The lowest surface capacitance at −0.6 V versus RHE suggested massive positive charges within IHP (Supplementary Fig. 29), contributing to the peaked NO3‒ concentration at that potential. While the excessive potential (above −0.6 V) triggered severe downward shift of EF into the valence band of Ag-MoS2 (Supplementary Fig. 30 and Supplementary Table 4, see details in the corresponding discussion), forming a degenerate semiconductor with metallic property, as proved by the decreased arc radius at high-frequency region. This could severely destroy the S-L junction to impact the enrichment of NO3‒. We also implemented finite element method simulations for the NO3‒ distribution on the three catalyst surfaces through COMSOL, which matched the experimental results demonstrated (Supplementary Fig. 31 and Supplementary Table 5).
Under the S-L junction-induced NO3‒ enrichment feedback, we evaluated the NO3RR performance in a standard three-electrode system at ambient temperature and pressure. In this reaction system, NH4+, NO3‒, and NO2‒ were monitored and quantified by coloration and 1H nuclear magnetic resonance (NMR) spectroscopy70 (Supplementary Figs. 32–35). A typical industrial and agricultural groundwater-relevant NO3‒ concentration of 10 mM was reasonably used in the electrolyte for the standard electrochemical tests35,37,38,69. Linear sweep voltammetry (Supplementary Fig. 36) curves of W-MoS2, MoS2, and Ag-MoS2 all presented a higher current density between −0.5 and − 1.65 V versus RHE in the NO3‒ electrolyte relative to NO3‒-free solutions. Particularly, the most significant current difference occurred on Ag-MoS2, revealing its promising NO3RR activity.
Following the chronoamperometry measurements at various applied potentials tested in a flow cell reactor (Supplementary Fig. 37), the NO3RR performance was assessed for the three catalysts with a 0.5 h duration. Of these, Ag-MoS2 displayed the superior performance (Fig. 5a, b), with a near unit NH3 Faradaic efficiency (FE) of 99.7% at − 0.6 V versus RHE (~ 200 mA cm−2) and an optimal NH3 yield rate of 20.1 mg h‒1 cm‒2 at − 1.0 V versus RHE (~ 340 mA cm−2). The optimal NH3 yield rate value was about 3.4-folds and 1.7-folds than those of W-MoS2 and MoS2, respectively. Similarly, the peak NH3 FE of Ag-MoS2 was approximately 3.2-folds and 1.8-folds than those of W-MoS2 and MoS2, respectively. These results displayed the superior NO3RR performance of Ag-MoS2 compared to its counterparts. Moreover, the performance surpassed most state-of-the-art NH3 activity ever reported at a low NO3‒ system (Fig. 5c and Supplementary Table 6)22,32,34,71,72. The electrochemically active surface area-normalized NH3 yield rate also clarified the excellent intrinsic activity of Ag-MoS2 (Supplementary Figs. 27 and 38). The blank experiments without adding NO3‒ to the electrolyte or working at OCP produced a negligible amount of NH3 (Supplementary Fig. 39), excluding the possible interference on the quantification results. Besides, the possible contribution of Ag nanoparticles on the NO3‒ enrichment and NO3RR performance of MoS2 has been eliminated (Supplementary Figs. 40–43).

a NH3 yield rate and NH3 FE of catalysts in 10 mM NO3– electrolyte at various applied potentials. b I–V plots of catalysts in 0.5 M K2SO4 with 10 mM NO3‒ electrolyte at various potentials for 0.5 h electrolysis. c A summary of recent NO3RR works on NH3 yield rate, NH3 FE and current density under ultralow NO3‒ concentrations (NO3‒ concentration ≤ 10 mM)22,32,34,71,72. d EIS tested at −0.6 V versus RHE in 10 mM NO3– electrolyte. e NO3– removal in initial 0.5 M K2SO4 with 100 mM NO3– electrolyte at − 0.6 V versus RHE in H-cell reactor. After 3 h of electrolysis, only 0.54 mM of NO3––N. Insert was the enlarged vision within 3 h. f NO3RR performance stability over Ag-MoS2 measured in a 0.5 M K2SO4 with 10 mM NO3– electrolyte at − 0.6 V versus RHE. g Schematic of the ammonia product synthesis process from 100 mM NO3– electrolyte to NH4Cl at − 1.0 V versus RHE. h The conversion efficiency of different steps for the ammonia product synthesis process. Numbers on the x-axis indicated the corresponding conversion steps in panel (g). Error bars indicate the relative standard deviations of the mean (n = 3).
To reveal the significance of NO3‒ enrichment for superior NO3RR performance under a low-NO3‒ concentration system, EIS measurements were conducted (Fig. 5d and Supplementary Fig. 16). In the Nyquist plots, semicircles with similar radii at the high-frequency region of the three samples indicated nearly equivalent conductivity. At the low-frequency region, W-MoS2 without S-L junction showed a straight line with a slope of 45°, suggesting that NO3‒ mass transfer was the rate control step for NO3RR. In contrast, the smaller arc radius over Ag-MoS2 illustrated the enhanced reaction rate of NO3RR (from 16.41 to 6.92 Ω cm‒2, Supplementary Table 7). The lowest phase angle of Ag-MoS2 in bode plots also testified its accelerated catalytic kinetics, ascribing to the distinct NO3‒ enrichment effect (Fig. 3e). In addition, from the NO3RR performance at a wide range of NO3‒ concentrations (Supplementary Fig. 44), the optimal Ag-MoS2 in K2SO4 system (Supplementary Figs. 45–46) exhibited its broad adaptability and achieved an excellent NH3 FE at − 0.6 V versus RHE (> 90%, in Supplementary Fig. 47), even at ultra-low concentration (1 mM). While the NH3 yield rate of Ag-MoS2 was significantly higher than those of W-MoS2 and MoS2 in low NO3‒ concentration (Supplementary Fig. 44, insert), it gradually became comparable as the concentration increased. These results highlighted the importance of NO3‒ enrichment to overcome the constraint of low-concentration NO3‒ electroreduction to NH3, via the S-L junction-mediated effect.
To examine the NO3‒ removal capability of Ag-MoS2, the conversion tests with an initial 100 mM NO3‒ were carried out in an H-cell reactor, and the remaining products were measured. Impressively, the NO3RR on the three samples followed typical quasi-first-order kinetics, and the NO3‒ to NH3 selectivity of Ag-MoS2 was up to 99.3% within only 3 h of electrolysis at − 0.6 V versus RHE (NO3‒ concentration decreased sharply from 100 mM to 0.54 mM, Fig. 5e and Supplementary Fig. 48), manifesting that nearly all the N sources were converted into NH3 (Supplementary Fig. 49). Comparatively, the NO3‒ to NH3 conversion efficiency of W-MoS2 and MoS2 were as low as ~ 16% and ~ 30%, and the residual NO3‒ still remained a relatively high level of 4.64 mM and 11.78 mM even after 48 h (Supplementary Fig. 50). Also, Ag-MoS2 behaved with stable performance and robust structure, as demonstrated by the steady eight-cycling NO3RR process at 25 °C (Fig. 5f), the negligible morphology change, tiny phase structure difference, and slight chemical valence state shift after the electrolysis (Supplementary Figs. 51, 52).
Furthermore, we demonstrated the practicality of the proposed setup for efficient NH3 collection. Here, we integrated electrocatalysis with an air-stripping method for the recovery of high-purity ammonia products at − 1.0 V versus RHE (Fig. 5g). After the NO3RR process, the cathodic electrolyte was transferred to a conical flask and adjusted to an alkaline state. Due to the high NH3 vapor pressure under alkaline conditions, the produced NH3 was efficiently extracted at 70 °C using an air stripping method (see details in Methods)73,74. Consequently, approximately 98.9% of the NH3 vapor was successfully stripped out from the electrolyte (Fig. 5h), indicating favorable NO3‒ conversion and simultaneous high-efficiency NH3 generation. Subsequently, around 97.3% of the outflowing NH3 gas was collected in an HCl solution, and approximately 97.1% of NH4Cl powder was finally obtained after rotary evaporation. The high-purity NH4Cl products were confirmed by XRD measurement (Supplementary Fig. 53), highlighting their potential as fertilizer for agricultural production.
Next, to investigate the practical application prospect, complex electrolyte (which included CO32‒, Na+, Cl‒, K+, NO3‒, and SO42‒) was prepared to simulate the nitrate-containing wastewater condition75. The NH3 Faradaic efficiency over Ag-MoS2 in simulated wastewater was 76.7%, while the NH3 yield rate reached 16.02 mg h−1 cm−2 (Supplementary Fig. 54a). The decreased NO3RR performance can be ascribed to the presence of interfering ions that influenced the targeted adsorption of NO3‒. Although the NO3RR performance in wastewater decreased to a certain extent, the NH3 yield rate and Faradaic maintained 80% and 76% of the optimal performance for simple electrolyte. In the recovery process, approximately 73.9% of NH4Cl powder was finally obtained (Supplementary Fig. 54b). Also, we have evaluated the preparation cost of Ag-MoS2 catalyst, whose price with 1 cm2 was as low as 0.475 ¥ (Supplementary Table 8). These results suggested that the Ag-MoS2 has the potential for the conversion of NO3‒ to ammonia product in actual industrial and agricultural wastewater.
To gain a deeper understanding of the NO3RR mechanism over Ag-MoS2 catalysts, we employed in situ attenuated total reflection infrared spectroscopy (ATR-IR) to capture intermediates and monitor the reaction. As the applied potential ranged from 0.4 to − 1.0 V versus RHE (Fig. 6a), the detected NO2 peaks (at ~ 1530 cm‒1)76 in the spectra of Ag-MoS2 indicated the deoxygenation of NO3‒. The pronounced characteristic peaks of N-H (at 3200–3380 cm‒1)77,78,79, -H (at ~ 2050 cm‒1)80, and NH4+ (at ~ 1450 cm‒1)81 verified the effective hydrogenation of nitrogen oxide intermediates to ammonia on a highly protonated surface. In comparison, the NO2, -H, and NH4+ signals were not prominent in the spectra of MoS2 and W-MoS2 (Supplementary Figs. 55, 56), indicating that the relatively slow reaction rates of the two catalysts resulted in less accumulation of related species on their surfaces. Online differential electrochemical mass spectrometry (DEMS) was further performed to detect molecular intermediates and products (Fig. 6b and Supplementary Figs. 57, 58). The mass-to-charge ratio (m/z) signals of NH3 (17), H2 (2), N2 (28), NO (30), NH2OH (33), and N2O (44) were detected and tracked. Among the three catalysts, the NH3 intensity was the strongest, while other signals (H2, N2, NO, and N2O) shrunk over Ag-MoS2, which demonstrated the promotion of NH3 generation under the S-L junction-induced NO3‒ enrichment on the Ag-MoS2 surface. Further, we calculated the electronic band structures and partial density of states (PDOS) through density functional theory (DFT). After Ag doping, the newly emerged band tail states at VBM extended the valence band of Ag-MoS2 (Fig. 6c, d and Supplementary Fig. 59)82,83, leading to the downward shift of the Fermi level. While, W doping induced upward shift of the Fermi level away from the VBM (Supplementary Fig. 60). These phenomena were in consistent with UPS and M-S results, unveiling that Ag doping benefited the downward band bending within S-L junction and thus the charge transfer between IHP and the catalyst. The optimal reaction pathways of Ag-MoS2 were also calculated (*NO3 → *NO2 → *NO → *N → *NH → *NH2 → *NH3) by comparing multiple possible branches (Fig. 6e and Supplementary Figs. 61–63). All three samples underwent the process of adsorption of NO3–, deoxygenation of the N species, and hydrogenation of the N species to synthesize NH3, in which the *NH2 → *NH3 process was the potential-determining step (PDS). The Gibbs free energy change (ΔG) of this step was 1.1, 0.95, and 1.02 eV for Ag-MoS2, MoS2, and W-MoS2 catalysts at pH = 7. The ΔG of Ag-MoS2 was comparable to those of W-MoS2 and MoS2, suggesting that Ag doping had little contribution to the ΔG of PDS. Although MoS2 and W-MoS2 exhibited reduced ΔG of PDS at corresponding optimal NO3– adsorption potentials (Supplementary Figs. 61 and 64, 65), while, their NO3RR performance were far below Ag-MoS2 (Fig. 5a). These results further indicated the decisive role of Ag doping-induced surface NO3– enrichment effect in promoting NO3RR activity in low concentration system.

a In situ ATR-IR spectra of Ag-MoS2. The Si-O signal was derived from the reduction of surface SiO2 on the Si semi-cylindrical prism substrate under the applied potentials. b Online differential electrochemical mass spectrometry (DEMS) measurements of NO3RR over Ag-MoS2 under the potential of −0.6 V versus RHE. Electronic band structures of (c) MoS2 and (d) Ag-MoS2. e Gibbs free energy diagram of various intermediates generated during NO3RR over Ag-MoS2 at pH = 7.
Discussion
In summary, we proposed a strategy to enrich charged chemicals in the vicinity o of the electrodes through catalyst bandstructure engineering, overcoming the bottleneck of mass transfer posed by intrinsic charge repulsion in electrocatalysis. We systematically demonstrated the S-L junction-mediated charge rearrangement effect on the IHP region, boosting electrocatalytic reduction of low-concentration NO3– to NH3. COMSOL simulations and experimental measurements, including in situ Raman spectra, IC, EIS, and ACV, elucidated that the construction of S-L junction induced positively charged IHP, significantly enriching NO3– anions concentration (by a factor of 27.6) to break the mass transfer limitation at the reduction potential.
As a proof of concept, an optimized Ag-MoS2 was introduced as a model platform for the NO3RR flow cell system. We evaluated the comprehensive performance through the decontamination of nitrate polluted solution into drinkable level, which requires efficient catalytic process within a broad range of concentration and high conversion rate. By virtue of the unparalleled performance at ultra-low concentrations (~ 20 mg h‒1 cm‒2, FE of nearly 100% under concentration of 10 mM), the catalyst with S-L junction succeeded in removing NO3– to low level within 3 h, while the counterpart without the junction cannot meet the level after treatment of 48 h. Considering the practicality of the utilization of the high-value-added ammonia product, we implement additional experimental efforts to show a near-unity efficiency by the conversion of NO3– to a high-purity NH4Cl, aiming to achieve sustainable development.
Efficient anion accumulation on the reaction interface is crucial for a wide range of renewable electricity-driven reduction applications. Thus, the strategy we proposed holds promise for various electrochemical anion reduction reactions, including NO3–, NO2–, AsO43–, and Cr2O72–, etc, inspiring innovative design for water treatment and remediation in an environmentally friendly way. Such investigations could illuminate avenues for both fundamental research and practical applications across various scientific domains, ranging from physics, chemistry to environmental sciences.
Methods
Chemicals
Sodium molybdate dehydrate (Na2MoO4 · 2H2O, ≥ 99.95%), thiourea (CH4N2S, ≥ 99%), silver nitrate (AgNO3, ≥ 99.8%), hydroxylamine hydrochloride (NH2OH·HCl, ≥ 99.99%), hexadecyl trimethyl ammonium bromide (C19H42BrN, CTAB, ≥ 99%), sodium tungstate dihydrate (Na2WO4 · 2H2O, ≥ 99.5%), salicylic acid (C7H6O3, ≥ 99%), trisodium citrate dihydrate (Na3C6H5O7·2H2O, ≥ 99%), sulfanilamide (C6H8N2O2S, ≥ 99.8%), p-dimethylaminobenzaldehyde ((CH3)2NC6H4CHO, PDAB, ≥ 98%), sodium nitroferricyanide (III) dihydrate (Na2Fe(CN)5NO · 2H2O, ≥ 99.98%), N-(1-Naphthyl) ethylenediamine dihydrochloride (C12H14N2·2HCl, ≥ 98%), potassium sulfate (K2SO4, ≥ 99%), potassium nitrate-15N (15KNO3, ≥ 99%), dimethyl sulfoxide (C2H6SO, DMSO-d6, ≥ 99.9%), maleic acid (C4H4O4, ≥ 99%), and ammonium chloride (14NH4Cl, ≥ 99.8%), ammonium-15N chloride (15NH4Cl, ≥ 98.5%) were purchased from Aldrich Chemical Reagent Co., Ltd. Sodium hydroxide (NaOH, ≥ 96%), sodium hypochlorite (NaClO, analytical pure), hydrochloric acid (HCl, 38%), potassium nitrate (KNO3, ≥ 99.999%), potassium nitrite (KNO2, ≥ 97%), and ethanol (C2H6O, ≥ 99.7%) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification. The water used in this research was purified through a Millipore system.
Preparation of Ag-MoS2
For the synthesis of Ag-MoS2 in a typical hydrothermal process, 4.27 mmol of Na2MoO4 · 2H2O, 18.40 mmol of CH4N2S, a certain amount of AgNO3, and 10.43 mmol of NH2OH · HCl were dissolved in 50 mL of deionized water under vigorous stirring for 0.5 h. 0.18 g of CTAB was then added to the mixed solution, and the pH value of the mixture was adjusted to 6. After that, carbon cloth with an area of 4 × 4 cm2 and a thickness of 0.35 mm was immersed into the homogeneous solution. The mixture was subsequently transferred into a 100 mL Teflon-lined stainless-steel autoclave and heated at 180 °C for 24 h in an oven. After cooling down to room temperature naturally, the products were washed with ethanol and deionized (DI) water repeatedly, followed by vacuum-freeze drying. The obtained sample was Ag-MoS2 grown on carbon cloth (catalyst mass of ~ 5 mg cm−2). Ag-MoS2 catalysts with various Ag-doped amounts were prepared according to the above procedure by changing the additional amount of AgNO3. Specifically, 0.213 mmol, 0.426 mmol, 0.639 mmol and 0.852 mmol AgNO3 were added to achieve 1.87 wt%, 3.49 wt%, 5.91 wt% and 6.91 wt% Ag doping amounts, respectively. These catalysts were denoated as Ag-MoS2-5, Ag-MoS2-10, Ag-MoS2-15 and Ag-MoS2-20 (Supplementary Table S1).
Preparation of MoS2
In a typical synthesis of MoS2, 4.27 mmol of Na2MoO4 · 2H2O, 18.40 mmol of CH4N2S, and 10.43 mmol of NH2OH · HCl were dissolved in 50 mL of deionized water under vigorous stirring for 0.5 h. 0.18 g CTAB was then added to the mixed solution, and the pH value of the mixture was adjusted to 6. After that, carbon cloth with an area of 4 × 4 cm2 and a thickness of 0.35 mm was immersed into the homogeneous solution. The mixture was subsequently transferred into a 100 mL Teflon-lined stainless-steel autoclave and heated at 180 °C for 24 h in an oven. After cooling down to room temperature naturally, the products were washed with ethanol and DI water repeatedly, followed by vacuum-freeze drying. The obtained sample was MoS2 grown on carbon cloth (catalyst mass of ~ 5 mg cm−2).
Preparation of W-MoS2
To systhesize W-MoS2, 4.27 mmol of Na2MoO4 · 2H2O, 18.40 mmol of CH4N2S, 0.426 mmol of Na2WO4 · 2H2O, and 10.43 mmol of NH2OH · HCl were dissolved in 50 mL of deionized water under vigorous stirring for 0.5 h. 0.18 g of CTAB was then added to the mixed solution, and the pH value of the mixture was adjusted to 6. After that, carbon cloth with an area of 4 × 4 cm2 and a thickness of 0.35 mm was immersed into the homogeneous solution. The mixture was subsequently transferred into a 100 mL Teflon-lined stainless-steel autoclave and heated at 180 °C for 24 h in an oven. After cooling down to room temperature naturally, the products were washed with ethanol and DI water repeatedly, followed by vacuum-freeze drying. The obtained sample was W-MoS2 grown on carbon cloth (catalyst mass of ~ 5 mg cm−2).
Electrochemical testing
Before the NO3RR tests, the pretreatment of Nafion 117 membrane (area: a circular shape with a diameter of 2 cm and a thickness of ~ 183 μm) was as follows: the membrane was first oxidized in 5% H2O2 solution at 80 °C for 1 h, next boiled in DI water for 1 h, then used 0.5 M H2SO4 at 80 °C for 1 h, finally washed with DI water.
The NO3RR tests were performed on an electrochemical workstation (PARSTAT 4000) with a typical three-electrode flow cell, including as-prepared catalyst electrode (area of 1 × 1 cm2, catalyst mass of ~ 5 mg cm−2), IrO2 electrode (area of 1 × 1 cm2), and a saturated calomel electrode as the working electrode, counter electrode, and reference electrode, respectively. Nafion 117 membrane was used to separate the anodic cell and cathodic cell. The flow rate of electrolyte (0.5 M K2SO4 with 10 mM NO3‒) was set at 2 mL min‒1 in cathodic and anodic chambers. All potentials reported in this work were referred to the RHE scale via calibration by the following equation: \({\rm{E}}({\rm{versus}}\,{\rm{RHE}})={\rm{E}}({\rm{versus}}\,{\rm{SCE}})+0.244+0.0591\times {\rm{pHvalue}}\). The three batches of catalysts (Ag-MoS2 (AgNO3 amount of 0.426 mmol), MoS2 and W-MoS2) were synthesized, respectively, and their NO3RR performance at various potentials were tested. The error bars were the mean values standard deviation according to the obtained data. For the chronoamperometry measurement, the potential was applied from − 0.2 to − 1.0 versus RHE. LSV was carried out in a voltage window from 0.25 to − 1.65 V versus RHE at scan rates of 10 mV·s−1. EIS of the samples was performed at the frequency region from 106 to 10−2 Hz with an amplitude of 20 mV at various applied potentials. During the electrochemical testing process, no iR compensation was performed. Mott-Schottky measurements were carried out at the frequency of 1000 Hz under dark conditions to obtain the flat band potential and carrier concentration of the samples, in which the dielectric constant of MoS2 was 7.675,84.
Detection and quantification of NH3 using UV-vis
The concentration of NH3 after chronoamperometry measurements with different potentials was detected by the salicylic acid method85. The cathodic electrolyte after electrolysis was collected and diluted to the detection range. Then, 2 mL of diluted liquid was added to 2 mL of 1 M NaOH solution containing 5 wt% C7H6O3 and 5 wt% Na3C6H5O7·2H2O. 1 mL of 0.05 M NaClO and 0.2 mL of C5FeN6Na2O solution (1 wt%) were subsequently mixed with the aforementioned solution and shaken well. After standing for 2 h, UV-vis spectrophotometer measurements were performed with the range from 500 to 800 nm, and the absorbance value at the wavelength of 655 nm was recorded. The concentration-absorbance curve was calibrated using standard ammonium chloride solution with concentrations of 0.5, 1.0, 3.0, 5.0, 10.0, 20.0 and 30.0 µg mL−1 in 0.5 M K2SO4 with NO3‒. The fitting curve \((y=0.10x+0.01,{{\rm{R}}}^{2}=0.999)\) exhibited a good linear relation of absorbance value with NH4+ concentration.
Detection and quantification of NO3 ‒ using UV-vis
The cathodic electrolyte after electrolysis was collected and diluted to the detection range. In the process, 5 mL of diluted sample solution was mixed with 0.1 mL of 1 M HCl. After standing for 20 min, the UV-vis absorbance at the wavelength ranging from 215 to 280 nm was recorded86,87. The absorbance difference between the wavelengths at 220 and 275 nm was calculated using the equation: \({\rm{A}}={{\rm{A}}}_{220\,{\rm{nm}}}-{{\rm{A}}}_{275\,{\rm{nm}}}\). The concentration-absorbance difference curve was calibrated using standard KNO3 solution with 1, 5, 10, 20, 30 and 50 µg mL−1 concentrations. The fitting curve \((y=0.034x-0.036,{{\rm{R}}}^{2}=0.999)\) exhibited a good linear relation of absorbance value with NO3‒ concentration.
Detection and quantification of NO2 ‒ using UV-vis
The preparation of the color reagent was as follows86,87: First, 0.5 g of C6H8N2O2S was dissolved in 50 mL of 2.0 M HCl solution, which was labeled as reagent A. 20 mg of N-(1-naphthyl) ethylenediamine dihydrochloride was then dispersed in 20 mL of DI water, which was labeled as reagent B. After 0.1 mL of reagent A mixed with 5 mL of standard or diluted sample solutions, 0.1 mL of reagent B was finally injected into the homogeneous solution, shaking up and standing for 30 min. The UV-vis absorbance at the wavelength ranging from 400 to 640 nm was recorded, in which the characteristic absorption peak of NO2‒ was identified at 540 nm. The concentration-absorbance difference curve was calibrated using standard KNO2 solution with concentrations of 0.2, 0.5, 1.0, and 2.0 µg mL−1. The fitting curve \((y=0.446x+0.039,{{\rm{R}}}^{2}=0.999)\) exhibited a good linear relation of absorbance value with NO2‒ concentration.
Detection and quantification of NH3 using 1H NMR
14NO3‒ and 15NO3‒ isotope labeling experiments were carried out on a Bruker AVANCE III HD NMR spectrometer (600 MHz) to support the UV-vis results12. After electrolysis, the pH value of the cathodic electrolyte was adjusted to 2 with 1 M HCl. 0.5 mL of the homogeneous solution was then mixed with 0.1 mL DMSO-d6 with 0.04% C4H4O4. 1H NMR was recorded to quantitatively determine NH3 concentration according to the corresponding standard curves.
Electrochemical in situ Raman spectroscopy
In situ Raman spectroscopy was measured by inVia Reflex (Renishaw, UK) with a 532 nm laser as the excitation source20. The electrochemical process was performed in the three-electrode custom-made Teflon reactor with a quartz window, including Ag/AgCl, Pt wire, and catalysts coated on the Au electrode as the reference electrode, counter electrode, and working electrode, respectively. The spectra were recorded by the potential from OCP to − 0.6 V versus RHE.
Electrochemical in situ ATR-IR spectroscopy
ATR-IR spectroscopy was tested on a Nicolet iS50 FT-IR spectrometer equipped with an MCT detector and cooled by liquid nitrogen20. The electrochemical process was performed in the three-electrode custom-made reactor, including Ag/AgCl and Pt wire as the reference electrode and counter electrode, respectively. The working electrode was prepared as follows: The Si semi-cylindrical prism was first polished with Al2O3 powder and sonicated in acetone and deionized water. The Si was placed in a piranha solution at 60 °C for 20 min to remove the organic contaminants. Then the reflecting surface of Si was deposited in the Au precursor mixture at 60 °C for 10 min, and the Au-coated Si conductive substrate was obtained. The catalyst ink was finally coated on the substrate-reflecting surface. After that, the spectra were recorded by the potential from 0.4 V to − 1.0 V versus RHE. The spectrum collected at OCP was as background subtraction.
Electrochemical online DEMS tests
The online DEMS tests21 were measured in the three-electrode customized reactors containing 0.5 M K2SO4 with 10 mM NO3‒ electrolyte, including catalysts coated on breathable film with Au plating layer, Pt wire, and saturated Ag/AgCl electrode as the working electrode, the counter electrode and the reference electrode, respectively. The potential of OCP and − 0.6 V versus RHE were applied alternately with an interval of 3 min after the baseline of the mass spectrometry kept steady. During the electrochemical process, the differential mass signals were recorded when the gaseous products formed on the electrode surface. The operation process was repeated five cycles to avoid accidental errors.
Direct ammonia product synthesis
By coupling NO3RR with the Ar stripping process, the cathodic electrolyte after electrolysis was adjusted to alkaline state, and sealed in a conical flask at 70 °C with flowing 100 sccm Ar gas to purge the NH3 gas out. The outlet stream was injected into 2 M HCl to capture the NH3 product. The NH3 amount in all the solutions was evaluated by the salicylic acid method mentioned above, and the removal efficiency and collection efficiency were calculated as following equations12, respectively:
To estimate the efficiency of the NH4Cl product, the HCl solution with the trapped NH3 was dried by rotary evaporator at 70 °C. The generated NH4Cl was measured by a balance and analyzed by XRD. The collection efficiency of NH4Cl from the acid trap was calculated by the following equation:
DFT calculations
All calculations were carried out by spin-polarized DFT with the Vienna Ab initio Simulation Package (VASP)88,89. Electron exchange-correlation was expressed by the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)90. To describe the ionic cores, the projector augmented wave (PAW) pseudopotential was applied91,92. The plane wave energy cutoff, and convergence criterion for electronic energy and forces were set as 450 eV, 10−5 eV, and 0.02 eV/Å. van der Waals (VDW) interactions were corrected using the D3 method of Grimme93. To compare the electronic structures of MoS2, Ag-MoS2 and W-MoS2, we constructed a 2 × 2 × 2 MoS2 supercell containing 16 Mo atoms and 32 S atoms. Substituting one Mo atom with an Ag atom is referred to as Ag-MoS2, while substituting one MoS2 atom with a W atom is referred to as W-MoS2. For investigating the reaction pathways of nitrate ions on three catalytic surfaces, we built a nanoribbon containing 20 Mo atoms and 40 S atoms. The bottom two layers were fixed to simulate the edge of a single-layer MoS2. For Ag-MoS2 and W-MoS2, we replaced one edge Mo atom with an Ag atom and a W atom, respectively. To avoid periodic interlayer interactions, we introduced a vacuum layer of 20 Å in the Z and Y directions. Aqueous phase H2O and NO3‒ were as the energetics references.
Data availability
Full data supporting the findings of this study are available within the article and its Supplementary Information, as well as from the corresponding author upon reasonable request. Source data are provided in this paper.
References
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
Chang, F., Gao, W., Guo, J. & Chen, P. Emerging materials and methods toward ammonia-based energy storage and conversion. Adv. Mater. 33, e2005721 (2021).
Guo, J. & Chen, P. Catalyst: NH3 as an energy carrier. Chem 3, 709–712 (2017).
Chatterjee, S., Parsapur, R. K. & Huang, K.-W. Limitations of ammonia as a hydrogen energy carrier for the transportation sector. ACS Energy Lett. 6, 4390–4394 (2021).
Kitano, M. et al. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat. Chem. 4, 934–940 (2012).
Han, G.-F. et al. Mechanochemistry for ammonia synthesis under mild conditions. Nat. Nanotechnol. 16, 325–330 (2021).
Service, R. F. New recipe produces ammonia from air, water, and sunlight. Science 345, 610 (2014).
Kyriakou, V., Garagounis, I., Vourros, A., Vasileiou, E. & Stoukides, M. An electrochemical Haber-Bosch process. Joule 4, 142–158 (2020).
John, J., MacFarlane, D. R. & Simonov, A. N. The why and how of NOx electroreduction to ammonia. Nat. Catal. 6, 1125–1130 (2023).
Chen, F.-Y. et al. Electrochemical nitrate reduction to ammonia with cation shuttling in a solid electrolyte reactor. Nat. Catal. 7, 1032–1043 (2024).
Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Liao, W. et al. Boosting nitrogen activation via Ag nanoneedle arrays for efficient ammonia synthesis. ACS Nano 17, 411–420 (2023).
Liao, W. et al. Interfacial engineering promoting electrosynthesis of ammonia over Mo/phosphotungstic acid with high performance. Adv. Funct. Mater. 31, 2009151 (2021).
Liao, W. et al. Triggering in-plane defect cluster on MoS2 for accelerated dinitrogen electroreduction to ammonia. J. Energy Chem. 62, 359–366 (2021).
Suryanto, B. H. R. et al. Nitrogen reduction to ammonia at high efficiency and rates based on a phosphonium proton shuttle. Science 372, 1187–1191 (2021).
Du, H.-L. et al. Electroreduction of nitrogen with almost 100% current-to-ammonia efficiency. Nature 609, 722–727 (2022).
Li, K. et al. Enhancement of lithium-mediated ammonia synthesis by addition of oxygen. Science 374, 1593–1597 (2021).
Fu, X. et al. Continuous-flow electrosynthesis of ammonia by nitrogen reduction and hydrogen oxidation. Science 379, 707–712 (2023).
Liao, W. et al. Sustainable conversion of alkaline nitrate to ammonia at activities greater than 2 A cm−2. Nat. Commun. 15, 1264 (2024).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402–414 (2023).
Wang, K. et al. Intentional corrosion-induced reconstruction of defective NiFe layered double hydroxide boosts electrocatalytic nitrate reduction to ammonia. Nat. Water 1, 1068–1078 (2023).
Daiyan, R. et al. Nitrate reduction to ammonium: From CuO defect engineering to waste NOx-to-NH3 economic feasibility. Energ. Environ. Sci. 14, 3588–3598 (2021).
Yang, X. et al. Achievements, challenges, and perspectives on nitrogen electrochemistry for carbon-neutral energy technologies. Angew. Chem. Int. Ed. 62, e202215938 (2023).
Burow, K. R., Nolan, B. T., Rupert, M. G. & Dubrovsky, N. M. Nitrate in groundwater of the united states, 1991−2003. Environ. Sci. Technol. 44, 4988–4997 (2010).
Smolders, A. J. P., Lucassen, E. C. H. E. T., Bobbink, R., Roelofs, J. G. M. & Lamers, L. P. M. How nitrate leaching from agricultural lands provokes phosphate eutrophication in groundwater fed wetlands: The sulphur bridge. Biogeochemistry 98, 1–7 (2009).
Ren, Y. et al. Microscopic-level insights into the mechanism of enhanced NH3 synthesis in plasma-enabled cascade N2 oxidation–electroreduction system. J. Am. Chem. Soc. 144, 10193–10200 (2022).
Sun, J. et al. Sustainable ammonia production via nanosecond-pulsed plasma oxidation and electrocatalytic reduction. Appl. Catal. B-Environ. 342, 123426 (2024).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, 2202952 (2023).
Xie, M. et al. Intermetallic single-atom alloy In-Pd bimetallene for neutral electrosynthesis of ammonia from nitrate. J. Am. Chem. Soc. 145, 13957–13967 (2023).
Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, e2204306 (2022).
Zhang, S., Li, M., Li, J., Song, Q. & Liu, X. High-ammonia selective metal–organic framework–derived Co-doped Fe/Fe2O3 catalysts for electrochemical nitrate reduction. PNAS 119, e2115504119 (2022).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Huang, Y. et al. Pulsed electroreduction of low-concentration nitrate to ammonia. Nat. Commun. 14, 7368 (2023).
Su, L. et al. Electrochemical nitrate reduction by using a novel Co3O4/Ti cathode. Water Res. 120, 1–11 (2017).
Chauhan, R. & Srivastava, V. C. Electrochemical denitrification of highly contaminated actual nitrate wastewater by Ti/RuO2 anode and iron cathode. Chem. Eng. J. 386, 122065 (2020).
Paredes, I., Otero, N., Soler, A., Green, A. J. & Soto, D. X. Agricultural and urban delivered nitrate pollution input to mediterranean temporary freshwaters. Agr. Ecosyst. Environ. 294, 106859 (2020).
Lee, C.-M. et al. Contribution of nitrate-nitrogen concentration in groundwater to stream water in an agricultural head watershed. Environ. Res. 184, 109313 (2020).
Sun, W. J. et al. Built-in electric field triggered interfacial accumulation effect for efficient nitrate removal at ultra-low concentration and electroreduction to ammonia. Angew. Chem. Int. Ed. 60, 22933–22939 (2021).
Luo, H. et al. Modulating the active hydrogen adsorption on Fe─N interface for boosted electrocatalytic nitrate reduction with ultra-long stability. Adv. Mater. 35, 2304695 (2023).
Hou, Y., Li, T., Yan, S. & Zou, Z. Oriented-growth Ta3N5/SrTaO2N array heterojunction with extended depletion region for improved water oxidation. Appl. Catal. B-Environ. 269, 118777 (2020).
Ding, C., Shi, J., Wang, Z. & Li, C. Photoelectrocatalytic water splitting: Significance of cocatalysts, electrolyte, and interfaces. ACS Catal. 7, 675–688 (2016).
Niu, H. et al. Structurally stable ultrathin 1T-2H MoS2 heterostructures coaxially aligned on carbon nanofibers toward superhigh-energy-density supercapacitor and enhanced electrocatalysis. Chem. Eng. J. 399, 125672 (2020).
Liu, R. et al. Highly efficient electrocatalytic N2 reduction to ammonia over metallic 1T phase of MoS2 enabled by active sites separation mechanism. Adv. Sci. 9, 2103583 (2021).
Xu, T. N. et al. Realization of Ag-S codoped p-type ZnO thin films. Appl. Surf. Sci. 316, 62–65 (2014).
Lim, J. W., Dong, W. J., Cho, W. S., Yoo, C. J. & Lee, J.-L. CuSx catalysts by Ag-mediated corrosion of Cu for electrochemical reduction of sulfur-containing CO2 gas to HCOOH. ACS Catal. 12, 13174–13185 (2022).
Xuan, N. et al. Single-atom electroplating on two dimensional materials. Chem. Mater. 31, 429–435 (2019).
Du, M. H., Yan, J., Cooper, V. R. & Eisenbach, M. Tuning fermi levels in intrinsic antiferromagnetic topological insulators MnBi2Te4 and MnBi4Te7 by defect engineering and chemical doping. Adv. Funct. Mater. 31, 2006516 (2020).
Li, M. et al. Controllable p-type doping of monolayer MoS2 with tantalum by one-step chemical vapor deposition. J. Mater. Chem. C. 10, 7662–7673 (2022).
Lu, X., Song, H., Cai, J. & Lu, S. Recent development of electrochemical nitrate reduction to ammonia: A mini review. Electrochem. Commun. 129, 107094 (2021).
Zhang, X. et al. Recent advances in non-noble metal electrocatalysts for nitrate reduction. Chem. Eng. J. 403, 126269 (2021).
Koval, C. A. & Howard, J. N. Electron transfer at semiconductor electrode-liquid electrolyte interfaces. Chem. Rev. 92, 411–433 (1992).
Mao, S. et al. Specific adsorption-oxidation strategy in cathode inner helmholtz plane enabling 4.6 V practical lithium-ion full cells. Nano Lett. 23, 7014–7022 (2023).
Wang, L., Shen, C., Huang, C., Chen, J. & Zheng, J. Regulating the electrical double layer with supramolecular cyclodextrin anions for dendrite-free zinc electrodeposition. ACS Nano 17, 24619–24631 (2023).
Yan, C. et al. Regulating the inner helmholtz plane for stable solid electrolyte interphase on lithium metal anodes. J. Am. Chem. Soc. 141, 9422–9429 (2019).
Luo, J. et al. Regulating the inner helmholtz plane with a high donor additive for efficient anode reversibility in aqueous Zn-ion. batteries. Angew. Chem. Int. Ed. 135, e202302302 (2023).
Herzog, G., Moujahid, W., Strutwolf, J. & Arrigan, D. W. M. Interactions of proteins with small ionised molecules: Electrochemical adsorption and facilitated ion transfer voltammetry of haemoglobin at the liquid|liquid interface. Analyst 134, 1608–1613 (2009).
Luo, J. et al. Regulating the inner helmholtz plane with a high donor additive for efficient anode reversibility in aqueous Zn-ion batteries. Angew. Chem. Int. Ed. 62, e202302302 (2023).
Luo, Z.-Z. et al. Extraordinary role of Zn in enhancing thermoelectric performance of Ga-doped n-type PbTe. Energ. Environ. Sci. 15, 368–375 (2022).
Li, C.-Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697–701 (2019).
Cai, C. et al. Atomically local electric field induced interface water reorientation for alkaline hydrogen evolution reaction. Angew. Chem. Int. Ed. 62, e202300873 (2023).
Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Zhao, R. et al. A Bi-Co corridor construction effectively improving the selectivity of electrocatalytic nitrate reduction toward ammonia by nearly 100%. Adv. Mater. 35, 2306633 (2023).
Kumar, A. et al. Efficient nitrate conversion to ammonia on f-block single-atom/metal oxide heterostructure via local electron-deficiency modulation. ACS Nano 16, 15297–15309 (2022).
Yang, C. et al. Large-scale synthetic Mo@(2H-1T)-MoSe2 monolithic electrode for efficient hydrogen evolution in all pH scale ranges and seawater. Appl. Catal. B Environ. 304, 120993 (2022).
Chen, W. et al. Deciphering the alternating synergy between interlayer Pt single-atom and NiFe layered double hydroxide for overall water splitting. Energ. Environ. Sci. 14, 6428–6440 (2021).
Bu, L. et al. Photolysis of nitrate by solar light in agricultural runoffs: Degradation of emerging contaminant vs. Formation of unintended products. Sep. Purif. Technol. 269, 118751 (2021).
Andersen, S. Z. et al. A rigorous electrochemical ammonia synthesis protocol with quantitative isotope measurements. Nature 570, 504–508 (2019).
Wang, Y. et al. Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022).
Lim, J. et al. Structure sensitivity of Pd facets for enhanced electrochemical nitrate reduction to ammonia. ACS Catal. 11, 7568–7577 (2021).
Liao, P. H., Chen, A. & Lo, K. V. Removal of nitrogen from swine manure wastewaters by ammonia stripping. Bioresour. Technol. 54, 17–20 (1995).
Yuan, M.-H., Chen, Y.-H., Tsai, J.-Y. & Chang, C.-Y. Ammonia removal from ammonia-rich wastewater by air stripping using a rotating packed bed. Process Saf. Environ. Prot. 102, 777–785 (2016).
Zhang, G. et al. Ammonia recovery from nitrate-rich wastewater using a membrane-free electrochemical system. Nat. Sustain. 7, 1251–1263 (2024).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
Wirz, R., Ferri, D. & Baiker, A. ATR-IR spectroscopy of pendant NH2 groups on silica involved in the knoevenagel condensation. Langmuir 22, 3698–3706 (2006).
Wiles, D. M. & Suprunchuk, T. The infrared absorption spectra of thiosemicarbazide and related compounds: NH2 and NH vibrations. Can. J. Chem. 47, 1087–1089 (1969).
Milligan, D. E. & Jacox, M. E. Matrix-isolation infrared spectrum of the free radical NH2. J. Chem. Phys. 43, 4487–4493 (1965).
Gong, Z. et al. Modulating metal-nitrogen coupling in anti-perovskite nitride via cation doping for efficient reduction of nitrate to ammonia. Angew. Chem. Int. Ed. 135, e202308775 (2023).
Chen, L., Li, J. & Ge, M. Drift study on cerium−tungsten/titiania catalyst for selective catalytic reduction of NOx with NH3. Environ. Sci. Technol. 44, 9590–9596 (2010).
Böker, T. et al. Band structure of MoS2, MoSe2, and α−MoTe2: Angle-resolved photoelectron spectroscopy and ab initio calculations. Phys. Rev. B 64, 235305 (2001).
Alidoust, N. et al. Observation of monolayer valence band spin-orbit effect and induced quantum well states in MoX2. Nat. Commun. 5, 4673 (2014).
Evans, B. L., Young, P. A. & Ditchburn, R. W. Optical absorption and dispersion in molybdenum disulphide. Proc. R. Soc. Lond. Ser. A 284, 402–422 (1997).
Searle, P. L. The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. Analyst 109, 549–568 (1984).
Carvalho, A. P., Meireles, L. A. & Malcata, F. X. Rapid spectrophotometric determination of nitrates and nitrites in marine aqueous culture media. Analusis 26, 347–351 (1998).
Polatides, C. & Kyriacou, G. Electrochemical reduction of nitrate ion on various cathodes – reaction kinetics on bronze cathode. J. Appl. Electrochem. 35, 421–427 (2005).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
We gratefully thank the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No. 52121004 for L.C.), National Natural Science Foundation of China (Grant No. 22376222 for M.L., 52372253 for J.F., 52202125 for C.C.), the Science and Technology lnnovation Program of Hunan Province (2023RC1012 for M.L.), Central South University Research Program of Advanced Interdisciplinary Studies (Grant No. 2023QYJC012 for M.L.), Central South University Innovation-Driven Research Program (Grant No. 2023CXQD042 for J.F.), and Hunan Provincial Natural Science Foundation (2025JJ60117 for W.L.). We would like to acknowledge the help from Beam Lines BL01C1 in the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan) for various synchrotron-based measurements. We also acknowledge funding and support from the Deutsche Forschungsgemeinschaft (DFG) under Germany´s Excellence Strategy – EXC 2089/1 – 390776260 cluster of excellence e-conversion, the Bavarian program Solar Technologies Go Hybrid (SolTech) and the Center for NanoScience (CeNS). L.Z. acknowledges the LMU-CSC program for a doctoral fellowship at LMU. We are grateful for resources from the High Performance Computing Center of Central South University.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
W.L., J.W., and M.L. conceived the research and supervised the project. W.L. and J.W. performed the experiments. W.L., J.W., M.L., X.Z., Q.W., C.K., T.C., H.L., Y.Z., C.C., J.F., L.Z., L.C., and B.X. analyzed the data. Y.T. and K.L. carried out the DFT simulations. W.L., J.W., C.L., and E.C. wrote the draft. All authors discussed the results and contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, W., Wang, J., Tan, Y. et al. Near-Unity Nitrate to Ammonia conversion via reactant enrichment at the solid-liquid interface. Nat Commun 16, 5715 (2025). https://doi.org/10.1038/s41467-025-60671-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60671-y
