
Abstract
Formalin-fixed paraffin-embedded (FFPE) samples represent a vast, untapped resource for epigenomic research, yet molecular tools for deep analysis of these specimens remain limited. We introduce spatial FFPE-ATAC-seq, an approach for in situ profiling chromatin accessibility within archived tissues. This approach overcomes formalin-induced crosslinking challenges, allowing high-resolution mapping of chromatin landscapes while preserving tissue architecture. Applying spatial FFPE-ATAC-seq to mouse and human tissues, including brain and thymus, reveals intricate spatial organization and distinct cell types in alignment with tissue morphology. Integration with single-cell RNA sequencing validates the precision of our chromatin profiles in identifying key cell types and regulatory elements. We further apply this method to human melanoma, comprehensively characterizing chromatin accessibility across both tumor and non-tumor regions. This method significantly expands the toolkit for epigenomic research, unlocking the potential of an extensive collection of archived FFPE samples for studying gene regulation and disease mechanisms with spatial context.
Similar content being viewed by others


Introduction
Formalin-fixed paraffin-embedded (FFPE) tissues are fundamental to clinical practice, forming the basis of histopathological diagnoses for human diseases. As the gold standard of surgical and pathological sample preservation, FFPE blocks maintain histological architecture and cellular features over time while offering cost-effective storage compared to fresh frozen specimens1. Even after long-term storage, FFPE sections remain viable for ongoing and retrospective studies, offering significant potential for genomic and molecular analysis. Accordingly, FFPE preservation has been used for over a century, with billions of cell blocks archived worldwide, creating an invaluable yet underutilized resource for human biology and translational research2.
While spatial transcriptomics has enabled the mapping of gene expression across FFPE-preserved tissues3,4,5, current spatial technologies are not compatible with other omics layers, such as epigenome profiling, which is crucial for understanding epigenomic changes that regulate gene expression. Chromatin profiling in FFPE samples could provide valuable insights into the mis-regulation of promoters and enhancers in disease, with significant implications for diagnostics and therapeutic interventions. However, performing chromatin accessibility profiling on FFPE tissues presents considerable challenges due to the extensive crosslinking induced by formalin fixation6, which can obscure chromatin structure and hinder the Tn5 transposome from efficiently accessing genomic DNA7. These challenges are further compounded by the need to preserve the spatial context of the chromatin accessibility, which can be disrupted by improper or incomplete tissue processing.
In this study, we extend our previously developed spatial ATAC-seq8,9—originally limited to fresh-frozen samples—to establish spatial FFPE-ATAC-seq, enabling chromatin accessibility profiling in FFPE tissues (Fig. 1a). Our approach maintains the core principles of spatial ATAC-seq while introducing minimal modifications to overcome the challenges associated with chromatin profiling in FFPE samples. Recent years have seen the development of several methods for detecting chromatin states in FFPE samples, including FAIRE10, FFPE-CUTAC2, FACT-seq11, and FFPE-ATAC12. In contrast, our spatial FFPE-ATAC-seq assay preserves tissue architecture, enabling spatially resolved chromatin accessibility profiling. This advancement opens opportunities for retrospective studies and clinical research by leveraging the vast collections of archived FFPE specimens.

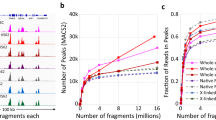
a Schematic workflow for spatially resolved chromatin accessibility profiling in FFPE tissues using spatial FFPE-ATAC-seq. b Distribution of transcription start site (TSS) enrichment score in different target retrieval (TR) conditions with bulk or 50-µm resolution. PK: proteinase K. c Comparison of TSS enrichment score and number of unique fragments in different TR conditions at 50-µm resolution. Box plots show the median (center line), the first and third quartiles (box limits) and 1.5x interquartile range (whiskers). Fastq files were downscaled to 50 million reads per sample for consistent comparison. d The fragment size distribution for each sample in (c). e The enrichment of ATAC reads around TSS for each sample in (c). f Fragments of samples listed in (b, c) mapping to the mouse genome (FF: n = 3; Bulk: n = 8; Spatial: n = 13). Data are presented as mean ± SD. g Heatmap of all-by-all Spearman correlations between all TR conditions in (c). Correlation was calculated using peak set of each sample. Source data are provided as a Source Data file.
Results
Spatial FFPE-ATAC-seq workflow
To maximize the utility and compatibility of spatial ATAC-seq for FFPE samples, we conducted extensive testing and implemented key modifications, specifically refining the target retrieval (TR) process to accurately capture open chromatin regions. The workflow of spatial FFPE-ATAC-seq is summarized in Fig. 1a and Supplementary Fig. 1, which includes three major steps: (1) FFPE tissues underwent deparaffinization, rehydration, and target retrieval to break crosslinks to facilitate Tn5 integration into accessible genomic regions. (2) In situ Tn5 transposition was performed, during which the universal ligation linker and adapters were inserted into accessible genomic regions, providing molecular handles for spatial barcodes. (3) Using microfluidic channels, two orthogonal sets of barcodes (A1–A50 and B1–B50) were successively delivered to the tissue. Their ligation to a universal linker created a spatial grid comprising 2500 individually barcoded locations8. Tissue slides were then imaged to link the spatial barcodes to tissue morphology. After tissue lysis, the barcoded DNA fragments were amplified for library preparation.
Evaluation of spatial FFPE-ATAC-seq
To enable spatial chromatin accessibility profiling in FFPE tissues, we implemented several key modifications to the target retrieval (TR) process: (i) optimizing heat-induced retrieval temperature, (ii) incorporating additional proteinase K (PK) treatment to break protein-DNA crosslinks, and (iii) evaluating different TR solutions, including Tris-EDTA buffer (pH 9.0), 0.8 M Tris-HCl buffer (pH 8.0) (as used in FFPE-CUTAC2; Supplementary Data 1), and citrate buffer (pH 6.0). To assess the effectiveness of these modifications, we generated both bulk and spatial FFPE-ATAC-seq libraries from three-year-old archived mouse brain FFPE tissues. We used the transcription start site (TSS) enrichment score and the number of unique fragments as key quality metrics, driving key insights into spatially resolved chromatin features. First, we observed increased TSS enrichment scores as the retrieval temperature was lowered from 99 °C to 65 °C. The highest TSS enrichment score (~4) was achieved using a combination of 1 × Tris-EDTA buffer (pH 9.0) at 65 °C and PK digestion at 10 ng/µl for 45 minutes (Fig. 1b). Second, while different TR reagents produced comparable TSS scores (~4), the average number of unique fragments per condition varied: 7695 (Tris-EDTA buffer), 5305 (citrate buffer), and 4340 (0.8 M Tris-HCl buffer) per 50-µm pixel resolution (Fig. 1c). Notably, only the treatment of Tris-EDTA buffer (pH 9.0) yielded the unique fragments comparable to those from spatial ATAC-seq in fresh-frozen samples (average: 7761). We further examined the impact of varying sodium chloride concentrations (0 nM, 250 nM, and 250 mM) across different TR reagents and found no obvious differences in the median number of fragments (Fig. 1c).
Although spatial FFPE-ATAC-seq preferentially yielded fragments less than 100 bp, it exhibited strong enrichment around transcription start site (TSS) regions (Fig. 1d, e). In contrast to standard ATAC-seq, which displays clear nucleosome periodicity, spatial FFPE-ATAC-seq produced smaller fragment sizes, likely reflecting DNA degradation introduced during the reverse crosslinking step (Supplementary Fig. 2a–d). Sequencing reads from FFPE samples showed high mapping rates (>85%) to the mouse genome (Fig. 1f). Utilizing the same 50 × 50 barcode scheme (50-µm pixel size, 5 × 5 mm region), spatial FFPE-ATAC-seq demonstrated a strong correlation (r = 0.61–0.89) with control profiles generated from spatial ATAC-seq on fresh-frozen samples (Fig. 1g).
Compared to published spatial ATAC-seq data from developing mouse embryos8,13, the spatial FFPE-ATAC-seq data of mouse brain displayed comparable TSS score, similar numbers of unique fragments, and high enrichment around the TSS (Supplementary Fig. 2e, f). However, the correlation between mouse embryo and brain datasets was moderate (Supplementary Fig. 2 g), suggesting dynamic changes in open chromatin structure from embryonic development to adulthood.
Spatial ATAC-seq profiling of mouse FFPEs
We next sought to identify cell types by using unsupervised clustering, which revealed 6–8 distinct clusters whose spatial projections closely aligned with tissue morphology (Fig. 2a and Supplementary Fig. 3). For example, clusters 1 and 2 corresponded to the mouse cerebellum, while cluster 5 was specific to the cortex region (Fig. 2a). To further validate these clusters, we compared the spatial distribution of known marker genes with results from spatial ATAC-seq of fresh-frozen mouse samples (Fig. 2b). The cerebellar clusters exhibited increased chromatin accessibility at genes involved in neuronal differentiation (e.g., Pax6, Car10, and Prox1). In contrast, the cortex cluster was characterized by enhanced accessibility at genes associated with neuronal fate specification (e.g., Kalrn, Bcl11b, and Tbr1). Interestingly, in the cortex, spatial FFPE-ATAC-seq revealed a sharper and more distinct chromatin accessibility pattern at the Pou6f2 locus than conventional spatial ATAC-seq. This suggests that spatial FFPE-ATAC-seq may provide additional resolution in detecting chromatin accessibility differences across specific brain regions.

a Spatial-FFPE-ATAC-seq on an FFPE mouse brain section (sample S4_2). Left: tissue scanning after microfluidic device barcoding. Middle: spatial distribution of unique fragments. Right: spatial pattern and UMAP of each cluster. b Spatial mapping of accessibility score of selected marker genes in different clusters for spatial FFPE-ATAC-seq (samples: S4_1 and S4_2) and fresh frozen spatial ATAC-seq (sample: FF_1). c Quantification of ATAC-seq peaks in genomic features extracted from cerebellar region, comparing FF and FFPE samples. (FF: n = 3; FFPE: n = 6; two-tailed Student’s t test). Data are presented as mean ± SD. d Comparison of ATAC-signals of selected genes between FFPE and FF cerebellar clusters. e Integration of scRNA-seq data17 with spatial-FFPE-ATAC-seq (sample S4_2). f Spatial mapping of selected cell types identified through label transfer. CNINH1: cerebellar inhibitory neuron 1; CBGRC: cerebellar granular cells; MOL1: Mature Oligodendrocyte 1; TEGLU7: telencephalon excitatory neurons 7. Scale bar: 500 μm. ns: not significant. Source data are provided as a Source Data file.
Tn5-accessible chromatin is typically enriched at gene promoters near TSS, as well as in intergenic regions associated with regulatory elements like enhancers14,15. To further evaluate the effects of FFPE processing on spatial chromatin accessibility profiles, we compared spatial FFPE-ATAC-seq to control spatial ATAC-seq performed on fresh-frozen cerebellum samples. We analyzed chromatin accessibility distributions across genomic features and assessed the localization of key marker genes. Spatial FFPE-ATAC-seq accurately captured the expected distribution of chromatin accessibility across genomic features, showing no significant differences in the proportions of promoters, introns, and intergenic regions compared to fresh-frozen spatial ATAC-seq data (Fig. 2c). By annotating ATAC-seq peaks with the closest genes, we identified 4817 common genes, and 1482 and 3079 unique genes in FF and FFPE groups, respectively (Supplementary Fig. 4a). GO analysis of common genes revealed enrichment for neuronal processes such as synapse organization and dendritic development, consistent with cerebellar functions (Supplementary Fig. 4b). In contrast, FFPE-unique GO terms were largely associated with general housekeeping processes, rather than specialized cerebellar functions (Supplementary Fig. 4c). Similarly, although some FF-unique GO terms, such as cell projection organization, related to neuronal features important for axon and dendrite formation, most were associated with general cellular processes, including protein degradation and organelle organization (Supplementary Fig. 4d).
We next applied STAMP16, a deep learning-based spatial topic modeling method, to decompose gene activity scores from FFPE samples into spatial topics. STAMP revealed distinct spatial patterns corresponding to brain regions such as the cortex and cerebellum (Supplementary Fig. 4e). To evaluate biological relevance, we performed GSEA on the top 50 genes from each topic using brain region markers from the Allen Brain Atlas. Significant enrichment (FDR < 0.1; Supplementary Fig. 4f) confirmed that the spatial gene activity captured brain region-specific organization, highlighting the capability of our FFPE-based protocol to preserve meaningful spatial epigenomic signals.
To further assess the preservation of spatial structure in FFPE samples, we computed two standard spatial autocorrelation metrics—Moran’s I and Geary’s C—for each gene and compared them to matched fresh-frozen (FF) samples. Both metrics showed moderate positive correlations between FFPE and FF (Moran’s I: PCC = 0.4021; Geary’s C: PCC = 0.4034), indicating that FFPE samples retained substantial spatial gene activity patterns (Supplementary Fig. 5a). Together, these results suggest that despite extensive chemical fixation and harsh paraffin embedding inherent to FFPE processing, our spatial FFPE-ATAC-seq protocol reliably captures biologically meaningful spatial relationships comparable to those observed in fresh-frozen tissue.
While previous FFPE-ATAC study has reported high Gabrb2 chromatin accessibility in the cerebellum12, our spatial FFPE-ATAC-seq data provided a more refined understanding of its localization (Fig. 2b). Specifically, we observed preferential enrichment of Gabrb2 accessibility in the cerebellar region, revealing a previously unappreciated spatial component of its regulatory landscape that was obscured in bulk FFPE-ATAC12 data. Furthermore, genome tracks for Gabrb2 and other marker genes exhibited strong concordance with fresh-frozen spatial ATAC-seq, further validating the accuracy of our spatial FFPE-ATAC-seq approach (Fig. 2d and Supplementary Fig. 5b).
Next, we mapped spatial FFPE-ATAC-seq data to specific cell types by integrating it with scRNA-seq data17 (Fig. 2e). For example, cerebellar inhibitory neurons (CBINH1) and granule cells (CBGRC) were enriched in the gray matter of the cerebellum, while telencephalon excitatory neurons 7 (TEGLU7) were localized in the cortex region (Fig. 2f). We also observed high reproducibility between replicates via correlation analysis (r > 0.9) (Supplementary Fig. 5c). Notably, the high concordance between spatial FFPE-ATAC-seq data and single-cell ATAC-seq data18 further validated the robustness and reliability of our FFPE-based protocol (Supplementary Fig. 5d). Taken together, our results demonstrate that spatial FFPE-ATAC-seq faithfully captures key features of chromatin accessibility in FFPE tissues, providing a robust approach for epigenomic profiling in archived samples.
Spatial mapping of human cerebellum FFPEs
Spatial FFPE-ATAC-seq offers considerable promises for analyzing the vast global repository of clinical FFPE samples by integrating spatial information with chromatin accessibility profiles. To assess its applicability in human tissues, we applied this method to a 5 mm × 5 mm region of interest within a human cerebellum FFPE tissue section, leveraging the conserved spatial architecture shared between mice and human cerebellum (Figs. 2a and 3a). Consistent with observations in mouse tissues, nucleosome-free fragments constituted most of the captured library, exhibiting a TSS enrichment score of 3.09 (Supplementary Fig. 6a and 6b). Analysis of this human cerebellum section (2500 pixels, 50-µm pixel size) yielded a median of 11,703 unique fragments per pixel, with a 19.2% duplication rate from a total of 107.4 million reads (Supplementary Data 2).

a Spatial-FFPE-ATAC-seq profiling of a FFPE human cerebellum section (n = 1). Left: tissue scanning after microfluidic device barcoding. Middle: spatial distribution of unique fragments. Right: spatial pattern and UMAP of each cluster. b H&E-stained image of an adjacent tissue section (n = 1). Arrows labeled the molecular layer, cerebellar medulla, and granular layer regions. c Integration of scRNA-seq data19 with spatial-FFPE-ATAC-seq from human cerebellum. d–f Spatial mapping of selected cell types identified through label transfer: oligodendrocytes (d) and granule cells (e). f, g Genome track visualization of selected marker genes across different clusters. Scale bar: 500 μm.
To further benchmark spatial FFPE-ATAC-seq, we compared our data to bulk ATAC-seq peaks from the ENCODE human cerebellum dataset (Supplementary Fig. 6c). The genome was divided into non-overlapping windows of 0.5 kb, 1 kb, and 10 kb, and each window was labeled as positive if it overlapped an ENCODE peak. We then counted the number of spatial FFPE-ATAC-seq reads per window and performed receiver operating characteristic (ROC) analysis by varying the read count threshold. The resulting area under the curve (AUC) values—0.927, 0.931, and 0.870 for 0.5 kb, 1 kb, and 10 kb windows, respectively—demonstrate strong concordance between spatial FFPE-ATAC-seq and bulk ATAC-seq profiles (Supplementary Fig. 6c). Aggregate peak annotations from spatial FFPE-ATAC-seq data exhibited a distribution similar to ENCODE human cerebellum ATAC-seq data, particularly at promoter regions (Supplementary Fig. 6d). Furthermore, nucleosome depletion regions (NDRs) identified via ENCODE bulk-ATAC significantly overlapped with spatial FFPE-ATAC-seq peaks, encompassing 68% of annotated genes (Supplementary Fig. 6e). A similar trend was observed for H3K4me3-marked genes identified by ChIP-seq from human cerebellum, with over 66% overlap with spatial FFPE-ATAC-seq peaks (Supplementary Fig. 6f). Genome browser tracks of representative loci demonstrated high concordance across these datasets (Supplementary Fig. 6g). These results demonstrate that our FFPE-based method reliably captures chromatin accessibility patterns.
Unsupervised clustering delineated six distinct clusters within the cerebellum (Fig. 3a), corresponding to structures such as the cerebellar medulla (cluster 4), granular layer (cluster 1), and molecular layer (clusters 2, 3). These clusters align well with H&E-stained tissue architecture (Fig. 3b). Integrating these data with single-cell RNA-seq datasets from human cerebellar cells19 revealed regionally specialized cell types, including oligodendrocytes and granule cells, consistent with observed tissue morphology (Fig. 3c–e). Th oligodendrocyte marker genes (e.g., OLIGO2, MBP, SOX2) exhibited high chromatin accessibility in the medulla (Fig. 3d), while the granular cell marker PAX6 was enriched in the granular layer (Fig. 3e, f). Consistent with mouse studies, increased accessibility at GABA-A receptor loci (e.g., GABRB2, GABRA1, GABRA6) was observed in the granular layer cluster, reinforcing the conserved role of GABAergic signaling in the vertebrate cerebellum (Figs. 2b, d and 3e, g).
Chromatin states of human thymus FFPEs
Building on the successful application of our method to human tissues, we next explored how spatial resolution influences data quality using a human thymus FFPE sample. Serial tissue sections were processed to generate ATAC-seq data at both 50-µm and high-resolution 10-µm scales, with an adjacent section stained using H&E to provide spatial and histological context (Fig. 4a–c). The 50-µm device captures a 5 × 5 mm region (2500 pixels), while the 10-µm device covers a 5.5 × 5.5 mm area (48,400 pixels), approaching single-cell resolution. Both resolutions exhibited similar fragment profiles and TSS enrichment scores (50-µm: 5; 10-µm: 4) (Supplementary Fig. 7a, b). Median unique fragments per pixel were 19,557 (50-µm) and 9309 (10-µm), with duplicate rates of 26% and 61.9%, respectively (Supplementary Data 2). MACS2 peak calling revealed 92% overlap between different resolutions, demonstrating a high degree of consistency in chromatin accessibility profiles across different spatial scales (Supplementary Fig. 7c).

a Schematic illustration of experimental design: 50-μm ATAC (n = 2), 10-μm ATAC (n = 1), and H&E-staining (n = 1). b H&E-stained image of an adjacent tissue section. Dashed circle indicated overlapping area of spatial FFPE-ATAC-seq. Arrows labeled the cortex, septum, medulla, and medulla-cortex (MC) boundary. c, d Spatial FFPE-ATAC-seq profiling of FFPE human thymus sections with different resolutions. Left: tissue scanning before microfluidic device barcoding. Middle: spatial distribution of unique fragments. Right: spatial pattern and UMAP of each cluster. e Integration of scRNA-seq data20 with spatial-FFPE-ATAC-seq with different resolutions. f Spatial mapping of selected cell types identified through label transfer. TECs: medullary thymic epithelial cells. g, Spatial mapping of gene scores for selected marker genes in different clusters. h–m, 50-μm ATAC (h, j, l) and 10-μm ATAC(i, k, m) data were used for genome track (h, i) and pseudotime analysis (j–m). h–i Genome track visualization of selected marker genes across different clusters. j, k Pseudotemporal reconstruction of T cell development from cortex to medulla region. l, m, Dynamics of gene scores of CD8A and KRT5 along the pseudotime shown in (j, k) respectively. Scale bar: 500 μm.
Unsupervised clustering identified six and eight distinct clusters at the 50-µm and 10-µm resolutions, respectively (Fig. 4c, d). Both resolutions revealed spatial patterns corresponding to tissue histology. For example, Clusters 1-3 (50-µm) and clusters 1,4,6 (10-µm) mapped to the cortex; cluster 4 (50-µm) and cluster 2 (10-µm) localized to the medulla; and clusters 5-6 (50-µm) and cluster 6 (10-µm) marked the cortex-medulla boundary. Notably, 50-µm resolution revealed distinct segregation among the three cortex clusters, each associated with different lobules, highlighting the tissue’s inherent heterogeneity (Fig. 4c). The 10-µm resolution, however, captured finer details, exhibiting higher similarity to the adjacent H&E-stained data, particularly in resolving the interlobular septa, which provide structural support and compartmentalization within the thymus (Fig. 4b, d).
Cell types were then assigned to each cluster by integrating these data with a scRNA-seq dataset20 (Fig. 4e). Cortical clusters were enriched with T cells, medullary clusters with medullary thymic epithelial cells (TECs), and cortex-medulla boundary clusters with mesenchymal cells (Fig. 4f). The observed chromatin accessibility patterns were validated by comparison with a spatial transcriptomics dataset21. For example, T cell markers CD8A and CD8B showed higher chromatin accessibility in the cortex, consistent with their spatial gene expression patterns (Fig. 4g–i and Supplementary Fig. 7d). The medullary TEC marker KRT5 exhibited high chromatin accessibility and gene expression in the medulla. Pseudotemporal analysis of CD8A and KRT5 further supported their chromatin accessibility changes along the cortical-to-medullary axis (Fig. 4j–m), consistent with the trajectories of T cell development22. Although the thymus is primarily a T-cell maturation site, B cells also reside in the thymus, particularly in the medulla. While spatial transcriptomics detected few B cells, we observed high chromatin accessibility of the B cell marker CD20 (MS4A1) in the medulla (Fig. 4g and Supplementary Fig. 7d), highlighting the enhanced sensitivity of spatial FFPE-ATAC-seq in detecting rare cell populations that may be underrepresented in transcriptome-based methods. Correlation analysis between replicates and resolutions demonstrated high reproducibility (r = 0.876 for 50-µm resolution; r = 0.941 for different resolutions; Supplementary Fig. 7e), emphasizing the technical robustness and consistency of spatial FFPE-ATAC-seq across experimental conditions. Taken together, these results highlight the utility of spatial FFPE-ATAC-seq for profiling chromatin accessibility in FFPE tissues, with the choice of resolution dependent on the desired level of anatomical detail.
Spatial chromatin heterogeneity in tumor FFPEs
To further evaluate its clinical applicability, particularly in tumor samples, we applied spatial FFPE-ATAC-seq to human melanoma, a highly aggressive skin cancer23. Adjacent tissue sections were processed for H&E staining and spatial ATAC-seq profiling (Fig. 5a, b). At 50-µm resolution, we achieved a median of 8,762 unique fragments per pixel, with a TSS enrichment score comparable to normal human tissues (4.12) (Supplementary Figs. 6b, 7b, and Fig. 5b–d). Unsupervised clustering delineated six distinct spatial clusters, subsequently categorized as non-tumor (clusters 1,2) and tumor (clusters 3-6) (Fig. 5c). Within non-tumor regions, we observed high chromatin accessibility at the CD274 (PD-L1) and PDCD1 (PD-1) loci. The tumor regions were further subdivided into four clusters, each characterized by unique marker gene accessibility. For instance, cluster 4 exhibited enrichments of accessibility at KIT and NRAS, two genes critical for melanoma progression24,25. The RNA-binding protein RBFOX1 showed increased accessibility across clusters 3, 5, and 6. High BRAF accessibility was observed throughout the tumor region (Fig. 5c), coinciding with BRAF as the most common early genetic alteration in melanoma26. Genome tracks at the BRAF locus and surrounding regions revealed distinct chromatin accessibility peaks with a high signal-to-noise ratio at the tumor boundary (cluster 4; Fig. 5f), highlighting its regulatory activity in this region. These spatially resolved ATAC profiles reveal substantial tumor heterogeneity within melanoma, providing insights that may ultimately inform H&E-based clinical decision-making.

a H&E-stained image of an adjacent tissue section (n = 1). Dashed circle indicated an overlapped area of spatial FFPE-ATAC-seq. Dashed lines indicated the boundary region of tumor and black square labeled the region of interest. b Spatial FFPE-ATAC-seq profiling of FFPE melanoma tissue section (50-μm resolution; n = 1). Left: tissue scanning before microfluidic device barcoding. Right: spatial pattern and UMAP of each cluster. Black square labeled the region of interest. c, d Insert fragments size distribution (c) and TSS enrichment profiles (d) of FFPE melanoma sample. e Spatial mapping of gene scores for selected marker genes in different clusters. f Genome track visualization of selected marker genes across different clusters. Scale bar: 500 μm.
Finally, we examined the fraction of fragments that mapped to the human genome which showed a similar mapping rate observed in mouse samples (Fig. 1f and Supplementary Fig. 5g). Spearman correlation analysis assessed the similarity of chromatin accessibility profiles across all human samples (Supplementary Fig. 5h). Strong positive correlations (r = 0.77–0.94) were observed within samples from the same tissue, indicating consistent chromatin accessibility patterns. In contrast, comparisons between samples from distinct tissues revealed substantially weaker correlations (r = 0.29–0.50), reflecting significant inter-tissue variability.
Discussion
Given the heavy crosslinking in FFPE samples, selecting optimal target retrieval (TR) conditions is essential for exposing the original chromatin states. We tested over 50 TR conditions to identify the most effective approach. The minimal modifications introduced in our method enhance its versatility, making it applicable across various platforms and easily integrated into other existing protocols.
We present spatial FFPE-ATAC-seq, an approach for spatially resolved chromatin accessibility profiling in FFPE tissues. By optimizing the target retrieval process—incorporating heat-induced retrieval, proteinase K treatment, and Tris-EDTA buffer—we adapted our previously developed spatial ATAC-seq8 for FFPE samples. This optimized protocol produced high-quality data, comparable to fresh-frozen samples, as shown by consistent TSS enrichment scores, similar unique fragment counts, high mapping rates, and accurate capture of open chromatin regions. In mouse brain, for example, spatial FFPE-ATAC-seq successfully captured distinct chromatin accessibility patterns across different brain regions and cell types, validated by comparison with spatial ATAC-seq data from fresh-frozen samples. The refined localization of Gabrb2 accessibility in the cerebellar region, which was missed by bulk FFPE-ATAC12, underscores the power of spatial resolution.
Application of spatial FFPE-ATAC-seq to human cerebellum and thymus tissues demonstrated its versatility across different tissue types. Multi-resolution analysis of the thymus revealed the trade-off between resolution and detail information, with 10-µm resolution resolving finer anatomical structures like interlobular septa. Critically, application to human melanoma revealed substantial tumor heterogeneity, identifying distinct subclusters with unique marker gene accessibility, including clinically relevant genes like KIT, NRAS, BRAF, CD274 (PD-L1), and PDCD1 (PD-1). The observed BRAF accessibility throughout the tumor region, coupled with high signal-to-noise ratio at the BRAF locus, underscores the method’s sensitivity.
In summary, spatial FFPE-ATAC-seq represents a transformative tool in epigenetic research, enabling high-resolution spatial profiling of chromatin accessibility in FFPE tissues. Our results demonstrate the technology’s capability to generate high-quality, spatially resolved epigenomic data from archival samples. As we continue to refine and expand its applications, spatial FFPE-ATAC-seq will play a crucial role in advancing precision medicine by linking epigenetic features to disease processes, improving diagnostic accuracy, and enabling the development of more targeted therapeutic interventions.
Methods
Ethics statement
All experimental procedures involving animals were conducted in strict accordance with international ethical standards. All procedures and protocols used were pre-approved by Animal Care and Use Committee of Pennsylvania Animal Care Facilities.
Preparation of paraffin tissue slides
The human cerebellum and thymus sections were purchased from Zyagen (HP-202, HF-702). The human melanoma tissue block was purchased from BioIVT. Adult mouse brain tissue (18 weeks) was obtained from the C57BL/6 mice housed in the University of Pennsylvania Animal Care Facilities. All mice were maintained in 12-h light/12-h dark cycle at room temperatures ranging between 20 and 25 °C and humidities between 40 and 60%. The adult mouse (18 weeks) was sacrificed by CO2, and brain was harvested, fixed in 10% Neutral Buffered formalin, and processed for embedding in low temperature melting paraffin. The brains were sagittal sectioned into 8-10 μm sections and collected on glass slides.
Microfluidic device fabrication and assembly
The molds for polydimethylsiloxane (PDMS) microfluidic devices were fabricated using standard photolithography. The manufacturer’s guidelines were followed to spin-coat SU-8-negative photoresist (nos. SU-2025, Microchem) onto a silicon wafer (no. C04004, WaferPro). The heights of the features were about 50 μm for 50-μm-wide devices, respectively. We mixed the curing and base agents in a 1:10 ratio and poured the mixture onto the molds. After degassing for 30 min the mixture was cured at 70 °C for 2 h. Solidified PDMS was extracted for further use. The fabrication and preparation of the PDMS device follow the published protocol27.
Preparation of the Tn5 transposome
Unloaded Tn5 transposase (C01070010) was purchased from Diagenode, and the transposome was assembled according to the manufacturer’s guidelines. The transposome was assembled by combination of Tn5MErev and Tn5ME-A or Tn5ME-B. The oligo sequences used for transposome assembly were as follows:
Tn5MErev: 5′-/Phos/CTGTCTCTTATACACATCT-3′
Tn5ME-A: 5′-/Phos/TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′
Tn5ME-B: 5’-/Phos/CATCGGCGTACGACTAGATGTGTATAAGAGACAG-3′
DNA oligos, DNA barcode sequences, and other key reagents
Lists of the DNA oligos that were used for sequencing library construction and PCR, DNA barcode sequences (A1-50, B1-50), and all other key reagents are provided in Supplementary Data 3. 10-μm FlowGel Inventor Kit was purchased from AtlasXomics (AXO-0464(03)).
Tissue deparaffinization, rehydration, and target retrieval
Tissue slide was immersed in CitriSolv for three changes, followed by rehydration in a series of ethanol dilutions, including two times of 100% ethanol, one time of 95% ethanol, 70% ethanol, and 30% ethanol, culminating in a final wash with PBS buffer. Each step was performed for 5 minutes. Subsequently, the tissue slide was submerged in modified target retrieval buffer (1x eBioscience™ IHC Antigen Retrieval Solution + 250 nM NaCl) at 65 °C for 15 minutes and subjected to 10 ng/μl proteinase K buffer at 37 °C for 45 minutes. After cooling down to room temperature and a brief dip in distilled water, tissue image was captured using Keyence Imaging System.
Spatial profiling of ATAC-seq with FFPE samples
After the target retrieval, tissue was washed twice with 1 ml of 1× DPBS and cleaned with ddH2O. The sequential order for spatial ATAC profiling is as follows: 1. ATAC-seq; 2. Ligation of barcode A; 3. Ligation of barcode B; 4. Reverse crosslink; 5. Library construction; 6. Library QC and sequencing.
-
1.
ATAC-seq: tissue was permeabilized with lysis buffer (3 mM MgCl2, 0.01% Tween-20, 10 mM Tris-HCl pH 7.4, 0.01% NP40, 10 mM NaCl, 1% bovine serum albumin (BSA), 0.001% digitonin) for 15 min and washed twice with wash buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.1% Tween-20) for 5 min. Transposition mixture (5 μl of home-made loaded Tn5 transposome, 33 μl of 1× DPBS, 50 μl of 2× Tagmentation buffer, 1 μl of 1% digitonin, 1 μl of 10% Tween-20, 10 μl of nuclease-free H2O) was added and incubated at 37 °C for 30 min. Next, 200 μl of 40 mM EDTA was added and incubated for 5 min at room temperature, to stop the transposition.
-
2.
Ligation of barcode A:: Barcode A was pre-annealed with ligation linker 1, briefly, 10 μl of 100 μM ligation linker, 10 μl of 100 μM individual barcode A (A1-50) oligo and 20 μl of 2× annealing buffer (20 mM Tris pH 7.5–8.0, 100 mM NaCl, 2 mM EDTA) was mixed and annealed (95 °C for 5 min and cycling from 95 °C to 12 °C, 0.01 °C per cycle). For the first barcode (barcode A) in situ ligation, the PDMS chip A covered the region of interest (ROI). For alignment purposes, a 10× objective lens (BZ-X800 Series, Keyence) was used to take a brightfield image. The PDMS device and tissue slide were clamped tightly with a homemade acrylic clamp. For each channel, 5 μl of ligation master mix containing individual barcode was loaded, it was prepared by mixing 2 μl of ligation mixture (27 μl of T4 DNA ligase buffer, 72.4 μl of RNase-free water, 5.4 μl of 5% Triton X-100, 11 μl of T4 DNA ligase), 2 μl of 1× NEBuffer 3.1 and 1 μl of each annealed DNA barcode A (A1 − A50 25 μM). Vacuum was used to load the ligation master mix into 50 channels of the device, followed by incubation at 37 °C for 30 min in a humidified container. The PDMS chip and clamp were removed after incubation and washed with 1× NEBuffer 3.1for 5 min. Then the slide was washed with water and dried with compressed air.
-
3.
Ligation of barcode B: Barcode B was pre-annealed with ligation linker 1, briefly, 10 μl of 100 μM ligation linker, 10 μl of 100 μM individual barcode B (B1-50) oligo and 20 μl of 2× annealing buffer (20 mM Tris pH 7.5–8.0, 100 mM NaCl, 2 mM EDTA) was mixed and annealed (95 °C for 5 min and cycling from 95 °C to 12 °C, 0.01 °C per cycle). For the second barcode (barcode B) in situ ligation, the PDMS chip B was covered to the ROI and a further brightfield image was taken with the 10× objective lens. An acrylic clamp was applied to clamp the PDMS, and the tissue slide together. Annealing of barcodes B (B1 − B50, 20 μM) and preparation of the ligation master mix were carried out as for barcodes A. The tissue was then incubated at 37 °C for 30 min in a humidified container. After incubation, the PDMS chip and clamp were removed, and tissue was washed with 1× DPBS for 5 min. The slide was then washed with water and dried with compressed air. A brightfield image covering each barcoding axis was then taken for further alignment.
-
4.
Reverse crosslink: lastly, the ROI on the tissue was digested with 100 μl of reverse crosslinking mixture (0.4 mg ml–1 proteinase K, 1 mM EDTA, 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 1% SDS) at 58 °C for 2 h in a wet box. The lysate was then collected in a 0.2 ml tube and incubated at 60 °C overnight.
-
5.
Library construction: The lysate was collected and purified with Zymo DNA Clean & Concentrator-5 and eluted with 20 μl of nuclease-free water for ATAC library construction. 30 μl of PCR mixture (25 μl of 2× NEBNext Master Mix, 2.5 μl of 10 μM indexed N7XX primer, 2.5 μl of 10 μM N501 PCR primer) was added to the eluted gDNA, total 50 ul. PCR reaction was first performed with the following program: 72 °C for 5 min, 98 °C for 30 s and then cycling at 98 °C for 30 s, 63 °C for 30 s, 72 °C for 1 min, 12 times. The final PCR product was purified by 1.0x SPRI beads (50 μl) and eluted in 20 μl of nuclease-free water.
-
6.
Library QC and sequencing: The Agilent D5000 Screentape was used to determine the size distribution and concentration of the library before sequencing. NGS was conducted on an Illumina NovaSeq X Plus sequencer (paired-end, 150-base-pair mode).
Data preprocessing
For ATAC-seq data processing, linkers 1 and 2 are used to filter read 2. The filtered reads are aligned to the mouse (GRCm38) or human (GRCh38) reference genomes using BWA, followed by sorting and indexing with Samtools for efficient data handling and retrieval. The resulting data is converted into fragment files using the sinto toolkit for downstream analysis.
Data clustering and visualization
Firstly, we identified the location of pixels on tissue from the brightfield image using a custom python script (https://github.com/PengfeiGuo0123/Spatial-FFPE-ATAC-seq).
The normalization and dimension reduction were conducted with SnapATAC228. For spatial data visualization, to facilitate the mapping of data onto the original tissue, the gene score matrix derived from ArchR was imported into Seurat as a Seurat object. Then we plotted spatial maps using SpatialPlot. The size of the pixels was adjusted for visualization by modifying the ‘pt.size.factor’ parameter within the Seurat package. Peaks were called with pseudo-bulk bam files using MACS2 with parameters ‘--keep-dup=1 --llocal 100000 --min-length 1000 --max-gap 1000 --broad-cutoff=0.1’.
Chromatin dynamics analysis and cell type identification
Pseudo-time analysis on RNA was performed using ArchR. For ATAC data, trajectory analysis was conducted using the addTrajectory function from the ArchR package. To analyze a specific gene, the ATAC signal for each pixel was calculated using the getGeneScore function, which identifies the subset of signals within the gene window, weighted by their distance from the gene. This gene score matrix was then utilized for cell type identification using the FindTransferAnchors and TransferData functions from Seurat.
Statistics and reproducibly
For spatial fresh-frozen and FFPE ATAC-seq experiments in mice, at least three biological replicates were included per individual, and the findings were reproducible across different animals. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to group allocation during experiments or outcome assessment.
Statistical analyses were performed using GraphPad Prism 9. An unpaired two-tailed Student’s t-test was applied for comparisons where the sample size was n ≥ 3. Details of the specific statistical tests, number of replicates, and measures of variation are provided in the corresponding figure legends. All key findings were confirmed in independent experiments, as indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw and processed data reported in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE278007. Resulting fastq files were aligned to the mouse (mm10) or human (hg38) reference genome. Published data for data quality comparison and integrative data analysis include: mouse brain cell atlas [http://mousebrain.org/adolescent/downloads.html], scATAC-seq data of mouse brain [http://catlas.org/mousebrain/#!], scRNA-seq data of human cerebellum GSE165657, ENCODE human cerebellum ATAC-seq [https://www.encodeproject.org/experiments/ENCSR802GEV], ENCODE human cerebellum ChIP-seq, H3K4me3 [https://www.encodeproject.org/experiments/ENCSR042ITN], scRNA-seq data of human thymus [https://developmental.cellatlas.io], and spatial transcriptome of human thymus [https://zenodo.org/records/13207776]. Source data are provided with this paper.
Code availability
Data analysis pipeline is available on GitHub [https://github.com/PengfeiGuo0123/Spatial-FFPE-ATAC-seq] and via Zenodo at [https://doi.org/10.5281/zenodo.15425611] (ref. 29).
References
Blow, N. Tissue issues. Nature 448, 959–960 (2007).
Henikoff, S. et al. Epigenomic analysis of formalin-fixed paraffin-embedded samples by CUT&Tag. Nat. Commun. 14, 5930 (2023).
Liu, Y., Enninful, A., Deng, Y. & Fan, R. Spatial transcriptome sequencing of FFPE tissues at the cellular level. Preprint at bioRxiv https://doi.org/10.1101/2020.10.13.338475 (2020).
Bai, Z. et al. Spatially exploring RNA biology in archival formalin-fixed paraffin-embedded tissues. Cell 187, 6760–6779.e6724 (2024).
Gracia Villacampa, E. et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genomics 1, 100065 (2021).
Cejas, P. et al. Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat. Med. 22, 685–691 (2016).
Fanelli, M. et al. Pathology tissue–chromatin immunoprecipitation, coupled with high-throughput sequencing, allows the epigenetic profiling of patient samples. Proc. Natl. Acad. Sci. 107, 21535–21540 (2010).
Deng, Y. et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 609, 375–383 (2022).
Guo, P. et al. Multiplexed spatial mapping of chromatin features, transcriptome and proteins in tissues. Nat. Methods 22, 520–529 (2025).
Marcel, S. S. et al. Genome-wide cancer-specific chromatin accessibility patterns derived from archival processed xenograft tumors. Genome Res. 31, 2327–2339 (2021).
Zhao, L. et al. FACT-seq: profiling histone modifications in formalin-fixed paraffin-embedded samples with low cell numbers. Nucleic Acids Res. 49, e125 (2021).
Zhang, H. et al. Profiling chromatin accessibility in formalin-fixed paraffin-embedded samples. Genome Res. 32, 150–161 (2022).
Llorens-Bobadilla, E. et al. Solid-phase capture and profiling of open chromatin by spatial ATAC. Nat. Biotechnol. 41, 1085–1088 (2023).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016).
Zhong, C., Ang, K. S. & Chen, J. Interpretable spatially aware dimension reduction of spatial transcriptomics with STAMP. Nat. Methods 21, 2072–2083 (2024).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e1022 (2018).
Li, Y. E. et al. An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136 (2021).
Zhong, S. et al. Single-cell epigenomics and spatiotemporal transcriptomics reveal human cerebellar development. Nat. Commun. 14, 7613 (2023).
Park, J.-E. et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 367, eaay3224 (2020).
Li, Y. et al. Unraveling the spatial organization and development of human thymocytes through integration of spatial transcriptomics and single-cell multi-omics profiling. Nat. Commun. 15, 7784 (2024).
Ueno, T. et al. Role for CCR7 ligands in the emigration of newly generated T lymphocytes from the neonatal thymus. Immunity 16, 205–218 (2002).
Robertson, B. M., Fane, M. E., Weeraratna, A. T. & Rebecca, V. W. Determinants of resistance and response to melanoma therapy. Nat. Cancer 5, 964–982 (2024).
Choi, Y. S. et al. Topical therapy for regression and melanoma prevention of congenital giant nevi. Cell 185, 2071–2085 (2022).
Larkin, J. et al. Nilotinib in KIT-driven advanced melanoma: results from the phase II single-arm NICAM trial. Cell Rep. Med. 5, 101435 (2024).
Dankort, D. et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 41, 544–552 (2009).
Su, G. et al. Spatial multi-omics sequencing for fixed tissue via DBiT-seq. STAR Protoc. 2, 100532 (2021).
Zhang, K., Zemke, N. R., Armand, E. J. & Ren, B. A fast, scalable and versatile tool for analysis of single-cell omics data. Nat. Methods 21, 217–227 (2024).
Guo, P. & Mao, L. PengfeiGuo0123/Spatial-FFPE-ATAC-seq: v1.0.0 (version). Zenodo https://doi.org/10.5281/zenodo.15425611. (2025).
Acknowledgements
We acknowledge support from the Packard Fellowship for Science and Engineering (to Y.D.), John Q. Trojanowski Research Scholar Award from the Penn Institute on Aging (to Y.D.), and the National Cancer Institute and National Institute on Aging of the National Institutes of Health (grant numbers R61CA287408 and R01AG085344 to Y.D.).
Author information
Authors and Affiliations
Contributions
Y.D. and P.G. conceived and designed the study; Experimental Investigation: P.G., Y.C., A.C., C.N.L., D.J., and Y.H.; Data Analysis: P.G., Y.C., L.M., Y.C., X.X., and Y.D.; Original Draft: P.G. and Y.D. All authors reviewed, edited, and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y.D., P.G., and Y.C. have submitted a provisional patent application to the United States Patent and Trademark Office related to the spatial profiling of chromatin accessibility in FFPE tissues described in this work (Application No. 63/675,580). Y.D. is the scientific advisor of AtlasXomics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xi Chen, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, P., Chen, Y., Mao, L. et al. Spatial profiling of chromatin accessibility in formalin-fixed paraffin-embedded tissues. Nat Commun 16, 5945 (2025). https://doi.org/10.1038/s41467-025-60882-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60882-3
