
Abstract
Autophagy classically functions to protect cells and organisms during stressful conditions by catabolizing intracellular components to maintain energy homeostasis. Lysosome-autophagosome fusion is a critical step in emptying degraded unwanted contents. However, the mechanism of autophagosome fusion with lysosomes is still not fully understood. Here, we report that DNA Damage-Regulated Autophagy Modulator 1 (DRAM1) interacts with Vesicle Associated Membrane Protein 8 (VAMP8) to mediate the fusion of autophagosomes with lysosomes. This DRAM1-VAMP8 interaction is enhanced upon stimulation of autophagy. However, DRAM1 preferentially mediates the fusion between autophagosomes and lysosomes by enhancing the assembly of the STX17-SNAP29-VAMP8 complex. Moreover, we reveal that DRAM1 specifically promotes the stability of lysosomal VAMP8 via inhibiting VAMP8 degradation by CHIP mediating ubiquitination. We also identify that DRAM1 inhibits the ubiquitination of VAMP8 at Lys 68,72, and 75 via competitively binding with CHIP. Furthermore, we demonstrate that DRAM1 promotes the extravasation of Hepatocellular Carcinoma (HCC) cells, and this process relies on enhanced autophagosome degradation. Our study reveals a mechanism for regulating autolysosome formation by DRAM1-VAMP8 association and suggests a potential strategy to inhibit the extravasation of HCC.
Similar content being viewed by others

Introduction
Autophagy is an evolutionary conserved lysosomal degradation pathway in eukaryotic cells that can degrade unwanted cytoplasmic materials and organelles to maintain cellular homeostasis1,2. The fusion of autophagosomes with lysosomes is a key step to complete autophagic degradation3,4,5. During this process, the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes precisely regulate the formation of autolysosomes4,6,7. It has recently been reported that autophagosomal SNARE syntaxin 17 (STX17) interacts with cytosolic SNARE SNAP29 and the lysosomal SNARE VAMP8, and all of these proteins are required for autophagosome-lysosome fusion3,7,8. However, the molecular mechanism for how the fusion of autophagosomes with lysosomes is regulated has not been fully understood.
VAMP8 was first identified as an endosomal SNARE that participates in diverse biological functions, including endosomal fusion8. Among the relevant SNAREs, VAMP8 is preferentially located on the lysosomal membrane and is also distributed on the plasma membrane, early endosomes, and late endosomes6. A recent study revealed that VAMP8 might play a role in guiding lysosomes toward autophagosomes9. However, only a few proteins that cooperate with VAMP8 to govern the fusion of autophagosomes-lysosomes have been identified. This raises intriguing questions about which additional proteins interact with VAMP8 to mediate this fusion process and how they act.
DNA Damage-Regulated Autophagy Modulator 1 (DRAM1) is an evolutionarily conserved trans-membrane protein, mainly located in lysosomes to induce autophagy and down-regulated in multiple types of cancer10,11. This protein was previously identified as the first direct molecular link between the tumor suppressor p53 and autophagy10. Since then, various roles for DRAM1 in several processes have been described, including autophagy, cell death, immunity, and cellular differentiation12. DRAM1 directs newly synthesized amino acid transporters to lysosomes and drives lysosomal amino acid efflux13. In addition to lysosomes, DRAM1 isoforms are also localized to peroxisomes, autophagosomes, and the endoplasmic reticulum (ER)10, and DRAM1 can promote binding between lysosomes and ER14. The most reported mechanisms of DRAM1-triggered autophagy are mainly focused on promoting the generation of autophagy and increasing the fusion between autophagosomes and lysosomes. However, the mechanism by which DRAM1 enables the fusion.
DRAM1 is a downstream target of p53 and plays an important role in autophagy, and aberrant expression of DRAM1 is associated with various tumors15,16,17. DRAM1 may exert a dual function in carcinogenesis. In fact, DRAM1 has been reported to play a tumor suppressor role in Non-small-cell lung cancer (NSCLC)18, Clear cell renal cell carcinoma (ccRCC)19, and Neuroblastoma (NB)20. However, DRAM1 also acts as a tumor promoter in Gastric cancer (GC)21, head and neck squamous cell carcinoma (HNSCC)22 and Hepatocellular carcinoma (HCC)23. From the TCGA database analysis, the high expression of DRAM1 negatively relates to the overall survival of HCC, Kidney renal clear cell carcinoma (KIRC), Sarcomas (SARC) and Skin cutaneous melanoma (SKCM) patients. Among these cancers, survival of HCC patients is the most significantly negatively correlated with DRAM1 expression level.
HCC represents the fourth most common cause of cancer-related deaths worldwide24. Metastasis is one of the leading causes contributing to the low survival rate of HCC patients24,25. Metastasis is a complex multi-step process and represents the most deadly aspect of cancer25. Substantial evidence demonstrates that only a minor fraction of tumor cells that enter the circulation survive and extravasate to form metastases26. There is also evidence that tumor cells in the circulation acquire resistance to environmental stress by upregulation of protective mechanisms, such as autophagy27,28. However, most HCC patients are only diagnosed at advanced stages29, where tumors have significant energy demands, which makes autophagy beneficial for the tumor30. Therefore, inhibiting autophagy is a promising strategy for restriction of metastasis. A comprehensive understanding of the mechanisms of DRAM1 in HCC will enhance our knowledge of the intrinsic mechanisms underlying HCC development and provide insights into the mechanisms and highlight potential therapeutic targets for HCC.
In the current study, DRAM1 was identified as a protein partner of VAMP8 on lysosomes. We found that the interaction between DRAM1 and VAMP8 was enhanced by the induction of autophagy. Additionally, our results revealed that DRAM1 can be induced in response to nutrient starvation, both in human cells in vitro and in the zebrafish model in vivo, thereby promoting autolysosome formation. Moreover, we identified that DRAM1 promotes the assembly of the STX17-SNAP29-VAMP8 complex to facilitate the fusion of autophagosomes with lysosomes. We also demonstrated that DRAM1 increases the stability of VAMP8 to ensure the formation of the STX17-SNAP29-VAMP8 complex. More importantly, we identified that DRAM1 is competitive with the arboxy-terminus of Hsc70 interacting protein (CHIP) to hinder the ubiquitination of VAMP8. Furthermore, our in vivo work in both mouse and zebrafish demonstrate that DRAM1 is required for the efficient extravasation of HCC, an essential step in cancer dissemination, and this function relies on enhanced autolysosome formation. Hence, our findings demonstrate that DRAM1 regulates the stabilization of VAMP8 to facilitate the assembly of the STX17-SNAP29-VAMP8 complex, further elucidating how exactly this protein acts as an autophagy regulator in response to stressful conditions.
Results
DRAM1 is a VAMP8 interacting partner and this interaction is enhanced upon autophagy stimulation
VAMP8 is key in mediating autophagosome fusion with lysosomes3,4,31. To identify proteins interacting with VAMP8 by mass spectrometry (MS), we performed immunoprecipitation (IP) with GFP-tagged VAMP8 as bait (Supplementary Fig. 1A). Interestingly, we identified DRAM1 as an interacting partner of VAMP8 (Fig. 1A). DRAM1 and VAMP8 largely localize on the lysosomal membrane3,10 and play a vital role in regulating autophagy. However, the interplay between these two proteins is unclear.

A Endogenous interaction between DRAM1 and VAMP8 as demonstrated with the co-immunoprecipitation (Co-IP) assay in 293 T cells. B The interaction of VAMP8 and DRAM1 was determined by GST pull-down assay. E.coli-expressed GST and GST-VAMP8 were used in GST pull-down assays with lysates of 293 T cells transfected with Flag-DRAM1. C, D Colocalization of VAMP8 with DRAM1 in normal and starvation conditions. Representative confocal images are shown, and the intensity of VAMP8 colocalized with DRAM1 was quantified by Image J (n = 20 cells/group). Scale bars: 10 μm. Data are shown as means ± SD representative for three independent experiments. E, F Relative levels of DRAM1 protein immunoprecipitated by VAMP8 in normal and starvation conditions, and quantification of relative DRAM1 protein level. The level of co-precipitated DRAM1 was quantified by Image J and normalized to the corresponding VAMP8 (set to 1 in 0 h). G Schematic representation of the developmental features of early zebrafish larvae used in autophagy activation assay. H Level of basal autophagy in zebrafish larvae at the indicated time points. Protein samples were extracted from 3, 5, 7, and 10 dpf zebrafish larvae (n = 30 larvae/sample). I Relative expression level of p62, Lc3 and Dram1 at indicated time points. The RNA samples were extracted from 3, 5,7 and 10 dpf zebrafish larvae (n = 30 larvae/sample). J Level of DRAM1 and autophagy activity in 293 T cells under the EBSS treatment at the indicated time points. K The workflow of the experiments shown in L examines the degradation of accumulating Lc3-II in zebrafish larvae after CQ treatment. L Level of p62 and Lc3 protein in WT and mCherry-Dram1 expressing 5 dpf larvae. Protein samples were extracted from zebrafish larvae (n = 30 larvae/sample). Data in (F,I) are presented as mean ± SD, n = 3 independent experiments. Data in (A, B, C, E, H, J, L) are representative of three independent experiments. P values were calculated by two-tailed Student’s t tests (D, F). P-values are indicated in the figure.
Furthermore, we examined whether VAMP8 physically interacted with DRAM1. A GST pull-down assay was performed using recombinant GST-tagged VAMP8 and the lysates of 293 T cells transfected with Flag-DRAM1. Results revealed that DRAM1 indeed directly interacts with VAMP8 (Fig. 1B). To determine whether this interaction was conserved in other vertebrates, we transfected zebrafish DRAM1 (Dram1) and human VAMP8 in 293 T cells to examine if zebrafish Dram1 also interacts with human VAMP8. The IP results also showed that zebrafish Dram1 interacts with human VAMP8 (Supplementary Fig. 1B). Next, we performed immunostaining to examine the colocalization between DRAM1 and VAMP8. Results showed that DRAM1 indeed colocalizes with VAMP8 (Fig. 1C). Intriguingly, this colocalization significantly increased with Earle’s balanced salt solution (EBSS) treatment (Fig. 1C, D). Additionally, the immunostaining results confirmed that DRAM1 colocalizes with VAMP8 on lysosomes (Supplementary Fig. 1C). Furthermore, the interaction between DRAM1 and VAMP8 in EBSS treatment or Torin1 (an inhibitor of mTOR) treatment was increased in IP assays (Fig. 1E, F, Supplementary Fig. 1D), suggesting that binding between DRAM1 and VAMP8 might facilitate autophagic activity.
Next, we studied the function of DRAM1/Dram1 in starvation. The zebrafish model is widely used in autophagy studies32. In the present study, the relationship between Dram1 expression level and autophagy under starvation conditions was evaluated using this animal model. Zebrafish embryos have a yolk that can provide a nutrient resource for growth at the early stage of embryos (1-3 dpf, day post-fertilization). Afterwards, the nutrient resources will be gradually exhausted. At this point, larvae begin to hunt for food (Fig. 1G). We noticed that autophagy is activated at 3 and 5 dpf larvae, as shown by high levels of Lc3-II by WB analysis. Instead, autophagy levels decrease in 7 and 10 dpf larvae (Fig. 1H). We also collected the same stage of larvae for transcriptional analysis. The results showed that Dram1, p62 and Lc3 expression levels were not significantly different at 3 and 5 dpf, while they were induced at 5 to 7 dpf (Fig. 1I). This suggests that starvation induces dram1 expression in the zebrafish model. Moreover, we examined the DRAM1 protein level in cells under starvation conditions by EBSS treatment. As expected, the DRAM1 level gradually increased with the time frame of EBSS treatment (Fig. 1J).
To investigate if the function of DRAM1/Dram1 is conserved in mediating autophagy in starvation conditions, we adopted the autophagy reporter Tg(CMV:GFP-map1lc3b) zebrafish line33. We injected human DRAM1 or zebrafish Dram1 mRNA into zebrafish embryos at one-cell stage and collected larvae at 6 dpf to image GFP-Lc3 punctae as a marker for autophagosome formation (Supplementary Fig. 1E). The results showed that DRAM1/Dram1 injected larvae display significantly decreased the number of GFP-Lc3 punctae compared to WT larvae (Supplementary Fig. 1F, G). In agreement, autophagy assay by WB showed that DRAM1/Dram1 injected larvae contain lower levels of Lc3-II and p62 than WT in starvation conditions (Supplementary Fig. 1H).
We also used the Tol2 system to generate a stable transgenic line overexpressing zebrafish Dram1 (Supplementary Fig. 1I). We genotyped the embryos to identify stable overexpression (OE) of mCherry-Dram1 by either fluorescence observation or PCR (Supplementary Fig. 1J). We pretreated zebrafish with CQ to block autophagosome fusion with lysosomes. Then, we removed the CQ and replaced the fresh embryos water to examine the speed of degradation of accumulated Lc3-II over a time course (Fig. 1K). We observed that the degradation of Lc3-II in WT zebrafish is slower than in Dram1 overexpressing zebrafish (Fig. 1L). Taken together, these results suggest that the DRAM1-VAMP8 association is specifically enhanced by autophagy induction.
DRAM1 promotes the degradation of autophagosomes via enhanced autolysosomes formation
DRAM1 regulation of different stages of autophagy has been investigated by several studies. It has been shown that DRAM1 affects mTOR activity to mediate autophagy initiation16 and promotes autolysosome formation via enhanced lysosome acidification11. However, the mechanism by which DRAM1 regulates the autophagosome-lysosome fusion process remains unclear.
To further investigate the function of DRAM1 in modulating autophagosome-lysosome fusion, we used the CRISPR/Cas9 method to generate DRAM1 KO 293 T and HeLa cells. We found that the protein levels of LC3-II and p62 were significantly higher in DRAM1 KO cells than in Control cells, either following EBSS treatment or in untreated conditions (Fig. 2A–C). Meanwhile, we investigated whether DRAM1 is involved in autophagosome-lysosome fusion using tandem fluorescent-tagged LC3 (RFP-GFP-LC3) in HeLa cells33. We found that DRAM1 KO cells have a significantly decreased percentage of GFP-RFP+/GFP+RFP+ + GFP-RFP+ compared to control cells (Supplementary Fig. 2A, B). In contrast, DRAM1 OE had the opposite effect (Supplementary Fig. 2A, B).

A Levels of p62 and LC3 protein were examined by WB in DRAM1 KO and Control 293 T cells. B, C Quantification of LC3-II/GAPDH and p62/GAPDH fold changes in Control and DRAM1 KO 293 T cells. D Schematic paradigm of in vitro fusion of GFP-LC3 and mCherry-LAMP1 vesicles in 293 T cells. E, F Confocal microscopy analysis of the in vitro autophagosome-lysosome fusion assay experiments and represented the percentage of mCherry-LAMP1 vesicles that fused with GFP-LC3 (n = 25 samples). Data are shown as means ± SD accumulated from two independent experiments. G Representative electron micrograph of autolysosome-like structures in the brain region of 6 dpf larvae. H The quantification of the number of autolysosome-like structures in the imaged region (n = 5 larvae). Scale bars: 2 μm. Data are shown as means ± SD representative for two independent experiments. I The workflow of the experiments is shown in (J) to examine the degradation of accumulating LC3-II under a standard medium after CQ treatment. J The degradation of LC3-II was examined by WB in DRAM1 KO and Control cells. K Quantification of LC3-II/GAPDH in indicated time point. The relative LC3-II protein levels were quantified by Image J and normalized with that of GAPDH, and the relative LC3-II levels are shown as the percentage of the initial LC3-II content. L–N Autophagosome fusion with lysosome was detected by Lysotracker staining in zebrafish larvae. Representative confocal images are shown. Scale bars: 10 μm. The percentage of GFP-LC3 punctae colocalizing with LysoTracker was quantified in a predefined region (n = 12 larvae). Data are shown as means ± SD accumulated from two independent experiments. Data in (B, C, K) are presented as mean ± SD, n = 3 independent experiments. Data in (A, J) and (E, G, M) are representative of three and two independent experiments, respectively. P values were calculated by two-tailed Student’s t tests (B, C, F, K, N) or One-way ANOVA with Tukey’s Multiple Comparison post hoc test (adjusted P-values for multiple comparisons) (H). P-values are indicated in the figure.
To verify the role of DRAM1 in the fusion process, we also performed autophagosome-lysosome fusion in vitro assays34,35 (Fig. 2D). We found that the fusion activity of GFP-LC3 autophagosomes from DRAM1 KO cells with mCherry-LAMP1 lysosomes was significantly lower than for GFP-LC3 autophagosomes from Control cells (Fig. 2E, F).
To examine how DRAM1 mediates the degradation of autophagosomes, we pretreated cells with CQ to block autophagosome fusion with lysosomes. Then, we removed CQ and replaced the normal medium to measure the speed of degradation of accumulated autophagosomes (LC3-II) over a time course (Fig. 2I). We observed that the degradation of LC3-II in Control cells requires about 6 h to show clear reduction (Fig. 2J, K), and DRAM1 KO led to a significant delay in the degradation of LC3-II compared with Control cells (Fig. 2J, K). However, in DRAM1 OE cells, the sharp decrease in LC3-II occurred within 3 h (Supplementary Fig. 2C, D and E). Taken together, DRAM1 OE led to a significant increase in the degradation of LC3-II compared to Control cells (Supplementary Fig. 2C–E).
To visualize whether Dram1 affects autolysosome formation in the zebrafish model, we used CRISPR/Cas9 technology to generate Dram1 Crispant zebrafish in the GFP-Lc3 transgenic background36 (Supplementary Fig. 2F, G). The effect of CRISPR/Cas9 was validated by sanger sequencing and WB (Supplementary Fig. 2H, I). Firstly, we used transmission electron microscopy (TEM) to detect autolysosome formation in zebrafish larvae37. Then, we sliced sections from the brain region and observed that the number of autolysosomes in stable Dram1 OE larvae was significantly higher than in WT. In contrast, we observed that the number of autolysosomes was lower in Dram1 crispant zebrafish than in WT (Fig. 2G, H). We also observed that Dram1 deficiency leads to decreased GFP-Lc3 colocalization with lysotracker-positive particles in zebrafish (Fig. 2L–N). Collectively, these data indicate that DRAM1/Dram1 increases the degradation of autophagosomes by promoting autophagosome fusion with lysosomes in cells and zebrafish.
DRAM1 facilities assembly of STX17-SNAP29-VAMP8 complex
Based on the observation that DRAM1 promotes the fusion of the autophagosome with lysosomes and interacts with VAMP8, substantial studies have demonstrated that STX17-SNAP29-VAMP8 complex acts as a key component in the fusion of the autophagosome with lysosomes38,39, we hypothesized that DRAM1 regulates the STX17-SNAP29-VAMP8 complex to promote autolysosome formation. Therefore, we determined whether DRAM1 promotes autolysosome formation via increasing assembly of the STX17-SNAP29-VAMP8 complex. As expected, Co-IP experiments with Flag-STX17 showed that DRAM1 OE increased the levels of GFP-SNAP29 and GFP-VAMP8 compared to Control (Fig. 3A, B). In contrast, DRAM1 KO significantly decreased the levels of GFP-SNAP29 and GFP-VAMP8 precipitated by Flag-STX17 (Supplementary Fig. 3A, B). We then assessed the colocalization of STX17 with VAMP8 in cells. This colocalization was remarkably elevated in DRAM1 OE cells (Fig. 3C, D) and decreased in DRAM1 KO cells (Supplementary Fig. 3C). These results indicate that DRAM1 promotes the formation of the STX17-SNAP29-VAMP8 complex. In agreement, the immunoprecipitation of STX17 resulted in co-precipitation of endogenous SNAP29 and VAMP8, and higher levels of SNAP29 and VAMP8 were co-immunoprecipitated by STX17 in DRAM1 OE cells compared to Control cells (Fig. 3E, F). Similarly, higher levels of STX17 and VAMP8 were co-immunoprecipitated by SNAP29 (Fig. 3G, H) and higher levels of STX17 and SNAP29 were co-immunoprecipitated by VAMP8 in DRAM1 OE compared to Control cells (Fig. 3I, J). In contrast, DRAM1 KO significantly decreased the level of endogenous SNAP29 and VAMP8 co-immunoprecipitated by STX17, and the level of STX17 and VAMP8 co-immunoprecipitated by SNAP29 (Supplementary Fig. 3D–G). Together, these results demonstrated that DRAM1 facilitates the formation of the SNAP29-STX17-VAMP8 complex.

A Levels of GFP-SNAP29 and GFP-VAMP8 co-immunoprecipitated by Flag-STX17 in Control and DRAM1 OE cells. 293 T cells transfected with the indicated constructs were immunoprecipitated using anti-Flag antibodies and subjected to WB analysis. B Quantification of relative levels of GFP-SNAP29 and GFP-VAMP8 that are co-immunoprecipitated by Flag-STX17 in Control and DRAM1 OE cells. The level of co-precipitated GFP-SNAP29 and GFP-VAMP8 were quantified by Image J and normalized to the corresponding Flag-STX17 (set to 1 in control cells). C, D Colocalization of Flag-STX17 with VAMP8 in Control or DRAM1 OE HeLa cells. Representative confocal images are shown, and the percentage intensity of STX17 colocalized with VAMP8 was quantified by Image J (n = 9 cells/group). Data are shown as means ± SD representative of two independent experiments. Scale bars: 10 μm. E Levels of endogenous SNAP29 and VAMP8 co-immunoprecipitated by STX17 in Control and DRAM1 OE 293 T cells. Cells transfected with the indicated constructs were immunoprecipitated using anti-STX17 antibodies and subjected to WB analysis. F Quantification of relative levels of SNAP29 and VAMP8 that are co-immunoprecipitated by STX17 in Control and DRAM1 OE cells. The level of co-precipitated SNAP29 and VAMP8 were quantified by Image J and normalized to the corresponding STX17 (set to 1 in control cells). G Levels of endogenous STX17 and VAMP8 immunoprecipitated in Control and DRAM1 OE cells. H Quantification of relative levels of STX17 and VAMP8 that are co-immunoprecipitated by SNAP29 in Control and DRAM1 OE 293 T cells. I Levels of endogenous STX17 and SNAP29 immunoprecipitated in Control and DRAM1 OE cells. 293 T cell extracts were immunoprecipitated with anti-VAMP8 and the resulting precipitants were analyzed by WB using the indicated antibodies. J Quantification of relative levels of STX17 and SNAP29 that are co-immunoprecipitated by VAMP8. Data in (B, F, H, J) are presented as mean ± SD, n = 3 independent experiments. Data in (A, E, G, I, C) are representative of three and two independent experiments, respectively. P values were calculated by two-tailed Student’s t tests (B, D, F, H, J). P values are indicated in the figure.
DRAM1 promotes STX17-SNAP29-VAMP8 formation via enhanced stability of lysosomal VAMP8
Next, we investigated how exactly DRAM1 increases the stability of the STX17-SNAP29-VAMP8 complex to facilitate autolysosome formation. It has been reported that the acetylation of STX17 and the O-GlcNAcylation of SNAP29 lead to decreased assembly of the STX17-SNAP29-VAMP8 complex34,40. Therefore, we performed Co-IP to detect the acetylation of STX17 and O-GlcNAcylation of SNAP29 level in DRAM1 OE and Control. Still, we noticed that the acetylation of STX17 and O-GlcNAcylation of SNAP29 was not affected by DRAM1 OE (Supplementary Fig. 4A, B). Next, we assessed if DRAM1 affects the STX17, SNAP29, and VAMP8 protein levels to facilitate the assembly of the STX17-SNAP29-VAMP8 complex. To explore this possibility, we first performed WB to measure STX17, SNAP29, and VAMP8 protein levels in DRAM1 KO and OE cells with and without Rapa and CQ treatments. The results showed that the protein level of VAMP8 clearly increased in DRAM1 OE cells compared to Control, both in CQ and Rapa treatment (Fig. 4A). Conversely, DRAM1 KO leads to decreased protein levels of VAMP8 (Fig. 4A). This suggests that DRAM1 might stabilize the VAMP8 protein.

A The protein levels of STX17, SNAP29, and VAMP8 were examined by WB in 293 T cells. B, C The protein levels of VAMP8 were measured by WB after transfection with different dosages of Flag-DRAM1. C The quantification of relative VAMP8 protein level. The level of VAMP8 were quantified by Image J and normalized to the corresponding GAPDH (set to 1 at 0 h). D Relative expression levels of VAMP8 were detected by quantitative PCR. E Protein levels of STX17, SNAP29, and VAMP8 were examined by WB with CHX treatment in 293 T cells. F The quantification of relative VAMP8 protein by Image J and normalized with GADPH in 293 T cells. G Localization of TMEM192-3xHA fusion protein with lysosomes in HeLa cells. Scale bars: 10 μm. H Schematic of the workflow for the Lyso-lP method. HA-Lyso cells refer to cells expressing 3xHA-tagged TMEM192. I The lysosomal VAMP8 was examined using the Lyso-lP method in 293 T cells. J The level of lysosomal VAMP8 was quantified by Image J and normalized with corresponding HA-TMEM192 (set to 1 in control cells). K, L Protein levels of VAMP8 in cytoplasmic and lysosomes with CHX treatment in Control and DRAM1 KO cells and quantification. The lysosomes fraction was isolated by a lysosome enrichment kit, and the purity of the fractions was also assessed using LAMP1 (Lysosome markers) and GAPDH (Cytoplasmic) antibodies. M The levels of LC3 and p62 were measured by WB with or without co-transfection of Flag-DRAM1 and GFP-VAMP8. N The protein levels of LC3 and p62 were measured by WB with or without co-transfection of Flag-DRAM1 and si-VAMP8 in 293 T cells. Data in (C, D, F, J, L) are presented as mean ± SD, n = 3 independent experiments. Data in (A, B, E, G, I, K, M, N) are representative of three independent experiments. P-values were calculated by two-tailed Student’s t tests (C, F, J, L) or One-way ANOVA with Tukey’s Multiple Comparison posthoc test (adjusted P-values for multiple comparisons) (D). P values are indicated in the figure.
To validate this possibility further, we transfected different dosages of Flag-DRAM1 to detect VAMP8 protein levels, and we noticed that VAMP8 protein levels gradually increased with the increasing dosages of DRAM1 (Fig. 4B, C). This effect was not due to the transcriptional change since VAMP8 levels were not significantly different in DRAM1 KO, DRAM1 OE and Control cells (Fig. 4D). These results suggest that DRAM1 promotes the formation of autolysosomes by stabilizing VAMP8.
Next, we performed Cycloheximide (CHX) chase experiments to detect the degradation of VAMP8. The results showed that the degradation rate of VAMP8 was slower in DRAM1 OE cells compared to the Control (Supplementary Fig. 4C, D). In contrast, the degradation rate of VAMP8 was significantly increased in DRAM1 KO (Fig. 4E, F).
Considering that DRAM1 and VAMP8 can colocalize on lysosomes, we asked if DRAM1 stabilizes lysosomal or cytosolic VAMP8. We utilized the previously reported Lyso-IP method to isolate the lysosome fraction from cells41,42,43. TMEM192 is a transmembrane protein that retains its lysosomal localization, even upon overexpression. Therefore, it represents an optimal lysosomal marker for Lyso-IP, outperforming other established markers such as LAMP142,44. The lysosomal transmembrane protein TMEM192-3xHA was stably expressed in 293 T cells to label lysosomes (Fig. 4G), and lysosomes were isolated by immunoprecipitation using anti-HA magnetic beads (Fig. 4H). Lysosome integrity and purity were evaluated by immunoblotting of lysosomal (TMEM192-HA, LAMP1, Cathepsin D), non-lysosomal (GORASP2, CALR and TOM20) markers, as well as lysosome-associated proteins (VAMP8 and DRAM1). Interestingly, the results showed that lysosomal VAMP8 was elevated upon DRAM1 overexpressing, indicating DRAM1 promotes the stability of VAMP8 on lysosomes (Fig. 4I, J). Additionally, we isolated lysosomes to examine the VAMP8 protein level in DRAM1 KO cells with Cycloheximide (CHX) treatment. Results showed that DRAM1 KO led to a significant reduction in the stability of the lysosomal VAMP8 compared to cytoplasmic VAMP8 (Fig. 4K, L). This indicates that DRAM1 mainly stabilizes lysosomal VAMP8.
Next, we asked if DRAM1 and VAMP8 have a cooperative function in mediating autophagy. The autophagy assay by WB results showed that co-overexpression of DRAM1 and VAMP8 produced an additive effect on mediating autophagy with lower p62 levels compared to individually overexpressed DRAM1 or VAMP8 (Fig. 4M). We continued to examine if DRAM1 regulates autophagy depending on VAMP8. Thus, we used specific siRNAs to silence VAMP8 to evaluate the effect of DRAM1 OE on autophagy. As expected, the impact of DRAM1 in modulating autophagy is diminished by knockdown of VAMP8 (Fig. 4N). This suggests that DRAM1 modulates autophagy depending on VAMP8.
Taken together, our results indicate that DRAM1 enhanced lysosomal VAMP8 stability, which leads to increased formation of STX17-SNAP29-VAMP8 complex, which eventually contributes to autolysosome formation.
DRAM1 reduces CHIP-mediated ubiquitination of VAMP8
We sought to elucidate the mechanisms by which DRAM1 modulates the stability of VAMP8. The two major intracellular protein degradation systems are the ubiquitin-proteasome system (UPS) and autophagy45. Therefore, we investigated the possible mechanisms of VAMP8 protein downregulation via CHX assay using autophagy and proteasome inhibitors. The results showed that DRAM1 mediates VAMP8 stabilization via the ubiquitin-proteasome pathway (Fig. 5A). Next, we analyzed the ubiquitination of VAMP8 under DRAM1 OE and deficient conditions, and we found that DRAM1 depletion significantly increased the ubiquitination of VAMP8 (Fig. 5B, C), while DRAM1 OE reduced its ubiquitination compared with Control cells (Fig. 5B, C). These results imply that DRAM1 stabilizes VAMP8 through the ubiquitin-protease pathway.

A The protein levels of VAMP8 were measured by WB in Control and DRAM1 KO cells. B Ubiquitination analysis of extracts of Control or sh-DRAM1 293 T cells transfected with the indicated plasmids. C The quantification of ubiquitination of GFP-VAMP8 from immunoprecipitation by GFP antibody. The bands intensity above 43 KDa corresponding to GFP-VAMP8 was quantified by densitometry analysis (using Image J), and band intensity normalized to the GFP bead band intensity (set to 1 in control cells). D The protein levels of VAMP8 were analyzed by WB. E Effect of CHIP on ubiquitination of VAMP8 from extracts of 293 T cells transfected with GFP-VAMP8 together with or without the Flag-CHIP or si-CHIP. F Effect of CHIP on ubiquitination of VAMP8 from extracts of 293 T cells. G The quantification of ubiquitination of GFP-VAMP8 from immunoprecipitation by GFP antibody. H The endogenous protein level of STX17, SNAP29 and VAMP8 were examined by Co-IP. I, J Representative confocal image showing colocalization between VAMP8 and LAMP1 and quantification (n = 11 cells/group). Data are shown as means ± SD representative of two independent experiments. Scale bars: 10 μm. K Workflow of generation of Chip crispant zebrafish in the background of GFP-Lc3 transgenic zebrafish and lysotracker staining. L Schematic representation of the Chip genetic and protein domain architecture and CRISPR target site. M, N Autophagosome fusion with lysosome was detected by Lysotracker staining in zebrafish larvae. Representative confocal images are shown. Scale bars: 10 μm. The percentage of GFP-LC3 punctae colocalizing with LysoTracker was quantified by Image J in a pre-defined region (n = 12 larvae), data are shown as means ± SD representative of two independent experiments. Data in (C, G) are presented as mean ± SD, n = 3 independent experiments. Data in (A, B, D, E, F, H) and (I, M) are representative of three and two independent experiments, respectively. P-values were calculated by two-tailed Student’s t tests (C and N) or One-way ANOVA with Tukey’s Multiple Comparison post hoc test (adjusted P-values for multiple comparisons) (G and J).
DRAM1 has not been reported to exert roles on protein ubiquitination. Therefore, to explore the potential mechanisms, we performed Co-IP with GFP-tagged VAMP8 as bait by using mass spectrometry to screen ubiquitin proteins interacting with VAMP8 (Supplementary Fig. 5A). We identified Ring Finger protein 126 (RNF126) and carboxyl terminus of Hsc70-interacting protein (CHIP) as potential E3 ligases targeting with VAMP8 (Supplementary Fig. 5B, C). To confirm our mass spectrometry results, we examined whether CHIP or RNF126 interacts with VAMP8 by Co-IP in 293 T cells. Results revealed that CHIP and RNF126 interact with VAMP8 (Supplementary Fig. 5D, E). We also observed that CHIP plays a vital role in mediating the ubiquitination of VAMP8, compared to RNF126 (Supplementary Fig. 5F). To clarify the extent at which different E3 ligases contribute to VAMP8 ubiquitination, we adopted short hairpin RNA (sh-RNA) to silence different E3 ligases. We silenced CHIP and RNF126, which were identified in this study, but also MARCHF9, which has been previously reported to ubiquitinate VAMP8 in a membrane proteome study in B cells46. As a negative control for E3 ligase targeting VAMP8, we also silenced TRIM27, which is known to directly polyubiquitinate ULK1 (a core protein to initiate the occurrence of autophagy in mammalian cells)47. Ubiquitination assays showed that silencing CHIP reduces the ubiquitination of VAMP8, but the silencing of other ligases did not affect the ubiquitination level of VAMP8 (Supplementary Fig. 5G). Next, we overexpressed these E3 ligases to detect if this affected the protein level of VAMP8. The results showed that CHIP OE significantly reduces the VAMP8 level compared with other candidates (Supplementary Fig. 5H). Additionally, to detect if RNF126 affects the stability of VAMP8, we conducted a CHX degradation assay of VAMP8 by silencing RNF126. The results showed that silencing of RNF126 does not affect the VAMP8 stability (Supplementary Fig. 5I). These data implicate that CHIP is the primary E3 ligase for the ubiquitination of VAMP8.
To determine whether CHIP affects the stability of VAMP8, we treated different dosages of siRNA to silence CHIP and monitor the change of VAMP8 protein. The silencing of CHIP leads to elevated VAMP8 protein levels in a siRNA dosage-dependent manner (Fig. 5D). In contrast, CHIP OE results in decreased VAMP8 protein levels in a Flag-CHIP transfected dosage-dependent manner (Supplementary Fig. 5J). This indicates that CHIP negatively correlates with the protein level of VAMP8.
Since we demonstrated that VAMP8 degradation depends on the ubiquitin-proteasome system, we examined if CHIP affects the ubiquitination of VAMP8. As expected, the results revealed that CHIP OE significantly enhanced the ubiquitination of VAMP8 (Fig. 5E). Conversely, the silencing of CHIP had the opposite effect on the ubiquitination of VAMP8 (Fig. 5E). Next, we assessed if DRAM1 affects CHIP-mediated ubiquitination of VAMP8. Co-IP results showed that DRAM1 competitively inhibits the CHIP-mediated ubiquitination of VAMP8 (Fig. 5F, G). Additionally, we found that DRAM1 competitively binds to VAMP8 and CHIP (Supplementary Fig. 5K) and that DRAM1 stabilizes VAMP8 in the presence of CHIP (Supplementary Fig. 5L, M).
We speculated that DRAM1 and CHIP could interact with the same amino acid region of VAMP8, leading to competitive binding. The results revealed that the amino acid region from residue 66 to 100 is responsible for the interaction of VAMP8 with DRAM1 and CHIP (Supplementary Fig. 5N, O). We also investigated if CHIP inhibits the assembly of the STX17-SNAP29-VAMP8 complex. As expected, CHIP decreases the assembly of the STX17-SNAP29-VAMP8 complex, and this reduced assembly is rescued by DRAM1 (Fig. 5H).
We then asked if CHIP affects the stability of lysosomal VAMP8, and the immunostaining results showed that CHIP OE reduces the VAMP8 colocalization to the lysosome (Fig. 5I, J). On the contrary, DRAM1 OE strongly rescued CHIP-mediated reduction of lysosomal VAMP8 (Fig. 5I, J).
Zebrafish larvae have an ortholog of human CHIP. Using the zebrafish model, we asked if CHIP/Chip affects fusion between autophagosomes and lysosomes. For this, we utilized CRISPR/Cas9 technology to generate Chip crispant zebrafish in the transgenic GFP-Lc3 background (Fig. 5K, L), and the effect of CRISPR/Cas9 was validated by sanger sequencing and WB (Supplementary Fig. 5P, Q). We incubated 6 dpf WT and Chip crispant larvae with Lysotracker to measure the GFP-LC3 fusion with lysosomes (Fig. 5K). The results showed that Chip crispants display significantly enhanced colocalization between GFP-Lc3 and Lysotracker positive particles (Fig. 5M, N). These results suggest that DRAM1 binds with VAMP8, leading to a competitive reduction of CHIP-mediated ubiquitination of VAMP8. The resulting stabilization of VAMP8 contributes to the enhanced fusion between autophagosomes and lysosomes by encouraging the formation of the STX17-SNAP29-VAMP8 complex.
DRAM1 inhibits the ubiquitination of VAMP8 on Lys 68, 72, and 75
To identify the exact VAMP8 ubiquitination sites, we generated 8 VAMP8 point mutants, each with different Lysines (K) replaced by an Arginine (R) (Fig. 6A). The ubiquitination assay results showed that a mutant with all the Lysines in the amino acid region 24–75 replaced with Arginines (VAMP8(24-75)7xK-R), as well as the single K-R mutants for Lysines in the amino acid region 66–100 (VAMP868K-R, VAMP872K-R, and VAMP875K-R), had significantly reduced levels of VAMP8 ubiquitination (Fig. 6B). We also generated mutations of all the Lysine residues in the amino acid region 24-64 (VAMP8(24-64)4xK-R) or the amino acid region 68-75 (VAMP8(68-75)3xK-R) (Fig. 6C). The results showed that both 3XKR and 4XKR mutants significantly reduced the ubiquitination compared with WT VAMP8 (Fig. 6D, E). Since we have demonstrated that CHIP binds with the region of VAMP8 in amino acid region 66–100, this region contains 68,72 and 75 Lysines (K). Therefore, we asked whether CHIP affects the stability of VAMP8(68-75)3xK-R, we performed WB to detect the protein level of GFP-VAMP8(68-75)3xK-R in the presence or absence of CHIP. The results revealed that CHIP does not decrease the protein level of VAMP8(68-75)3xK-R (Fig. 6F).

A Schematic structure of human VAMP8 and its mutants used in this study. Positions of the SNARE motif, transmembrane domain and the seven Lys residues are indicated. B The ubiquitination of GFP-VAMP8 and its mutants were examined by Co-IP. GFP-tagged VAMP8 and its mutants were transfected into 293 T cells for 48 h, and immunoprecipitated with anti-GFP antibody. C Schematic structure of mutation of human VAMP8 4×K-R and VAMP8 3×K-R used in this study. D The ubiquitination of GFP-VAMP8, VAMP8 4×K-R and VAMP8 3×K-R were examined by Co-IP. GFP-tagged VAMP8, VAMP8 4×K-R and VAMP8 3×K-R were transfected into 293 T cells for 48 h, and cell lysates were immunoprecipitated with anti GFP antibody. Transfected cells were then treated with MG132 (10 μM) for 6 h before collection. E The quantification of ubiquitination of GFP-VAMP8 from immunoprecipitation by GFP. The bands intensity above 43KDa corresponding to GFP-VAMP8 were quantified by densitometry analysis (using Image J), and band intensity normalized to the GFP bead band intensity (set to 1 in control cells). Data are shown as means ± SD from three independent experiments. F The protein level of GFP-VAMP8 in the presence or absence of Flag-CHIP by WB assay. 293 T cells were co-transfected with the indicated vector for 48 h, then, the cell lysate was used for WB analysis. G The effect of CHIP on assembly of STX17-SNAP29-VAMP8 complex in the presence of VAMP83×K-R. 293 T cells were co-transfected with the indicated vector for 48 h, and cell lysates were immunoprecipitated with anti STX17 or SNAP29 antibody, respectively. H The effect of DRAM1 on assembly of STX17-SNAP29-VAMP8 complex in the presence of VAMP83K-R. 293 T cells were co-transfected with the indicated vector for 48 h, and cell lysates were immunoprecipitated with anti STX17 or SNAP29 antibody, respectively. Data in (B, D, F–H) are representative of three independent experiments. P-values were calculated by One-way ANOVA with Tukey’s Multiple Comparison post hoc test (adjusted P-values for multiple comparisons) (E). P-values are indicated in the figure.
We further examined the effect of CHIP in VAMP8(68-75)3xK-R assembly in the STX17-SNAP29-VAMP8 complex. We found that VAMP8(68-75)3xK-R eliminates the CHIP inhibition on STX17-SNAP29-VAMP8 formation (Fig. 6G, H). However, DRAM1 still promoted the assembly of the STX17-SNAP29-VAMP8 complex in the presence of VAMP8(68-75)3xK-R. These findings are consistent with the fact that the amino acid region 66-100 is responsible for the interaction of VAMP8 with DRAM1 and CHIP. Collectively, these results demonstrate that DRAM1 reduces CHIP-mediated ubiquitination of VAMP8 Lysine residues at positions 68, 72 and 75.
DRAM1 promotes extravasation of HCC in mouse and zebrafish models
We analyzed high-throughput sequencing data from 33 different tumor types available in The Cancer Genome Atlas (TCGA) database, and found that survival of Hepatocellular carcinoma (HCC) patients is the most significantly negatively correlated with DRAM1 expression level (Supplementary Fig. 6A). Firstly, to explore the function of DRAM1 in HCC progression, we adopted well-established HCC models, in which myr-AKT1 and N-RasV12 or Myc and myr-AKT1 proto-oncogenes are stably integrated into the genome of hepatocytes by Sleeping Beauty (SB) transposase through hydrodynamic injection to induce HCC tumorigenesis48,49. DRAM1 overexpression plasmid or empty vector plasmid was co-injected with myr-AKT1/N-RasV12 plasmids in 8 weeks old C57 mouse via tail vein (Supplementary Fig. 6B). At 5 weeks post hydrodynamic injection, we observed that DRAM1 OE led to significantly increased tumor burden, as indicated by an increased abdominal size (Supplementary Fig. 6C), liver tumor foci (Supplementary Fig. 6D), liver weight (Supplementary Fig. 6E), and the ratio of liver weight to body weight (Supplementary Fig. 6F). In addition, hematoxylin and eosin (HE) staining revealed that DRAM1 OE could increase tumor densities (Supplementary Fig. 6G). We also observed that DRAM1 overexpression promotes autophagosome degradation in liver tumor compared with control liver tumor (Supplementary Fig. 6H).
Furthermore, we also used the Myc and myr-AKT1-induced HCC model to validate the function of DRAM1 in the progression of HCC. The Myc plasmid containing the firefly luciferase as a reporter allows us to monitor the progression of HCC (Supplementary Fig. 6I)50. DRAM1 overexpression plasmid or sh-Dram1 plasmid was co-injected with Myc-Luc/myr-AKT1 plasmids in 8-week-old C57BL/6 J mouse. After 5 weeks, we measured the bioluminescence (BLI) of Myc as an indicator for progression of HCC. The DRAM1 OE group showed a stronger BLI signal and higher tumor burden than the control group (Supplementary Fig. 6J–L). Conversely, the strengths of the BLI signal and tumor burden were lower in the sh-Dram1 group compared to the control group (Supplementary Fig. 6J–L). Collectively, these data demonstrate that DRAM1 plays a significant role in HCC progression.
The predominant cause of death in HCC patients is tumor progression with metastasis24. Next, to directly test if DRAM1 affects the metastasis capacity of HCC, we generated DRAM1 KO and DRAM1 OE in the widely used HCC cell lines HepG2 and MHCC97H (97H)51,52. The metastasis ability was evaluated by transwell migration and invasion assays53. The results showed that DRAM1 OE increases the migration and invasion ability of HCC cells (Fig. 7A, B, Supplementary Fig. 6M). Conversely, DRAM1 KO delays the migration and invasion of HCC cells (Fig. 7A, B, Supplementary Fig. 6M). This indicates that DRAM1 promotes the migration and invasion of HCC cells in vitro.

A Representative image of Transwell migration assay (up) and transwell invasion assay (down) using HepG2 cells. Scale bar: 100 μm. B The quantification of migrated cells (upper) and invading cells (down) per field is shown in the bar graphs. Data are presented as mean ± SD from three independent experiments. C Scheme representing the metastatic assay for (D–H). D Representative lung tissue from post 5 weeks vein injection. E Bar plot showing the calculated tumor nodules in lungs, n = 5 mice. Data are shown as means ± SD representative of two independent experiments. F Representative H&E staining of lung sections post 5 weeks of injection. The arrows indicate histologically confirmed lung lesions. Scale bar: 1000 μm. G Representative macroscopic images of liver metastases via tail vein injection of 97H. The arrow indicates the visible tumor in the liver. H Representative H&E-stained liver sections. Scale bar: 1000 μm. I Scheme representing the metastatic assay. J The bioluminescence imaging of five representative mice with M3 cells injection. Luc, firefly luciferase (n = 5 mice). K Lung metastasis incidence in C57BL/6 mice after M3 injection. Data were combined from two independent experiments (n = 12 mice). L The representative image shows the immigration of 97H-mCherry in the tail region at 3 dpi. Scale bar: 100 μm. M Quantification of the number of cell clumps in the tail region by Image J, n = 9 larvae/group. N Representative image showing the extravasated 97H-GFP cells out of blood vessels in the tail region at 2 dpi. Scale bar: 100 μm. O Quantification of the percentage of extravasated 97H-GFP in the tail region. The extravasated 97H-GFP cell in the tail region was calculated by Image J. n = 9 larvae/group. Data in (A, L–O) and (D–H, J, K) are representative of three and two independent experiments, respectively. P values were calculated by two-tailed Student’s t tests (E) or One-way ANOVA with Tukey’s Multiple Comparison post hoc test (adjusted P-values for multiple comparisons) (B, M, O) and two-tailed Chi-square test for (K).
Autophagy is essential for maintenance and metabolic adaptations that facilitate tumor metastasis20,54. Therefore, to further explore DRAM1’s role in metastasis in vivo, we utilized a mouse tail vein metastasis model to validate if DRAM1 promotes metastasis in vivo55. Around 3 × 106 MHCC97H cells were injected in 5-week-old nude mice (BALB/C-NU) (Fig. 7C). Mice were euthanized 5 weeks later to analyze HCC metastasis in the lung by gross observation and HE staining. The tail vein lung metastasis model demonstrated that mice injected with DRAM1 OE cells enhanced tumor lung metastasis (Fig. 7D–F). Meanwhile, we also can observe that tumor cells exhibited muti-macrometastases in the livers with injected DRAM1 OE cells (Fig. 7G, H). In contrast, few or no macrometastases were found in mice’s liver that received control cells (Fig. 7G, H). In parallel, around 3×106 HCCLM3 (M3) cells with luciferase were injected in 5-week-old nude mice (BALB/C-NU), and examined metastasis by bioluminescence after 3 weeks injection (Fig. 7I). The results showed that DRAM1 overexpression increases the incidence of lung metastasis compared with the control group (Fig. 7J, K). These results from the mouse models provide compelling evidence that DRAM1 exerts a vital role in enhancing extravasation in HCC in vivo.
Furthermore, we also applied a well-described zebrafish tumor xenograft model to evaluate the importance of DRAM1 in HCC metastasis in vivo56. 3 dpf zebrafish larvae were injected with ~300 HCCLM3 or MHCC97H cells stably labeled with mCherry or GFP via the Duct of Cuvier (DoC) (Supplementary Fig. 6N). We examined the volume of cells in the injected region shortly after injection and ensured that the fluorescence intensity of cells was similar in each group (Supplementary Fig. 6O, P). In zebrafish larvae, circulating cancer cells can extravasate into the tail region. At 3 days post-injection (dpi), we detected the immigrated cells in the tail region. The results showed that DRAM1 enhanced HCC cell migration (Fig. 7L and M, and Supplementary Fig. 6Q, R). To visually trace the metastatic extravasation of HCC cells, we also employed the zebrafish xenograft model by intravenously (IV) injecting ~100 GFP-labeled 97H cells into 2 dpf Tg(Kdrl:mCherry) zebrafish embryos (with red fluorescent blood vessels)57. We observed that most larvae injected with DRAM1 OE cells had a higher percentage of extravasated cells in tail region. Larvae injected with DRAM1 KO cells had a significantly reduced percentage of extravasated cells in tail region (Fig. 7N, O). Furthermore, we also injected HCCLM3 cells into the hindbrain ventricle of 2 dpf zebrafish embryos (Supplementary Fig. 6S). The results showed that DRAM1 OE significantly increased HCCLM3 cells emigrating from the hindbrain (Supplementary Fig. 6T). In contrast, DRAM1 deficiency led to the opposite effect (Supplementary Fig. 6T). Collectively, our data indicate that DRAM1 promotes the extravasation of HCC both in mouse and zebrafish models.
Defect in DRAM1-mediated autolysosome formation leads to inhibited HCC extravasation
The mechanism by which DRAM1 promotes HCC extravasation is unclear. Based on our observation that DRAM1 promotes the fusion of the autophagosome with lysosomes via enhanced stability of VAMP8, we analyzed the TCGA data. We found that high expression of VAMP8 significantly reduces the survival period of hepatocellular carcinoma (HCC) patients (Supplementary Fig. 7A). We further identified a correlation between the expression levels of these 2 proteins in HCC patients (Supplementary Fig. 7B).
Hence, we speculated that DRAM1 increases metastasis via enhanced autolysosome formation. Firstly, we examined the autophagy level by DRAM1 gain-or-loss function in HepG2 and MHCC97H cells. The results showed that overexpression of DRAM1 promotes autolysosome formation in HCC cells, as indicated with lower p62 and LC3-II levels (Fig. 8A). In contrast, DRAM1 deficiency inhibited autolysosome formation in HCC cells with accumulated p62 and LC3-II (Supplementary Fig. 7C). Next, we detected if DRAM1 promotes VAMP8 stability in HCC cells by CHX degradation assay. The results showed that DRAM1 deficiency decreased the stability of VAMP8 in HepG2 and MHCC97H cells (Fig.8B and Supplementary Fig. 7D). These data demonstrate a correlation between DRAM1 and VAMP8 levels in HCC cells and suggest that DRAM1 may promote metastasis of HCC cells by mediating autolysosome formation.

A Levels of p62 and LC3 protein were examined by WB. B The WB shows the protein stability of VAMP8 in the CHX chase assay at the indicated time points. C Levels of p62 and LC3 protein were examined by WB in HepG2 cells. D Levels of p62 and LC3 protein were examined by WB in ATG7 KO and Control cells. E, F Representative images of cell colonies in transwell assays with the indicated group. Transwell migration assay (Upper) and transwell invasion assay (Down) in HepG2 cells. Representative Figures from three independent experiments are shown. The quantification of migrated cells per field is shown in the bar graphs. Data are shown as means ± SD from three independent experiments. Scale bar: 100 μm. G The representative image shows the immigration of Control and sh-ATG5 HepG2-GFP cells, with or without stable overexpression DRAM1, the images were acquired at 3 dpi at the tail region in WT (AB) zebrafish embryos. The cells were infected with lentivirus encoding GFP and approximately 300 HepG2-GFP cells were injected into the Duct of Cuvier (DoC) at 2 dpf (top image). Micro-metastases developed at 3 dpi (5 dpf) in the tail region (GFP, HepG2 cells). Scale bar: 100 μm. H Quantification of the number of tumor clumps in the tail region at 3 dpi. The number of tumor clumps was calculated in tail region by Image J, n = 9 larvae. I Representative images showing the migration of Control and ATG7 KO 97H-GFP cells, with or without stable overexpression DRAM1, in the hindbrain region at 2 dpi. The white cycle indicates the hindbrain region. Scale bar: 100 μm. (J) Quantification of the number of migrated tumor cells out of the hindbrain region at 2 dpi, n = 6 larvae. Data are shown as means ± SD. Data in (A–E, G–J) are representative of three independent experiments, P-values were calculated by two-tailed Student’s t tests (F, H, J). P-values are indicated in the figure.
To test our hypothesis, we performed knockout and knockdown experiments in HepG2 and MHCC97H cells to ablate ATG5 or ATG7, which are required for autophagosome formation58,59. As expected, deficiency of ATG5 or ATG7 leads to defects in autophagosome formation (Fig. 8C, D). We also observed that DRAM1 lost its function in mediating autophagy in ATG5 or ATG7 deficiency HCC cells (Fig. 8C, D). Autophagy is an essential mechanism for cell survival1. Hence, we examined the effect of DRAM1 on cell proliferation ability in ATG5 or ATG7 deficiency HCC cells. The results showed that DRAM1 promotes the proliferation of HCC cells. However, this promotion ability was abolished in ATG5 or ATG7 deficient cells (Supplementary Fig. 7E–H). We further asked if DRAM1 affects the metastasis in ATG5 or ATG7 deficiency HCC cells in vitro. Therefore, we conducted transwell migration and invasion assays to determine the metastasis ability. The results showed that overexpression of DRAM1 promotes the migration and invasion of HCC cells, and this ability was diminished in ATG5 or ATG7-deficient HCC cells (Fig. 8E, F, Supplementary Fig. 7I, J). These data suggest that DRAM1 promotes metastasis via enhanced autolysosome maturation.
Furthermore, we tested if DRAM1 promotes metastatic extravasation of HCC via enhanced autolysosome formation in vivo in the zebrafish model. The results from DoC and hindbrain injection approaches showed that DRAM1 does not promote extravasation when ATG5 or ATG7 is deficient (Fig. 8G–J). Collectively, these data from cells and the zebrafish model demonstrated that DRAM1 promotes extravasation of HCC via enhanced autolysosome formation.
Discussion
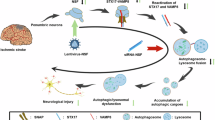
In this study, we identified that DRAM1 acts as an interactive partner of VAMP8 and this interaction is enhanced by the induction of autophagy. This interaction was demonstrated to be involved in regulating autolysosome formation. More specifically, DRAM1 promotes the stability of lysosomal VAMP8 via inhibiting ubiquitination of VAMP8, which results in enhanced assembly of the STX17-SNAP29-VAMP8 complex, eventually contributing to driving the fusion of the autophagosome with lysosomes. We also identified that CHIP is a vital E3 ligase that mediates the ubiquitination of VAMP8, which leads to its proteasome degradation. DRAM1 competes with CHIP for binding to VAMP8 on lysosomes. Therefore, induction of autophagy and DRAM1 hinders the CHIP-mediated ubiquitination of VAMP8 on lysosomes. Moreover, we identified that the Lysine residues 68, 72 and 75 of VAMP8 are required for its ubiquitination by CHIP and, thereby, VAMP8 targeting for proteasome degradation. More importantly, we uncovered that DRAM1 is important for HCC metastasis and extravasation (Fig. 9).

In the absence of DRAM1: DRAM1 deficiency leads to CHIP directly binding with VAMP8, which contributes to ubiquitination and degradation of VAMP8, eventually causing the decreased assembly of STX17-SNAP29-VAMP8 complex and inhibition of the autolysosome formation, which leads to inhibiting the extravasation of HCC cells. In the presence of DRAM1: DRAM1 interacts with VAMP8 on the lysosome membrane and inhibits CHIP binding with VAMP8, which results in reduced ubiquitination of VAMP8 and enhances the stabilization of the STX17-SNAP29-VAMP8 complex, facilitating the autolysosome formation and eventually promotes the extravasation of HCC cells.
DRAM1 interacting partners remain poorly characterized. A direct interaction between DRAM1 and p62, Bax, EPS15, SCAMP3, SLC1A5, and LAT1 have been demonstrated13,60. In this study, we employed MS to demonstrate that DRAM1 interacts with VAMP8. DRAM1 and VAMP8 both colocalize on lysosomal membranes and function in the fusion of autophagosomes with lysosomes11,61. Interestingly, we observed that interaction between DRAM1 and VAMP8 was enhanced by activation of autophagy with EBSS or Torin treatment. This suggests that the interaction of these two proteins has a role in regulating the process of autophagy.
The reported mechanism of DRAM1-triggered autophagy is mainly focused on promoting the generation of autophagy and increasing the fusion between autophagosomes and lysosomes10,60. However, the exact molecular mechanism of how DRAM1 modulates autophagosome-lysosome fusion has not been completely elucidated. Here, we demonstrated that DRAM1 promotes the stability of lysosomal VAMP8 to facilitate the assembly of the STX17-SNAP29-VAMP8 complex. VAMP8 is a component of the autophagic SNARE protein complex. This complex has been previously identified to drive autophagosome-lysosome fusion62,63. VAMP8 phosphorylation contributes to the formation of unfused lysosome clusters on autophagosomes and works as a switch between membrane contact and fusion to control the autophagic flux9,31. Conversely, upon stimulation, dephosphorylation to form new fully zippered SNAREs may resume lysosome-autophagosome fusion to promote autophagy31,61. However, the significance of VAMP8 ubiquitination in autophagy is unknown. We showed that CHIP leads to the ubiquitination of VAMP8 and its subsequent proteasomal degradation, which provides experimental evidence that VAMP8 ubiquitination affects the assembly of the STX17-SNAP29-VAMP8 complex. CHIP has been known as an E3 ubiquitin ligase and regulates the Transcription Factor EB (TFEB) induced autophagy lysosome pathway64. In this study, we demonstrated that CHIP is a vital E3 ubiquitin ligase for modulating the stability of VAMP8, which is responsible for the stability of STX17-SNAP29-VAMP8.
Our data demonstrate that VAMP8 can be targeted for proteasome degradation. However, VAMP8 is typically recycled after mediating the fusion between autophagosomes and lysosomes since the lysosomal membrane components on autolysosomes are recycled through autophagic lysosome reformation (ALR)65. The current study has not investigated whether the interaction between DRAM1 and VAMP8 also affects the efficiency of VAMP8 recycling. However, we speculate that DRAM1 might facilitate the re-using of VAMP8, since DRAM1 protects VAMP8 from ubiquitination.
We adopted a hydrodynamic injection-induced HCC model to demonstrate that DRAM1 promotes the progression of HCC, which is consistent with data extracted from database resources indicating that high levels of DRAM1 are closely associated with shorter overall survival in HCC patients. Afterwards, using a mouse and zebrafish xenograft model, we demonstrated that DRAM1 promotes metastasis of HCC cells. Given the many cellular functions DRAM1 is involved in, it is unsurprising that this autophagy and cell death regulator has been implicated in cancer. DRAM1-mediated autophagy contributes to starvation-induced apoptosis in normal hepatocytes but not in HCC cells, which suggests that the vast majority of cancer-related deaths are due to metastasis rather than primary tumors66. The blood circulation facilitates the spread of tumors to distant tissues and organs28. It has been reported that the extravasated tumor cells have a very short half-life (2–10 min) in the blood circulation67. This causes a low percentage of extravasating cells to arrive successfully in remote areas. Enhanced autophagy is beneficial for the survival of extravasating cancer cells. In agreement, when autophagosome formation is impaired, the extravasating ability of HCC cells is significantly inhibited. Our results partially explain that DRAM1 promotes metastasis via enhanced autolysosome formation. Our study also suggests potential strategies for investigating the mechanism of autophagy-based therapy in hepatocellular cancer.
Autophagy is a double-edged sword and plays different roles in the progression of tumors54,68. DRAM1, a key regulator of autophagy, also shows a dual role across different tumor types and at different progression stages. In non-small cell lung cancer (NSCLC), DRAM1 acts as a tumor suppressor by facilitating the endocytosis and lysosomal degradation of the epidermal growth factor receptor (EGFR)18. This cascade of events ultimately leads to accelerated EGFR degradation, inhibiting tumor growth, migration, and invasion. Similarly, in breast cancer, DRAM1-induced autophagy has been shown to promote cell apoptosis, thereby exerting a negative impact on cancer cell survival69. However, the role of DRAM1 is not universally suppressive across all cancers. For example, un glioblastoma multiforme (GBM), DRAM1 promotes the migration and invasion of GBM stem cells through p62-mediated autophagy20. In agreement, we found that DRAM1 promotes the metastatic extravasation of HCC both in mouse and zebrafish models. These findings suggest that DRAM1 can act as a metastasis promoter in certain cancers. Future research should focus on elucidating the specific contexts and mechanisms that drive the dual roles of DRAM1 in different cancers. This may involve identifying biomarkers that can predict the predominant function of DRAM1 in individual patients or exploring targeted therapies that can selectively modulate DRAM1 activity based on the specific tumor context. This detailed understanding will be crucial for the development of effective and safe cancer therapies targeting DRAM1.
Collectively, our findings identify and characterize the physiological function of DRAM1 mediating stability of lysosomal VAMP8 for regulating autophagosome fusion with lysosomes. Additionally, given that DRAM1 has been implicated in the increased metastatic ability of HCC, it might represent a suitable target for restricting HCC dissemination.
Methods
Antibodies and reagents
Commercially available antibodies for Western blot, co-immunoprecipitation, and immunostaining are listed in Supplementary Table 1. Lipofectamine 2000 transfection reagent (Thermo Fisher, 11668019) was used for transient transfection, according to the manufacturer’s instructions. Cells were analyzed 36-48 hours after transfection.
Immunoprecipitation and Western blot
After transfection or treatment, whole cell extracts were lysed in RIPA lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Triton X-100 (Sangon Biotech, 9002-93-1), and 1 mM EDTA), supplemented with a proteinase inhibitor cocktail (Biomake, B14001). The lysates were centrifuged at 12,000 × g for 30 min at 4 °C, and the supernatants were incubated overnight with primary antibodies and protein A/G beads (Bimake, B23202). The beads were washed at least 3 times in RIPA lysis buffer. The immunoprecipitate was eluted from the beads by adding and boiling in 1 × SDS loading buffer (100 mM Tris-HCl, pH 6.8, 4% [wt/vol] SDS, 200 mM dithiothreitol, 0.2% [wt/vol] bromophenol blue, and 20% [vol/vol] glycerol) for 10 min at 98 °C. Samples were then subjected to SDS-PAGE and Western blotting. The proteins were transferred to 0.45 mm PVDF membranes (Millipore, IPFL85R). Transferred membranes were blocked in TBST 5% skimmed milk (Coolaber, CN7861), TBST (TBST, 1XTBS contains 0.1% Tween 20) buffer or TBST 3% BSA (Sangon Biotech, A600903-0005) for phospho-specific antibodies for 2 h. Primary antibodies were diluted in TBST buffer and incubated for 2 h at room temperature or 4 °C overnight. Secondary antibodies were also diluted in TBST buffer with proper dilution. Restore stripping buffer (Thermo Fhisher, 46430) was used for stripping antibodies from membranes. The membranes were visualized with SuperSignal West Pico Chemiluminescent substrate (Thermo Fisher, 34579). Western blotting images were captured using the Tanon image system (Tanon, 1600). Band intensities were quantified by densitometric analysis using FIJI/Image and values were normalized against GAPDH as a loading Control.
Zebrafish lines and handling of embryos
Zebrafish (Danio rerio) were handled in compliance with the Hubei University of Technology animal welfare regulations and maintained according to standard protocols (http://ZFIN.org). Embryos were grown at 28.5 °C in egg water (60 μg/ml Instant Ocean sea salts). The zebrafish lines used in this study are listed in Supplementary Table 2. Larvae used for stereo fluorescence or confocal imaging were kept under anesthesia in egg water containing 200 μg/ml tricaine (3-aminobenzoic acid ethyl ester, Sigma-Aldrich).
Generation of Dram1 overexpressing transgenic zebrafish line
For the generation of the Dram1 overexpressing transgenic zebrafish line, the Tol2 transposase mRNA was transcribed using the SP6 transcription Kit (AM1340, Thermo Fisher) from a Tol2 plasmid (31831, Addgene). A mixture of Tol2 transposase mRNA (100 pg/embryo) and a plasmid containing the CMV-mCherry-Dram1 construct flanked by the Tol2 recombination sites (100 pg/embryo) was injected into one-cell stage embryos. The effect of transgenesis was examined by fluorescent microscopy or by PCR.
GST-protein purification
GST-VAMP8 were expressed in Escherichia coli BL21 (Tsingke, TSV-A09). Bacteria were induced with 0.1 mM IPTG (Sangon Biotech, A600168-0025) for 16 h at 26 °C to induce protein expression. The recombinant proteins were purified using glutathione-Sepharose 4B beads (GE Healthcare Life Science, 17-0756-01), eluted with glutathione (Beyotime, S0073) at 4 °C for 4 h to release the proteins.
Transmission electron microscopy and image quantification
The 6 dpf zebrafish larvae were fixed overnight in PBS buffer (pH 7.4) containing 2.5% glutaraldehyde at 4 °C, post-fixed in 1% OsO4 at 4 °C for 2.5 h, dehydrated with graded ethanol and acetone, infiltrated in a mixture of acetone and epoxy resin (1:1 for 1 h, and then 1:2 for 8 h) and finally embedded in SPI-PON 812 at 60 °C for 48 h. Semithin sections (1.5 µm) and ultrathin sections (72 nm) were obtained using a Leica (Wetzlar, Germany) EM UC7 ultramicrotome. The semithin sections were stained with methylene blue and observed using a conventional light microscope. The ultrathin sections were observed and photographed using an HT7700 transmission electron microscope (Hitachi High-Tech, Tokyo, Japan) after being stained with uranyl acetate and lead citrate.
Mass spectrometry analysis
The GFP-VAMP8 and GFP-Vector immunoprecipitated in starved and control conditions were collected from three biological replicates (n = 3), and were examined by WB, then rest of the sample was run on 8% acrylamide gels for 50 min with 120 V, stained with Coommassie Blue staining reagent, whereupon an area of between 25 and 50 kDa (roughly 1 cm × 1.5 cm) region’s Coomassie-stained was excised and destained and subjected to in-gel proteolytic digestion with trypsin (Promega) for 20 h at 37 °C. Then, the peptides were detected by tandem mass spectrometry (MS/MS) in Q ExactiveTM (Thermo) coupled online to the UPLC. Intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using NCE setting as 27; ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 1E4 in the MS survey scan with 30.0 s dynamic exclusion. The electrospray voltage applied was 2.0 kV. Automatic gain control (AGC) was used to prevent overfilling of the ion trap; 1E5 ions were accumulated for the generation of MS/MS spectra. For MS scans, the m/z scan range was 350 to 1800 m/z. Fixed first mass was set as 100 m/z. Protein identification were performed with MASCOT (http://www.matrixscience.com/software) by searching Uniprot Aedis Aegypti. The Mass Spectrometry was performed and analyzed by YanXin Wuhan Protein Technology Co. Ltd (www.bioyx.com). The processed mass spectrometry results is provided in Supplementary Data 1.
In vitro autophagosome-lysosome fusion assay and image quantification
GFP-LC3 and mCherry-LAMP1 constructs were overexpressed separately in either 293 T or DRAM1 KO 293 T cells with EBSS treatment. Cells were cracked 36 h after transfection using 1 ml disposable syringes in homogenization buffer (250 mM sucrose [Sangon Biotech, A502792], 20 mM HEPES-KOH, pH 7.4) followed by centrifugation at 1200 g for 10 min. Post-nuclear supernatant (PNS) was prepared. The PNS from GFP-LC3 expressing cells containing GFP-LC3-positive autophagosomes was mixed with the PNS from mCherry-LAMP1 expressing cells containing mCherry-LAMP1-positive lysosomes in the presence of the ATP-regeneration system (10 mM creatine phosphate [Sangon Biotech, A610821], 1 mM ATP [Selleck, S1985], 20 μg/ml creatine kinase [BioVision Technologies, P1301-1]). The samples were then incubated at 37 °C by gently shaking for 2 h. After incubation, all the samples were centrifuged at 100,000 g for 15 min and immobilized on glass coverslips for confocal microscopy analysis. Acquired images were analyzed by FIJI/Image J to count the number of GFP-Lc3 punctae and double-labelled vesicles. More than 5000 GFP-LC3 punctae were analyzed.
Immunofluorescence and fluorescence microscopy in vitro
Cells were fixed with 3.7% paraformaldehyde (PFA; Sigma Aldrich, 441244) or 3.7% formaldehyde (Merck, 1040031000) or 100% methanol, washed and blocked with blocking buffer (PBS, 1% bovine serum albumin, 0.1% saponin [Sigma-Aldrich, S4521]). Primary and secondary antibodies were diluted in the blocking buffer and incubated for 1 h. Nuclei were stained with DAPI during the incubation with the secondary antibody for automated image acquisition and fluorescence microscopy, respectively. To quantify LysoTracker staining within GFP-Lc3 punctae, the mean intensity of LysoTracker overlapping with GFP-LC3 signal was measured using FIJI/ImageJ quantification tools.
Drug treatments
The following drugs were used: Chloroquine diphosphate salt (CQ, 10 μM for cells or 20 μM for zebrafish larvae, Sangon Biotech, A506569), Rapamycin (10 nM; Selleck, S1039), Cycloheximide (CHX, 20 μg/mL, MCE, HY-12320), Torin 1 (100 nM, MCE, HY-13003). Absolute dimethyl sulfoxide (DMSO, Sangon Biotech, A100231) was used as a solvent to make stock solutions, which were typically diluted at 1:1000 to obtain the final desired concentration. Drugs were administered via egg water or culture medium to zebrafish embryos and human cells, respectively.
q-RT-PCR
Total RNA was extracted from different stages of zebrafish or cells using Trizol reagent (ThermoFisher, 15,596,026) according to the manufacturer’s instructions and purified with Rneasy Min Elute Clean up kit (TIANGEN, DP412). RNAs were quantified using a NanoDrop 2000c instrument (Thermo Fisher). cDNA was reverse transcribed using a HiScript cDNA synthesis kit (Vazyme, R323-01). Quantitative PCR was performed using the Universal SYBR-qPCR Master Mix (Vazyme, Q511-02). The primers used in this study are listed in Supplementary Table 3.
Tandem RFP-GFP-LC3 in HeLa cells
HeLa cells were transfected with mRFP-GFP-LC3 or co-transfected with the indicated constructs for 48 h. Then, cells were fixed with 4% PFA (dissolved in PBS) at 37 °C for 15 min, sealed with nail polish and observed under a Leica SP8 (Leica) confocal fluorescent microscope equipped with a 63×/1.42 numerical aperture (NA) objective lens. Cells were randomly selected, and the numbers of GFP punctae and mRFP punctae were counted manually in a two-blinded manner. The number of GFP+ RFP+ punctae and GFP-RFP+ punctae were calculated based on the numbers of GFP punctae and RFP punctae.
Confocal laser scanning microscopy and image quantification
Fixed or live embryos were mounted with 1.5% low melting agarose (140727, SERVA) and imaged using a Leica SPE8 confocal microscope. For quantification of basal autophagy, fixed 6 dpf larvae were imaged by confocal microscopy with a 63x oil immersion objective (NA 1.2) in a predefined region of the tail fin to detect GFP-Lc3-positive vesicles. The number of GFP-Lc3 vesicles per condition was quantified using Fiji/ImageJ software. GFP-Lc3 fluorescence intensity was assessed using FIJI/ ImageJ quantification tools.
LysoTracker staining
Embryos were incubated for 1 h in egg water with 10 μM LysoTracker Red (Solarbio, L8010) and rinsed several times before imaging.
Lysosome immunoprecipitation (Lyso-IP)
Lysosomes were isolated as previously described41. In brief, 4 × 106 293 T cells stably expressed TMEM192-HA, compared to no TMEM192-HA as a negative Control. Cells were washed 3 times with PBS and scraped in 1 mL of ice-cold KPBS buffer (136 mM KCl, 10 mM KH2PO4, 2 mM EDTA, pH 7.25) supplemented with Complete EDTA-free Protease inhibitor. Cells were centrifuged at 1000 x g for 2 min at 4 °C, resuspended in 1 mL ice-cold KPBS, and homogenized by 30 strokes with a 2 mL homogenizer. Cell homogenate was centrifuged at 1000 x g for 2 min at 4 °C, 100 μL of supernatant was collected (whole cell lysate), and the remaining supernatant was incubated with 100 μL of Pierce Anti-HA magnetic beads (Thermo Fisher Scientific, 88837) for 5 min on an end-over-end rotator at 40 °C. Beads were collected on the side of the tube with a magnet and washed five times with KPBS and five times with KPBS+ buffer (170 mM KCl, 10 mM KH2PO4, 2 mM EDTA, pH 7.25) supplemented with Complete EDTA-free protease inhibitor, followed by two gentle washes with KPBS+ on the vortex. The beads were collected with a magnet, the supernatant was discarded, and the beads were resuspended in 100 μL of KPBS buffer. The purity of isolated lysosomes was validated by immunoblotting of lysosome markers (TMEM192-HA, LAMP1, CTSD) and non-lysosome markers (CALR, ER; Golgi, GORASP2; TOM20, mitochondria).
Hydrodynamic tail vein Myc/AKT, N-RAS/AKT HCC mouse model
Eight-week-old male WT C57BL/6 mice were used and the procedure was performed as described previously48. In brief, 15 mg of plasmids encoding human AKT1 (myristylated AKT1 or myr-AKT1) and human neuroblastoma Ras viral oncogene homolog (N-RasV12) or mouse Myelocytomatosis oncogene (Myc) and human AKT1 (myristylated AKT1 or myr-AKT1along with sleeping beauty (SB) transposase with in a ratio of 10:1 were diluted in 2 mL saline (0.9% NaCl), filtered through 0.22 mm filter and injected into the lateral tail vein of C57BL/6 mice in 5–7 s. The constructs used in this study showed the long-term expression of genes via hydrodynamic injection. To examine the role of DRAM1 in liver carcinogenesis using this model, the pT3-DRAM1 were constructed by cloning, pT3-sh-Dram1 [shRNA targeting DRAM1 regions conserved in both human and mouse (sh1: CCAGAGTGTCACTCTAAGGAT, sh2:TGATAAACTTCTCTGCATTTC) will also be mixed and injected together with the AKT1 and N-Ras or AKT1 and My and SB transposase. The study protocol was approved by and performed in accordance with the Committee of the Use of Live Animals in Teaching and Research at the Hubei University of Technology. The maximal tumor burden was permitted by HBUT and the maximal tumor burden in the experiments was not exceeded the standard. Hydrodynamic transfection of the oncogenes into the mouse liver resulted in liver tumor formation by 4 weeks post-injection, and with lethality occurring after 8 weeks post-injection. The liver enlargement was monitored through weekly Bioluminescence Imaging (Caliper Life Sciences, Alameda, CA, USA).
Lung metastasis assays in nude mice
Four-week-old female BALB/c-nu nude mice were purchased from the Vital River Laboratory (Beijing). Single-cell suspensions of HCCLM3-Luc or MHCC97H cells (3 × 106 per 100 μL PBS) were injected into the tail vein of 5-week-old male BALB/c mice. The development of metastasis for HCCLM3-Luc cells was monitored by bioluminescent reporter imaging. All mice were euthanized by cervical dislocation and lung metastases were analyzed. Pulmonary tumor nodules were counted under a stereoscopic microscope.
Organ harvesting, tissue processing, and H&E staining
After euthanizing mice, their livers or lungs were removed and rinsed in PBS. Samples collected from the livers or lungs were fixed overnight in freshly prepared neutral-buffered formalin. Fixed tissue samples were embedded in paraffin. Five-micron sections were placed on slides and stained with hematoxylin and eosin (H&E) to observe cell morphology.
Bioluminescence Imaging
The abdominal area of skin was depilated using a depilatory cream 1 day before imaging. On the day of imaging, mice were intraperitoneally injected with D-luciferin (150 mg/kg) and placed in a light-tight mouse imaging chamber following anesthesia. A photographic (gray-scale) reference image was obtained 10 min after the administration of D-luciferin, and bioluminescence images were captured immediately thereafter. Images were obtained with a CCD camera, using the IVIS Imaging System (Caliper Life Sciences, Alameda, CA, USA). Regions of interest were drawn in the abdominal area and total counts (photons) in all areas were summed. The signal intensities of each defined region of interest were quantified as photon count rate per unit body area per unit solid angle subtended by the detector (photons/s/cm2/steradian).
Transwell migration and invasion assays
Cell migration and invasion were performed as previously described70. For migration assay, 5 × 103 HepG2 or MHCC97H cells expressing different sgRNAs were plated in 24-well plate inserts (8.0 μm pore polycarbonate membrane, Costar). Cells in the upper part of the transwells were removed with a cotton swab; migrated cells were fixed in paraformaldehyde 4% and stained with crystal violet 0.5%. Filters were photographed and the total number of cells were counted. Every experiment was repeated at least three times independently. For the invasion assay, 5 × 103 HepG2 or MHCC97H cells were seeded in a Matrigel (354234, BD Biocoat)-coated upper chamber. After 24–48 h, invading cells were fixed, stained according to the manufacturer’s instructions and counted. These experiments were performed three times.
Vector construction
DNA fragments encoding DRAM1, VAMP8, CHIP and RNF126 amplified by PCR were gifts from Prof. Jiahuai Han (Xiamen University, China). These were cloned into the pcDNA3.1(Invitrogen, V79020), p3× FLAG-CMV-7.1 (Sigma-Aldrich, E7533), pEGFP-N3 (Clontech, 6080–1), pmCherry-N1 (Clontech, 632,523) and pcDNA3.0-3×HA (Miaolingbio, P9394) vectors for the different experiments. The constructs of Flag-VAMP8, GFP-VAMP8, and Flag-STX17 were a gift from Prof. Junlin Teng (Peking University). All complete constructs were confirmed by Sanger sequencing.
Cell culture, siRNAs, and transfection
All cell lines were originally purchased from the China Center for Type Culture Collection (CCTCC, Wuhan, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, SH30022.01) containing 10% heat-inactivated fetal bovine serum (Biological Industries, 04-001-1ACS), 100 units/ml penicillin, 100 μg/ml streptomycin (Biosharp, BL505A) and 4 mM L-glutamine at 37 °C with 5% CO2 in a humidified atmosphere. The medium was replaced every 2–3 days, and the cells were sub-cultured and used for experiments at 80–90% confluence. DNA transfection was performed using GenJetVer.II (SignaGen, SL100489) and RNA interference was conducted with lipofectamine 2000 (Invitrogen, 11,668,019) according to the manufacturer’s instructions. siRNAs used were: DRAM1#1 5ʹ-CCACAGAAAUCAAUGGUGATT-3ʹ, DRAM1#2 5ʹ-CUCCCGUAUAUCAGUGAUAUU-3ʹ, CHIP#1 5ʹ-GGAGAUGGAGAGCUAUGAU-3ʹ, CHIP#2 5ʹ-CUGUGAAGGCGCACUUCUU-3ʹ, VAMP8#1 5ʹ- VAMP8#2 5ʹ-CAGGGACAACCCUCCAUAAAU-3ʹ, these oligos were purchased from Genscript (China, Nanjing). For knockdown experiments, 293 T cells or HeLa cells were transfected with Lipofectamine 2000 (Invitrogen). Briefly, cells were seeded and grown to 60% confluency; siRNA and Lipofectamine 2000 reagent were diluted into Opti-MEM medium and mixed at a 1:1 ratio. The mixture was incubated for 20 min at room temperature and added to the cells. Starvation was carried out by switching the culture medium from complete medium to low glucose (1000 mg/L) DMEM medium, without fetal bovine serum or EBSS (Hyclone, SH30024.01).
Generation of knockout cell lines
To generate DRAM1 or ATG7 knockout cell lines, two pairs of short guide RNAs (sgRNA; sgRNA1 and 2) targeting exon1 of the human DRAM1 or exon1 of human ATG7 were cloned into pLenticrisprV2 (Addgene5296) to obtain lenti-DRAM1-sgRNA-Cas9 and lenti-ATG7-sgRNA-Cas9 constructs, following the depositor’s protocol. The primer sequences for the CRISPR plasmids are as follows: DRAM1 KO1 Forward: 5’-CACCGACGTGGTCGCCGTGCTCTCC-3’, KO1 Reverse: 5’-AAACGGAGAGCACGGCGACCACGTC-3’; DRAM1 KO2 Forward: 5’-CACCGGTTGACGTGCCCGGAGAGCA-3’, KO2 Reverse: 5’-AAACTGCTCTCCGGGCACGTCAACC-3’. ATG7 KO1 Forward: 5’-CACCGAGAAGAAGCTGAACGAGTAT-3’, KO1 Reverse: 5’-AAACTCTTCTTCGACTTGCTCATAC-3’. KO2 Forward:5’-CACCGCAATGTAGCTAGGACGTTGA-3’, KO2 Reverse: 5’-AAACGTTACATCGATCCTGCAACTC-3’.
The pooled knockout cells were generated by transduction with lentiviruses obtained abovefor 48 h, then the cells were selected by puromycin (2 μg/mL) for 1 weeks. To isolate single clonal DRAM1 or ATG7 knockout cells, the pooled DRAM1 or ATG7 KO cells were subcloned using limiting dilutions in 96-well plates. After clonal expansion, the cells were stored at −80 °C and Western blot assays were used to verify the knockout efficiency of DRAM1 in HeLa, 293 T, HepG2 and HCCLM3 cells, and ATG7 in MHCC97H cells.
Generation of stable knockdown cell Lines
Lentiviruses were produced by transfecting HEK293T cells with shRNA-targeting plasmids and the helper plasmids pSPAX2 (gag/pol), and pMD2G. The cell supernatants were harvested 48 h after transfection and were either used to infect cells or stored at 80 °C. To obtain stable knockdown cell lines, cells were infected at low confluence (20%) for 24 h with lentiviral supernatants diluted 1:1 with normal culture medium in the presence of 5 ng/mL of polybrene (Sigma). Forty-eight hours after infection, cells were placed under puromycin selection for one week and then passaged before use. Puromycin was used at 2 mg/mL to maintain 293 T, HepG2 and MHCC97H derivatives. Lentiviral-expressed sh-RNAs were obtained from Sigma (Mission shRNA). Typically, mixed sh-RNAs targeted were identified and tested, and the effect of knockdowns were verified by WB analysis. The sequence of target genes were listed in the supplementary Table 4.
Injection of HCC tumor cells into zebrafish
Either AB (ZFIN ID: ZDB-GENO-960809) or Tg(Kdrl:mCherry) (ZFIN ID: ZDB-TGCONSTRCT-110127-23) fish were bred prior to the start of an experiment, and Petri dishes containing larvae were cleaned every day, removing dead and malformed larvae after harvesting up to 2 days post-fertilization. Approximately 300 cells labeled with either mCherry or GFP were injected into the Duct of Cuvier (DoC) of 2 dpf zebrafish embryos using a Pneumatic Picopump and a manipulator (WPI). The injected embryos were further maintained in a 34 °C incubator until the end of the experiments. Images were acquired with a Leica M165 FC stereo fluorescent microscope.
Statistics and reproducibility
All experimental data (Mean ± SD) was analyzed using unpaired, two-tailed t-tests for comparisons between two groups and one-way ANOVA with Tukey’s multiple comparisons post hoc test (adjusted P-values for multiple comparisons) for comparisons between more than two groups. The incidence of lung metastasis was analyzed using two-tailed Chi-square (ns, no significant difference; *p < 0.05; **p < 0.01; ***p < 0.001). n values and P values are indicated in the figure. Purposeful statistical subjects varied across experiments, and the cell experiments was independently repeated three times with the similar results, the zebrafish and mouse experiments were done at least two independent times with the similar results. No data were excluded from the analysis. Source data are provided as a Source Data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the supplementary materials. Survival analyses comparing hepatocellular carcinoma (HCC) patients with low vs. high DRAM1 or VAMP8 expression were performed using RNA-seq and clinical data from The Cancer Genome Atlas (TCGA) liver hepatocellular carcinoma cohort (TCGA-LIHC). Processed data are available via the Genomic Data Commons (GDC) portal (https://portal.gdc.cancer.gov/). The Pearson’s correlation analysis between DRAM1 and VAMP8 expression was performed using bulk RNA-seq data from 364 HCC patients, obtained from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/).The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the iProX partner repository71,72 with the dataset identifier (PXD064129). The remaining data are available within the article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Mizushima, N. & Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 383, 1564–1576 (2020).
Tian, X., Teng, J. & Chen, J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 17, 2680–2688 (2021).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012).
Zhang, R. et al. RUNDC1 inhibits autolysosome formation and survival of zebrafish via clasping ATG14-STX17-SNAP29 complex. Cell Death Differ. 30, 2231–2248 (2023).
Moreau, K., Renna, M. & Rubinsztein, D. C. Connections between SNAREs and autophagy. Trends Biochem Sci. 38, 57–63 (2013).
Stroupe, C. Autophagy: cells SNARE selves. Curr. Biol. 21, R697–R699 (2011).
Lorincz, P. & Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 432, 2462–2482 (2020).
Chen, Q. et al. Prefused lysosomes cluster on autophagosomes regulated by VAMP8. Cell Death Dis. 12, 939 (2021).
Crighton, D. et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134 (2006).
Zhang, X. D., Qi, L., Wu, J. C. & Qin, Z. H. DRAM1 regulates autophagy flux through lysosomes. PLoS One 8, e63245 (2013).
Mah, L. Y., O’Prey, J., Baudot, A. D., Hoekstra, A. & Ryan, K. M. DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 8, 18–28 (2012).
Beaumatin, F. et al. mTORC1 Activation Requires DRAM-1 by Facilitating Lysosomal Amino Acid Efflux. Mol. Cell 76, 163–76 e8 (2019).
Wang, X. et al. The p53 target DRAM1 modulates calcium homeostasis and ER stress by promoting contact between lysosomes and the ER through STIM1. Proc. Natl Acad. Sci. USA 121, 1091–6490 (2024). (Electronic)).
Wu, X. et al. Increased expression of DRAM1 confers myocardial protection against ischemia via restoring autophagy flux. J. Mol. Cell. Cardiol. 124, 70–82 (2018).
Lu, T. et al. DRAM1 regulates autophagy and cell proliferation via inhibition of the phosphoinositide 3-kinase-Akt-mTOR-ribosomal protein S6 pathway. Cell Commun. Signal 17, 28 (2019).
Zhang, R. et al. Deficiency in the autophagy modulator Dram1 exacerbates pyroptotic cell death of Mycobacteria-infected macrophages. Cell Death Dis. 11, 277 (2020).
Geng, J. et al. DRAM1 plays a tumor suppressor role in NSCLC cells by promoting lysosomal degradation of EGFR. Cell Death Dis. 11, 768 (2020).
Feng, Q. et al. DRAM1 plays a tumor suppressor role in clear cell renal cell carcinoma through modulating Akt signaling. Acta Histochemica 124, 151874 (2022).
Galavotti, S. et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 32, 699–712 (2013).
Wu, X. et al. DRAM1 enhances the proliferation and metastasis of gastric cancer through the PI3K/AKT/mTOR signaling pathway and energy metabolism. Sci. Rep.15, (2025).
Xin, Y., Jiang, Q., Liu, C. & Qiu, J. Plumbagin has an inhibitory effect on the growth of TSCC PDX model and it enhances the anticancer efficacy of cisplatin. AGING 15, 1945–4589 (2023).
Chen, C. et al. DRAM1 regulates the migration and invasion of hepatoblastoma cells via autophagy‑EMT pathway. Oncol. Lett. 16, 2427–2433 (2018).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers.7, (2021).
Chambers, A. F., Groom, A. C. & MacDonald, I. C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572 (2002).
Lin, D. et al. Circulating tumor cells: biology and clinical significance. Sig. Transduct. Target. Ther.6, (2021).
Mowers, E. E., Sharifi, M. N. & Macleod, K. F. Autophagy in cancer metastasis. Oncogene 36, 1619–1630 (2016).
Hosseini, H. et al. Early dissemination seeds metastasis in breast cancer. Nature 540, 552–558 (2016).
Brown, Z. J., et al. Management of Hepatocellular Carcinoma: A Review. AMA Surg 158, 2168–6262 (2023).
Hashemi, M. et al. Targeting and regulation of autophagy in hepatocellular carcinoma: revisiting the molecular interactions and mechanisms for new therapy approaches. Cell Commun. Signal. 21, (2023).
Huang, H. et al. mTOR-mediated phosphorylation of VAMP8 and SCFD1 regulates autophagosome maturation. Nat. Commun. 12, 6622 (2021).
Fleming, A. & Rubinsztein, D. C. Zebrafish as a model to understand autophagy and its role in neurological disease. Biochim Biophys. Acta 1812, 520–526 (2011).
Zhou, C. et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 8, 1215–1226 (2012).
Shen, Q. et al. Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy. 1–13 (2020).
Zhang, X. et al. GRASP55 Senses Glucose Deprivation through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev. Cell 45, 245–61 e6 (2018).
Masselink, W. Crispants take the spotlight. Lab Anim. 50, 95–96 (2021).
van der Vaart, M. et al. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense [corrected]. Cell Host Microbe 15, 753–767 (2014).
Nakamura, S. & Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 130, 1209–1216 (2017).
Diao, J., Yip, C. K. & Zhong, Q. Molecular structures and function of the autophagosome-lysosome fusion machinery. Autophagy Rep. 3, (2024).
Guo, B. et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 16, 1215–1226 (2014).
Abu-Remaileh, M. et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017).
Laqtom, N. N. Studying lysosomal function and dysfunction using LysoIP. Nat. Rev. Mol. Cell Biol. 24, 773 (2023).
Shilatifard, A. & Ben-Sahra, I. Purifying and profiling lysosomes to expand understanding of lysosomal dysfunction–associated diseases. J. Clin. Investig.135, (2025).
Fernandes, S. A. et al. Spatial and functional separation of mTORC1 signalling in response to different amino acid sources. Nat. Cell Biol. 26, 1918–1933 (2024).
Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu Rev. Biochem 86, 123–128 (2017).
Hör, S., Ziv, T., Admon, A. & Lehner, P. J. Stable Isotope Labeling by Amino Acids in Cell Culture and Differential Plasma Membrane Proteome Quantitation Identify New Substrates for the MARCH9 Transmembrane E3 Ligase. Mol. Cell. Proteom. 8, 1959–1971 (2009).
Yang, Y. et al. TRIM27 cooperates with STK38L to inhibit ULK1-mediated autophagy and promote tumorigenesis. EMBO J. 41, (2022).
Chen, X. & Calvisi, D. F. Hydrodynamic Transfection for Generation of Novel Mouse Models for Liver Cancer Research. Am. J. Pathol. 184, 912–923 (2014).
Xu, Z. et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 70, 1600–1613 (2019).
Kim, M. J. & Ahituv, N. The Hydrodynamic Tail Vein Assay as a Tool for the Study of Liver Promoters and Enhancers. Pharmacogenomics. Methods Mol. Biol. 1015, 279–289 (2013).
Arzumanian, V. A., Kiseleva, O. I. & Poverennaya, E. V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 22, (2021).
Tian, J. et al. New human hepatocellular carcinoma (HCC) cell line with highly metastatic potential (MHCC97) and its expressions of the factors associated with metastasis. Br. J. Cancer 81, 814–821 (1999).
Justus, C. R., Marie, M. A., Sanderlin, E. J. & Yang, L. V. Transwell In Vitro Cell Migration and Invasion Assays. Methods Mol. Biol. 2644, 1940–6029 (2023).
Debnath, J., Gammoh, N. & Ryan, K. M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 24, 560–575 (2023).
Speak, A. O. et al. A high-throughput in vivo screening method in the mouse for identifying regulators of metastatic colonization. Nat. Protoc. 12, 2465–2477 (2017).
He, S. et al. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J. Pathol. 227, 431–445 (2012).
Lawson, N. D. & Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307–318 (2002).
Komatsu, M. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 (2005).
Nishimura, T. & Tooze, S. A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov.6, (2020).
Guan, J. J. et al. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 6, e1624 (2015).
Wang, L. & Diao, J. VAMP8 phosphorylation regulates lysosome dynamics during autophagy. Autophagy Rep. 1, 79–82 (2022).
Jian, F. et al. The STX17-SNAP47-VAMP7/VAMP8 complex is the default SNARE complex mediating autophagosome–lysosome fusion. Cell Res. 34, 151–168 (2024).
Zheng, D. et al. Human YKT6 forms priming complex with STX17 and SNAP29 to facilitate autophagosome-lysosome fusion. Cell Rep. 43, (2024).
Ballinger, C. A. et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. (0270-7306 (Print)).
Zhou, CA-O. et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. (1476–4679 (Electronic)).
Gupta, G. P. & Massague, J. Cancer metastasis: building a framework. Cell 127, 679–695 (2006).
Follain, G. et al. Hemodynamic Forces Tune the Arrest, Adhesion, and Extravasation of Circulating Tumor Cells. Dev. Cell 45, 33–52 e12 (2018).
Mulcahy Levy, J. M. & Thorburn, A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 27, 843–857 (2019).
Cui, L. I. et al. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol. Rep. 35, 3639–3647 (2016).
Zhou, C. et al. B-lymphoid tyrosine kinase-mediated FAM83A phosphorylation elevates pancreatic tumorigenesis through interacting with β-catenin. Sig. Transduct. Target. Ther.8, (2023).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2022).
Acknowledgements
We are grateful to Prof. Daniel Klionsky and Prof. Annemarie Meijer for sharing the GFP-Lc3 transgenic zebrafish line. We also thank Dr. Quanchi Chen for his kind assistance with the zebrafish tumor injection. We sincerely thank the Hubei University of Technology for financial and equipment support for this research. This work was supported by the National Natural Science Foundation of China (82273970 to J.F.T., 32270768 to C.F.Z., 82370715 to X.Z.C., 32125011 to Z.Y.S.). Wuhan Science and Technology Project (2023020201020376 to R.Z.). The Hubei Province Science Foundation (2024AFB765 to R.Z.). The Innovation Group Project of Hubei Province (2023AFA026 to J.F.T.) and the National Key R&D Program of China (2023YFC2507900 to J.F.T.). The Key Cultivation Project of Hubei Province for Science and Technology (2024DJA037 to J.F.T.). The National Natural Science Foundation of Hubei (2025AFA085 to C.F.Z.).
Author information
Authors and Affiliations
Contributions
R.Z., X.Z., and H.B. performed molecular biology and zebrafish experiments and wrote the main manuscript. J.T., C.Z. and Z.S. designed the whole project and supervised all experiments. R.Z., X.Z. and H.B. conducted all experiments and analyzed the data. Q.C., X.Y., S.L., V.T., C.Y., Y.W., S.M., X.H., Y.Y., H.T., X.C., S.C.L., H.L., S.X., D.G., Q.Z. and X.Z.C.; contributed with critical feedback on the presented work. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thierry Galli, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, R., Zhang, X., Bai, H. et al. DRAM1 promotes the stability of lysosomal VAMP8 to enhance autolysosome formation and facilitates the extravasation. Nat Commun 16, 5826 (2025). https://doi.org/10.1038/s41467-025-60887-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60887-y
This article is cited by
-
SNX-3 confers lysosomal fusion-competence to sustain basal autophagy
Cellular and Molecular Life Sciences (2026)
