
Abstract
Constructed wetlands are used to clean domestic wastewater via phytoremediation, commonly involving the use of reeds. The process results in the production of large amounts of polluted plant tissues, which are then considered unusable waste products. In this study, the reusability of reeds and nettle-polluted tissues is investigated. Fermenting contaminated plant tissues to produce liquid fertilizer is a sustainable means to remove 87-95% of persistent organic pollutants. A multiomics approach combining metabolomics and amplicon metagenomics is used to analyze the mechanisms that occur during fertilizer production from polluted plant tissues and identify the microbes that are likely key for this transformation. A consortium of bacteria and fungi with cellulolytic activity is identified. In addition, the obtained liquid fertilizer positively impacts plant growth in the presence of pathogens and therefore exhibits potential application in farming. This approach may be a simple, commercially attractive solution for the management of contaminated plant tissues originating from constructed wetlands, which are currently considered problematic, useless waste products.
Similar content being viewed by others

Introduction
The removal of persistent organic pollutants (POPs) is a key goal of wastewater treatment strategies, as POPs can be harmful to both humans and the environment. However, this is challenging as they are not easily degraded via conventional means. One means that has achieved success is the use of constructed wetlands (CWs), which are typically used to treat domestic wastewater discharged from small towns. CWs are usually composed of reed beds, sometimes followed by tertiary treatment by spontaneous plants1. CWs are natural solutions for treating wastewater and rely on ecosystemic functions such as phytoremediation. Phytoremediation is described as the ability of plants and associated microbes to remove pollutants. Several mechanisms are involved in phytoremediation, including microbial metabolism of pollutants and pollutant sequestration within plant tissues2.
The common reed (Phragmites australis) is used worldwide in CWs because it is able to grow in a wide variety of climates and survives in polluted environments where it sequesters pollutants within its tissue3,4. Indeed, CW management strategies typically involve cutting the aerial parts of the reeds at least once a year to allow for continuous reed growth and concomitant POPs sequestration. These cut plant tissues are dried on a pile to reduce their weight, but then they are considered waste, as they contain sequestered POPs. These piles simply accumulate, and the plant tissues are left to compost, or they are burned if the piles become unmanageable.
The annual amount of reed biomass produced in constructed wetlands is high; for example, 107 Mt in China5 and 42.1 kt in Lake Winnipeg6. Considering a mean biomass value of 4 kg/m27,8 and 6,178,094 m2 of reed beds in France9, we estimate that 25 kt of reed biomass is produced in CWs per year. This does not consider the numerous CWs that have been recently built near commercial areas and roads to manage rain runoff in the context of “sponge cities”. In 2024, according to the World Health Organization, only 56.6% of the world’s population has access to water treatment services, which means that the reed biomass generated each year will increase in the future as we improve access to clean water, given that CWs potentially represent a simple and cheap solution for wastewater treatment. Unfortunately, there is no realistic management strategy for the plant tissues discarded from CWs. The lack of a commercial use for these plant cuttings represents a key limitation for the wider usage of CWs for wastewater treatment.
Common nettle (Urtica dioica L.) is another plant that is often naturally found in close proximity to reeds in CW beds, where it appreciates the elevated nitrogen content. It is typically considered invasive8,10 and is often removed by controlled flooding while maintaining the reed populations. Although nettle is treated as an unwanted colonizer in the context of CWs, it is appreciated since centuries by gardeners11 as a means to produce liquid fertilizer known as “nettle water”. Nettle water is a prototypical “liquid plant manure” that is produced from the fermentation of plant material and is commonly sold throughout Europe. These liquid plant manures are mostly studied for their beneficial effects on nutrient content, plant growth, and pathogen resistance in the context of crop production in organic farming12,13,14,15. Given the well-established benefits and commercial utility of nettle water, the potential for producing nettle and reed fertilizer from the aerial parts collected during the annual cutting of an active CW is investigated.
The hormones and metabolic profiles of nettle and reed fertilizers are assessed via mass spectrometry. Since these plants are expected to contain POPs prior to fermentation, POP levels are followed throughout the fermentation process. Strikingly, POP levels are dramatically reduced during fertilizer production, whereas the levels of beneficial metabolites increase significantly. The effects of these fertilizers on plant fitness are assessed, which reveals that they can also be used to promote plant growth and pathogen resistance. Finally, the microbial consortia that are responsible for these changes during fertilizer fermentation are investigated.
This work is beneficial for reconsidering the management strategies for plant cuttings originating from wastewater processing in CWs. Instead of being treated as end-product waste, these cuttings can now be considered a means to produce safe liquid fertilizer to promote plant growth in agriculture or gardening and reduce the amounts of pesticides needed. The obtained fertilizer can also be used locally by cities to maintain green spaces and create a circular economy. The aim of this work is to propose a cost-effective solution to transform plant cuttings from constructed wetlands, which are considered waste, into a solution for plant protection and growth promotion.
Results
Analysis of persistent organic pollutants and plant metabolites in plants and fertilizers
The mass spectrometry analysis revealed 979 features in nettle and 963 features in reed, among which 508 and 515 were annotated, respectively, and were categorized into metabolites, lipids and POPs (Fig. 1 and Supplementary Data 1). During the fertilizer production process, a consortium of microorganisms (bacteria and fungi) develops and breaks down plant tissues to feed (Fig. 1a), which leads to a reduction in the number of original plant metabolites that can be found in the fertilizers after 1 month (Fig. 1b, c). This reduction applies to all of the diverse metabolic compounds found in plants, as every family is impacted (Fig. 1d). In particular, a targeted analysis was used to show that the sugars were completely consumed (Supplementary Fig. 1), presumably as these sugars are often preferentially used for microbial growth. Interestingly, the microbial consortium that developed was also able to cope with the POPs that are released from the polluted plant tissues when the cells are destroyed. Indeed, the number of POPs originally detected in the plants decreased with respect to their respective fertilizers and across all POP classes (Fig. 1d). The abundance of the remaining pollutants significantly decreased in fertilizer treatment, with average removal rates of 95% for nettle and 87% for reed (Fig. 2), which were calculated for POPs identified as level 1 according to the Schymanski classification. Among the remaining POPs, none are regulated in terms of maximum residue limits16,17,18,19.

a Summary of the study design and major results; (b), total number of features and number of annotated metabolites, lipids and POPs in nettle and nettle fertilizer; (c), total number of features and number of annotated metabolites, lipids and POPs in reed and reed fertilizer; (d), number of annotated compounds in nettle, reed and their respective fertilizers sorted into families. The metabolite and lipid families are shown according to the NP Classifier classification at the pathway level. POP families are given according to the Norman Substance Database classification. Annotations are at levels 3, 2 and 1 of the Schymanski classification. n = 3 biological replicates for panels (b, c, d). For further details, see Supplementary Data 1. The removal rate presented in panel a was calculated on the basis of the abundance of persistent organic pollutants (Schymanski level 1) in nettle, reed and their respective fertilizers. Source data are provided as a Source Data file. Panel a Created in BioRender. Heintz, D. (2025) https://BioRender.com/1un3pdy.

The relative abundance of POPs (Schymanski level 1) is given for the plant tissues (100%, light green) and their respective fertilizers to visualize the removal rate for each POP. The efficiency of fertilizers removing POPs from diverse families is depicted for (a) nettle fertilizer and (b) reed fertilizer (n = 3 biological replicates). Average removal rates of 95% (nettle) and 87% (reed) were obtained for POPs identified as level 1 according to the Schymanski classification. Absolute quantification is given in the Supplementary Table 1. Source data are provided as a Source Data file.
Effects of nettle and reed fertilizers on plant growth and pathogen resistance
Plant fertilizers are commonly used in gardens either to promote plant growth or to reduce the impact of pathogens. The effects of nettle and reed fertilizers on tomato plant height, biomass and disease severity in the presence of Rhizoctonia solani were tested (Fig. 3 and Supplementary Fig. 2). R. solani is an important soil-borne fungal pathogen responsible for root and stem rot in crop plants with a worldwide distribution, which results in economic losses for approximately 200 host plant species, especially the Solanaceae family, which includes eggplant, pepper, potato, tobacco and tomato cultivated under both greenhouse and field conditions. Nettle and reed fertilizers increased the height of tomato plants exposed to R. solani (Fig. 3a). Nettle fertilizer also increased root biomass and reduced disease severity compared with those of nontreated inoculated plants (Fig. 3b, c). The beneficial impacts of the plant fertilizers could be due to effects on the plant itself, direct effects on R. solani growth or a combination of both.

The effects of fertilizers on (a) tomato plant height, (b) the biomass weights of aerial tissues and roots, and (c) disease severity in aerial tissues and roots were measured (n = 12 biological replicates). d Efficacy of 10% nettle fertilizer on the radial growth of the R. solani AG3 strain cultivated on 1.5% malt agar medium supplemented or not with antibiotics after 4 days of incubation at 25 °C in the dark. Statistical analysis was performed through a pairwise Mann-Whitney test with Bonferroni correction. Different letters above the bars indicate statistically significant differences between the groups for the considered observation, exact p-values are given in a Source Data file. Pictures of the plants at 2, 4 and 6 weeks are provided in Supplementary Fig. 2. In boxplots, the center line represents the median, the box limits correspond to the upper and lower quartiles, the whiskers show upper and lower values excluding outliers. Upper and lower limits to determine outliers are calculated based on 1.5 times the interquartile range. Individual data points are presented as dots. Source data are provided as a Source Data file.
In the absence of pathogens, reed fertilizer increased the aerial length, whereas nettle fertilizer increased the root length (Supplementary Fig. 3). Next, the impact of nettle fertilizer on Rhizoctonia solani growth was assessed, as this fertilizer had the greatest impact on in vivo conditions. Nettle fertilizer inhibited R. solani mycelial growth in vitro (Fig. 3d). Interestingly, microbial growth was observed at the site of nettle fertilizer inoculation. To assess the role that this microbial growth could play in inhibiting R. solani, these assays were repeated in the presence of antibiotics (chloramphenicol and streptomycin). Strikingly, antibiotic supplementation eliminated the inhibitory effect of nettle fertilizer on Rhizoctonia solani. These findings suggest that the bacterial community associated with nettle fertilizer is involved in the protective effects observed in tomato plants exposed to R. solani and could have a direct effect on the pathogen, in addition to possibly enhancing plant innate immunity.
Bacterial and fungal communities in nettle and reed fertilizers
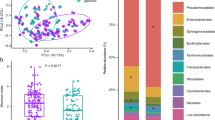
Nettle and reed fertilizers were spread onto malt agar (supplemented with chloramphenicol and streptomycin) and LB agar Petri dishes, which revealed the presence of both fungal and bacterial populations with distinct colony morphologies, suggesting the presence of unique microbial communities (Supplementary Fig. 4). To further identify bacteria involved in the nettle and reed fertilizer production process, a taxonomic analysis was conducted via the V4 16S rRNA gene and ITS amplicon analysis to identify specific bacterial/fungal/yeast amplicon sequence variants (ASVs) associated with the liquid fertilizers. Each fertilizer contained strikingly different microbial profiles (Fig. 4). Nettle fertilizer was characterized by abundant Alcaligenes parafaecalis, Microvirgula curvata, Pseudomonas phenolilytica, Anaeromicrophila herbiliytica and Bacteroides reticulotermitis. Reed fertilizer was instead characterized by relatively high levels of Delftia acidovorans, Clostridium guangxiense and uncharacterized Acinetobacter, Delftia, Dysgonomonas, Shinella and Alcaligenes genera. Interestingly, these communities were highly specific, with little similarity in the relative abundances of the major bacterial species (Fig. 4c). Whereas numerous bacterial species were identified in the nettle/reed fertilizers, the corresponding yeast/fungal communities were almost exclusively dominated by a single ASV. The reed fertilizer was almost entirely composed of Aspergillus fumigatus, whereas the nettle fertilizer contained Scedosporium minutisporum (Fig. 4b).

a Composition of the bacterial communities in the nettle and reed fertilizers (n = 4 biological replicates each across two experiments); b, composition of the fungal communities in the nettle (n = 3) and reed fertilizers (n = 2). c Relative abundance of selected major differentially abundant 16S rRNA gene ASVs between nettle and reed fertilizers (DESeq2 foldchange > 2 and adjusted p < 0.01). Boxplots represent the data interquartile range, with the center line denoting the median and samples requiring a pseudocount for logarithmic plotting are highlighted with transparency; *** = adjusted p-value < 0.001. ASVs not present at >2% in multiple fertilizer samples were merged into the other category, see Supplementary Data 2 and 3 for further details. Source data are provided as a Source Data file.
The bacterial and fungal communities were further characterized by assessing their reported growth phenotypes, including their oxygen requirements, cellulolytic/sugar fermentation activity, affinity for nitrogen and resistance to POPs (Fig. 5a, b). The two communities were predicted to grow under anaerobic conditions and either use sugars or degrade cellulose for their growth. The physicochemical characteristics of the nettle fertilizer were monitored over 30 days (Fig. 5c) and revealed a severe decrease in oxygen availability after 2 days, which is consistent with the establishment of anaerobic bacterial and fungal communities.

a Growth conditions and metabolic capacities of the nettle fertilizer microbial community; (b) growth conditions and metabolic capacities of the reed fertilizer microbial community; (c) physicochemical characteristics measured in the nettle fertilizer over 30 days. Only ASVs present at >2% in multiple fertilizer samples are displayed. See Supplementary Data 3 for further details. Source data are provided as a Source Data file.
Metabolic activity of microbial communities in nettle and reed fertilizers
The links between the metabolic changes observed during fertilizer generation (plant tissue degradation, POP release/degradation and the production of plant signaling compounds) and the microbial consortia identified by assessing microbial genetic potential (Fig. 6) were investigated. Cellobiose and D-glucose levels were severely reduced in both nettle/reed fertilizers (98%-100% reduction), and the microbial communities in these fertilizers are predicted to encode cellulases and beta-glucosidases, with the nettle community predicted to have greater levels of the latter (Supplementary Fig. 5). Therefore, the microbial communities in fertilizers have the genetic capacity to break down the cell walls of plant tissues made of cellulose. This tissue disruption would likely lead to POPs release and allow for direct microbial access for further degradation.

The combination of metabolomics and microbiome datasets allows the reconstruction of (a) plant tissue disruption (cellulose degradation) and POPs release and degradation (anthracene, biphenyl) and (b) microbial production of plant signaling compounds that are beneficial to plant growth and pathogen resistance (indole-3-acetate, salicylic acid). c Taxon functional abundance of indole-3-acetate and salicylic acid. Pathways were reconstructed on the basis of the pathway maps available in KEGG. Metabolites detected in the datasets are identified with colors indicating their abundance in fertilizers (red, up; blue, down; yellow, fertilizer-only). Enzymes predicted to be present in fertilizer microbial communities are displayed with an asterisk and are listed in Supplementary Fig. 5. Source data are provided as a Source Data file. Illustrations created in BioRender. Heintz, D. (2025) https://BioRender.com/rdtop5m.
Interestingly, anthracene and biphenyl, which disappeared from the fertilizers, can be degraded into salicylate and benzoate, respectively, both of which were detected in high amounts in the fertilizers (Supplementary Figs. 6 and 7) and can be used by plants. Salicylic acid (SA), a plant hormone and signal transduction compound, can also be produced from chorismate and naphthalene, and the enzymes involved in salicylic acid production and further transformation to catechol and gentisate were detected in the sequencing data (Fig. 6 and Supplementary Fig. 5). Notably, chorismate/iso-chorismate feeds into nonribosomal peptide synthesis and polyketide synthesis (NRPS/PKS) pathways, which are used by bacteria/fungi to produce siderophores for iron acquisition.
Another plant hormone, indole-3-acetate (IAA), which can be produced from tryptophan, was also increased in the fertilizers, and the enzymes involved in IAA production were predicted to be present in the taxa identified in these fertilizers (Fig. 6), although the microbial communities predicted to be involved in this process differed substantially between nettle/reed. The nettle fertilizer community was dominated by Alcaligenes parafecalis and Microvirgula curvata, whereas the reed fertilizer community had a mix of different microbes that could participate in this process. Interestingly, A. parafecalis and M. curvata were also predicted to encode for the pathway that would generate salicylate from chorismate but would also encode for the final step of the conversion of salicylic acid to catechol. Indole-3-acetic acid, salicylic acid and benzoic acid were quantified at 739.74 ± 63.69 ng/mL, 6.74 ± 0.27 ng/mL and 18.78 ± 0.46 ng/mL in nettle fertilizer, respectively, and at 185.83 ± 1.62 ng/mL, 6.67 ± 0.25 ng/mL and 32.96 ± 1.00 ng/mL in reed fertilizer, respectively.
To supplement the microbial pathway predictions, we also performed shotgun metagenomic sequencing on nettle fertilizer samples and assigned taxonomy/gene presence on the assembled contigs (Supplementary Data 10 and 11). This analysis confirmed that the nettle had been fully digested during fermentation as Urtica-associated reads were almost indetectable (<0.1% relative abundance). In contrast, we were able to identify numerous microbial species in the dataset, with Stenotrophomonas rhizophila having the highest N50 (623Kb) amongst species with >1% relative abundance. We next identified and annotated potential microbial genes to assess the functional potential of the nettle fertilizer microbial communities. We were able to identify at least 40 species with >1000 annotated KO terms/species and thus were able to confirm the presence of our PICRUSt2-inferred pathways within these communities. As these pathways would likely be intact within a single organism, their co-expression would allow for plant tissue/POP breakdown to produce the mass spectrometry-detected metabolites.
Discussion
Two distinct cellulolytic microbial communities responsible for the bioremediation processes that occur in nettle and reed fertilizers were identified. To degrade the pollutants stored in plant tissues and presumably originating from the inlet water (Supplementary Data 4), microbes first have to degrade the plant tissues where these POPs are sequestered. Nettle and reed tissues contain high levels of cellulose, hemicellulose and lignin: reed tissues contain 33–36% cellulose, 20–22% hemicellulose and 18.25% lignin20, whereas nettle tissues contain 42.5% cellulose, 18.7% hemicellulose and 15.2% lignin21. Single microbial strains are unable to efficiently degrade these lignocellulosic substrates; instead, guilds of microbial consortia work together to break down plant tissues22. Indeed, a heterogeneous microbial consortium is needed to transform lignocellulose through the lactate platform23. The microbial populations identified in the nettle and reed fertilizers were distinct despite the fertilizers having developed at similar pH values (nettle 5.82–6.21; reed 6.08–6.50) and temperature profiles (22.6–30.5 °C). The analysis of the microbial community in tap water was performed, and did not show the presence of microbes. Therefore, different starting microbial sources, different metabolic profiles of the plants used to make the fertilizers, and the sequestration of different POPs within reeds vs nettle might be responsible for the development of these distinct microbial communities. However, both were composed of microbial species that have been reported to display cellulolytic capacities. These include the major members of each fertilizer, such as M. curvata, A. parafaecalis, A. herbiliytica and C. guangxiense. The type strain of M. curvata (sharing 99% rRNA sequence identity with M. aerodenitrificans), which was originally isolated from hydrocarbon-contaminated soils (along with Clostridium and Pseudomonas spp), has been detected in groundwater contaminated with polycyclic aromatic hydrocarbons24 (PAHs). These findings suggest that Microvirgula may possess inherent resistance to POPs and other industrial contaminants. In our study, it also showed high potential for producing salicylic acid through iso-chorismate. The closely related species M. aerodenitrificans, a known cellulose degrader25, has been isolated and characterized in woodchip bioreactors and has been highlighted for its potential for nitrate pollution removal25. A. parafaecalis (a recently upgraded species originally known as Alcaligenes faecalis subsp. parafaecalis) displays a natural tendency for the consumption of cellulosic and lignin waste, resulting in high glucose yields26. In addition, a functional prediction suggests that it may be one of the major contributors to indole-3-acetate production, as it was predicted to use either indole-3-acetamide or indole-3-acetonitrile as substrates. While A. parafaecalis was originally named due to its initial identification from feces, it was later found to be widely distributed in soil, water, and human-associated environments27. It has also been described as resistant to micropollutants27,28,29, and several studies have highlighted its pollutant bioremediation capacity30,31,32. These include a variety of pollutants, such as phenols33, PAHs34, drug residues35, arsenic36, isoprene37 and pesticides38,39. Interestingly, A. parafecalis was identified predominantly in nettle fertilizer, whereas reed fertilizer instead was characterized by a distinct, undefined Alcaligenes species, highlighting that each fertilizer community likely possesses a unique microbial guild that works together to degrade the specific plant polysaccharides and POPs present.
A. herbilytica has been reported to possess numerous polysaccharide-degrading genes and utilize several complex carbohydrates, such as cellulose, xylan and hemi-cellulose glucomannan. Clostridium guangxiense was also identified, and Clostridia species are known for their ability to degrade cellulose40,41 via active cellulase complexes called cellulosomes42, which might explain their presence in fertilizers, although their polyphyletic nature makes it difficult to ascribe specific roles without a more complete genome sequencing approach.
The nettle fertilizer fungal/yeast community was almost entirely composed of Scedosporium minutisporum, which is able to degrade PAHs43 and is primarily isolated from industrial settings44. The fungal/yeast community in reeds is instead dominated by Aspergillus fumigatus. A. fumigatus has been frequently tested in industrial settings to remove contaminants45,46 and has the ability to degrade plant cell wall polysaccharides47,48. Strains of A. fumigatus have also been reported to produce gibberellins49, which can improve plant growth; however, more comprehensive analyses, such as shotgun metagenomic sequencing, are needed to confirm the presence of these pathways in the strain recovered from reed fertilizer.
During the microbial bioremediation of POPs, a high number and diversity of transformation products can be formed50,51,52, which also occur in planta53,54. These transformation products are sometimes described to be more hazardous than their parent compounds53. POPs transformation products are not well represented in databases. Currently, the regulations applied to POPs do not consider the transformation products, and knowledge is lacking regarding their toxicity. Collective work from the scientific community is still needed to advance this topic. To investigate further this question, in silico prediction of POPs transformation products was performed, using 79 biological rules (Supplementary Data 5), as already described55. This resulted in suspect lists of POPs transformation products, further used to annotate the unidentified features of the dataset (Supplementary Data 6). In order to cover a diversity of mechanisms, transformation products were predicted for POPs which were totally and partially degraded in fertilizers: bendiocarb, fluacrypyrim, mevinphos, biphenyl, gabapentin, nandrolone, tramadol, vigabatrin, bis(2-ethylhexyl)phthalate, triphenyl phosphate, anthracene, fluoranthene and phenanthrene. None of the unidentified features in the dataset corresponded to predicted POPs transformation products. For POPs totally degraded in fertilizers, this indicates that degradation is complete, and no transformation products remain at detectable levels. An explanation for the partial degradation of POPs might be the pH of the medium or the presence of elements such as fluor in the structure of the POPs. As examples, fluacrypyrim is not fully degraded, most probably due to the presence of fluor in its composition, and the degradation rate of gabapentin has been described to be minimum at an approximate pH of 656, which corresponds to the pH of reed and nettle fertilizers.
In addition, some POPs were detected in fertilizers, but not in the corresponding plant tissues, which is surprising. One could think that these POPs originate from the tap water used to produce the fertilizers, but except for oxadixyl, this was not the case (Supplementary Data 7). This difference might be explained by the different extraction procedures used to analyze POPs from either plant tissues or fertilizers. Specifically, 300 mg of fresh plant tissues was ground for methanolic extraction, while 50 mL of fertilizer was dried and solubilized in 1 mL of methanol/water (90/10). These 50 mL of fertilizers corresponded to 5 g of the corresponding plant tissues used for fermentation. Therefore, there is a concentration effect between fertilizers and plant tissues, which was taken into account throughout the manuscript by normalizing the peak areas to the original mass of plant tissues for each type of sample (see the methods section). However, this might allow some POPs to be detected in fertilizers but not in fresh plant tissues, in which they are below the detection limit of the mass spectrometer.
In addition to cellulolytic activity and their ability to degrade POPs, the microbial communities that develop in nettle and reed fertilizers also produce compounds that promote plant growth and pathogen resistance, either by promoting plant innate immunity57,58 or by direct interactions59. In our study, the nettle and reed fertilizers promoted increased plant height and root biomass in the presence of microbial pathogens. The presence of compounds such as indole-3-acetic acid, gibberellins, salicylic acid or benzoic acid, all of which act as plant hormones, might play a role in these growth-promoting effects, although the exact mechanisms involved were not investigated here. The additional nutrients in the nettle and reed fertilizers (nitrogen, phosphorus, copper, and zinc; Supplementary Fig. 8) could also explain the increase in plant growth that we observed.
Liquid plant fertilizers have always been used in agriculture for their biostimulant and phytoprotective properties. The metabolic content of these natural bioactive products has been analyzed and found to be rich in flavonoids, phenolics, essential oils, saponins, tannins, terpenes and alkaloids60. The reduction in disease severity when plants are treated with nettle fertilizer may thus be due to either stimulating plant innate immunity or direct competition between the microbial communities of the fertilizers and pathogens, as described in other studies61.
Among the microbial communities found in nettle fertilizer, A. parafaecalis62,63 has been shown to play a role in stimulating plant innate immunity, has potential as a bionematocide39,64 and protects plants against the plant pathogen Erwinia carotovora65. Our results also suggest the presence of Stenotrophomonas rhizophila in nettle fertilizer; a microbe with known beneficial impacts on plant health66 and which has been reported to inhibit R. solani growth67. Consequently, we hypothesize that the microbial communities that develop during fermentation, and their combined effort, may be key in helping plants combat pathogen invasion68,69.
A strength of this study is the use of multi-omics to assess both the composition of microbes involved in nettle/reed fermentation into fertilizer and untargeted metabolomics to identify specific metabolites being produced/depleted during this process. We were also able to confirm the presence of the microbial pathways that would be required to complete these processes and demonstrated the practical utility of the final liquid fertilizer to suppress a common plant pathogen in vivo. However, we recognize that our study has several weaknesses. We primarily employed targeted, PCR-based approaches for assessing microbial community composition/function that may lack the resolution to fully characterize the microbial community present. Future work using both long and short-read shotgun metagenomics to comprehensively assemble the microbial genomes present in nettle fertilizer would provide more information on the exact pathways employed by the microbial community to degrade POPs, enhance plant growth and suppress pathogens. We also only assessed the endpoint of fertilizer fermentation, so it is unclear whether the final microbial community was stable throughout fermentation or whether there were successive changes in the community composition/function as fermentation progressed and POPs from nettle/reed were released. In addition, without directly profiling microbial protein/gene expression, it is unclear whether there are specific members of the community that are key for POP degradation, as not all microbes with the genetic capacity to degrade POPs would actively participate in this process, or whether there are specific microbial guilds that cooperatively work to breakdown specific POPs. It is also unclear whether single microbes on their own, or several microbes acting as a community, serve to protect against R. solani (Fig. 3). As our data suggests that the microbial community responsible for the fermentation process is mainly anaerobe (Fig. 5), and the pathogen suppressing effect occurs in presence of oxygen, the fermentation microbial community we profiled may shift to a new state in planta. Future experiments incorporating metaproteomic and/or metatranscriptomic analysis alongside untargeted metabolomics with sampling throughout the one-month fermentation period and during application in planta would help answer these questions.
While this study only examined the impact of nettle fertilizer on a single crop and pathogen, we expect that these findings would be generally applicable to a wide range of commercially important crops and their associated microbial pathogens. Indeed, the numerous studies mentioned above all revealed roles for different beneficial microbes contributing to pathogen suppression in different crops. As such, future work should focus on characterizing the effects of these fertilizers on a wide range of crops and their associated pathogens, with a focus on crops that are not only commonly affected by microbial pathogens but that also typically require greater amounts of synthetic fungicides (e.g., potato)70. This would also advance the concept of “pesticide-free agriculture”71, which is increasingly recognized as a key long-term aspirational goal in the agricultural field to promote sustainable, ecologically friendly methods to control pests and increase crop yields.
Nettle fertilizer is already commonly sold for agriculture, but also in garden centers to stimulate plant growth and pathogen protection. The industrial landscape exists and could benefit from additional raw material to prepare fertilizers from plant cuttings originating from CWs. Moreover, nettle fertilizer can also be used locally to maintain green spaces by the cities that own the CWs. This promotes a circular economy in which the goods produced locally benefit local populations. For example, this work allowed the creation of a company, Holositech (www.holositech.com), which manages plant cuttings from the CWs of Montiers-sur-Saulx and surrounding villages to produce nettle fertilizer, which is used locally.
In this study, a multiomics approach combining metabolomics and amplicon metagenomics was used to decipher the mechanisms that occur during fertilizer production from polluted plant tissues. We propose fermenting nettle and reed to produce liquid fertilizers as an efficient means to remove POPs from these polluted plant tissues. In addition, these fertilizers were shown to positively impact plant growth in the presence of pathogens and therefore have commercial applications in farming and gardening. Thus, these approaches may be simple, commercially attractive solutions for the management of contaminated plant tissues originating from constructed wetlands, which are currently considered problematic, useless waste products.
Methods
Experimental design and sampling strategy
The inlet water samples and all of the plant samples were collected at a full-scale constructed wetland study site located in Montiers-sur-Saulx, France (48.541192, 5.276331). This site receives domestic municipal wastewater and does not collect rain runoff. It is composed of two vertical flow reed beds used for primary treatment. The CW has a capacity of approximately 160 people equivalents, an inlet flow rate of 190 m3/d and was set up in 2015. Three biological replicates of reed (Phragmites australis) and nettle (Urtica dioica L.) aerial parts were collected from the same bed. Five sampling points were dispatched over the bed to cover its whole surface and mixed together to obtain a mean sampling. The plants were cut at their base, and the whole aerial parts were chopped into small pieces using a manual pruner. Fertilizers were made onsite from 600 g fresh weight of plant aerial cuttings (stems and leaves from reeds and nettles) in 6 L of tap water. The tap water used for fertilizer fermentation was obtained from the municipal water tower of Montiers-sur-Saulx (48.31500, 5.16011) and was not subjected to any additional treatment. The mixture was macerated for one month in the dark at room temperature. Three 50 mL aliquots of each fertilizer were subsequently collected for further analysis. The samples were transported at 4 °C until arrival to the laboratory and frozen at −20 °C before preparation. Samples of chopped plant aerial parts and tap water were taken before maceration, transported at 4 °C until arrival to the laboratory and frozen at −20 °C before preparation.
Chemicals
Deionized water was obtained from a Direct-Q UV station (Millipore, Massachusetts, USA). Analytical-grade methanol (MeOH), ethyl acetate and isopropanol were purchased from Fisher Chemicals (New Hampshire, USA). Acetic and formic acids were purchased from Sigma-Aldrich (Missouri, USA), ammonium formate was purchased from Fluka Analytical (Missouri, USA), and NaOH was purchased from Agilent Technologies (California, USA). Deuterated abscisic acid (2H6 ABA) was obtained from OlChemIm. Analytical standards of abscisic acid (ABA), 6-benzylaminopurine (BAP), indole-3-butyric acid potassium salt (IBA), salicylic acid (SA), trans zeatin (t-zea), sucrose, fructose, glucose, arabinose, maltose, trehalose, raffinose, melezitose, cellobiose, sorbose, mannose, melibiose, methyl glucopyranoside, stachyose and tryptophan were purchased from Sigma; mannotetraose was purchased from MEGAzyme; benzoic acid (BA), and gibberellic acid (GA3) were purchased from Fluka; brassinolide (BR), cis-12-oxo-phytodienoic acid (cis-OPDA), castasterone (CS), cathasterone (CT), dihydrozeatin (dhZEA), gibberellins A1, A4, A7 and GA20 (GA1, GA4, GA7, GA20), jasmonic acid (JA), jasmonic acid-isoleucine (JA-ILE), 12-hydroxy-jasmonic acid (12-OH-JA), and orobanchol (Oro) were purchased from OlChemIm; indole-3-acetic acid (IAA) was purchased from Serva, and 6-furfurylaminopurine (kinetin, KIN), 6-(γ, γ-dimethylallylamino)purine (2iP) were purchased from Duchefa. 12-hydroxy-jasmonic acid-isoleucine (12-OH-JA-Ile), 12-carboxy-jasmonic acid (12-COOH-JA) and 12-carboxy-jasmonic acid-isoleucine(12-COOH-JA-Ile) were generous gifts from Doctor Patrick Wehrung. Analytical performance quality checks were performed with a mix of pesticides obtained from Restek (ref. 32563 for gas chromatography and refs. 31972 and 31978 for liquid chromatography).
Sample preparation for metabolite, lipid and POP analysis
The nettle and reed aerial parts were weighed to 300 mg fresh weight, ground in liquid nitrogen and extracted in 1.5 mL of cold MeOH (4 °C) spiked with 0.2 µg/mL 2H6 ABA. The samples were vortexed and left for 16 h at −20 °C. Then, the samples were centrifuged (15 min, 18,506 x g, 4 °C) and the supernatant was recovered in a glass vial. Two additional extraction steps with 1.5 mL of cold MeOH were performed with immediate recovery of the supernatant in the same glass vial. The supernatants were freeze-dried under vacuum, and the samples were recovered in 300 µL of MeOH. The inlet and tap water, nettle and reed fertilizers were measured to 50 mL, freeze-dried under vacuum and suspended in 1 mL of MeOH:H2O (90:10). Aliquots of plant and fertilizer extracts were freeze-dried and recovered in the same volume of ethyl acetate for gas chromatography coupled with mass spectrometry targeted analysis of POPs.
LC-TQ-MS/MS targeted analysis of hormones and sugars
Hormone and sugar analysis was performed via ultrahigh-performance liquid chromatography (UHPLC) on an UltiMate 3000UHPLC system (Thermo) coupled to an EvoQ Elite (Bruker) triple quadrupole (TQ) mass spectrometer equipped with an electrospray ionization (ESI) source in multiple reaction monitoring (MRM) mode to obtain fragmentation of the compounds (MS/MS) as described by Zumsteg et al., 202372. Chromatographic separation was achieved on an AcquityUPLC ® HSS T3 C18 column (2.1 × 100 mm, 1.8 μm, Waters) coupled to an Acquity UPLC HSS T3 C18 precolumn (2.1 × 5 mm, 1.8 μm, Waters). Five microliters of the samples were injected and separated using a 15 min gradient of solvents A (H2O, 0.1% formic acid) and B (MeOH, 0.1% formic acid) at 35 °C and 0.300 mL.min-1, starting with 5% B for 2 min, reaching 100% B at 10 min, holding 100% B for 3 min, returning to 5% B in 1 min and holding for initial conditions for 1 min. The cone gas (30 L/h), heated probe gas (30 L/h) and nebulizing gas (35 L/h) used was nitrogen. The cone and heated probe temperatures were 350 and 300 °C, respectively, and the capillary voltage was set at 3.5 kV. Hormones were analyzed by multiple reaction monitoring (MRM). The cone voltage, daughter ion and collision energy were determined using the MRM builder function on commercial standards. A mixture of the commercial standards of the hormones described in the “Chemicals” section served as a positive control (Supplementary Fig. 9).
For sugar analysis, chromatographic separation was performed on an AcquityUPLC ® BEH Amide column (2.1 × 100 mm, 1.7 μm, Waters) coupled to an Acquity UPLC BEH Amide precolumn (2.1 × 5 mm, 1.7 μm, Waters). Five microliters of the samples were injected and carried through the column following a 24.5 min gradient of solvent A (ACN/H2O 98/2, 0.1% triethylamine) and B (ACN/H2O 30/70, 0.1% triethylamine) at 45 °C at a flux of 0.200 mL min, starting with 2% B for reaching 58% B at 20 min, returning to 2% B in 0.5 min and holding initial conditions for 4 min. Nitrogen was used as the cone gas (30 L h), heated probe gas (30 L h) and nebulizing gas (35 L h). The cone and heated probe temperatures were 350 and 300 °C, respectively, and the capillary voltage was set at 4 kV. Sugars were analyzed by multiple reaction monitoring (MRM). A mixture of the commercial standards of the sugars described in the “Chemicals” section served as a positive control.
Data processing was performed with MS Data Review 8.2 software (Bruker). Hormones and sugars were identified to be at level 1 of the Schymanski classification73 on the basis of their retention time, mass of the parent ions and presence of at least 1 daughter ion. The signal-to-noise ratio (S/N) was set to 5 (considered the limit of detection (LOD)), the retention time window was set to 0.25 min, and the mass variation was set to 0.7 Da. For quantification, an S/N ratio of 5 was considered the limit of quantification (LOQ) as already published74.
GC-TQ-MS/MS targeted analysis of POPs
Gas chromatography (GC) was performed on a triple quadrupole detector (TQ, SCION 436-MS, Bruker) in multiple reaction monitoring (MRM) mode as described by Villette et al., 202375. An Rxi®−5Sil MS column (30 m × 0.25 mm, 0.25 µm, Restek) with a constant flow of helium gas (1 mL/min, Alphagaz 2, Air Liquide) was used. The injector was operated at 280 °C, the oven was set at 70 °C for 0.7 min, and the temperature was increased to 180 °C at 30 °C/min and then to 300 °C at 10 °C/min, with a stabilization time of 2 min. The electron impact energy for fragmentation was 70 eV, and 328 POPs (pesticides, toxicants, and polycyclic aromatic hydrocarbons) were targeted (Supplementary Data 8). Analytical quality checks were performed using a mixture of pesticides to assess the retention times (ref 32563, Restek) (Supplementary Fig. 10).
Data processing was performed with MS Data Review 8.2 software (Bruker). POPs were identified to be at level 1 of the Schymanski classification73 on the basis of their retention time, mass of the parent ions and presence of at least 2 daughter ions. The signal-to-noise ratio (S/N) was set to 3 (considered the limit of detection (LOD)), the retention time window was set to 0.25 min, and the mass variation was set to 0.5 Da. For quantification, an S/N ratio of 5 was considered the limit of quantification (LOQ) as already published74.
LC-HRMS/MS targeted analysis of POPs
Targeted analysis of POPs was performed using liquid chromatography (LC) coupled with high-resolution mass spectrometry (HRMS) on a Dionex Ultimate 3000 (Thermo, Massachusetts, USA) coupled to a Q-TOF Impact II (Bruker, Germany) as described by Maurer et al., 202176. This method allows for the identification of POPs based on their retention time, exact mass and fragments using a database containing 2072 compounds (848 pesticides and 1224 toxicants; Bruker Pesticides&Tox Screener, Supplementary Data 9). Briefly, a C18 column (AcclaimTM RSLC 120 C18, 2.2 µm, 120 A, 2.1 × 100 mm, Dionex bonded silica products) equipped with an Acquity UPLC- BEH C18 precolumn (2.1 × 5 mm, 1.7 µm) was operated at 35 °C, with a gradient of solvent A (H2O/MeOH 90/10, 0.01% formic acid, 314 mg.L−1 ammonium formate) and solvent B (MeOH, 0.01% formic acid, 314 mg.L−1 ammonium formate). The spectrometer was operated at 2 Hz in positive ion mode using broad-band collision-induced dissociation (bbCID) fragmentation mode over a mass range from 30 to 1000 Da. Analytical quality checks were performed using a mixture of pesticides obtained from Restek (refs 31972 and 31978, Restek) (Supplementary Fig. 11). The spectrometer was calibrated on the [M + H]+ form of 57 reference ions of a calibration solution in high-precision calibration (HPC) mode with a standard deviation below 1 ppm. Recalibration of each raw data was performed during data analysis using the calibration segment. The data processing was performed using TASQ 1.4 (Bruker) to identify POPs to the level 1 of the Schymanski classification73. The limit of detection (LOD) was considered as a signal-to-noise (S/N) ratio greater than 3. The parameters used for identification were a retention time range of 0.3 min, mSigma value less than 30, mass variation less than 3 ppm and matching fragment ions. For quantification, a S/N ratio of 5 was considered the limit of quantification (LOQ) as already published74.
LC-HRMS/MS nontargeted analysis of metabolites, lipids and POPs
Nontargeted analysis of metabolites, lipids and POPs was performed using liquid chromatography (LC) coupled with high-resolution mass spectrometry (HRMS) on a DioneX Ultimate 3000 (Thermo, Massachusetts, USA) coupled with a Q-TOF Impact II (Bruker, Germany) as described by Zumsteg et al., 202372. Briefly, the samples were ran through an AcquityUPLC ® HSS T3 C18 column (2.1 × 100 mm, 1.8 μm, Waters) equipped with an Acquity UPLC HSS T3 C18 precolumn (2.1 × 5 mm, 1.8 μm, Waters) using a gradient of solvent A (H2O, 0.1% formic acid) and solvent B (MeOH, 0.1%) as described above for hormone analysis, starting with 2% solvent B. The spectrometer was equipped with an electrospray ionization (ESI) source and operated in positive ion mode over a mass range from 20 to 1000 Da with a spectra rate of 8 Hz in AutoMS/MS fragmentation mode. The calibration of the instrument was performed as described above for POPs analysis. The data processing was performed using Metaboscape 4.0 (Bruker) to obtain a feature list corresponding to level 5 (exact mass) in the Schymanski classification, using a minimum intensity threshold of 10000, a minimum peak length of 3 spectra, a signal-to-noise ratio (S/N) of 3 (considered the limit of detection, LOD) and a correlation coefficient threshold of 0.8. The [M + H]+, [M+Na]+, [M + NH4]+ and [M + K]+ ions were authorized as possible primary ions. Only the features detected in at least 80% of the samples in at least one group were kept. The features were annotated using analyte lists (Schymanski level 3) derived from publicly available databases: the Norman Substance Database (https://www.norman-network.com/nds/susdat/), KNApSAcK (http://www.knapsackfamily.com/KNApSAcK_Family/), Natural Products Atlas (https://www.npatlas.org/), Phenol Explorer (http://phenol-explorer.eu/), T3DB, PlantCyc (https://plantcyc.org/), FoodDB (https://foodb.ca/), YMDB, ECMDB, LipidMaps (https://www.lipidmaps.org/), HMDB. Spectral libraries were used to achieve Schymanski level 2 annotations: Bruker MetaboBASE Personal Library 3.0, Mass Bako of North America (MoNA_LCMSMS), and MSDIAL-LipidsDBs VS34. The parameters used for annotation were a 3ppm deviation on the mass, a mSigma value (assessing good fitting of the isotopic patterns) below 30, and an MS/MS score below 800 for the spectral libraries. The NP Classifier classification was used at the pathway level77 to determine metabolite and lipid families. POP families were determined according to the Norman Substance Database classification. POPs transformation products were predicted in silico following 79 biological rules in the MetabolitePredict software (Bruker) as already published55.
Effects of fertilizers on plant growth and pathogen resistance
Tomato cv. Marmande seeds were sown in vermiculite moistened with tap water in a 30 × 20 cm container and then transferred to a climatic chamber: 23 °C day/17 °C night - photoperiod of 16 h light/8 h dark. One week after sowing, the seedlings were transplanted into 5.5 cm round pots in a 50/50 - v/v potting soil (Floradur B seed, Floragard) - vermiculite (Floragard vermiculite, 2-3 mm) mixture. Pots were watered once a week with tap water and transferred to a climatic chamber under the same conditions as described previously. Two weeks after transfer to round pots, each pot was drenched either with 15 mL of water (control) or with each of the fertilizers (nettle fertilizer, reed fertilizer, commercial fertilizer Algoflash Naturasol) prepared at 10% in tap water. A second treatment was carried out 6 days after the 1st treatment with 15 ml of tap water (control) or each 10% liquid fertilizer solution. Twenty-four hours after the 2nd treatment, the plants were transplanted into 8 × 8 x 8 cm pots containing a potting soil-vermiculite mixture (50/50 - v/v) contaminated by adding 2.5% (w/w) of a mixture of millet and barley seeds (1/1 – w/w) previously inoculated with R. solani AG3. A control condition was prepared by adding 2.5% (w/w) of a mixture of millet and barley seeds not inoculated with R. solani AG3 to the same potting soil-vermiculite mixture. After transplanting, the tomato plants (12 biological replicates per condition) were watered with tap water and placed in a climatic chamber under the same conditions as those described above. Disease assessment of the aerial system was performed 4 weeks after the last transplantation via a 0–5 scale as previously published78 (0 = no lesion, 1 = disease affecting ≤ 10% of total leaf area, 2 = disease affecting 11–25% of total leaf area, 3 = disease affecting 26–50% of total leaf area, 4 = disease affecting 51–75% of total leaf area and 5 = completely necrotic leaves of total leaf area). Six weeks after the last transplantation, the roots were thoroughly washed to remove any soil adhering to them. The fresh root biomass of each plant was then determined, and disease assessment of the roots was performed via a 0-6 scale as previously published78 (0 = no lesion, 1 = small lesions on primary roots, 2 = discoloration up to 50% of primary root, 3 = discoloration 51–75% of primary root, 4 = discoloration > 75% and necrosis covering up to 30% of primary roots, 5 = necrosis covering up to 31–60% of primary roots and 6 = necrosis covering > 61% or dead root).
To assess the impact of the products on plant growth in the absence of pathogens, the entire growth and transplanting phase were the same, right up to the transplantation into soil inoculated with R. solani. After being transplanted into 8 x 8 x 8 pots, the plants were grown under the same climatic conditions as those described above. Twelve days after transplanting, the following observations (nondestructive and destructive) were made: number of leaves, chlorophyll content, length and fresh weight of the aerial tissues and roots of the tomato plants in the absence of pathogens. The number of leaves corresponds to the number of fully developed leaves and the sum of the latter with those in development; the length of the plant corresponds to the length between the collar and the apical part of the plant, for both the aerial and the root parts. Aerial and root biomass measurements were carried out with a precision weighing scale (Precisa XT2200 C; 0. 5 g - 2200 g; e = 0.1 g; d = 0.01 g); the chlorophyll content was measured via a SPAD 502 Plus® (Konica Minolta).
In vitro microbial analysis and confrontation test
For in vitro observation of microbial communities in nettle and reed fertilizers, 10% nettle and reed fertilizers were spread onto 1.5% malt agar (15 g/L malt extract, 15 g/L agar, Difco) and LB (Lennox) agar (10 g/L Bacto peptone, 5 g/L yeast extract, 5 g/L NaCl, and 12 g/L agar, Merck, Sigma Aldrich) to allow fungal and bacterial communities to develop in the dark at 25 °C for 7 days or 28 °C for 3 days, respectively. The in vitro confrontation test was performed in 90 mm Petri dishes on 1.5% malt agar supplemented or not with 50 µg/mL chloramphenicol (Merck, Sigma Aldrich) and 50 µg/mL streptomycin sulfate (Merck, Sigma Aldrich). Fifty microliters of sterile distilled water (control) or 10% nettle fertilizer was added to four symmetrical 8 mm diameter wells, and a 5 mm mycelial plug of R. solani AG3 was inoculated at the center of each Petri dish. Pictures were taken after four days of incubation at 5 °C in the dark.
DNA extraction and sequencing library construction for microbial community profiling
DNA extraction and isolation for targeted bacterial (16S rRNA) and fungal (ITS) sequencing were performed from two batches of liquid fertilizers generated from nettle/reed using a ZymoBIOMICS DNA Mini Kit by Microsynth AG. Two batches were processed to obtain sufficient samples for subsequent downstream analyses in both the 16S rRNA and ITS datasets. Extraction, lysis and DNA isolation were performed according to the manufacturer’s recommendations with bead beating using a FastPrep-24 instrument (MP Biomedicals; 4 cycles of 45 s at speed 4 followed by 1 cycle of 45 s at speed 6.5). A total of 400 µl of raw extract was prepared for DNA isolation, and we also attempted extractions from the same volume of unprocessed tap water. The concentration of the isolated DNA was assessed via PicoGreen measurement (Quant-iT™ PicoGreen™ dsDNA Assay Kit, Thermo Fisher). To sequence the V3-V4 region of the bacterial 16S rRNA gene, two-step, Nextera barcoded PCR libraries were generated using 341 F (5′- CCT ACG GGN GGC WGC AG -3′) and 805 R (5′- GAC TAC HVG GGT ATC TAA TCC -3′) with 20 PCR cycles for the first step and 10 PCR cycles for the second step. To sequence the internal transcribed spacer (ITS2) regions of the fungal 18S rRNA gene, two-step, Nextera barcoded PCR libraries were generated using ITS3 (5′- GCA TCG ATG AAG AAC GCA GC -3′), and ITS4 (5′- TCC TCC GCT TAT TGA TAT GC -3′) with 20 PCR cycles for the first step and 15 PCR cycles for the second step. The amplicon libraries were subsequently sequenced on an Illumina MiSeq using a V2 500-cycle kit. Extraction for shotgun metagenomic profiling was done in triplicate on nettle fertilizer samples using an MP Biomedicals FastDNA SPIN Kit for Soil and a FastPrep 5 G instrument. Metagenomic DNA was fragmented and sequencing libraries constructed using an Ion Xpress Plus Fragment Library kit, with fragmentation timings optimized to generate read inserts of 150 bp. Fragment profiles were confirmed on an Agilent Bioanalyzer. The replicates were each sequenced on separate 540 chips using an Ion S5 sequencer with the addition of an external calibration standard to generate high-quality sequencing data for subsequent analysis.
Metagenomics data analysis
The 16S rRNA gene and ITS datasets were analyzed separately as detailed below. For the multiplexed reads, the locus-specific primers were removed using cutadapt. Reads <200 bp or without an identified primer sequence were removed from further analysis. Subsequent analysis was performed in R using the dada2 and phyloseq packages. The reads were trimmed by 5 bp at the 3’ end, screened for potential phiX contamination and filtered to remove reads with N bases or with an expected error rate >1. The reads were denoised and merged, and chimeras were removed using dada2 with pooling to identify amplicon sequence variants (ASVs). The taxonomy of ASVs was assigned via the RDP naive Bayesian classifier algorithm against either the Microbial Database for Activated Sludge (MiDAS; https://www.midasfieldguide.org/guide) 5.1 database (16S rRNA gene) or the UNTIE 9.0 (general release 27.10.2022) fungal database (ITS). ASVs with perfect, full-length alignments matching a single species in either the NCBI 16S rRNA gene or ITS (downloaded on April 11th, 2024) had their species annotation updated to match NCBI taxonomies. The resulting ASVs were also screened for potential off-target hits to the reed and nettle ITS regions. ASVs with BLAST alignments at >90% identity and lengths >300 were considered off-target amplicons and removed from further analysis. Microbial phylogenetic distances were determined by placing 16S rRNA gene ASV sequences within the overall MiDAS guide tree using SEPP. The datasets were normalized to 28,000 reads where appropriate for analysis, and samples with <28,000 reads were not considered further. To note, the tap water datasets resulted in few reads passing quality control and off-target filtering (<1650 reads/replicate for 16S, <1200 reads/replicate for ITS) and thus they were discarded from further consideration. DESeq2 was used to determine the differentially abundant 16S rRNA gene/ITS ASVs and control for the experiment (Supplementary Data 2). The bacterial genetic potential and each ASV’s predicted contribution were calculated using PICRUSt2 and mapped to KEGG terms and enzyme classification (EC). Shotgun metagenomic reads from the replicates were quality filtered and merged prior to contig assembly with SPAdes79 using the recommended parameters for Ion Torrent reads. Resulting contigs had their taxonomy assigned using Kraken280 with the PlusPFP (standard plus protozoa, fungi and plant) database (December 2024 version). (Supplementary Data 10). To improve gene predictions from fungal communities, we identified taxa with existing gene prediction models from related species as previously proposed81. Thus, gene prediction from contigs belonging to the orders Sordariales, Hypocreales, and Magnaporthales was performed with AUGUSTUS82 using models from Neurosporsa crassa, Fusarium graminearum, and Magnaporthe grisea, respectively. Contigs from the phylum Basidiomycota were predicted using the AUGUSTUS Coprinus cinereus model, and contigs from the genus Urtica (fertilizer source) used the Arabidopsis model. All other contigs (bacterial/unclassified/remaining Eukaryotes) had their gene predictions made using Prodigal83 in anonymous (metagenomic) mode. Raw sequencing reads from each replicate were mapped back to the contigs using bwa-mem284, and gene coverage was quantified using HTSeq85 (Supplementary Data 11). Predicted proteins were further annotated with GhostKOALA86 to assign KEGG terms and EC identifiers. ECs predicted to be involved in the metabolism of specific metabolites were identified via a literature review, and enzymes involved in compound metabolism were identified via KEGG. Information on microbial growth phenotypes (e.g., oxygen requirements, cellulolytic activity, sugar fermentation, nitrogen affinity and POP resistance) was obtained from the Bacterial Diversity Metadatabase (BacDive, https://bacdive.dsmz.de/) or manually curated from the literature if not available in this database.
Nutrient analysis
The nutrient contents (nitrogen NTK, phosphorus, CaO, MgO and K2O) were analyzed via the French standards NF EN 25663, NF EN ISO 6878, NF 13346 and NF 11885, respectively.
Statistics & reproducibility
The effects of fertilizers on plant growth and pathogen resistance were statistically analyzed. Twelve replicates were used to conduct the experiment and check reproducibility. Owing to the small number of replicates, a nonparametric statistical test was used: statistical analysis was performed through a Kruskal-Wallis test. The effects were considered significantly different when the p value was < 0.05. DESeq2 was used to determine the differentially abundant 16S rRNA gene/ITS ASVs and control for the experiment with fold changes >2 and Benjamini-Hochberg corrected p-values < 0.001 considered significant.
To allow a direct comparison of the abundance of the features detected in plants and fertilizers, the peak areas obtained from mass spectrometry analysis were normalized to the original amounts of plant material used for each sample preparation (300 mg for plant aerial parts, 5 g for 50 mL of fertilizers).
No statistical method was used to predetermine sample size. No data were excluded from the analyses except for 16S/ITS sequencing experiments with final filtered reads <28,000. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The LC-HRMS/MS raw data generated in this study have been deposited in the MassIVE database under accession code MSV000094525 [https://doi.org/10.25345/C5K64B45Q]. LC-HRMS/MS processed data are available in Supplementary Data 1. The raw demultiplexed reads from the 16S rRNA, ITS and metagenomic sequencing experiments generated in this study have been deposited to NCBI SRA under accession code PRJNA1107565. Source data are provided in this paper.
References
Vymaza, J. The historical development of constructed wetlands for wastewater treatment. Land 11, https://doi.org/10.3390/land11020174 (2022).
Carvalho, P. N., Basto, M. C. P., Almeida, C. M. R. & Brix, H. A review of plant–pharmaceutical interactions: from uptake and effects in crop plants to phytoremediation in constructed wetlands. Environ. Sci. Pollut. Res. 21, 11729–11763 (2014).
Maurer, L., Villette, C., Zumsteg, J., Wanko, A. & Heintz, D. Large scale micropollutants and lipids screening in the sludge layers and the ecosystem of a vertical flow constructed wetland. Sci. Total Environ. 746, 141196 (2020).
Kalu, C. M., Rauwane, M. E. & Ntushelo, K. Microbial spectra, physiological response and bioremediation potential of phragmites australis for agricultural production. Front. Sustain. Food Syst. 5, 1–13 (2021).
Cui, X. et al. Biochar from constructed wetland biomass waste: A review of its potential and challenges. Chemosphere 287, 132259 (2022).
Wang, Y. C. et al. Bioenergy production potential for aboveground biomass from a subtropical constructed wetland. Biomass Bioenergy 35, 50–58 (2011).
Barbera, A. C. et al. Growth and biomass production of different plant species in two different constructed wetland systems in Sicily. Desalination 246, 129–136 (2009).
Vymazal, J. Vegetation development in subsurface flow constructed wetlands in the Czech Republic. Ecol. Eng. 61, 575–581 (2013).
Ministère de la transition écologique, de la biodiversité, de la forêt, de la mer et de la pêche. Portail sur l’assainissement collectif. https://assainissement.developpement-durable.gouv.fr/pages/data/listeSteu.php (2024).
Zhu, S. X. et al. Effects of plant diversity on biomass production and substrate nitrogen in a subsurface vertical flow constructed wetland. Ecol. Eng. 36, 1307–1313 (2010).
Li, T. S. C. Use of stinging nettle as a potential organic fertilizer for herbs. J. Herbs, Spices Med. Plants 2, 93–98 (1994).
Domenico, P. Bat guano and Nettle slurry (Urtica dioica L.) used as biostimulants on Delosperma cooperi and Sedum rubrotinctum plants. World J. Adv. Res. Rev. 03, 017–023 (2019).
Maričić, B. et al. Stinging nettle (Urtica dioica l.) as an aqueous plant-based extract fertilizer in green bean (phaseolus vulgaris l.) sustainable agriculture. Sustainability 13, https://doi.org/10.3390/su13074042 (2021).
Peterson, R. & Jensen, P. Effects of nettle water on growth and mineral nutrition of plants. I. composition and properties of nettle water. Biol. Agric. Hortic. 2, 303–314 (1985).
Nygaard Sorensen, J. & Thorup-Kristensen, K. Plant-based fertilizers for organic vegetable production. J. Plant Nutr. Soil Sci. 174, 321–332 (2011).
Parlement européen et Conseil de l’Union Européenne Directive 2013/39/UE du Parlement européen et du Conseil du 12 août 2013 modifiant les directives 2000/60/CE et 2008/105/CE en ce qui concerne les substances prioritaires pour la politique dans le domaine de l’eau. J. des. Communautés Eur. du 2013, 1–17 (2013).
EUROPEAN UNION Regulation (Eu) 2019/1009 of the European Parliament and of the Council of 5 June 2019. J. Eur. Union 2019, 1–114 (2019).
Ministère de l’Environnement. Note technique du 12 août 2016 relative à la recherche de micropolluants dans les eaux brutes et dans les eaux usées traitées de stations de traitement des eaux usées et à leur réduction. (2016).
Ministère de la transition écologique. Note technique du 29 septembre 2020 relative aux objectifs nationaux de réduction des émissions, rejets et pertes de substances dangereuses dans les eaux de surface et à leur déclinaison dans les SDAGE 2022−2027 (2020).
Dolores Gómez-Sánchez, M., Sánchez, R., Espinosa, E., Rosal, A. & Rodríguez, A. Production of cellulosic Pulp from reed (Phragmites australis) to produce paper and paperboard. Bioprocess Eng. 1, 65–68 (2017).
Smuga-Kogut, M., Szymanowska-Powałowska, D., Markiewicz, R., Piskier, T. & Kogut, T. Ionic liquid pretreatment of stinging nettle stems and giant miscanthus for bioethanol production. Sci. Rep. 11, 1–11 (2021).
Jiménez, D. J., Korenblum, E. & Van Elsas, J. D. Novel multispecies microbial consortia involved in lignocellulose and 5-hydroxymethylfurfural bioconversion. Appl. Microbiol. Biotechnol. 98, 2789–2803 (2014).
Shahab, R. L. et al. A heterogeneous microbial consortium producing short-chain fatty acids from lignocellulose. Science 369, 80 (2020).
Toledo Ramos, D., Luis Busi da Silva, M., Wolfgang Nossa, C., Alvarez, P. J. J. & Corseuil, H. X. Assessment of microbial communities associated with fermentative-methanogenic biodegradation of aromatic hydrocarbons in groundwater contaminated with a biodiesel blend (B20). Biodegradation 5, 681–691 (2014).
Anderson, E. L., Jang, J., Venterea, R. T., Feyereisen, G. W. & Ishii, S. Isolation and characterization of denitrifiers from woodchip bioreactors for bioaugmentation application. J. Appl. Microbiol. 129, 590–600 (2020).
Iqtedar, M. et al. Bioethanol production from urban cellulosic waste employing alcaligenes faecalis hi-1 isolated from gut of termite heterotermes indicola. Biosci. J. 37, 1–10 (2021).
Lang, J. et al. Genomic and resistome analysis of Alcaligenes faecalis strain PGB1 by Nanopore MinION and Illumina Technologies. BMC Genom. 23, 1–14 (2022).
Magiorakos, A. P. et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281 (2012).
Nouri, F. et al. Prevalence of common nosocomial infections and evaluation of antibiotic resistance patterns in patients with secondary infections in Hamadan, Iran. Infect. Drug Resist. 13, 2365–2374 (2020).
Bharali, P., Das, S., Konwar, B. K. & Thakur, A. J. Crude biosurfactant from thermophilic Alcaligenes faecalis: Feasibility in petro-spill bioremediation. Int. Biodeterior. Biodegrad. 65, 682–690 (2011).
Singha, L. P., Kotoky, R. & Pandey, P. Draft genome sequence of alcaligenes isolated from crude oil-contaminated soil. 5, https://doi.org/10.1128/genomea.01346-17 (2017).
Shah, P. D., Dave, S. R. & Rao, M. S. Enzymatic degradation of textile dye Reactive Orange 13 by newly isolated bacterial strain Alcaligenes faecalis PMS-1. Int. Biodeterior. Biodegrad. 69, 41–50 (2012).
Kumar, A. et al. Optimization of culture condition for growth and phenol degradation by Alcaligenes faecalis JF339228 using Taguchi Methodology. Desalin. Water Treat. 51, 3153–3163 (2013).
John, R. C., Essien, J. P., Akpan, S. B. & Okpokwasili, G. C. Polycyclic aromatic hydrocarbon-degrading bacteria from aviation fuel spill site at Ibeno, Nigeria. Bull. Environ. Contam. Toxicol. 88, 1014–1019 (2012).
Li, X., Xu, Q.-M., Cheng, J.-S. & Yuan, Y.-J. Improving the bioremoval of sulfamethoxazole and alleviating cytotoxicity of its biotransformation by laccase producing system under coculture of Pycnoporus sanguineus and Alcaligenes faecalis. Bioresour. Technol. 220, 333–340 (2016).
Arthon, S. & Yi-Tin, W. Arsenite oxidation by alcaligenes faecalis Strain O1201. J. Environ. Eng. 131, 1293–1301 (2005).
Uttarotai, T. et al. Genome characterisation of an isoprene-degrading alcaligenes sp. isolated from a tropical restored forest. Biology 11, 1–15 (2022).
Quiroz-Castañeda, R. E. et al. Identification of a New Alcaligenes faecalis Strain MOR02 and Assessment of Its Toxicity and Pathogenicity to Insects. Biomed. Res. Int. 2015, https://doi.org/10.1155/2015/570243 (2015).
Ju, S. et al. Alcaligenes faecalis ZD02, a Novel Nematicidal bacterium with an extracellular serine protease virulence factor. 82, 2112–2120 (2016).
Xing, D. et al. Ethanoligenens harbinense gen. nov., sp. nov., isolated from molasses wastewater. Int. J. Syst. Evol. Microbiol. 56, 755–760 (2006).
Van Dyke, M. I. & McCarthy, A. J. Molecular biological detection and characterization of Clostridium populations in municipal landfill sites. Appl. Environ. Microbiol. 68, 2049–2053 (2002).
Desvaux, M., Guedon, E. & Petitdemange, H. Cellulose catabolism by Clostridium cellulolyticum growing in batch culture on defined medium. Appl. Environ. Microbiol. 66, 2461–2470 (2000).
Venice, F., Spina, F., Davolos, D., Ghignone, S. & Varese, G. C. The genomes of Scedosporium between environmental challenges and opportunism. IMA Fungus 14, https://doi.org/10.1186/s43008-023-00128-3 (2023).
Rougeron, A. et al. Ecology of scedosporium species: Present knowledge and future research. Mycopathologia 183, 185–200 (2018).
Elzakey, E. M., El-Sabbagh, S. M., Eldeen, E. E. S. N., Adss, I. A. A. & Nassar, A. M. K. Bioremediation of chlorpyrifos residues using some indigenous species of bacteria and fungi in wastewater. Environ. Monit. Assess. 195, 779 (2023).
Matúš, P., Littera, P., Farkas, B. & Urík, M. Review on Performance of aspergillus and penicillium species in biodegradation of organochlorine and organophosphorus Pesticides. Microorganisms 11, https://doi.org/10.3390/microorganisms11061485 (2023).
de Vries, R. P. & Visser, J. Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol. Mol. Biol. Rev. 65, 497–522 (2001).
Miao, Y. et al. Genome-wide transcriptomic analysis of a superior biomass-degrading strain of A. fumigatus revealed active lignocellulose-degrading genes. BMC Genom. 16, 1–20 (2015).
Khan, A. L. et al. Gibberellins producing endophytic Aspergillus fumigatus sp. LH02 influenced endogenous phytohormonal levels, isoflavonoids production and plant growth in salinity stress. Process Biochem. 46, 440–447 (2011).
Mohsin, H., Shafique, M., Zaid, M. & Rehman, Y. Microbial biochemical pathways of arsenic biotransformation and their application for bioremediation. Folia Microbiol. 68, 507–535 (2023).
Narayanan, M., Ali, S. S. & El-Sheekh, M. A comprehensive review on the potential of microbial enzymes in multipollutant bioremediation: Mechanisms, challenges, and future prospects. J. Environ. Manag. 334, 117532 (2023).
Megharaj, M., Ramakrishnan, B., Venkateswarlu, K., Sethunathan, N. & Naidu, R. Bioremediation approaches for organic pollutants: A critical perspective. Environ. Int. 37, 1362–1375 (2011).
Marsik, P. et al. Metabolism of ibuprofen in higher plants: A model Arabidopsis thaliana cell suspension culture system. Environ. Pollut. 220, 383–392 (2017).
He, Y. et al. Metabolism of ibuprofen by Phragmites australis: uptake and phytodegradation. Environ. Sci. Technol. 51, 4576–4584 (2017).
Villette, C., Maurer, L., Wanko, A. & Heintz, D. Xenobiotics metabolization in Salix alba leaves uncovered by mass spectrometry imaging. Metabolomics 15 , https://doi.org/10.1007/s11306-019-1572-8 (2019).
Zour, E., Lodhi, S. A., Nesbitt, R. U., Silbering, S. B. & Chaturvedi, P. R. Stability studies of gabapentin in aqueous solutions. Pharm. Res. Off. J. Am. Assoc. Pharm. Scientists 9, 595–600 (1992).
Pieterse, C. M. J. et al. Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 52, 347–375 (2014).
Chialva, M. et al. Native soils with their microbiotas elicit a state of alert in tomato plants. N. Phytol. 220, 1296–1308 (2018).
Danquah, C. A., Minkah, P. A. B., Junior, I. O. D., Amankwah, K. B. & Somuah, S. O. Antimicrobial Compounds from Microorganisms. Antibiotics 11, 1–20 (2022).
Gwinn, K. D. Chapter 7 - Bioactive Natural Products in Plant Disease Control. (Elsevier, 2018).
Vannier, N., Agler, M. & Hacquard, S. Microbiota-mediated disease resistance in plants. PLoS Pathog. 15, 1–7 (2019).
Neerackal, G. M. et al. Potential application of Alcaligenes faecalis strain No. 4 in mitigating ammonia emissions from dairy wastewater. Bioresour. Technol. 206, 36–42 (2016).
Amer, O. A. et al. Exploring new marine bacterial species, Alcaligenes faecalis Alca F2018 valued for bioconversion of shrimp chitin to chitosan for concomitant biotechnological applications. Int. J. Biol. Macromol. 196, 35–45 (2022).
Basharat, Z., Yasmin, A., He, T. & Tong, Y. Genome sequencing and analysis of Alcaligenes faecalis subsp. phenolicus MB207. Sci. Rep. 8, 1–10 (2018).
Liu, X. et al. The genome sequence of Alcaligenes faecalis NBIB-017 contains genes with potentially high activities against Erwinia carotovora. Genome Announc. 4, 10–11 (2016).
Ryan, R. P. et al. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525 (2009).
Raio, A. et al. Stenotrophomonas rhizophila Ep2.2 inhibits growth of Botrytis cinerea through the emission of volatile organic compounds, restricts leaf infection and primes defense genes. Front. Plant Sci. 14, 1–17 (2023).
Wei, Z. et al. Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 6, (2015).
Durán, P. et al. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 175, 973–983.e14 (2018).
Zhang, Y. et al. Pesticide use is affected more by crop species than by crop diversity at the cropping system level. Eur. J. Agron. 159, https://doi.org/10.1016/j.eja.2024.127263 (2024).
Jacquet, F. et al. Pesticide-free agriculture as a new paradigm for research. Agron. Sustain. Dev. 42, 1–24 (2022).
Zumsteg, J., Bossard, E., Gourguillon, L., Villette, C. & Heintz, D. Comparison of nocturnal and diurnal metabolomes of rose flowers and leaves. Metabolomics 20, 1–12 (2023).
Schymanski, E. L. et al. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol. 48, 2097–2098 (2014).
Villette, C. et al. In situ localization of micropollutants and associated stress response in Populus nigra leaves. Environ. Int. 126, 523–532 (2019).
Villette, C., Maurer, L., Zumsteg, J. & Heintz, D. Mass spectrometry imaging for biosolids characterization to assess ecological or health risks before reuse. Nat. Commun. 14, 4244 (2023).
Maurer, L. et al. Towards a model for road runoff infiltration management. Npj Clean Water 4, (2021).
Kim, H. W. et al. NPClassifier: A deep neural network-based structural classification tool for natural products. J. Nat. Prod. 84, 2795–2807 (2021).
Drizou, F., Graham, N. S., Bruce, T. J. A. & Ray, R. V. Development of high-throughput methods to screen disease caused by Rhizoctonia solani AG 2-1 in oilseed rape. Plant Methods 13, 1–14 (2017).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De novo assembler. Curr. Protoc. Bioinforma. 70, e102 (2020).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 1–13 (2019).
Belliardo, C. et al. Improvement of eukaryotic protein predictions from soil metagenomes. Sci. Data 9, 1–14 (2022).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. Nat. Commun. 6, 1–8 (2010).
Vasimuddin, M., Misra, S., Li, H. & Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. in2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) 314–324 (2019).
Anders, S., Pyl, P. T. & Huber, W. HTSeq — a Python framework to work with high-throughput sequencing data. 31, 166–169 (2015).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Acknowledgements
DH is supported by grants 251174 and 199015 from Agence de l’Eau Seine Normandie/Codecom Portes de Meuse/European Research Council. A.S. is supported by Genome Canada and Ontario Genomics Institute (OGI-149), the Canadian Institutes of Health Research (ECD-144627), and the Ontario Ministry of Economic Development and Innovation (Project 13440). The metagenomic bioinformatic analysis was enabled in part by Compute Ontario and the Digital Research Alliance of Canada RRG allocation to A.S. We thank Alexandre Laflotte and Maxime Duhamel from providing the nettle and reed fertilizers. We thank Dr Patrick Saindrenan for his input about salicylic acid functions in plants. Figure 1a and illustrations in Fig. 6 were created with BioRender.com; Fig. 5a, b was created with SankeyMATIC.
Author information
Authors and Affiliations
Contributions
S.B. and D.H. conceived and designed the experiments; C.V., J.Z., L.M., T.B., R.R., K.F., A.C., J.B., and D.H. performed the experiments; J.B., C.V., J.Z., L.M., T.B., R.R., K.F., and A.C. analyzed the data; S.B., A.S., and D.H. contributed materials/analysis tools; J.B., C.V., L.M., A.S., and D.H. wrote the paper. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Atef Nassar, Tanvi Singh and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Butcher, J., Villette, C., Zumsteg, J. et al. Microbial bioremediation of persistent organic pollutants in plant tissues provides crop growth promoting liquid fertilizer. Nat Commun 16, 5768 (2025). https://doi.org/10.1038/s41467-025-60918-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60918-8
This article is cited by
-
Metagenomics and its impact on environmental and therapeutic microbiology
Archives of Microbiology (2026)
-
Performance evaluation and technology adoption communication of cucumber in hydroponic system with liquid fertilizer from Arachis hypogea biomass
Discover Applied Sciences (2025)
