
Abstract
Rehmannia chingii (2n = 2x = 28) is an important folk medicinal plant with high therapeutic value, particularly due to its richness in iridoid glycosides. However, research on its evolution and gene functional identification has been hindered by the lack of a high-quality genome. Here, we present the 1.169 Gb telomere-to-telomere (T2T) genome sequence of R. chingii. Phylogenetic analysis confirms that Rehmannia belongs to the Orobanchaceae family. We find that structural genes of the 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway and the iridoid pathway are predominantly expressed in R. chingii leaves. Further analyses reveal a cytochrome P450 gene cluster localized on chromosome 8, and identify RcCYP72H7 within this cluster as an aucubin epoxidase, capable of catalyzing aucubin epoxidation to form catalpol. The genome offers valuable resources for studying iridoid glycoside biosynthesis and the evolutionary history of Rehmannia, and will help to faciliate genetic improvement of R. chingii for pharmaceutical and health-related applications.
Similar content being viewed by others
Introduction
The Rehmannia chingii plant has been adopted as a folk medicine and health food for long time for treating hematemesis, bleeding, sore throat, and empyrosis1. The genus Rehmannia consists of seven species: R. chingii, R. glutinosa, R. piasezkii, R. henryi, R. solanifolia, R. chrysantha, and R. elata, which are primarily distributed in China2,3. Among them, R. glutinosa, R. solanifolia, and R. chrysantha are tetraploids (2n = 4x = 56), whereas R. chingii, R. piasezki, R. elata, and R. henryi are diploids (2x = 28). Previous study has suggested that R. chingii may be one of the parental species of R. glutinosa4.
Chemical studies have revealed that the roots of Rehmannia plants are rich in iridoid glycosides (including catalpol)1,5, phenylethanol glycosides6,7, ionone glycosides8,9, and saccharides10. Iridoid glycosides are the most abundant monoterpenoid components in Rehmannia11, with catalpol being the predominant iridoid glycoside. Catalpol exhibits several pharmacological properties, including neuroprotective, anti-inflammatory, anti-apoptotic, antitumor, antioxidant, and antifibrotic effects12,13,14. It has been selected as an indicator for evaluating the quality of Rehmanniae radix and has been included in the Chinese Pharmacopoeia since the 2005 edition. The biosynthesis pathways of several important iridoid glycosides, such as aucubin15, catalpol16,17, and picrosides18,19, have been identified in plants using precursor feeding, transcriptome analyses, and functional genomics analyses.
Studies have shown that iridoid glycoside biosynthesis may occur via the 2-C-methyl-d-erythritol 4-phosphate (MEP), mevalonate (MVA), and iridoid pathway17,20. The MVA pathway occurs in the cytoplasm and primarily provides the precursors isoprenoid pyrophosphate (IPP) and dimethylallyl diphosphate (DMAPP) for sesquiterpene and triterpene biosynthesis21. The MEP pathway occurs in plastids and mainly provides IPP and DMAPP for the synthesis of monoterpenes, diterpenes, and tetraterpenes22. The MEP and MVA pathways have been well studied in the model plant Arabidopsis thaliana and other organisms23. Iridoid glycosides are derived from geraniol, which is synthesized from geranyl pyrophosphate (GPP) and catalyzed by geraniol synthase (GES)24. Geraniol is further catalyzed by geraniol 10-hydroxylase (G10H), 10-hydroxygeraniol dehydrogenase (10HGO), iridoid synthase (IS), iridoid oxidase (IO), and 7-deoxyloganetic acid glucosyl transferase (7DLGT), ultimately forming 7-deoxyloganic acid17,25. This compound is a key biosynthetic precursor for seco-iridoids and iridoids, and its biosynthesis pathway has been recently elucidated in monoterpenoid indole alkaloid (MIA)-producing plants, such as Catharanthus roseus26 and Camptotheca acuminata27. However, several key steps in forming iridoids from 7-deoxyloganic acid remain largely unknown in flowering plants.
Cytochrome P450 (CYP) is a vast multigene superfamily widely present in animals, plants, and microorganisms. As one of the most powerful biological catalysts in nature, P450 was named for its characteristic absorption peak at 450 nm exhibited by its carbon-monoxide-binding form28. P450s contain four conserved key domains: the heme-binding domain, PERF/W region, I-helix, and K-helix motif29. Most plant P450s are membrane-bound enzymes anchored in the endoplasmic reticulum membrane via an N-terminal domain30. In plants, P450 genes extensively participate in the biosynthesis of biomolecules such as flavonoids, isoflavones, fatty acids, plant hormones, lignins, signaling molecules, and other related compounds31. Both the biosynthesis and post-biosynthetic modification of terpenoids require the involvement of P450 enzymes32. The major P450 enzymes involved in monoterpenoid biosynthesis belong to the CYP71, CYP72, and CYP76 subfamilies. Representative examples include CYP71A32 (menthofuran synthase), which catalyses the conversion of (+)-pulegone to (+)-menthofuran33; G10H (CYP76B6)34; and IO/7-deoxyloganetic acid synthase (7DLS, CYP72A26)35, which participates in iridoid synthesis. Recently, three members of the CYP72 subfamily, VaAS, CaAS, and PtAS—were cloned from Vitex agnus-castus, Callicarpa americana, and Paulownia tomentosa, respectively, and identified as aucubin synthases (AS) that hydroxylate bartsioside at C6 to form aucubin36. Aucubin is the direct biosynthetic precursor of catalpol through epoxidation16,17. However, the specific P450 members involved in catalpol biosynthesis remain unclear.
High-quality genomes greatly facilitate functional genomic studies in Rehmannia species. In 2021, the genome of R. glutinosa ‘Qinhuai’ (tetraploid, 4x = 56) was sequenced using a combination of Illumina, Nanopore, and Hi-C platforms. Only one set of genomes was assembled at the chromosome level, and ~ 52.61% of the assembled scaffolds were anchored on 14 pseudochromosomes37. This draft genome is incomplete and highly fragmented. Due to the high chromosomal ploidy, heterozygosity, and poor cytogenetic characterization, obtaining a high-quality telomere-to-telomere (T2T) genome for R. glutinosa remains a challenge. As a diploid Rehmannia species rich in iridoid glycosides1, phenylethanoid glycosides38, and anthocyanins39, R. chingii possesses an established, efficient genetic transformation and gene editing system39, making it ideal for assembling a high-quality T2T genome and serving as a model plant for functional genomics research in the Rehmannia genus.
In this study, we present a T2T gap-free genome assembly of R. chingii obtained by combining ultra-long Oxford Nanopore Technology (ONT), PacBio HiFi, Illumina, and high-throughput chromosome conformation capture (Hi-C) technologies. The gap-free genome of R. chingii provides opportunities to analyze its centromere and telomere regions, clarify genome evolution and whole-genome duplications, and identify key genes involved in iridoid glycoside biosynthesis. The genome and transcriptome resources are of great scientific significance for further functional research and the breeding of Rehmannia.
Results
The T2T gap-free reference genome for R. chingii
A robust individual from TMS11, a population of R. chingii distributed on the West Tianmu Mountain of Lin’an County, Zhejiang Province, was selected for T2T gap-free reference genome assembly (Fig. 1a, b). Chromosome counts from root tips confirmed that R. chingii is diploid (2n = 28) (Fig. 1c). K-mer distribution analysis (k = 17, depth = 89) revealed an estimated genome size of 1.139 Gb for R. chingii, with a heterozygosity rate of 1.93% (Supplementary Fig. 1 and Supplementary Table 1). To achieve a high-quality genome assembly, an assembly strategy that integrated Illumina, PacBio HiFi, Oxford Nanopore Technologies (ONT), and Hi-C sequencing was employed. In total, 121.34 Gb (~106.49× coverage) of Illumina reads, 74.02 Gb (~64.96× coverage) of PacBio HiFi reads, 114.01 Gb (~100.06× coverage) of ultra-long Nanopore reads, and 126.95 Gb (~111.42× coverage) of Hi-C reads were generated (Supplementary Table 2). The assembly process resulted in 651 contigs with an N50 value of 82.2 Mb (Table 1), which is 124.92-fold higher than the contig N50 value of R. glutinosa37. Contigs were anchored using Hi-C data, and 14 contigs were assembled at the chromosomal level, accounting for 95.31% of the genome (Table 1). Most of the remaining 637 unanchored contigs were mitochondrial genomic DNAs (88.31%) (Supplementary Data 1). A heat map of the Hi-C-assembled chromosomes indicated a complete genome assembly (Fig. 1d). The final high-quality genome was 1.169 Gb, gap-free across all 14 chromosomes, with a contig N50 length of 82.2 Mb (Fig. 1e and Table 1).

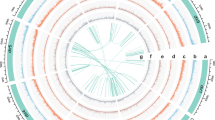
a Plant morphology of R. chingii. b Morphology of the anther, stigma, fruit, seed, seedling, root, corolla, ovary, and calyx of R. chingii. Scaled bars: white, 1 cm; yellow, 1 mm. c Chromosome number determined from root tip cell of R. chingii. d Hi-C interaction map of R. chingii, with blue boxes representing individual chromosomes. The x- and y-axes indicate the ordered positions of the chromosomes of the genome. e Circos plot of the R. chingii genome. Tracks from a to g represent chromosomes, gene number, GC content, repeat density, LTR density, LTR/Copia density, and LTR/Gypsy density, respectively. Source data are provided as a Source Data file.
The quality and completeness of the R. chingii genome assembly were evaluated using multiple methods. Reads from next-generation sequencing were mapped to the assembled genome to assess accuracy, yielding a mapping rate of 98.5% and a coverage rate of 99.96% (Supplementary Table 3), indicating a high level of genome completeness. Benchmarking Universal Single-Copy Orthologs (BUSCO) and Core Eukaryotic Genes Mapping Approach (CEGMA) analysis revealed 99.07% and 94.35% genome completeness, respectively (Supplementary Fig. 2 and Table 1). Furthermore, LTR Assembly Index (LAI) score was 22.22 (Table 1), and the consensus quality value (QV) of the genome was 46.02. Collectively, these results confirm that the final R. chingii T2T gap-free genome assembly exhibits exceptional reliability and quality.
Finally, telomere and centromere were detected. Using the seven-base telomere repeat sequence (‘CCCTAAA’) as a query, 28 telomeres were identified across all 14 chromosomes of the R. chingii genome (Supplementary Fig. 3a and Supplementary Table 4). Using Tandem Repeats Finder, centromeric sequences were identified in all 14 chromosomes of the assembled genome (Supplementary Fig. 3b). The locations of these centromeric regions are listed in Supplementary Table 5. Further analysis identified 199 protein-coding genes within the centromere regions (Supplementary Data 2). GO term enrichment analysis revealed that many of these genes were associated with cellular lipid catabolic processes, squalene monooxygenase (SQM) activity, and fatty acid catabolic processes (Supplementary Fig. 4a). KEGG pathway enrichment analysis indicated that these genes were involved in ribosome function, steroid biosynthesis, and sesquiterpenoid and triterpenoid biosynthesis pathways (Supplementary Fig. 4b).
Annotation of the R. chingii genome
Repetitive sequences comprise a substantial proportion of eukaryotic genomes40. To annotate the R. chingii genome for repetitive sequences, de novo and homology-based predictions were applied. A total of 1,225,434 repetitive sequences were identified in the TMS11 genome, spanning 947,376,855 bp (~77.21% of the genome) (Fig. 1d and Supplementary Table 6). The predominant repeat type was long terminal repeats (LTRs), accounting for 49.14% of the genome, followed by unknown repeats (22.82%), DNA transposons (2.84%), long interspersed nuclear elements (2.10%), and short interspersed nuclear elements (0.3%) (Supplementary Table 6).
Gene prediction was conducted using three approaches: de novo prediction, homology-based prediction, and transcriptome RNA-seq analysis (next-generation and third-generation transcriptomes). A total of 29,322 genes were predicted in the R. chingii genome (Supplementary Fig. 5), with an average gene length of 3,928.48 bp and an average exon count of 4.94 per gene. Comparative analysis of gene, CDS, exon, and intron lengths revealed that R. chingii had longer introns than other plants (Supplementary Table 7). Functional annotation was performed using the six databases—NR, SwissProt, KEGG, InterPro, Pfam, and GO—resulting in the annotation of 29,007 (98.93%) out of the 29,322 predicted genes in the R. chingii genome (Supplementary Table 8). For the noncoding RNAs of the R. chingii genome, a total of 414 microRNAs, 6687 transfer RNAs (tRNAs), 22,925 ribosomal RNAs (rRNAs), and 3977 small nuclear RNAs were predicted (Supplementary Table 9).
Evolution of gene families in R. chingii
To investigate the genome evolution of R. chingii, annotated protein-coding genes from 12 Lamiales species (R. glutinosa, S. asiatica, Phtheirospermum japonicum, Orobanche cernua var. cumana (O. cumana), Lindenbergia luchunensis, Buddleja alternifolia, Olea europaea, Paulownia fortunei, Antirrhinum majus, Sesamum indicum, Erythranthe guttata, and M. guttatus), one Gentianales species (C. roseus), one Vitales species (Vitis vinifera), two Solanales species (Solanum lycopersicum and S. tuberosum), and the model species (A. thaliana), as well as one monocot (Oryza sativa), were clustered into 48,535 gene families, including 2013 common gene families and 272 common single-copy gene families (Fig. 2a, b). Within R. chingii, 1399 genes were assigned to 751 unique gene families (Supplementary Fig. 6). GO and KEGG enrichment analyses revealed that these gene families were involved in biological processes such as homologous recombination, photosynthesis–antenna proteins, sesquiterpenoid and triterpenoid biosynthesis, and transferase activity (Supplementary Fig. 7). Additionally, 90 genes belonging to 66 families were found to be unique in two Rehmannia species (R. chingii and R. glutinosa) compared to the other 17 species. Functional enrichment analysis indicated that these unique gene families were primarily involved in glutathione metabolism, ubiquinone and other terpenoid−quinone biosynthesis, and carotenoid biosynthesis (Supplementary Fig. 8).

a Copy number distribution of all gene families in 19 selected angiosperm species. b Gene family cluster petal diagram, with the central circle representing common gene families and the outer petals depicting species-specific gene families. c Phylogenetic analysis, divergence times, and gene family expansions and contractions among 19 plant species. The phylogenetic tree was constructed based on 104 single-copy orthologous genes, with O. sativa as the outgroup. Divergence times (MYA) are shown in blue numbers next to branch nodes. Gene family expansions and contractions are represented by green and red numbers, respectively. Source data are provided as a Source Data file.
The 272 common single-copy gene families were used to construct a maximum likelihood phylogenetic tree (Fig. 2c). Traditional taxonomy classifies Rehmannia within the Scrophulariaceae family41. However, recent evolutionary analyses of single-copy genes suggest that Rehmannia belongs to Orobanchaceae42,43, though genomic evidence is needed. In this study, R. chingii was identified as a sister lineage to R. glutinosa, with a divergence time of approximately 10.08 million years ago (MYA). The two Rehmannia species were clustered with the four Orobanchaceae species (S. asiatica, O. cumana, L. luchunensis, and P. japonicum), diverging from them around 33.49 MYA. These findings align with a previous phylogenetic tree based on nuclear gene sequences43. Subsequently, R. chingii was found to be more closely related to P. fortunei (Paulowniaceae, ~50.17 MYA), the two Phrymaceae species (M. guttatus and E. guttata, ~53.99 MYA), and S. indicum (Pedaliaceae, ~57.05 MYA). The divergence time between B. alternifolia (Scrophulariaceae) and R. chingii was estimated to be around 61.45 MYA. These results support the classification of R. chingii within Orobanchaceae rather than Scrophulariaceae.
During species evolution, the expansion and contraction of gene families play an important role in plant-specific trait formation and phenotype differentiation44. Expanded gene families can provide new traits and pathways that enhance environmental adaptation45. Analysis of gene family expansion and contraction in the 19 plant species revealed that 141 gene families were expanded in R. chingii, whereas 74 gene families were contracted (Fig. 2c). KEGG analysis showed that the contracted genes were primarily involved in anthocyanin biosynthesis (e.g., anthocyanidin 3-O-glucoside 6”-O-acyltransferase), monoterpenoid biosynthesis, and diterpenoid biosynthesis (Supplementary Fig. 9a). Conversely, the expanded gene families in R. chingii were primarily associated with phenylpropanoid, flavonoid, flavone, and flavonol biosynthesis (Supplementary Fig. 9b). Furthermore, 129 gene families (1283 genes) were expanded in both R. chingii and R. glutinosa compared with the other 17 species. GO and KEGG enrichment analyses indicated that these expanded gene families were mainly involved in monoterpenoid biosynthesis, sesquiterpenoid and triterpenoid biosynthesis, anthocyanin biosynthesis, flavone and flavonol biosynthesis, and carotenoid biosynthesis (Supplementary Fig. 10).
Whole-genome duplications of R. chingii
Whole-genome duplications (WGDs) have long been considered an important evolutionary force in plants46. Synonymous substitution rate (Ks) analysis revealed a peak at 0.646 (Fig. 3a), demonstrating a recent WGD in R. chingii ~ 57.69 MYA. Interestingly, the Ks distribution of intragenomic paralogs in R. glutinosa showed two distinct peaks at Ks values of approximately 0.074 and 0.699, suggesting that R. glutinosa experienced two lineage-specific WGD events. The most recent WGD event in R. glutinosa likely occurred 6.63 MYA and may have resulted from chromosome duplication. The Ks peak for orthologs between R. glutinosa and R. chingii was 0.105, with a divergence time estimated at 9.37 MYA. This suggests that a WGD event occurred in R. glutinosa after its divergence from R. chingii.

a Ks distribution analysis. b Dot plots of paralogous gene pairs in the R. chingii genome (3872 gene pairs). c Schematic representation of synteny between different chromosomes, with lines connecting orthologous genes. d Syntenic comparison of the homologous chromosomes between R. chingii and R. glutinosa genomes. e Dot plots of syntenic blocks comparing R. chingii vs. R. glutinosa, R. chingii vs. O. cumana, R. chingii vs S. asiatica, and R. chingii vs. S. indicum. In (c, d), Gray lines show collinear blocks between species. Color lines highlight the major syntenic blocks spanning the genomes. Source data are provided as a Source Data file.
Chromosome structure and collinearity within R. chingii were analyzed using dot plots, which illustrated the presence of homologous blocks among different chromosomes (Fig. 3b). Notably, fragments in Chr1, Chr5, Chr6, Chr7, and Chr8 exhibited strong synteny with Chr3, Chr12, Chr13, Chr11, and Chr9, respectively (Fig. 3c and Supplementary Fig. 11), indicating remnants of a single round of WGD. Chromosome rearrangements play an important role in species formation47. To further understand the evolutionary history of R. chingii, a genomic collinearity analysis was conducted with R. chingii, R. glutinosa, S. indicum, O. cumana, and S. asiatica. The results demonstrated that the R. chingii and R. glutinosa had the highest degree of collinearity, particularly between R. chingii Chr5, Chr11, and Chr12 and R. glutinosa Chr13, Chr8, and Chr1, respectively (Fig. 3d). Additionally, Chr9 in R. chingii may have originated from the fusion of Chr4 and Chr5 in R. glutinosa (Fig. 3d).
Higher chromosomal collinearity was observed between R. chingii and the two Orobanchaceae plants (O. cumana, and S. asiatica), whereas lower collinearity was found in S. indicum (Pedaliaceae) (Supplementary Fig. 12). This further illustrates that R. chingii diverged early from S. indicum but more recently from O. cumana and S. asiatica. Comparative genomic analyses of R. chingii with R. glutinosa, O. cumana, S. asiatica, and S. indicum revealed syntenic depth ratios of 2:2, 2:2, 2:3, and 2:2, respectively (Fig. 3e and Supplementary Fig. 13). These results suggest that these species experienced similar WGD events.
Genomic variation between R. chingii and R. glutinosa genomes
Despite the high degree of genomic synteny between R. chingii and R. glutinosa, their assembly structures show inconsistencies (Fig. 3d). To investigate genomic variations and structural differences between these species, Syri v1.63 was used for a detailed collinearity analysis. The results identified 2989 syntenic regions (113–120 Mb) shared between the R. chingii and R. glutinosa genomes (Supplementary Table 10). However, significant interspecific genomic variation was observed, with chromosomal distribution heterogeneity (Supplementary Fig. 14). Notably, poor collinearity was detected between Chr01 of R. chingii and Chr05 of R. glutinosa, and extensive non-collinear regions were found between Chr03 and Chr10, Chr08 and Chr03, and Chr14 and Chr06 of R. chingii and R. glutinosa, respectively.
A large number of structural rearrangements were identified, including 440 inversions, 6914 translocations, and 2019 duplications in R. chingii and 4779 duplications in R. glutinosa (Supplementary Table 10). Additionally, 2,555,560 SNPs were detected between the two genomes, further highlighting their genetic divergence.
Identification of enzyme genes involved in iridoid glycoside biosynthesis
Iridoid glycosides are among the most abundant medicinal compounds found in plants of the Rehmannia genus. All parts of the R. chingii plant are rich in iridoid glycosides1. The expression of catalytic enzyme genes is strongly correlated with the accumulation of corresponding secondary metabolites in plant tissues39. The roots, stems, leaves, corollas, veinless leaves, leaf veins, root cortex, root xylem, and tender shoots of R. chingii were analyzed for total iridoid glycoside content and gene expression (Fig. 4a). Initial analysis of the roots, stems, leaves, and corollas revealed the highest accumulation in the leaves (Fig. 4b). Further comparisons among tender shoots, veinless leaves, leaf veins, root cortex, and root xylem showed that tender shoots exhibited the highest content, followed by veinless leaves > leaf veins and root cortex > root xylem (Fig. 4c).

a Phenotype of the selected tissues used for transcriptome analysis. b Total iridoid contents in roots, stems, leaves, and corollas. Values are means ± SD of three biological replicates. c Total iridoid contents in veinless leaves, leaf veins, root cortices, root xylems, and tender shoots. Values are means ± SD of three biological replicates. d Expression patterns of enzyme genes involved in the iridoids biosynthetic pathway, identified via BLAST alignment with C. roseus amino acid sequences. Transcript levels from RNA-seq data are visualized as heatmaps and normalized with log10(FPKM + 1). DXR 1-deoxy-D-xylulose 5-phosphate reductoisomerase, DXS 1-deoxy-D-xylulose-5-phosphate synthase, CMS 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase, CMK 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase, MCS 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase, HDS (E)−4-hydroxy-3-methylbut-2-enyl-diphosphate synthase, HDR 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase, AACT acetyl-CoA C-acetyltransferase, HMGS hydroxymethylglutaryl-CoA synthase, HMGR hydroxymethylglutaryl-CoA reductase, MVK mevalonate kinase, PMK phosphomevalonate kinase, MVD diphosphomevalonate decarboxylase, IPI isopentenyl-diphosphate Delta-isomerase, GPPS geranyl diphosphate synthase, GES geraniol synthase, G10H geraniol 10-hydroxylase, 10HGO 10-hydroxygeraniol dehydrogenase, IS iridoid synthase, IO iridoid oxidase. In (b, c), Different letters above the bars indicate significant differences (P < 0.05) as determined by one-way ANOVA with Tukey’s tests. Source data are provided as a Source Data file.
Based on the structural gene sequences involved in iridoid glycoside synthesis in C. roseus35 and A. thaliana, candidate genes were identified in the assembled R. chingii genome using BLAST. A total of 66 genes were identified, including 16, 18, and 41 genes belonging to the MVA, MEP, and iridoid pathways, respectively (Supplementary Table 11). To explore the key enzyme genes involved in iridoid glycoside biosynthesis, transcriptome sequencing was first performed on the roots, stems, leaves, and corollas of R. chingii. In total, 87.37 Gb of raw reads and 84.81 Gb of clean reads were obtained (Supplementary Table 12), with 85.33%–86.93% of the clean reads mapped to R. chingii genes (Supplementary Table 13). The correlation coefficients of three replicates of the same sample exceeded 0.98 (Supplementary Fig. 15a). A principal component analysis (PCA) clustered the three replicates together (Supplementary Fig. 15b), demonstrating the reproducibility of the RNA-seq results. The expression patterns of structural genes in the MEP pathway were analyzed, revealing that nine genes exhibited the highest expression levels in R. chingii leaves. These included two genes encoding 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MCS) (Rch09d0354 and Rch08d1491); two genes encoding 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (HDR) (Rch04d0517 and Rch04d1570); and five genes encoding 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (CMS), 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase (CMK), (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS), and 1-deoxy-D-xylulose-5-phosphate synthase (DXS) (Fig. 4d). These findings are consistent with those indicating the highest total iridoid glycoside content in the leaves of R. chingii. In the iridoid pathway, twelve structural genes exhibited the highest expression levels, including four genes encoding geranyl diphosphate synthase (GPPS) (Rch09d0946, Rch06d0093, Rch04d0681, and Rch07d0358); two genes encoding 10-hydroxygeraniol dehydrogenase (10HGO) (Rch06d1809 and Rch13d1523); two genes encoding iridoid synthase (IS) (Rch08d0091 and Rch08d0092); and four genes encoding GES, G10H, and isopentenyl-diphosphate delta-isomerase (IPI) (Fig. 4d). These genes exhibit expression patterns similar to those of the nine MEP genes implicated in iridoid glycoside biosynthesis. Notably, eighteen catalytic enzyme genes in the MVA pathway displayed expression patterns that differed from those in the MEP and iridoid pathways (Fig. 4d), suggesting that they might not regulate iridoid glycoside biosynthesis in R. chingii.
Next, transcriptome sequencing was performed on the veinless leaves, leaf veins, root cortices, root xylems, and tender shoots of R. chingii. A total of 98.53 Gb of raw reads and 96.32 Gb of clean reads were obtained (Supplementary Table 14), with 71.25%–81.98% of the clean reads mapped to R. chingii genes (Supplementary Table 15). Correlation analysis and PCA revealed high consistency among the three biological replicates (Supplementary Fig. 16a). Tissue pairs from the same organs, namely, the root xylem and root cortex and the veinless leaf and leaf vein, clustered closely in PCA results (Supplementary Fig. 16b), likely due to their similar expression profiles. By analyzing the expression characteristics, it was determined that structural genes involved in the iridoid glycoside biosynthesis displayed tissue-specific expression across the five tissues (Fig. 4d). Specifically, candidate genes showed higher expression levels in the veinless leaves compared to leaf veins, consistent with the observation that total iridoid glycoside content was higher in the R. chingii veinless leaves. Conversely, candidate gene expression was nearly undetectable in the root xylem and root cortex, suggesting that catalpol biosynthesis primarily occurs in the leaves rather than in the roots.
Squalene epoxidase (SE) catalyzes the epoxidation of squalene, forming 2,3(S)-oxidosqualene 4, which undergoes subsequent hydroxylation, glycosylation, and cyclization to yield triterpenes and phytosterols48,49. It has been speculated that SE or SQM may play a role in the epoxidation of aucubin to produce catalpol19. In the R. chingii genome, two SE genes and five SQM genes were identified; however, their expression levels were low across all examined tissues (Supplementary Table 16), differs from the expression patterns of candidate structural genes in the MEP and iridoid pathways. An SQM gene, Rch08d0024 (designated RcSQM), exhibited relatively higher expression levels in R. chingii leaf tissues, roots, and corollas (Supplementary Table 16). To investigate its function, the coding sequence of RcSQM was cloned into a pMDC83 overexpression vector and transformed into A. tumefaciens strain GV3101. RcSQM was transiently expressed in N. benthamiana leaves using aucubin as a substrate. However, no catalpol was detected (Supplementary Fig. 17), indicating that RcSQM is unlikely to be involved in the epoxidation of aucubin to form catalpol.
Identification of CYP450s involved in catalpol biosynthesis
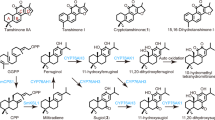
Catalpol is the most abundant iridoid glycoside in Rehmannia species plants17. The MEP and iridoid pathways have been well studied; however, relatively few studies have focused on the downstream pathway of catalpol biosynthesis, which begins with cis-trans-nepetalactol. Based on the structural characteristics of catalpol and the limited research on the biosynthesis pathways of catalpol and other iridoid glycosides15,16,35,36, it was speculated that catalpol synthesis begins with cis-trans-nepetalactol and ends with catalpol (Fig. 5a). In summary, cis-trans-nepetalactol undergoes three successive oxidation steps catalyzed by IO/7-deoxyloganetic acid synthase (7DLS) to form 7-deoxyloganetic acid. Subsequently, catalpol is synthesized through three additional steps: hydroxylation, dehydration, and decarboxylation, followed by epoxidation. Oxidation and hydroxylation are presumed to be primarily catalyzed by cytochrome P450 (CYP)50.

a Proposed biosynthetic pathway of catalpol in R. chingii. G10H geraniol 10-hydroxylase, 10HGO 10-hydroxygeraniol dehydrogenase, IS iridoid synthase, IO iridoid oxidase, 7DLGT 7-deoxyloganetic acid glucosyl transferase, AS aucubin synthase. b Expression patterns of the 15 cytochrome P450 genes in Group I are shown in Supplementary Fig. 18. c Phylogenetic tree of cytochrome P450 proteins in Group I of R. chingii and their homologous proteins from other plants. Species abbreviations: Rc Rehmannia chingii, At Arabidopsis thaliana, Cr Catharanthus roseus, Ca Callicarpa americana, Caa Camptotheca acuminate, Va Vitex agnus-castus, Pt Paulownia tomentosa. d GC-MS analysis of products in S. cerevisiae expressing RcG10H. Extracted ion chromatograms show geraniol (m/z [M + H]+ = 154.14) and 10-hydroxygeraniol (m/z [M + H]+ = 170.13). Source data are provided as a Source Data file.
A total of 320 candidate P450 genes were identified in the R. chingii genome through screening with the Conserved Domain Database (CDD), SMART, and Pfam databases. These genes were clustered into three groups based on their expression patterns across nine R. chingii tissues. Group I contained fifteen genes, including RcG10H and RcIO (Supplementary Fig. 18). Most of these genes were predominantly expressed in the leaf, veinless leaves, and leaf veins, with lower expression levels in the roots, stems, root cortices, and root xylems (Fig. 5b). A phylogenetic analysis revealed that these fifteen RcP450s were clustered into five subfamilies, with the largest number belonging to the CYP72 group (including ten RcP450s). RcG10H (Rch13d0510) and RcIO (Rch07d1582) were found to belong to the CYP76 group (Fig. 5c).
G10H is a key enzyme in the iridoid pathway51, responsible for hydroxylating the monoterpenoid geraniol at the C-10 position to produce 10-hydroxygeraniol34,52. To verify the molecular function of RcG10H, its coding sequence was cloned into a pYeDp60 vector and introduced into Saccharomyces cerevisiae strain WAT11. The geraniol feeding method was then used to analyze 10-hydroxygeraniol content in yeast cultures. Preliminary HPLC analysis revealed that an oxidation product was produced upon RcG10H expression, with a retention time (RT = 10.5 min) corresponding to that of standard 10-hydroxygeraniol (Supplementary Fig. 19). No product was observed in the empty vector control. GC-MS analysis confirmed that the oxidation product was indeed 10-hydroxygeraniol, as the retention time (RT = 14.7 min) and ion mass spectra (m/z = 170.13) matched those of the standard 10-hydroxygeraniol (Fig. 5d and Supplementary Fig. 20). These results demonstrate that RcG10H possesses geraniol 10-hydroxylase activity.
P450 gene cluster on chromosome 8 participates in catalpol biosynthesis
Further analysis revealed that eight members of the CYP72A family are distributed as a P450 gene cluster on chromosome 8 of the R. chingii genome (Fig. 6a). Similar P450 gene clusters were also identified in the genomes of the model plant A. thaliana and the related species S. indicum (Fig. 6a), suggesting that this cluster may have formed before the divergence of R. chingii from A. thaliana and S. indicum. A synteny analysis of these three species revealed strong synteny between the P450 genes of R. chingii and S. indicum, whereas no synteny was observed between R. chingii and A. thaliana (Fig. 6a). Specifically, the gene pair RcCYP72H7 and LOC105171621 exhibited synteny, while the remaining seven P450 genes in R. chingii were syntenic with LOC105171626, suggesting that these seven P450 genes may have formed by recent tandem duplication. Most RcP450 genes in the CYP72A subfamily showed high expression in the leaf, including the veinless leaf and leaf vein (Figs. 5b and 6a), suggesting that this P450 gene cluster may participate in the same iridoid biosynthesis pathway. RcCYP72E13 and RcCYP72E14, members of the CYP72A subfamily cluster on chromosome 8, are homologous to VaAS, CaAS, and PtAS36 (Supplementary Fig. 21) and may catalyze the conversion of bartsioside to aucubin in R. chingii.

a Genomic organization and syntenic relationships of cytochrome P450 gene clusters in R. chingii and their orthologs in S. indicum and A. thaliana. Green lines indicate the evolutionary trajectory of P450 homologs, and the red-blue gradient represents expression levels (FPKM values). b Screening of aucubin 7,8-epoxidase activity in selected P450 candidates via Agrobacterium-mediated transient expression in N. benthamiana leaves. MRM chromatograph showing aucubin (m/z 364 → 167/149) and catalpol (m/z 380 → 183/165). c Western blot analysis of N. benthamiana leaves transiently expressing RcCYP72H7-GFP, with Actin used as a loading control. d Catapol content in R. chingii leaves transiently expressing RcCYP72H7. Data are shown as mean ± SD (n = 5 biological replicates). **p < 0.01; Student’s t-test. e RcCYP72H-GFP expression in N. benthamiana leaf cells. Bar = 20 μm. Source data are provided as a Source Data file.
Given that the epoxidation of aucubin to catalpol occurs adjacent to aucubin synthase activity, it was hypothesized that specific enzymes of the CYP72A subfamily cluster may be involved in catalpol synthesis. To test this hypothesis, the seven candidate genes were cloned in R. chingii and heterologously expressed in N. benthamiana leaves. The coding sequence of seven highly expressed CYP72 genes (FPKM value > 10) was cloned into a pMDC83 overexpression vector and transformed into A. tumefaciens strain GV3101. Upon reaching high expression levels four days after infiltration, Aucubin was injected into the corresponding leaf infiltration region. One day post-infection, leaf metabolites were analyzed using liquid chromatography–mass/mass (LC-MS/MS). A new prominent peak was detected, exhibiting the same retention time (RT = 2.93 min) and MS spectra (m/z = 380) as catalpol, specifically in leaves expressing RcCYP72H7 (Fig. 6b and Supplementary Fig. 22). This suggests that RcCYP72H7 possesses aucubin epoxidase activity and therefore was designated R. chingii catalpol synthase (RcCS). To investigate these proteins expression in vivo, fusion expression vectors of the seven CYP72 genes with GFP were constructed and transiently expressed in N. benthamiana leaves. Single bands were detected in protein extracts from N. benthamiana leaves transiently expressing these vectors using western blot analysis with GFP antibodies (Fig. 6c and Supplementary Fig. 23). Furthermore, RcCYP72H7-GFP exhibited aucubin epoxidase activity and catalyzed catalpol formation (Supplementary Fig. 24).
To further examine the role of RcCYP72H7 in catalpol biosynthesis in vivo, Agrobacterium-mediated transformation was performed in R. chingii leaves. This resulted in a 55.07% increase in catalpol content (Fig. 6d and Supplementary Fig. 25), indicating that catalpol accumulation in plants can be effectively modulated through RcCYP72H7 expression regulation.
The subcellular localization of RcCYP72H7 was examined by observing fluorescence in N. benthamiana leaves transiently expressing RcCYP72H7-GFP. The fluorescence signal was distributed in the endoplasmic reticulum (ER) (Fig. 6e). Phylogenetic analysis revealed that orthologs of RcCYP72H7 were present in R. glutinosa, P. fortunei, and Handroanthus impetiginosus, as well as in the model plant A. thaliana (Supplementary Fig. 26). However, RcCYP72H7 shares low sequence identity with A. thaliana CYP72A13 (44.2%) (Supplementary Figs. 26 and 27), suggesting that the evolution of catalpol synthase has remained relatively conserved throughout the Lamiales family.
Discussion
R. chingii is a medicinal plant distributed in China that has gained increasing attention for its therapeutic and ornamental value1,39. Its close relative, R. glutinosa, has been widely used in traditional Chinese medicine due to its significant medicinal value7. However, due to its tetraploid nature, large genome size, numerous repetitive sequences, and high genomic heterozygosity, obtaining a high-quality genome for R. glutinosa has been highly challenging. This has hindered functional genomic research and constrained its germplasm innovation, variety improvement, and industrial development. In contrast, R. chingii has a smaller genome and well-developed genetic transformation and gene-editing systems39, making it an ideal model plant for functional genomic research in the Rehmannia genus.
In this study, a gap-free T2T genome of R. chingii was assembled by combining Illumina, PacBio HiFi, Nanopore ONT, and Hi-C data, achieving a contig N50 of 82.2 Mb. Quality assessments confirmed the genome’s high integrity. Compared to R. glutinosa37, the R. chingii genome exhibits superior integrity, longer contig N50 length, fewer contigs and scaffolds, and a higher proportion of genes annotated to chromosomes. Comparative genomic analysis revealed substantial structural variation between the two species (Supplementary Fig. 14). Few collinear regions were observed between R. chingii Chr01 and R. glutinosa Chr05, and significant non-collinear regions were identified between R. chingii Chr03, Chr08, Chr14 and R. glutinosa Chr10, Chr03, Chr06. Additionally, large chromosomal inversions were detected, such as between R. chingii Chr06 and R. glutinosa Chr07, R. chingii Chr07 and Chr11. These structural differences may underlie phenotypic and biological variations between the species. A high-quality reference genome of R. chingii facilitates molecular breeding and functional genomics studies. It serves as a robust framework for identifying functional genes and understanding metabolite biosynthetic pathways within the Rehmannia genus. Furthermore, it offers a crucial reference for genomic analysis of other Rehmannia species, advancing genetic and biotechnological applications in medicinal plant research.
The taxonomic classification of the Rehmannia genus has been debated. Traditionally, it was placed within Scrophulariaceae based on its morphological characteristics41. However, recent molecular studies have since demonstrated that Scrophulariaceae is polyphyletic, and at least five monophyletic groups exist in Scrophulariaceae53. Consequently, Rehmannia was first reassigned to Plantaginaceae54, before being incorporated in Orobanchaceae as a second nonparasitic branch55. A previous study reconstructed a phylogenetic tree using complete plastid genome sequences and compared floral organ development56. The results suggested that Rehmannia and Triaenophora clades should be classified within the Orobanchaceae tribe Rehmannieae rather than forming an independent family within Lamiales56. Further phylogenetic analyses using genomic and transcriptome data supported the placement of photoautotrophic Rehmannia as a sister group of all other Orobanchaceae species43. Comparative genomic analysis indicated that R. glutinosa is most closely related to E. guttata (Phrymaceae) and assumed that R. glutinosa belonged to Orobanchaceae37. However, the precise evolutionary relationships between R. glutinosa and other Orobanchaceae and Scrophulariaceae species remain unclear. In this study, a phylogenetic tree constructed using published genome data confirmed that R. chingii and R. glutinosa cluster with four Orobanchaceae species (O. cumana, S. asiatica, P. Japonicum, and L. luchunensis) rather than Buddleja alternifolia (Scrophulariaceae). These findings confirm that Rehmannia belongs to Orobanchaceae. However, given the significant morphological and ecological differences between Rehmannia and other Orobanchaceae species, we propose that Rehmannia and Triaenophora should be treated as an independent family, namely, Rehmanniaceae.
Plant-derived terpenoids have extensive structural diversity, with over 40,000 terpenoids identified from plants and other organisms57. These compounds originate from the universal precursors isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP), which are generated via two distinct pathways: the MVA pathway and the MEP pathway58. The MEP pathway primarily contributes to the biosynthesis of monoterpenes, diterpenes, and tetraterpenes, whereas the MVA pathway is mainly involved in the production of sesquiterpenes and triterpenes in plant cytoplasm21. Geranyl diphosphate (GPP) is synthesized through the condensation of one IPP and one DMAPP molecule59. Both iridoids and monoterpenes are derived from GPP60,61, with iridoid synthesis in most Lamiales species significantly correlated with the expression levels of GES, 8HGO, IS, and IO62. In C. roseus, in situ hybridization studies have shown that DXS, DXR, MECS, and G10H are specifically expressed in the internal phloem parenchyma of the adaxial phloem region63. Furthermore, single-cell RNA sequencing has revealed that the MEP and iridoid biosynthetic pathways are related to structural genes enriched in the internal phloem-associated parenchyma (IPAP) cells of C. roseus leaves, indicating that MIA biosynthesis initiates in IPAP cells64. In R. chingii, the expression patterns of iridoid pathway enzyme genes closely resemble those of the MEP pathway but notably differ from those of structural genes in the MVA pathway (Fig. 4d). This finding suggests that iridoid glycoside biosynthesis mainly occurs via the MEP pathway in R. chingii. The iridoid pathway genes, including GES, 10HGO, IS, and IO, exhibit preferential expression in R. chingii leaves, veinless leaves, and leaf veins, while showing weaker expression in the roots and stems. These results indicate that iridoid glycosides are mainly synthesized in R. chingii leaves and subsequently transported to the stems, roots, and other organs.
The P450 enzyme family is the largest in plants, with over 5100 annotated sequences65. P450 enzymes play important roles in hydroxylation, epoxidation, and other modification processes within terpenoids and MIA biosynthesis66. However, only a few reports have specifically examined the epoxidation of terpenoids and MIAs. For example, the CYP71 protein tabersonine 3-oxidase (T3O) catalyzes the conversion of 16-methoxytabersonine into 16-methoxytabersonine epoxide/imine alcohol, as well as the epoxidation of tabersonine to tabersonine epoxide67. In C. roseus, TEX1 (CYP71D521) and TEX2 (CYP71D347) catalyze the epoxidation of tabersonine to form lochnericine68. In Taxus mairei, the CYP725A member Taxane Oxetanase 1 (TOT1) catalyzes the conversion of taxadiene hexa-acetate into 1-dehydroxybaccatin IV and baccatin I, facilitating epoxide and oxetane ring formation69. Similarly, in Ophiorrhiza pumila, OpCYP716E111 catalyzes strictosamide 1 epoxidation, forming strictosamide epoxide 270. Additionally, C. acuminata CYP71BE206 exhibits strictosamide epoxidase activity when transiently expressed in N. benthamiana71.
To the best of our knowledge, epoxidation functions of the CYP72 enzyme family in plant terpenoid metabolism have not been characterized. In this study, we identified RcCYP72H7, a CYP72A family member, which catalyzes the epoxidation of aucubin at the C7-C8 position of the iridoid skeleton, converting it into catalpol (Fig. 6b). The epoxidation site catalyzed by RcCYP72H7 differs from previously reported epoxidases. T3O catalyzes tabersonine epoxidation at the C2-C3 position67, TEX1 and TEX2 catalyze epoxidation at the C6-C7 position to form lochnericine68, OpCYP716E111 and CaCYP71BE206 catalyze the epoxidation of strictosamide at the C2–C7 position70,71, and TOT1 could catalyze the formation of epoxide and oxetane rings at the C4 and C20 positions69. This study reveals epoxidase activity within the CYP72 family, expanding the known functions of P450 enzymes and providing a foundation for further research on the iridoid glycosides biosynthetic pathways in plants.
The CYP72 gene family is one of the largest enzyme families within the CYP450 superfamily, playing roles in secondary metabolism30. The CYP72A subfamily, a major branch of CYP450, catalyzes numerous critical reactions in specialized metabolite biosynthesis, including sterols and terpenoids. For example, in A. thaliana, CYP72A9 functions as a hydroxylase, converting C-13-H-type gibberellins (GAs) into C-13-OH-type counterparts72. Similarly, CYP72A154 catalyzes three consecutive oxidation steps at the C-30 position of 11-oxo-β-amyrin to produce glycyrrhetinic acid73. In monoterpenoid biosynthesis, CYP72A1 (secologanin synthase) from C. roseus catalyzes the oxidative cleavage of loganin to produce secologanin74, while CrCYP72A224 (7-deoxyloganic acid 7-hydroxylase, DL7H) hydroxylates 7-deoxyloganic acid to form loganic acid75. In R. chingii, Rch01d1353 and Rch10d1615, both members of the CYP72A subfamily, may contribute to iridoid glycoside biosynthesis. A CYP72 family gene cluster located on chromosome 8 of R. chingii includes RcCYP72E13 and RcCYP72E14, which are orthologs of aucubin synthases from C. americana, V. agnus-castus, and P. tomentosa (Fig. 5c). These enzymes may catalyze the C-6 hydroxylation of bartsioside in R. chingii.
This study further demonstrates that RcCYP72H7 within the gene cluster catalyzes the epoxidation of aucubin at the C-7 and C-8 positions, leading to catalpol biosynthesis. These findings highlight the essential role of CYP72 family genes in iridoid glycoside biosynthesis in Rehmannia species. Future functional studies of these genes will provide deeper insights into the complete iridoid glycosides biosynthetic pathway.
Methods
Plant materials, DNA extraction, library construction, and sequencing
A healthy individual from TMS11, a wild population of R. chingii distributed in the West Tianmu Mountain of China, was used in this study. TMS11 was cultivated in a solar greenhouse (~25 °C; 14 h light/10 h dark) at Henan Agricultural University in Zhengzhou, China. Fresh, young, healthy leaves were collected, immediately frozen in liquid nitrogen, and then preserved at −80 °C for DNA extraction. High-quality DNA was isolated from fresh young leaves using the cetyl-trimethylammonium bromide method. The extracted DNA was evaluated using an Agilent 2100 Bioanalyzer (Agilent, USA) and a NanoDropTM 2000 spectrophotometer (Thermo Fisher Scientific, USA), respectively.
For Illumina sequencing, a short-read (2 × 150 bp) library was prepared and sequenced on a DNBSEQ − T7 platform (BGI, China) to assess genome size, GC content, and heterozygosity. For PacBio HiFi sequencing, a standard SMRTbell library was constructed following the SMRTbell Express Template Prep Kit 2.0 manual (Pacific Biosciences, USA) and sequenced on a PacBio Sequel II system (Pacific Biosciences, USA). For ONT ultra-long sequencing, the library was prepared using the SQK-ULK001 kit (Circulomics, USA) per the manufacturer’s instructions and sequenced on a PromethION platform (ONT, UK). For HiC sequencing, genomic DNA was deep sequenced on an Illumina HiSeq X-Ten platform (Illumina, USA) in 150-bp paired-end reads mode, following the manufacturer’s standard protocols.
Genome de novo assembly and quality control
Genome size and heterozygosity were estimated using K-mer frequency analysis based on the Lander-Waterman theory76. The R. chingii genome was assembled using PacBio HiFi reads. Firstly, the raw data were corrected with CCS software (https://github.com/PacificBiosciences/ccs), and high-accuracy (99%) HiFi reads were assembled using Hifiasm (v0.16.1) with default parameters to generate a draft contig genome77. Illumina reads were then used for error correction with Pilon78. The ONT long reads were assembled using NextDenovo (https://github.com/Nextomics/NextDenovo) with the following parameters: genome size = 1.1 Gb, read cutoff = 50,000, seed cutoff = 55,959, and seed depth = 45.
Hi-C data were used to anchor contigs to chromosomes and remove short contigs. Hi-C valid interaction pairs were filtered using HiC-Pro79, and chromosome level assembly was performed with ALLHiC (v0.9.8)80 and Juicebox (v1.11.08)81 to cluster, order, and orient the contigs. The ONT genome was then used to fill gaps in the Hifiasm-assembled genome, which served as the backbone. A heatmap of genomic interactions was generated using HiCPlotter82.
Telomeres were identified by searching for the characteristic seven-base telomeric repeat (CCCTAAA at 5′ end or TTTAGGG at 3′ end) within 50 kb of each chromosome terminus, (http://telomerase.asu.edu/sequences_telomere.html). Regions with at least five motif repeats were considered telomeric. Centromeres were detected using Centromics software (https://github.com/ShuaiNIEgithub/Centromics), with approximate locations estimated based on the frequency of candidate centromeric tandem repeats.
Genome completeness was assessed using BUSCO and CEGMA. LTR_retriever (v2.9.0) was used to calculate the LAI for assembly quality assessment. Illumina and Hi-C reads were mapped to the final assembly with BWA (version 0.7) (https://github.com/lh3/bwa) to estimate mapping rates. Consensus quality and completeness of the T2T genome were evaluated using Merqury (https://github.com/marbl/merqury).
RNA sequencing for genome annotation
Mixed samples from four healthy tissues (root, stem, leaf, and corolla) were collected for RNA extraction using the TRIzol Universal Reagent (Takara, China). RNA sequencing (RNA-seq) libraries were prepared with the NEBNext Ultra RNA Library Prep Kit (NEB, USA) and sequenced on the Illumina Novaseq platform. Clean reads were obtained by removing adapters and low-quality reads from the raw data. For Single Molecule Real-Time (SMRT) sequencing, cDNA synthesis was performed using KAPA HiFi PCR kits (Kapa Biosystems, USA), followed by purification with the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA). The libraries were sequenced on the PacBio Sequel II platform.
Genome annotation
Repeat elements were identified using a combination of homology-based alignment and de novo prediction. Homology-based annotation was performed by aligning the genome assembly to the Repbase database (v21.12) (http://www.girinst.org/repbase) with the genome assembly using RepeatMasker (v4.1.4) (http://www.repeatmasker.org/). For de novo detection, a custom repetitive element library was constructed through ab initio prediction using RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html).
Gene models for R. chingii were generated by integrating de novo prediction, transcript-based assembly, and homology-based searches. For homology-based prediction, protein sequences from A. thaliana, E. guttata, S. indicum, and S. asiatica were downloaded and mapped to the R. chingii genome using TBLSTN83. Exon-intron boundaries were inferred with Exonerate (v2.2.0)84. Two ab initio gene-prediction software tools, Augustus (v3.2.3)85 and SNAP86, were used for de novo gene model prediction. To refine the gene structure annotation, unigenes derived from Illumina reads and PacBio reads were aligned to the genome assembly using BLAT87, and filtered using PASA. A final, non-redundant consensus gene set was generated by integrating de novo, homology-based, and transcript-based using EvidenceModeler88.
Functional annotation of predicted protein sequences was conducted through BLAST searches (E-value ≤ 1e-5) against the Swiss-Prot (http://www.uniprot.org), NR (http://www.ncbi.nlm.nih.gov/protein), Pfam (http://pfam.xfam.org), KEGG, and InterPro (https://www.ebi.ac.uk/interpro) databases. tRNA genes were identified using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE). rRNA fragments were detected through BLAST alignment against the rRNA database. snRNA and microRNA sequences were annotated with INFERNAL based on the Rfam database.
Comparative genomic analysis
Comparative genome analysis was conducted using R. chingii and 19 additional plant species: A. thaliana, R. glutinosa, S. asiatica, P. japonicum, O. cumana, L. luchunensis, B. alternifolia, O. europaea, P. fortunei, A. majus, S. indicum, E. guttata, M. guttatus, C. roseus, V. vinifera, S. lycopersicum, S. tuberosum, and O. sativa. OrthoFinder (v2.4)89 was used to classify protein sequences into gene families based on DIAMOND alignments (E-value < 0.001). Functional annotations of gene families were obtained using the Panther (v15) database90. Unique gene families in each species were determined through GO and KEGG enrichment analyses. The 272 single-copy gene protein sequences were aligned using MAFFT (v7.205), and a phylogenetic tree was generated with IQ-TREE (v1.6.11)91. O. sativa served as the outgroups. Divergence times were estimated using the MCMCTree92 program within the PAML package under default parameters. Gene family expansions and contractions were detected with CAFE (v4.2)93. Orthologous protein sequences were aligned between species using Diamond (v0.9.29.130)94 (E-value < 1e − 5, C-score > 0.5), and syntenic blocks were identified using MCScanX95 (-m 100). To investigate the evolution of the R. chingii genome, the synonymous substitution rate (Ks) for collinear gene pairs was calculated with wgd (v1.1.1)96, and a density map was generated using ggplot2.
Genomic variation detection
Pairwise genome alignments were conducted using MUMmer (v4.0.0rc1) with the parameters -mum -mincluter -500. Alignments were filtered with delta-filter (-1 -i 90 -l 100) and processed for structural variation detection using the SyRI pipeline (v1.6.3). Genomic alignment visualization was performed using plotsr (v0.5.5).
Identification of genes involved in iridoid biosynthesis
To identify candidate genes involved in the MEP, MVA, and iridoid biosynthesis pathways, protein sequences from A. thaliana and C. roseus were used as queries in a BLAST search against the R. chingii genome assembly using (E-value ≤ 1e-5). The gene sequences encoding 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), 1-deoxy-D-xylulose-5-phosphate synthase (DXS), 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (CMS), 4-(cytidine 5′-diphospho)−2-C-methyl-D-erythritol kinase (CMK), 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MCS), (E)−4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS), 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (HDR), acetyl-CoA C-acetyltransferase (AACT), hydroxymethylglutaryl-CoA synthase (HMGS), hydroxymethylglutaryl-CoA reductase (HMGR), mevalonate kinase (MVK), phosphomevalonate kinase (PMK), diphosphomevalonate decarboxylase (MDV), isopentenyl-diphosphate Delta-isomerase (IPI), geranyl diphosphate synthase (GPPS), GES, G10H, 10-hydroxygeraniol dehydrogenase (10HGO), and iridoid synthase (IS) were retrieved from NCBI. These gene sequences were used as bait to identify homologous genes in the R. chingii genome. Candidate genes were subsequently analyzed using the Pfam and SMART databases to verify conserved domains and motifs. Proteins with identical conserved domains were considered homologous.
Comparative transcriptome analysis
For transcriptome analysis, RNA sequencing (RNA-seq) was performed by Novogene (Beijing, China). To identify iridoid biosynthesis genes, additional tissue samples, including veinless leaf, leaf vein, root cortex, root xylem, and tender shoot, were collected at the seedling stage. Veinless leaf tissue was obtained by excising the central vein and isolating the regions between secondary veins using a surgical scalpel. Approximately 0.5 g of each tissue was used for RNA extraction. Three biological replicates were sequenced for each sample. RNA-seq reads were mapped to the R. chingii genome using HISAT2 (v2.1.0)97 with default parameters. Gene expression levels were quantified using the featureCounts software package and expressed as fragments per kilobase of exon model per million mapped fragments (FPKM). Differentially expressed genes (DEGs) were identified using the DESeq2 package in R (|log2(FoldChange)| > 1, padj < 0.05). GO and KEGG enrichment analyses of DEGs were conducted using the IMP software package98. Heatmaps of gene expression profiles were generated using MeV (v4.90)99.
Total iridoid extractions and measurements
Tissue samples, including leaves, veinless leaves, corolla, roots, stems, leaf veins, root cortices, root xylems, and tender shoots were cut into 0.5 × 0.5 cm sections and dried in an electric blast drying oven at 55 °C for 48 h. Air-dried samples (0.1 g) were powdered and then extracted with 50 mL 70% ethanol under sonication for 45 min. The extract was centrifuged at 1800 × g for 5 min, and 5 mL of the supernatant was further centrifuged at 11,300 × g for 5 min. A 2 mL aliquot of the final supernatant was diluted with 70% ethanol and used as the test sample. For iridoid quantification, 1 mol/L HCl was added to each test sample, followed by incubation at 90 °C for 30 min. The samples were cooled to room temperature, and 2,4-dinitrophenylhydrazine ethanol solution was added. The reaction mixture was incubated at 90 °C for 25 min and then cooled to room temperature. Next, 3 mL of 1 mol/L NaOH in 70% ethanol was added, and the mixture was allowed to react at room temperature for 1 h. The absorbance of the reaction solution was measured at 462 nm using a spectrophotometer. A blank control using 70% ethanol was prepared for calibration.
Heterologous expression of RcG10H in yeast
To construct the expression vector, the full-length CDS of RcG10H (Rch13d0510) was amplified using the specific primers listed in Supplementary Data 3, and subsequently inserted into the pYeDP60 vector, which had been digested with Eco RI and Sac I (New England Biolabs). The sequenced vectors carrying RcG10H were transformed into S. cerevisiae strain WAT11 using a thermal stimulation method. The empty pYeDP60 vector was used as the negative control.
For RcG10H expression in S. cerevisiae, the strain containing RcG10H was cultivated in 50 mL YPGEA (Glu) liquid culture medium for 15 h until an OD600 of 0.6–0.8 was reached. The cultures were centrifuged at 2800 × g for 5 min to collect the cells, which were then resuspended in 50 mL YPGEA (Gal) liquid culture medium. After 8 h of growth, 1 mM geraniol was added to the cultures, followed by incubation for an additional 72 h. The reaction was stopped by adding an equal volume (50 mL) of ethyl acetate. After 3 min of shaking, the supernatant fluid was collected and dried. The remaining cells were re-extracted using twice the volume (100 mL) of ethyl acetate for 3 min, and the extract was dried in a rotary evaporator. The dried extracts were then re-dissolved in 1 mL of methanol and filtered through 0.22 μm PTFE filters before GC-MS and HPLC analysis.
Identification of P450 family members
To identify P450 proteins in R. chingii, the Pfam 37.0 (http://pfam.xfam.org/) seed file (PF00067) for the P450 gene was downloaded. HMMER 3.0 and the local BLAST program were used to search for sequences containing the P450 protein domain. The identified protein sequences were aligned to remove redundant sequences. Additional verification of P450 family members was performed using the online tools SMART and CDD.
HPLC detection
Chromatographic separation was performed using a reversed-phase Diamonsil C18(2) column (250 mm × 0.42 mm × 5 μm; Dikma, China). The mobile phase consisted of (A) 0.1% methanoic acid in water and (B) methanol, with a gradient of 50% B from 0 to 20 min and 70% B from 20 to 45 min. The flow rate was set at 1 mL/min, with an injection volume of 5 μL. The detection wavelength was set at 210 nm, and the column temperature was maintained at 35 °C. The total analytical runtime was 45 min.
GC-MS analysis of geraniol and 10-hydroxygeraniol
GC-MS analysis was conducted using an Agilent 7890B gas chromatography system coupled with an Agilent 7000D mass spectrometer (Agilent, USA). An Agilent HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm) was used for the isolation of volatile compounds. The mass spectrometer was operated in electron impact ionization mode (EI, 70 eV). The injection volume was 1 μL in non-split mode, and the injection port temperature was set at 250 °C.
The column temperature was programmed as follows: the initial temperature was set at 50 °C, increased to 100 °C at a rate of 10 °C/min, then ramped to 170 °C at 5 °C/min where it was held for 1 min. High-purity helium was used as the carrier gas, with a flow rate of 1 mL/min. The mass spectrometer parameters were as follows: ion-source temperature was 230 °C, mass scan range was 20–500 amu, and a scan range time of 300 ms. Compound identification was performed based on the retention times of pure substances, library comparison with the NIST.17 library, and literature analysis.
Screening candidate CYP450 genes in aucubin epoxidation
For the construction of overexpression vectors, the full-length CDS of RcCYP72G20 (Rch08d0799), RcCYP72E13 (Rch08d0800), RcCYP72E14 (Rch08d0805), RcCYP72E15 (Rch08d0806), RcCYP72G19 (Rch08d0804), RcCYP72G21 (Rch08d0807), and RcCYP72H7 (Rch08d0811) were amplified using the corresponding primers listed in Supplementary Data 3. The amplified products were inserted into the pMDC83 vectors, which had been digested using the corresponding restriction enzymes from New England Biolabs. The sequenced vectors carrying candidate genes were transformed into the Agrobacterium strain GV3101 using a freeze-thaw method. The empty pMDC83 vector was used as the negative control.
For exogenous genes transient expression in N. benthamiana, the Agrobacterium strains containing the target genes were cultivated on LB solid culture medium for two days and subsequently resuspended in liquid MES culture medium (10 mM MES, 10 mM MgCl2, pH = 5.7, 100 μM acetosyringone) to reach an OD600 of 0.5–0.8 before infiltration. Four-week-old leaves were used for the transient expression of candidate genes. Agrobacterium GV3101 strains were infiltrated into N. benthamiana leaves using a 1 mL syringe without a needle. After infiltrations, the plants were kept in darkness for 24 h, and then grown under a 14-h light / 12-h dark cycle at 26 °C.
For substrate-feeding experiments, N. benthamiana leaves were infiltrated with 2.5 mM of aucubin (dissolved in an aqueous solution containing 10 mM MES) on the fourth day following Agrobacterium infiltration. After 24 h of incubation, N. benthamiana leaves were harvested, flash-frozen in liquid nitrogen, and extracted for LC-MS analysis.
LC-MS/MS detection
To extract metabolites from the N. benthamiana leaves, frozen tissues were ground into a fine powder using a Tissuelyser-48 L, and extracted with 5 mL of methanol, followed by sonication for 20 min at room temperature. The samples were centrifuged at 1800 × g for 10 min, and the supernatant was filtered through 0.22 μm PTFE filters before LC/MS analysis.
For LC-MS analysis, samples were analyzed using an EXPEC 5210 triple quadrupole mass spectrometer connected to the liquid chromatography system. Chromatographic separation was performed using a CNW Athena C18 column (2.1 × 100 mm, 1.8 μm), with a mobile phase consisting of methanol and 10 mM ammonium formate at a flow rate of 0.3 mL/min under a linear gradient elution. The gradient was set as follows: from 0 to 3.2 min, 3% methanol; 3.2–9.5 min, methanol was increased to 60%. The injection volume was 10 μL, and the column temperature was maintained at 35 °C. The mass spectrometer was operated in multiple reaction monitoring (MRM) mode with electrospray ionization (ESl) in positive ionization mode, monitoring the protonated precursor → product ion transitions at m/z 364 → 167 and m/z 364 → 149 for aucubin, m/z 380 → 183 and m/z 380 → 165 for catalpol. The capillary voltage was set at 4.5 kV, the desolvation gas temperature was 600 °C, the nebulizer gas flow rate was 1.5 L/min, and the counter gas flow rate was 1.2 L/min. The total analysis time was 8 min.
Transient expression of RcCYP72H7 in R. chingii
The Agrobacterium strain GV3101 carrying pMDC83-RcCYP72H7 was cultivated on LB solid culture medium for two days and resuspended in liquid MES culture medium to reach an OD600 of 0.5–0.8 before infiltration. Two-month-old R. chingii leaves were used for the transient expression of RcCYP72H7. Agrobacterium was infiltrated into the leaves using a 1 mL syringe without a needle. Following infiltrations, the light was turned off for 24 h, after which plants were grown under a 14-h light /12-h dark cycle at 26 °C. After five days of incubation, R. chingii leaves were harvested, flash-frozen in liquid nitrogen, and extracted for HPLC analysis.
Western blotting assays
The coding sequences of RcCYP72G2, RcCYP72E13, RcCYP72E14, RcCYP72E15, RcCYP72G19, RcCYP72G21, and RcCYP72H7 were cloned into the pBWA(A)HS-ccdb-Glosgfp vector under the control of the cauliflower mosaic virus promoter 35S promoter to generate fusion expression vectors containing the GFP gene and these CYP72 genes. The primers used for vector construction are listed in Supplementary Data 3. Agroinfiltrated N. benthamiana leaves transiently expressing CYP72 genes with GFP tags were ground into a fine powder in liquid nitrogen and suspended in plant protein extraction buffer containing proteinase inhibitor (Roche, cat# 04693132001). The mixture was centrifuged at 25,400 × g at 4 °C for 10 min, and the supernatant was collected and denatured in SDS sample buffer (GenScript, cat# M00676) at 99 °C for 5 min. Equal amounts of total protein per sample were separated on 8% SDS-PAGE gels, and proteins were transferred onto a polyvinylidene fluoride membrane (Millipore, cat# IPVH00010). The membrane was blocked with 5% nonfat milk (Solarbio, cat# D8340) to prevent nonspecific binding. An anti-GFP antibody (Roche, cat# 11814460001, clone# 13.1) was used as the primary antibody at a 1:1000 dilution, followed by incubation with a goat anti-mouse IgG H + L (HRP) secondary antibody (Abclonal, cat# AS003). Actin protein were detected using anti-Actin antibody (ABclonal, cat# AC009, clone# AMC0496). Protein detection was performed using a LI-COR Odyssey Infrared Imaging System (Tanon 5200).
Subcellular localization analyses
The plasmid of the constructed vector 35S-RcCYP72H7-GFP was introduced into the A. tumefaciens strain GV3101 for transient expression in N. benthamiana epidermal cells. After 72 h of infiltration, N. benthamiana epiderma cells were subjected to confocal laser scanning microscopy analysis.
Chromosome counts
After 50 days of growth, root tips (1−2 mm in length) of R. chingii were collected for chromosome counting. The root tips were submerged in 1 mol/L HCl for 5–10 min, followed by incubation on a slide warmer at 60 °C for 8 min. After washing three times with distilled water, the root tips were stained with carbol fuchsin on a glass slide, covered with a coverslip, and gently pressed to ensure even cell scattering. Chromosomes were observed and counted under a light microscope at magnifications of 100× and 400× magnification.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing data generated for this study, including ONT data, PacBio HiFi data, RNA sequencing reads, Illumina short reads and Hi-C data, have been deposited in the Genome Sequence Archive (GSA) database at the National Genomics Data Center under BioProject PRJCA032868. The assembled genome was submitted to Genome Warehouse (GWH, https://ngdc.cncb.ac.cn/gwh/) under accession GWHFIKC00000000.1 [https://ngdc.cncb.ac.cn/gwh/Assembly/88090/show]. Source data are provided with this paper.
References
Liu, Y. F. et al. Bioactive iridoid glycosides from the whole plants of Rehmannia chingii. J. Nat. Prod. 79, 428–433 (2016).
Yang, M. Q. et al. Rehmannia chrysantha (Rehmanniaceae), a new species from Inner Mongolia, northern China. Phytotaxa 265, 177 (2016).
Miao, C. et al. De novo transcriptome analysis identifies RpMYB1 as an activator of anthocyanin biosynthesis in Rehmannia piasezkii. Plant Physiol. Biochem. 215, 108964 (2024).
Xia, Z. et al. Identification of DNA barcoding in plants of Rehmannia Libosch. ex Fisch. et Mey. and origin of cultivated Rehmannia glutinosa (in Chinese). Chin. Tradit. Herb. Drugs 47, 648–654 (2016).
Liu, Y. F. et al. Four new iridoid glycosides from the roots of Rehmannia glutinosa. J. Asian Nat. Prod. Res. 10, 1–9 (2023).
Wang, F. et al. Transcriptome analysis of salicylic acid treatment in Rehmannia glutinosa hairy roots using RNA-seq technique for identification of genes involved in acteoside biosynthesis. Front. Plant Sci. 8, 787 (2017).
Wang, F. et al. Transcriptome-wide identification of WRKY transcription factor and functional characterization of RgWRKY37 involved in acteoside biosynthesis in Rehmannia glutinosa. Front. Plant Sci. 12, 739853 (2021).
Li, M. et al. A new ionone glycoside and three new Rhemaneolignans from the roots of Rehmannia glutinosa. Molecules 20, 15192–15201 (2015).
Zhou, J. et al. Four ionones and ionone glycosides from the whole plant of Rehmannia piasezkii. J. Asian Nat. Prod. Res. 24, 955–962 (2022).
Bian, Z. et al. Extraction, structure and bioactivities of polysaccharides from Rehmannia glutinosa: a review. J. Ethnopharmacol. 305, 116132 (2023).
Liu, C. et al. Rehmanniae Radix in osteoporosis: a review of traditional Chinese medicinal uses, phytochemistry, pharmacokinetics and pharmacology. J. Ethnopharmacol. 198, 351–362 (2017).
Jiang, B. et al. Catalpol: a potential therapeutic for neurodegenerative diseases. Curr. Med. Chem. 22, 1278–1291 (2015).
Liu, J. et al. Anti-inflammatory effect and mechanism of catalpol in various inflammatory diseases. Drug Dev. Res. 84, 1376–1394 (2023).
Zhang, Q. et al. Catalpol ameliorates liver fibrosis via inhibiting aerobic glycolysis by EphA2/FAK/Src signaling pathway. Phytomedicine 135, 156047 (2024).
Damtoft, S. et al. Late stages in the biosynthesis of aucubin in Scrophularia. Phytochemistr 33, 1089 (1993).
Damtoft, S. Biosynthesis of catalpol. Phytochemistry 35, 1187–1189 (1994).
Zhi, J. et al. Molecular regulation of catalpol and acteoside accumulation in radial striation and non-radial striation of Rehmannia glutinosa tuberous root. Int. J. Mol. Sci. 19, 3751 (2018).
Kumar, V. et al. A proposed biosynthetic pathway of picrosides linked through the detection of biochemical intermediates in the endangered medicinal herb Picrorhiza kurroa. Phytochem. Anal. 24, 598–602 (2013).
Shitiz, K. et al. NGS Transcriptomes and enzyme inhibitors unravel vomplexity of picrosides niosynthesis in Picrorhiza kurroa Royle ex. Benth. PLoS ONE 10, e0144546 (2015).
Van Moerkercke, A. et al. The bHLH transcription factor BIS1 controls the iridoid branch of the monoterpenoid indole alkaloid pathway in Catharanthus roseus. Proc. Natl. Acad. Sci. USA 112, 8130–8135 (2015).
Liao, P. et al. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol. Adv. 34, 697–713 (2016).
Krause, T. et al. HDR, the last enzyme in the MEP pathway, differently regulates isoprenoid biosynthesis in two woody plants. Plant Physiol. 192, 767–788 (2023).
Vranová, E. et al. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 64, 665–700 (2013).
Smit, S. J. et al. The genomic and enzymatic basis for iridoid biosynthesis in cat thyme (Teucrium marum). Plant J. 118, 1589–1602 (2024).
Alagna, F. et al. Identification and characterization of the iridoid synthase involved in oleuropein biosynthesis in olive (Olea europaea) fruits. J. Biol. Chem. 291, 5542–5554 (2016).
Miettinen, K. et al. The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 5, 3606 (2014).
Li, Y. et al. Functional characterization of Camptotheca acuminata 7-deoxyloganetic acid synthases and 7-deoxyloganetic acid glucosyltransferases involved in camptothecin biosynthesis. Plant Physiol. Biochem. 218, 109305 (2025).
Werck-Reichhart, D. & Feyereisen, R. Cytochromes P450: a success story. Genome Biol. 1, REVIEWS3003 (2000).
Vasav, A. P. & Barvkar, V. T. Phylogenomic analysis of cytochrome P450 multigene family and their differential expression analysis in Solanum lycopersicum L. suggested tissue specific promoters. BMC Genomics 20, 116 (2019).
Hansen, C. C. et al. Plant cytochrome P450 plasticity and evolution. Mol. Plant. 14, 1244–1265 (2021).
Schuler, M. A. & Werck-Reichhart, D. Functional genomics of P450s. Annu. Rev. Plant Biol. 54, 629–667 (2003).
Zhang, Y. et al. Cytochrome P450s in plant terpenoid biosynthesis: discovery, characterization and metabolic engineering. Crit. Rev. Biotechnol. 43, 1–21 (2023).
Bertea, C. M. et al. Demonstration that menthofuran synthase of mint (Mentha) is a cytochrome P450 monooxygenase: cloning, functional expression, and characterization of the responsible gene. Arch. Biochem. Biophys. 390, 279–286 (2001).
Collu, G. et al. Geraniol 10-hydroxylase, a cytochrome P450 enzyme involved in terpenoid indole alkaloid biosynthesis. FEBS Lett. 508, 215–220 (2001).
Salim, V. et al. 7-deoxyloganetic acid synthase catalyzes a key 3 step oxidation to form 7-deoxyloganetic acid in Catharanthus roseus iridoid biosynthesis. Phytochemistry 101, 23–31 (2014).
Rodríguez-López, C. E. et al. Phylogeny-aware chemoinformatic analysis of chemical diversity in Lamiaceae enables iridoid pathway assembly and discovery of aucubin synthase. Mol. Biol. Evol. 39, msac057 (2022).
Ma, L. et al. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput. Struct. Biotechnol. J. 19, 3954–3963 (2021).
Liu, Y. F. et al. Chemical constituents from whole plants of Rehmannia chingii (in Chinese). Chin. Tradit. Herb. Drugs 47, 1830–1833 (2016).
Zuo, X. et al. Purple Rehmannia: investigation of the activation of R2R3-MYB transcription factors involved in anthocyanin biosynthesis. Physiol. Plant. 175, e13920 (2023).
Liu, X. et al. Transposable element expansion and low-level piRNA silencing in grasshoppers may cause genome gigantism. BMC Biol. 20, 243 (2022).
Zhang, R. X. et al. Rehmannia glutinosa: review of botany, chemistry and pharmacology. J. Ethnopharmacol. 117, 199–214 (2008).
Li, X. et al. Phylogenetic relationships in Orobanchaceae inferred from low-copy nuclear genes: consolidation of major clades and identification of a novel position of the non-photosynthetic Orobanche clade sister to all other parasitic Orobanchaceae. Front. Plant Sci. 10, 902 (2019).
Xu, Y. et al. Comparative genomics of orobanchaceous species with different parasitic lifestyles reveals the origin and stepwise evolution of plant parasitism. Mol. Plant. 15, 1384–1399 (2022).
Renny-Byfield, S. & Wendel, J. F. Doubling down on genomes: polyploidy and crop plants. Am. J. Bot. 101, 1711–1725 (2014).
Chen, Y. et al. A reference-grade genome assembly for Astragalus mongholicus and insights into the biosynthesis and high accumulation of triterpenoids and flavonoids in its roots. Plant Commun. 4, 100469 (2023).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Rieseberg, L. H. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358 (2001).
Rasbery, J. M. et al. Arabidopsis thaliana squalene epoxidase 1 is essential for root and seed development. J. Biol. Chem. 282, 17002–17013 (2007).
Han, J. Y. et al. Regulation of ginsenoside and phytosterol biosynthesis by RNA interferences of squalene epoxidase gene in Panax ginseng. Phytochemistry 71, 36–46 (2010).
Mizutani, M. Impacts of diversification of cytochrome P450 on plant metabolism. Biol. Pharm. Bull. 35, 824–832 (2012).
Singh, S. K. et al. BHLH IRIDOID SYNTHESIS 3 is a member of a bHLH gene cluster regulating terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Direct 5, e00305 (2021).
Sung, P. H. et al. Functional expression of geraniol 10-hydroxylase reveals its dual function in the biosynthesis of terpenoid and phenylpropanoid. J. Agric. Food Chem. 59, 4637–4643 (2011).
Olmstead, R. G. et al. Disintegration of the Scrophulariaceae. Am. J. Bot. 88, 348–361 (2001).
The Angiosperm Phylogeny Group An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141, 399–436 (2003).
Xia, Z. et al. Familial placement and relations of Rehmannia and Triaenophora (Scrophulariaceae s.l.) inferred from five gene regions. Am. J. Bot. 96, 519–530 (2009).
Xia, Z. et al. 2021. Rehmannieae or Rehmanniaceae? Evidence from plastome sequences and floral morphology. Bot. J. Linn. Soc. 196, 145–162 (2021).
Withers, S. T. & Keasling, J. D. Biosynthesis and engineering of isoprenoid small molecules. Appl. Microbiol. Biotechnol. 73, 980–990 (2007).
Roberts, S. C. Production and engineering of terpenoids in plant cell culture. Nat. Chem. Biol. 3, 387–395 (2007).
Kumar, K. et al. Precursor feeding studies and molecular characterization of geraniol synthase establish the limiting role of geraniol in monoterpene indole alkaloid biosynthesis in Catharanthus roseus leaves. Plant Sci. 239, 56–66 (2015).
Zhang, L. et al. Chassis and key enzymes engineering for monoterpenes production. Biotechnol. Adv. 35, 1022–1031 (2017).
Kouda, R. & Yakushiji, F. Recent advances in iridoid chemistry: biosynthesis and chemical synthesis. Chem. Asian J. 15, 3771–3783 (2020).
Mint Evolutionary Genomics Consortium Phylogenomic mining of the mints reveals multiple mechanisms contributing to the evolution of chemical diversity in Lamiaceae. Mol. Plant 11, 1084–1096 (2018).
Burlat, V. et al. Co-expression of three MEP pathway genes and geraniol 10-hydroxylase in internal phloem parenchyma of Catharanthus roseus implicates multicellular translocation of intermediates during the biosynthesis of monoterpene indole alkaloids and isoprenoid-derived primary metabolites. Plant J. 38, 131–141 (2004).
Sun, S. et al. Single-cell RNA sequencing provides a high-resolution roadmap for understanding the multicellular compartmentation of specialized metabolism. Nat. Plants 9, 179–190 (2023).
Nelson, D. & Werck-Reichhart, D. A P450-centric view of plant evolution. Plant J. 66, 194–211 (2011).
Hamberger, B. & Bak, S. Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120426 (2013).
Carqueijeiro, I. et al. A BAHD acyltransferase catalyzing 19-O-acetylation of tabersonine derivatives in roots of Catharanthus roseus enables combinatorial synthesis of monoterpene indole alkaloids. Plant J. 94, 469–484 (2018).
Carqueijeiro, I. et al. Two tabersonine 6,7-epoxidases initiate lochnericine-derived alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 177, 1473–1486 (2018).
Jiang, B. et al. Characterization and heterologous reconstitution of Taxus biosynthetic enzymes leading to baccatin III. Science 383, 622–629 (2024).
Zhang, T. et al. Chemoproteomics reveals the epoxidase enzyme for the biosynthesis of camptothecin in Ophiorrhiza pumila. J. Integr. Plant Biol. 66, 1044–1047 (2024).
Pu, X. et al. Proteomics-guided mining and characterization of epoxidase involved in camptothecin biosynthesis from Camptotheca acuminata. ACS Chem. Biol. 18, 1772–1785 (2023).
He, J. et al. CYP72A enzymes catalyse 13-hydrolyzation of gibberellins. Nat. Plants 5, 1194 (2019).
Seki, H. et al. Triterpene functional genomics in licorice for identification of CYP72A154 involved in the biosynthesis of glycyrrhizin. Plant Cell 23, 4112–4123 (2011).
Irmler, S. et al. Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 24, 797–804 (2000).
Salim, V. et al. Virus-induced gene silencing identifies Catharanthus roseus 7-deoxyloganic acid-7-hydroxylase, a step in iridoid and monoterpene indole alkaloid biosynthesis. Plant J. 76, 754–765 (2013).
Lander, E. S. & Waterman, M. S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2, 231–239 (1988).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Servant, N. et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Zhang, X. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 16, 198 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 421 (2009).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 6, 31 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Korf, I. Gene finding in novel genomes. BMC Bioinforma. 5, 59 (2004).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Mi, H. et al. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019).
Nguyen, L. T. et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Puttick, M. N. MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 35, 5321–5322 (2019).
Han, M. V. et al. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Buchfink, B. et al. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Zwaenepoel, A. & Van de Peer, Y. wgd-simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 35, 2153–2155 (2019).
Kim, D. et al. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Chen, T. et al. IMP: bridging the gap for medicinal plant genomics. Nucleic Acids Res. 52, D1347–D1354 (2024).
Howe, E. A. et al. RNA-Seq analysis in MeV. Bioinformatics 27, 3209–3210 (2011).
Acknowledgements
This project was supported by National Science Foundation of China (82325049, 82373985 & 81872950), the Key Project at Central Government Level for the Ability Establishment of Sustainable Use for Valuable Chinese Medicine Resources (2060302). We thank Dr. Chengchao Xu from Institute of the Chinese Materia Medica of China Academy of Chinese Medical Sciences for providing assistance with data analysis.
Author information
Authors and Affiliations
Contributions
F.W., Y.Y., and L.H. conceived and designed the research. J.G., C.M., C.S., Y.-H.Y., P.Z., and Q.H. performed the experiments; F.W., C.M., J.G., and Y-H.Y. analyzed the data; F.W. and Z.J. drafted the manuscript; N.D., L.G., B.Z., and C.X. provided technical assistance and critical questions; F.W., H.S., and Y.L. performed the bioinformatics analyses and statistical analyses; Y.Y. and Z.Z. revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jiaru Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, F., Jiang, Z., Gao, J. et al. Analysis of the Rehmannia chingii geneome identifies RcCYP72H7 as an epoxidase in iridoid glycoside biosynthesis. Nat Commun 16, 6035 (2025). https://doi.org/10.1038/s41467-025-60909-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60909-9
