
Abstract
Almost all individuals with Down Syndrome (DS) develop Alzheimer’s disease (AD) by mid to late life. However, the degree to which AD in DS shares pathological changes with sporadic late-onset AD (LOAD) and autosomal dominant AD (ADAD) beyond core AD biomarkers such as amyloid-β (Aβ) and tau is unknown. Here, we used proteomics of cerebrospinal fluid from individuals with DS (n = 229) in the Down Alzheimer Barcelona Neuroimaging Initiative (DABNI) cohort to assess the evolution of AD pathophysiology from asymptomatic to dementia stages and compared the proteomic biomarker changes in DS to those observed in LOAD and ADAD. Although many proteomic alterations were shared across DS, LOAD, and ADAD, DS demonstrated more severe changes in immune-related proteins, extracellular matrix pathways, and plasma proteins likely related to blood-brain barrier dysfunction compared to LOAD. These changes were present in young adults with DS prior to the onset of Aβ or tau pathology, suggesting they are associated with trisomy 21 and may serve as risk factors for DSAD. DSAD showed an earlier increase in markers of axonal and white matter pathology and earlier changes in markers potentially associated with cerebral amyloid angiopathy compared to ADAD. The unique features of DSAD may have important implications for treatment strategies in this population.
Similar content being viewed by others

Introduction
Down syndrome (DS), caused by triplication of chromosome 21, is the most common genetic form of intellectual disability, affecting approximately 1 in 1000 live births1. Advances in health care for individuals with DS have significantly extended life expectancy in this population. However, the increase in life expectancy up to the seventh decade is now limited by Alzheimer’s disease (AD) dementia, which affects more than 90% of individuals with DS by this age, and has become the leading cause of mortality in this population2,3,4. Trisomy 21 leads to triplication of the APP gene, resulting in overproduction of the amyloid-β (Aβ) peptide and the development of Aβ plaques. By age 40, all individuals with DS develop extensive AD neuropathology5,6.
Early research on DS neuropathology was pivotal in the development of the AD amyloid hypothesis. Glenner and Wong first purified the Aβ peptide from DS cerebrovasculature and found it homologous to the Aβ protein from late-onset AD (LOAD) brains, linking chromosome 21 to AD neuropathology7. Later, mutations in APP on chromosome 21, which increase Aβ42 peptide production, were identified as a cause of ADAD8,9, establishing a shared pathophysiology of Aβ dyshomeostasis among LOAD, ADAD, and DSAD. Mutations in the APP, PSEN1, and PSEN2 genes in ADAD also lead to early β-amyloidosis and subsequent dementia5,6. Existing fluid and imaging biomarkers for AD show strikingly similar changes between ADAD and DSAD5,10. However, despite these similarities in core AD biomarkers between DSAD and ADAD, DS is associated with neurodevelopmental abnormalities and other conditions, resulting in differences compared to the general population in brain structure5,6, immune function11,12, and even in standard biochemical and hematological parameters13. Therefore, it remains unclear whether, and to what extent, the biological pathways associated with dementia in DSAD are similar to LOAD and ADAD beyond those reflected by the core AD biomarkers Aβ and tau. With the advent of effective anti-Aβ therapies for LOAD and considering the significant drug development efforts targeting other pathways, understanding the pathophysiology and natural history of DSAD and its relationship to other forms of AD has become an urgent priority to advance therapeutic opportunities for this important clinical population.
One way to advance understanding of the pathological changes associated with neurodegenerative disease is through proteomics. Proteomics analyzes disease-related changes at the level of proteins, which are the effectors of most biological functions and the source of most molecular disease biomarkers. Proteomic analysis of LOAD brain tissue and cerebrospinal fluid (CSF) over the past decade has illustrated the complex molecular pathology related to Aβ plaques, tau neurofibrillary tangles (NFTs), and cognitive decline in LOAD14,15,16,17,18,19,20, the most common form of AD. More recently, targeted proteomic analysis of ADAD CSF in the dominantly inherited Alzheimer network (DIAN) has shown how multiple biological pathways are altered after the onset of cerebral Aβ deposition21. A powerful characteristic of ADAD is the ability to place any measurement within a longitudinal framework due to the predictability of symptom onset—often operationalized as the estimated year of onset (EYO) metric—which enables study of the natural history of the disease using cross-sectional data. DSAD has similar predictability of symptom onset compared to ADAD4,5,6, and this predictability has been leveraged to study the evolution of imaging measures and standard AD CSF and plasma biomarkers in DSAD over the course of decades5,6.
To better understand the constitutive or neurodevelopmental features in DS and those features related to DSAD, here we report proteomic changes in DS CSF and how they compare to changes in established AD CSF biomarkers. We used tandem mass tag mass spectrometry (TMT-MS) to measure hundreds of proteins and analyzed the data using a systems biology approach to identify biological pathways influenced by trisomy 21 and DSAD. We compared the proteomic findings in DS to LOAD and to those reported in a previous study on ADAD. We found similarities in several biological processes between DSAD, LOAD, and ADAD, but also unique constitutive features in DS that occur prior to AD biomarker abnormalities, and differences in the temporal progression of many proteins compared to ADAD. These differences might have important implications for therapeutic development and clinical trial design in DSAD.
Results
Most proteomic changes in DS CSF occur prior to the onset of AD symptoms
In this study, we analyzed CSF samples from a total of 365 participants using mass spectrometry-based proteomics. Cohort characteristics are provided in Table 1. The cohort included euploid controls (n = 72), euploid individuals with late-onset sporadic preclinical, prodromal, or AD dementia (LOAD; n = 64), asymptomatic DS (asymDS; n = 96), individuals with DS and cognitive decline not due to AD (oDS = 14), individuals with DS and prodromal AD (proDS; n = 47), and individuals with DS and AD dementia (demDS; n = 72). Detailed demographic and clinical characteristics are provided in Supplementary Data 1. Proteomic TMT-MS-based analysis yielded a total of 1122 protein measurements across 365 cross-sectional cases. After correction for batch effects and filtering for proteins with abundance measurements in at least 80% of samples balanced across case groups, we identified 838 proteins for individual protein analyses. To determine whether a given protein was significantly altered in DS compared to controls, we employed a modeling framework previously used in ADAD to estimate protein levels in DS and control cases across estimated year of onset (EYO) using a Bayesian statistical approach21,22. We used age 50.2 for symptom onset (EYO = 0) in DS based on previous estimates5. Both DS and control cases were placed within this EYO framework to estimate the difference between protein levels in DS and controls from EYO –32 to 24 in 0.5 EYO intervals. The model allowed for non-linear protein level changes (Supplementary Fig. 1A). Examples of two altered proteins, amyloid precursor protein (APP) and neuronal pentraxin-2 (NPTX2), are shown in Fig. 1. Given the distribution of DS and control cases across EYO, the confidence of our estimates was greatest between EYO –20 and 10. Out of the 838 proteins analyzed, we observed 556 that were either increased or decreased in DS compared to controls at any EYO (Fig. 2, Supplementary Figs. 2,3, Supplementary Data 2, and Supplementary Information). A large majority (87%) of these proteins were altered prior to AD symptom onset and mapped to multiple biological pathways affected in the AD brain previously identified through AD brain protein co-expression analysis (Supplementary Fig. 2 and Supplementary Data 2)17. Although proteins increased and decreased in DS showed a similar pattern of change across EYO, there was a slight bias towards earlier changes in proteins that were increased in DS compared to those that were decreased in DS (Supplementary Fig. 3A). Five proteins had mixed direction of change (Supplementary Fig. 3B). We next assessed for effects of APOE and sex on the DS CSF proteome by first filtering for proteins significantly altered in DS and then separately for sex and APOE ε4 and ε2 effects on these filtered proteins (Supplementary Fig. 1B, C). We observed 18 proteins that were both significantly altered in DS and influenced by APOE ε4 across EYO (Supplementary Fig. 4 and Supplementary Data 3), and six proteins that were influenced by APOE ε2 (Supplementary Fig. 5 and Supplementary Data 3), although the APOE ε2 analysis was less powered. There was no overlap between proteins significantly altered in DS and influenced by APOE ε4 and ε2. We observed a greater number of proteins (79) significantly different in DS that were also significantly influenced by sex (Supplementary Fig. 6 and Supplementary Data 4). In summary, we observed 556 proteins that were significantly altered in DS across the EYO continuum, the vast majority of which were altered prior to AD symptom onset, illustrating early changes across multiple CSF protein measures in DS.

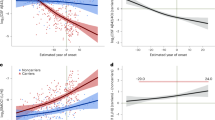
A, B Levels of the amyloid precursor protein (APP), located on chromosome 21, in DS (red) and control (blue) cerebrospinal fluid (CSF) (A), and the difference in APP levels between DS and controls (B), by estimated year of symptom onset (EYO). One outlier was removed from A for visualization purposes. C, D CSF levels of neuronal pentraxin-2 (NPTX2), a synaptic protein, in DS and controls (C), and the difference between NPTX2 levels in DS and controls (D). Two outliers were removed from C for visualization purposes. Solid lines indicate the mean protein level; shaded areas indicate the 99% credible interval. Periods of significant difference between DS and controls are highlighted in (B, D) (red indicates significantly increased levels in DS, blue indicates significantly decreased levels in DS). Shaded EYO values on the x-axis indicate periods of lower confidence estimates due to the smaller number of participants over the indicated EYO range. Plots for other proteins are provided in the Supplementary Information.

556 out of 838 CSF proteins analyzed had different levels in DS at any EYO. Differences were assessed in 0.5 EYO intervals. Red indicates increased levels in DS and blue indicates decreased levels in DS. Heat indicates the significance of the difference between DS and controls, with scale provided in the arrows on the right. The vertical black line highlights EYO = 0 (age 50.2). Shaded EYO values on the x-axis indicate periods of lower confidence estimates, including after EYO = 10, where DS data were sparse and therefore only proteins with strong differences are considered significant. Periods of significant change and direction of change for all 556 significant proteins and other CSF ELISA measurements are provided in Supplementary Fig. 2 and Supplementary Data 2. About 266 proteins were increased and 294 proteins were decreased across EYO, with five proteins showing mixed direction of change. Visualization of proteins separated by direction of change in DS is provided in Supplementary Fig. 3. Empirical p values were computed within a Bayesian analysis framework. All tests were two-sided with a pre-specified significance level of 0.01, corresponding to tail probabilities of 0.005 and 0.995. No multiple testing correction was applied.
Identification of systems pathological changes in DS using protein co-expression
To more clearly identify the biological processes altered in DS, we used protein co-expression network analysis to reduce the dimensionality of the CSF proteomic data. Protein co-expression analysis is a powerful technique to identify groups of proteins related by their common changes in abundance across individuals. Protein groups, or “modules,” can then be interrogated for the biological pathways they represent through ontology analysis. We constructed a protein co-expression network from the CSF of individuals with DS, identifying 29 distinct modules reflecting multiple different biological processes or pathways (Fig. 3, Supplementary Data 5, and Supplementary Information). Each module eigenprotein, or the first principal component of module protein expression, was assessed for correlation with established CSF AD biomarkers as well as cognitive function, age, sex, and APOE ε4. Module levels were also assessed across case groups, and for overlap with brain cell-type specific markers (Supplementary Data 6 and Supplementary Information). We observed a group of related modules strongly positively correlated with CSF total-tau (tTau), pTau181, pTau217, and pTau231, neurofilament light polypeptide (NFL, referred to subsequently as NEFL for consistency in nomenclature across proteins), and chitinase-3-like protein 1 (CHI3L1, also known as YKL-40) levels, and negatively correlated with Aβ42/40 ratio and cognitive function. These modules included 14-3-3 proteins and microtubule-associated protein tau (MAPT), glucose/energy metabolism, immune response, and actin cytoskeleton pathways. Notably, the M8 14-3-3/MAPT/Mixed module (Fig. 3B) was most strongly correlated with AD traits and AD-related cognitive impairment and was strongly elevated in DS with symptomatic AD (Fig. 3C). Modules that showed opposite relationships with these measures included M22 glycoprotein biosynthesis/endoplasmic reticulum (ER), M10 neuron migration/organization, and M6 cell migration. In addition, we identified a module, M23 Chr21/APP/SOD1/leukocyte migration, that was strongly elevated in DS and contained approximately half (8 out of 15) of the chromosome 21 proteins measured in our dataset, including superoxide dismutase (SOD1) and leukocyte migration markers (Supplementary Fig. 7). This module was not elevated in LOAD. Other modules that were strongly elevated in DS and not LOAD but were less strongly correlated to pTau included M18 extracellular matrix, M5 collagen, M25 complement, and M3 immunoglobulins. M2 plasma proteins was also elevated in DS, although to a lesser degree. In summary, we identified groups of proteins related by their co-abundance patterns that were both elevated and decreased in DS, some of which were highly correlated with LOAD biomarkers such as M8 14-3-3/MAPT/mixed and M4 glucose/energy metabolism, and some of which were unique to DS such as M23 Chr21/APP/SOD1/leukocyte migration and M3 immunoglobulins.

A–C About 1116 proteins measured across control, DS, and AD cases were used to construct a CSF protein co-expression network (A). Modules were annotated with their primary ontologies. Module eigenproteins were correlated to CSF total tau (tTau), pTau181, pTau217, pTau231, Aβ42/40, Aβ42/tTau, neurofilament light polypeptide (NEFL), chitinase-3-like protein 1 (CHI3L1), CamCog score (higher scores reflect better cognitive function in DS), age of controls (CT), age of DS cases, sex in CT (1 = male), sex in DS (1 = male), and APOE ε4 risk (ε2/2 = –2, ε4/4 = +2). Red indicates positive correlation; blue indicates negative correlation. Differences in module eigenprotein levels were assessed between AD and control (AD-CT), asymptomatic DS and control (AsymDS-CT), demented DS and control (DemDS-CT), demented DS and asymptomatic DS (DemDS-AsymDS), all DS and control (allDS-CT), and symptomatic DS (prodromal and demented) and AD (SymDS-AD) using a two-sided t-test without correction for multiple comparisons. Green indicates increased levels; blue indicates decreased levels. Brain cell type enrichment in each module was performed for neurons, oligodendrocytes (oligo), astrocytes (astro), microglia (micro), and endothelia (endo) using one-tailed Fisher’s exact test with Benjamini–Hochberg correction. Only cell type overlaps that reached statistical significance are colored. Module ontologies highlighted in bold demonstrated strong associations with AD traits, with modules highlighted in red showing the strongest associations. Module protein memberships are provided in Supplementary Data 5. Heatmap values are provided in Supplementary Data 6. B Protein members of the M8 14-3-3/MAPT/Mixed module, which was the module most strongly correlated to CSF AD biomarkers. Circle size indicates the strength of correlation to the module eigenprotein. Transparent blue lines represent human protein-protein interactions as provided in the BioGRID database. Gray lines represent top-ranked co-expression network edges. C Differences in M8 eigenprotein levels among groups (boxplot; control n = 72, preclinical AD n = 8, AD n = 56, other DS n = 14, Asym DS n = 96, prodromal DS n = 47, dementia DS n = 72), and correlation of the M8 eigenprotein to age in DS cases, CSF tau phosphorylated at residue 217 (pTau217), CSF amyloid-β 42/40 ratio (Aβ42/40), CSF neurofilament light polypeptide (NEFL) levels, and CSF chitinase-3-like protein 1 (CHI3L1, also known as YKL-40) levels. The difference between groups was assessed by one-way ANOVA and adjusted for age and sex. Correlations were performed using midweight bicorrelation. Boxplots represent the median, 25th, and 75th percentile extremes; thus, hinges of a box represent the interquartile range of the two middle quartiles of data within a group. The farthest data points up to 1.5 times the interquartile range away from box hinges define the extent of whiskers (error bars). Plots for other modules are provided in the Supplementary Information.
Many protein co-expression modules in DS are altered prior to decreases in CSF Aβ42/40
To assess the temporal progression of protein module changes in DS, we modeled module eigenprotein levels in DS and controls across EYO in a similar fashion to individual protein levels (Fig. 4 and Supplementary Data 7). Out of the 29 modules, 22 showed significant differences in DS at any EYO. Of these 22 modules, 11 were significantly altered in DS prior to changes in the Aβ42/40 or Aβ42/tTau ratio—commonly used to assess Aβ plaque deposition—and included M5 collagen, M18 extracellular matrix, and M25 complement. Modules demonstrating the earliest change in DS were M23 Chr21/APP/SOD1/leukocyte migration, as expected, and M13 Golgi/glycosylation. Modules that were strongly correlated to LOAD biomarkers—M8 14-3-3/MAPT/mixed and M9 actin cytoskeleton—were elevated approximately 10 years prior to symptom onset, whereas modules altered closer to symptom onset included M24 immune response and M11 neurogenesis/synapse. Interestingly, NEFL was elevated in DS CSF prior to elevations in tTau and pTau, suggesting early white matter pathology and axonal degeneration prior to Aβ-related elevations in pTau23,24,25. CHI3L1, a proposed biomarker of astrocytosis26, was elevated about 5 years prior to symptom onset at approximately the same time as M24 immune response, demonstrating consistent change in measures of inflammation preceding cognitive decline. Lastly, modules M20 semaphorins/lysosome, M26 endosome/lysosome and M4 glucose/energy metabolism were altered after symptom onset, with M4 glucose/energy metabolism demonstrating the largest progressive change among these modules affected late in the disease continuum. In summary, we identified multiple modules in DS CSF that were elevated prior to changes in Aβ and tau, with immune and synaptic modules temporally associated with the onset of cognitive symptoms.

Module eigenproteins, representing the first principal component of module protein abundance, were assessed for changes in DS by EYO. Differences were assessed in 0.5 EYO intervals. 22 out of 29 modules were significantly different in DS at any EYO. Module changes were compared to standard amyloid, tau, and neurodegeneration (AT(N)) CSF AD biomarkers. Red indicates increased levels in DS and blue indicates decreased levels in DS. The vertical black line highlights EYO = 0 (age 50.2). Shaded EYO values on the x-axis indicate periods of lower confidence estimates. Periods of significant change and direction of change for each module and CSF ELISA measurement are provided in Supplementary Data 7. Empirical p values were computed within a Bayesian analysis framework. All tests were two-sided with a pre-specified significance level of 0.01, corresponding to tail probabilities of 0.005 and 0.995. No multiple testing correction was applied.
Comparison of individual protein measures in DS to ADAD highlights common alterations but unique temporal patterns
We have previously illustrated the temporal progression of key AD biomarkers in ADAD CSF over the course of approximately 60 years21. To assess how and when these biomarkers are altered in DS, we applied the same modeling approach used in ADAD (Fig. 5). Measurements in the ADAD cohort were performed using a targeted mass spectrometry approach, whereas measurements in the DS cohort were performed using an untargeted mass spectrometry approach. We selected synaptic markers to validate measurements between the two mass spectrometry-based techniques, and observed good agreement (Supplementary Fig. 8). Because of the differences in power between the ADAD and DS cohorts, absolute EYO changes between measures in ADAD and DS are not directly comparable; however, relative temporal changes among markers can be compared. Taking this consideration into account, we observed very similar changes in Aβ42 and Aβ42/40 levels in ADAD and DS, with significant differences occurring approximately 15 years prior to symptom onset. In DS, similar to ADAD, multiple protein changes occurred prior to alterations in pTau181 and pTau217 (Supplementary Fig. 9). However, the relative temporal progression of these changes and direction of change was unique in DS for several proteins. SMOC1 and SPON1—members of the brain M42 matrisome module that show early elevation in ADAD—were elevated closer to the EYO in DS and around the time of changes in pTau levels, with stronger elevation of SPON1 in DS than observed in ADAD. NEFL demonstrated the earliest increase in DS, prior to increases in the 14-3-3 proteins YWHAZ and YWHAG (Supplementary Fig. 9). Proteins with notably earlier relative decreases in DS compared to ADAD included secretogranin-2 (SCG2), neurosecretory protein VGF (VGF), neuronal pentraxin-2 (NPTX2), and lactadherin (MFGE8). MFGE8 is the precursor to the protein fragment medin, which has previously been shown to be a significant protein component within the cerebrovasculature in cerebral amyloid angiopathy (CAA)27. Of the overlapping protein measurements with ADAD, MFGE8 demonstrated the earliest decrease in protein levels that remained decreased throughout the EYO timeframe (Supplementary Fig. 9). Interestingly, a select number of proteins mapping to brain metabolic modules were noted to change in the opposite direction to that observed in ADAD, including l-lactate dehydrogenase B chain (LDHB), alpha-enolase (ENO1), Parkinson disease protein 7 (PARK7), phosphatidylethanolamine-binding protein 1 (PEBP1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Some of these proteins such as LDHB, ENO1, PARK7, and MFGE8 were altered prior to changes in CSF Aβ42/40 levels. In summary, we observed many common protein changes between ADAD and DS, but unique temporal ordering of certain proteins related to Aβ plaques (SMOC1 and SPON1), synaptic changes (SCG2, VGF, and NPTX2), axonal and white matter changes (tTau and NEFL), and cerebral amyloid angiopathy (MFGE8) in DS, and differences in some metabolic proteins that may be related to trisomy 21.

Protein level differences between ADAD mutation carriers and non-carriers as described in ref. 21 (left) and DS and euploid individuals (right) were modeled in the same fashion across EYO. Red indicates increased levels in ADAD and DS and blue indicates decreased levels in ADAD and DS. The vertical black line highlights EYO = 0. Shaded EYO values on the x-axis in DS indicate periods of lower confidence estimates. Each protein was mapped to its corresponding brain co-expression network module as described in ref. 17 and DSAD CSF co-expression network module as shown in Fig. 3A. Colors for each module correspond to module numbers in each network (for example, M4 is yellow). Biomarker measurements that do not map to a module were measured using immunoassays. The progression of protein changes in DS were not the same as in ADAD. Empirical p values were computed within a Bayesian analysis framework. All tests were two-sided with a pre-specified significance level of 0.01, corresponding to tail probabilities of 0.005 and 0.995. No multiple testing correction was applied. ALDOA fructose-bisphosphate aldolase A, CHI3L1 chitinase-3-like protein 1, ENO1 alpha-enolase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, GDA guanine deaminase, GDI1 rab GDP dissociation inhibitor alpha, GMFB glia maturation factor beta, GOT1 aspartate aminotransferase, cytoplasmic, LDHB L-lactate dehydrogenase B chain, MDH1 malate dehydrogenase, cytoplasmic, MFGE8 lactadherin, NEFL neurofilament light polypeptide, NPTX2 neuronal pentraxin-2, NPTXR neuronal pentraxin receptor, PARK7 Parkinson disease protein 7, PEBP1 phosphatidylethanoamine-binding protein 1, PGAM1 phosphoglycerate mutase 1, PKM pyruvate kinase PKM, PPIA peptidyl-prolyl cis-trans isomerase A, SCG2 secretogranin-2, SMOC1 SPARC-related modular calcium-binding protein 1, SPON1 spondin-1, SPP1 osteopontin, THY1 thy-1 membrane glycoprotein, TPI1 triosephosphate isomerase, VGF neurosecretory protein VGF, YWHAG 14-3-3 protein gamma, YWHAZ 14-3-3 protein zeta/delta.
Comparison of DS CSF and brain proteomes reveals concordant and discordant alterations between compartments
To better understand the relationship between protein alterations in DS CSF and DS brain tissue, we compared CSF proteomic changes in our study to those recently reported from a localized proteomics study in DS brain on both plaque and non-plaque tissue using laser-capture microdissection28. A total of 376 matching gene symbols were present between the two proteomic datasets. In DS brain tissue without plaque, we observed significant increases in extracellular matrix (ECM) proteins including collagens and fibrinogens (such as COL6A3, COL6A1, COL1A1, and FGA), blood proteins such as hemoglobin (HBB and HBA1) and haptoglobin (HP), and Chr21 proteins (such as APP and NCAM2), concordant with alterations in these proteins observed in CSF (Fig. 6A). The prominent increase in immunoglobulins such as IGHG3 and IGHA1 in DS CSF was less notable in brain. Markers of myelination and white matter such as myelin-oligodendrocyte glycoprotein (MOG) and myelin-associated glycoprotein (MAG) were concordantly decreased in DS brain and CSF, as well as some neuronal markers such as VGF and parvalbumin (PVALB). As we have observed previously when comparing brain and CSF proteomic changes14,16, not all neuronal protein alterations were concordant between compartments. For instance, neuronal proteins such as YWHAG, neuromodulin (GAP43), and brain acid soluble protein 1 (BASP1) were increased in CSF but decreased in brain, whereas proteins such as NPTX1, chromogranin-A (CHGA), and pro-neuropeptide Y (NPY) were decreased in CSF but increased in DS brain. When the analysis was restricted to plaques, plaque-associated proteins such as serine protease HTRA1 (HTRA1), midkine (MDK), and SMOC1 were more significantly altered in DS brain compared to control, as expected17.

CSF proteomic data were compared to localized brain proteomic data from Aβ plaque and non-plaque tissue in DS and LOAD28. A CSF proteomic changes in all participants with DS (allDS) compared to control versus DS non-plaque tissue (left) and DS plaques (right) compared to control. B CSF proteomic changes in participants with dementia due to DSAD compared to LOAD versus DS non-plaque tissue (left) and DS plaques (right) compared to LOAD. Proteins that were significantly different in either CSF or brain at FDR <0.05 in each contrast are colored by the CSF network module in which they reside. (C) CSF network module eigenproteins were tested for differences in DS and LOAD brain tissue in both plaque and non-plaque regions (control n = 72, preclinical AD n = 8, AD n = 56, other DS n = 14, Asym DS n = 96, prodromal DS n = 47, dementia DS n = 72; n = 20 for each brain group). Differences in module eigenprotein by case status were assessed by Kruskal-Wallis one-way ANOVA. Boxplots represent the median, 25th, and 75th percentile extremes; thus, hinges of a box represent the interquartile range of the two middle quartiles of data within a group. The farthest data points up to 1.5 times the interquartile range away from box hinges define the extent of whiskers (error bars). Plots for other CSF network modules are provided in the Supplementary Information.
We also examined differences between DSAD and LOAD brain and CSF (Fig. 6B). Compared to LOAD, DSAD had higher levels of ECM and Chr21 proteins, and lower levels of white matter proteins and PVALB in brain and CSF compartments. Immunoglobulins were prominently increased in DSAD CSF compared to LOAD CSF, but this difference was not observed in brain tissue. Except for immunoglobulins, these alterations were noted in both non-plaque and plaque tissue. Some plaque-associated proteins, such as SPON1, were significantly increased in DSAD compared to LOAD, consistent with the more prominent elevation in SPON1 in DSAD CSF compared to ADAD CSF. In a separate analysis, we tested for differences in DS CSF protein network module eigenproteins in brain tissue for 19 modules that had sufficient brain protein coverage (Fig. 6C and Supplementary Information). Consistent with individual protein changes, we observed concordant changes in a number of module,s such as M23 Chr21/APP/SOD1/leukocyte migration, M5 collagen, M9 actin cytoskeleton, and M14 cell-cell adhesion. We also observed discordant changes in the 14-3-3/MAPT/mixed module heavily influenced by 14-3-3 proteins such as YWHAG, and in the M7 neurexins/neuropentraxins/synapse module, consistent with the discordant changes in module driver proteins noted above.
Given the concordant and prominent decrease in PVALB levels in DS brain and CSF, as well as the importance of this protein as a marker of PV+ inhibitory interneurons that are critical for proper excitation/inhibition balance in the brain29, we performed IHC for PVALB in control, LOAD, and DS brain tissue (Supplementary Fig. 10). PVALB was lower in both DSAD and LOAD brain compared to controls. Although we had few cases of young DS brain, PVALB levels in DS individuals in their twenties and thirties were the same levels as controls, whereas PVALB levels in DS individuals in their fifties and sixties were lower than those in controls or LOAD. The observed age dependence in PVALB levels in DS brain was consistent with the decline in PVALB levels in DS CSF with increasing EYO (Supplementary Fig. 10C).
In summary, we identified concordant changes in ECM, plasma, Chr21, myelin, and some neuronal proteins between DS brain and CSF compartments, whereas other neuronal proteins demonstrated discordant levels between brain and CSF as previously observed. PVALB, a marker of inhibitory interneurons, was prominently decreased in both brain and CSF compared to both control and LOAD, suggesting that potential disruption of excitatory/inhibitory balance may be a key feature of DS.
Discussion
In this study, we used proteomics in the CSF of individuals with DS to understand the pathological changes that are associated with trisomy 21 and how they evolve over the course of the syndrome prior to, during, and after the onset of AD-related cognitive symptoms. We compared these pathological changes to those observed in LOAD and ADAD and observed both common and unique features. DS exhibits constitutive marked elevations in CSF levels of immunoglobulins, complement, collagen and ECM proteins before changes in markers of Aβ plaque formation, suggesting that they could be linked to the triplication of genes encoded in chromosome 21. Moreover, the marked elevations of plasma proteins in DS CSF, perhaps suggesting loss of blood–brain barrier (BBB) integrity, and early evidence of CAA suggest that cerebrovascular dysfunction is an important early pathological change in DS. Moreover, DSAD was associated with earlier decreases in synaptic proteins, neurofilament light, and MFGE8—a proposed protein marker of CAA—than ADAD, and more prominent changes in ECM, plaque-associated, myelin-associated, and PV+ inhibitory neuron-associated proteins than LOAD. These findings indicate that although many pathway alterations are shared between DSAD, ADAD, and LOAD, DSAD has unique features compared to other forms of AD.
One of the key findings from our study is the clear elevation of individual proteins and protein modules related to the ECM in the CSF of individuals with DS. The M5 Collagen and M18 Extracellular Matrix modules showed increased levels prior to decreases in Aβ. These ECM changes may be related to trisomy 21 but could also be risk factors for DSAD or DSAD-associated complications. These closely related modules contained the two matrix metalloproteinases measured in our dataset—MMP2 and MMP14. MMP activity has been implicated in loss of arteriole integrity and consequent cerebral microhemorrhage30,31,32. Interestingly, metalloproteinase inhibitors 1 and 2 (TIMP1 and TIMP2) were also present in these modules. Elevated levels of TIMPs have been found to be increased in the leptomeningeal arteries in CAA32. Although atherosclerotic and arteriosclerotic vascular pathologies are uncommon in individuals with DS, CAA is more frequent in DS than in sporadic LOAD, likely due in part to APP overexpression and associated Aβ accumulation in blood vessels33. Neuropathological studies have identified CAA in individuals with DS as early as age 42, with severity increasing with age34,35. Furthermore, we observed a striking decrease in levels of MFGE8 in DS CSF, more prominently than observed in ADAD. One of the protein cleavage products of MFGE8—the medin fragment—accumulates in the smooth muscle of arterioles and catalyzes the aggregation of Aβ and subsequent development of CAA. Whether medin deposition is a principal driver of CAA in DS is currently unclear, as APP duplication itself can cause ADAD with prominent CAA36.
In addition to increased levels of APP and Aβ production caused by trisomy 21, other proteins on chromosome 21 may also be involved in causing cerebrovascular dysfunction. The collagens COL6A1, COL6A2, and COL18A1 located on chromosome 21, were found to be elevated in the CSF of individuals with DS, and other collagens such as COL6A1 and COL1A1 were also noted to be elevated in DS brain tissue compared to controls and LOAD28. Increased levels of COL6A2 have been observed in vessels affected by CAA37. Other proteins that can form amyloids, such as serum amyloid P-component (APCS), which has also been observed in vessels affected by CAA37, were found to be elevated early in DS, well before changes in Aβ levels. APCS mapped to the M2 Plasma Protein module that was elevated in DS, suggesting the abnormal presence of plasma proteins in DS CSF. Other classes of proteins elevated in the CSF of individuals with DS that may indicate leakage across the BBB included complement and immunoglobulins. As mentioned, alterations in proteins and protein modules related to the ECM reflect the importance of considering trisomy-related (constitutive of the syndrome) and DSAD-related (associated with the AD process) changes. While the two might be differentiated, the two can also interact to converge in a specific phenotype, in this case CAA, that might have relevant implications for anti-Aβ immunotherapy administration, such as potential increased risk of amyloid-related imaging abnormality (ARIA) side effects38.
Interestingly, M16 cell-substrate adhesion and M14 cell-cell adhesion modules were decreased early in DS, perhaps in part reflecting vascular endothelial dysfunction. Loss of BBB integrity may lead to leakage of plasma proteins such as fibrinogen into the brain parenchyma in DS28,39. Brain deposition of fibrinogen and its cleavage product fibrin is a pathological feature of loss of BBB integrity in multiple sclerosis and has been shown to promote neuroinflammation, loss of myelin integrity, and neurodegeneration40,41. Other plasma proteins, such as hemoglobin, that are released into the brain parenchyma in DS may also lead to progressive neurodegeneration. Notably, some of the trisomy-related modules were not strongly correlated to AD phenotypes but could function as risk factors for the development of AD-related cognitive impairment or for specific phenotypes such as CAA. Additional mechanistic studies and potential therapeutic trials targeting ECM proteins in DS are required to further test this hypothesis and determine their relevance for disease onset and effect on therapeutic strategies.
When examining the standard amyloid, tau, and neurodegeneration (AT(N)) CSF biomarkers for AD, NEFL emerged as the first marker to exhibit alterations following reductions in Aβ levels among individuals with DS. Elevations in NEFL and tTau occurred before elevation in pTau181 and pTau217, in contrast to what is observed in LOAD, but consistent with previous results in DSAD5. Increases in NEFL and tTau occurred relatively earlier in DS compared to ADAD in the DIAN cohort. In the large Colombian kindred of PSEN1 mutation carriers, plasma NEFL levels were found to be different from those of non-carriers 22 years before symptom onset42 with a similar relative temporal change to other core AD biomarkers compared to DS. The matrix (plasma versus CSF), cohort size, and specific ADAD mutations may affect direct comparisons for a given protein between DSAD and ADAD. NEFL has been shown to be associated with axonal and myelin integrity in both DSAD and ADAD23,24,25, and although it is not an AD-specific marker for neurodegeneration, both CSF and plasma concentrations of NEFL have shown outstanding diagnostic and prognostic performance for symptomatic DSAD with yearly augmented rates of change evident along the AD continuum43. White matter pathology is a key feature of DS25,44,45,46 and may be a predisposing factor for the development of DSAD. We observed decreased levels of the myelin markers MAG and MOG in DS brain and CSF compared to LOAD brain and CSF, and both markers were decreased in DS CSF compared to controls at least 20 years before estimated symptom onset. Individuals with DS may have increased susceptibility to myelin pathology, given that abnormalities and delays in myelination can be observed from birth47. Changes in MAG and MOG occurred after the earliest changes reflected in DS Chr21, ECM, complement, and immunoglobulin modules, but before changes in NEFL, pTau, and tau levels. It is therefore possible that the earlier elevations in NEFL levels in DSAD may be attributed to pronounced ECM and other early pathologies leading to dysfunction in myelination and subsequent axonal loss.
Previous studies have indicated no disparities in NEFL, tTau, or pTau levels among DS individuals when stratified by APOE genotype48,49 or sex50,51, two common risk factors for LOAD52,53. In our study, we also did not observe differences in these biomarkers by APOE genotype or sex; however, we identified other proteins different in DS and influenced by APOE ε4 (18), ε2 (6), and sex (79). The effects of APOE on the proteins assessed in this study were weak and transient within the EYO window of highest confidence, suggesting that APOE does not have a strong effect in DSAD. More proteins were affected by sex. The strongest differences in males were observed in proteins such as complement C1s subcomponent (C1S), versican core protein (VCAN) and the M16 Cell-Substrate Adhesion module involved in ECM interaction, APCS, and fibronectin (FN1), suggesting that males with DS may have more problems with the BBB than females. Conversely, females had higher levels of peptidyl-prolyl cis-trans isomerase A (PPIA, a marker of brain metabolism), phosphoglycerate kinase 1 (PGK1), ENO1, and GAPDH, similar to what is observed in LOAD CSF, suggesting females with DS may share more of a classical AD metabolic phenotype than males with DS. Because our DS cohort had slightly more males than females, in contrast to the DIAN cohort, which had slightly more females than males, this may be one reason why we observed a slight decrease in some metabolic markers compared to ADAD. Further study of sex differences surrounding the AD metabolic phenotype in DS and ADAD is needed.
Synaptic markers SCG2, VGF, and NPTX2 demonstrated markedly earlier relative changes in DS compared to ADAD. These markers have also been shown to change closer to the time of symptom onset in LOAD15,54. This observation suggests that pathologic processes leading to synaptic changes as reflected by these markers in ADAD and LOAD are occurring earlier in DSAD, yet are not proximately associated with AD-related cognitive decline. We observed changes in NPTX2 14 years prior to symptom onset in DSAD, consistent with a previous observation that NPTX2 levels in DS are not strongly associated with AD-related cognitive changes55. Other synaptic changes, as reflected in the M1 nervous system development and M11 neurogenesis and synapse modules, were more closely associated with AD-related cognitive decline, illustrating the complex evolution of neuronal and synaptic changes in DSAD. Elevations in the M24 Immune Response module also occurred proximate to the onset of cognitive decline. Inflammation is widely recognized as a pivotal factor in AD pathogenesis; interestingly, individuals with DS have been shown to have abnormal immune systems compared to euploid individuals from birth, as well as unique neuroinflammatory responses compared to individuals with LOAD56,57. Individuals with DS have elevations in pro-inflammatory markers including tumor necrosis factor α (TNFα), vascular endothelial growth factor A (VEGF-A), monocyte chemoattractant protein 1 (MCP-1), interleukin-1 beta (IL-1β), interstitial collagenase (MMP1), stromelysin-1 (MMP3), interleukin-22 (IL-22), and c-reactive protein (CRP)11,57,58. An important finding in our study was the early elevation in CSF complement levels. Complement has been shown to be involved in synaptic pruning processes by microglia, a process that can go awry in models of AD prior to Aβ plaque formation59,60. Complement activation in the brain is also associated with Aβ and tau NFT deposition61. Given the genetic evidence for the complement pathway as a risk factor for LOAD62,63, the combination of altered immune function, elevated CSF complement levels, and overproduction of Aβ may be a particularly important feature in the pathophysiology of DSAD.
Processes that were observed to change the latest in the disease course in DSAD were M20 Semaphorins/Lysosome, M26 Endosome/Lysosome, and M4 Glucose/Energy Metabolism. Multiple studies have identified various morphological and molecular abnormalities in the endo-lysosomal pathway in DS due to the causative role of APP triplication in endo-lysosomal dysfunction. Overproduction of the APP beta c-terminal fragment (β-CTF, also known as C99) causes dysfunctional endosomal recycling resulting in aberrant endosome accumulation in synapses64, and potentially catalyzes the formation of Aβ plaques65,66,67. Although we observed late changes in endosomal proteins in CSF, enlargement of endosomes in DS brains is observed very early, preceding Aβ deposition65,68. Interestingly, while conventional confocal microscopy has revealed larger EEA1-positive endosomes, super-resolution and electron microscopy has indicated these endosomes are normal in size but clustered69. We did not measure most vacuolar protein sorting (VPS) and RAS-related protein Rab (rab) proteins involved in the endosome recycling pathway, and further proteomic characterization of these proteins in DS CSF is needed. We did observe a decrease in the M13 Golgi/glycosylation module in DS, which, along with the M23 chromosome 21 module, were the earliest changes noted in the DS network. Because endosomes can be trafficked through the trans-Golgi network70, this early change may represent pathological changes in endosome recycling.
We noted a decrease in CSF PVALB levels 15 years before symptom onset—approximately the time of increases in NEFL—and PVALB was also the most decreased protein measured in non-plaque DS brain tissue compared to control and LOAD. IHC for PVALB showed decreased levels in older individuals with DS, consistent with previous findings71. PVALB is a marker of a class of fast-spiking inhibitory interneurons critical to the generation of gamma frequencies in the brain29, and loss of PV+ interneuron function has been associated with cognitive impairment and abnormal excitatory network activity72,73. Studies in animal models and clinical trials of the anti-epileptic medication levetiracetam have shown promise in LOAD74,75,76, and this medication may be particularly helpful in DSAD given the prevalence of epileptiform activity in DS. Other therapeutic strategies to target PV+ interneuron function, such as gamma entrainment, that are being tested in LOAD may also be a promising therapeutic strategy in DSAD, given our findings77,78. CSF PVALB could serve as a biomarker of target engagement and efficacy in DSAD clinical trials targeting this pathophysiology.
Our study has some limitations. There were few controls at younger ages in our cohort, leading to large uncertainty in the protein level estimates between DS and controls at younger ages. For instance, we did not observe expected elevated levels of Aβ42 in younger DS CSF as we have previously observed in ADAD CSF21, and as others have observed in DS CSF79, likely due to lack of power at earlier EYO. Future studies that include a larger number of young control and DS CSF samples will help to reduce this uncertainty. No other cohorts with sufficient CSF samples across the age range were available for longitudinal analysis, but analysis of such cohorts once they become available will be important for validation of our findings. Our proteomic depth was partly limited by the exclusion of proteins with higher levels of missing values. Future studies that incorporate multiple proteomic platforms and methods of measurement can help to increase the depth of coverage while minimizing missing values, as well as provide additional validation beyond the TMT to SRM MS validation performed in this study. Finally, analysis of even larger DS cohorts could provide more statistical power to develop a fourth model to incorporate the main and interaction effects of both sex and APOE genotype in relation to EYO.
In summary, the results from this study illustrate the multiple pathological alterations in DS that evolve over many years prior to AD onset and related cognitive decline. The early pathological alterations in immune function, blood–brain barrier, myelin, and inhibitory interneurons associated with trisomy 21 may contribute to the onset of AD pathology in DS. While many pathological alterations are shared among DSAD, ADAD, and LOAD, the temporal ordering of these changes in DS exhibits several unique features that may have important implications for therapeutic strategies in this population.
Methods
Study design and participants
We conducted a study at Hospital de Sant Pau in Barcelona, Spain, on adults with Down syndrome (DS) across the AD continuum. Our participants were enrolled through a population-based healthcare program that involves annual neurological and neuropsychological evaluations. Those who expressed interest in research were included in the Down Alzheimer Barcelona Neuroimaging Initiative (DABNI) cohort. We administered a semi-structured adapted health questionnaire, the Cambridge Examination for Mental Disorders of Older People with Down syndrome and other Intellectual Disabilities (CAMDEX-DS), to caregivers. This assessment tool was originally developed in Cambridge and has been adapted for use in Spanish. Information was gathered through family interviews and the review of medical or educational records containing past assessment results. To classify participants with DS along the AD continuum, we conducted consensus meetings involving neurologists/psychiatrists and neuropsychologists, who assessed participants blindly to biomarker data, as described previously, to classify individuals in asymptomatic (asymDS), those with prodromal AD (proDS) and AD dementia (demDS)5. We also had a subset of DS participants with cognitive impairment not due to AD (oDS) but rather from other non-degenerative causes such as psychiatric disease. Euploid participants underwent a structured neurological assessment along with a comprehensive neuropsychological battery. Genetic screening for trisomy 21 was conducted in adults with Down syndrome, and APOE ε4 carrier status was determined following previously published protocols80. Additionally, we recruited control participants without Down syndrome and individuals with sporadic preclinical AD or AD dementia from the Sant Pau Initiative on Neurodegeneration (SPIN) cohort80. SPIN participants underwent a structured neurological assessment and a comprehensive neuropsychological battery, brain imaging, and lumbar puncture for AD biomarkers. Controls had normal neuropsychological results for their age and education, a clinical dementia rating scale score of 0 and normal levels of core AD biomarkers in CSF81. Those with preclinical AD were cognitively unimpaired but had positive CSF AD biomarkers, and those with AD were cognitively impaired with positive CSF AD biomarkers. Recruitment took place between November 2008 and May 2022. For brain proteomic analyses, postmortem formalin-fixed and paraffin-embedded (FFPE) brain tissues from DS, LOAD, and cognitively normal age-matched controls were obtained from the National Institutes of Health NeuroBioBank (Maryland and Mt. Sinai brain banks), UK Brain Bank Network (South West Dementia brain bank), IDIBAPS Biobank from Barcelona, University of Pennsylvania, and NYU Grossman School of Medicine, including autopsy tissues from NYU Alzheimer’s Disease Research Center (ADRC), Center for Biospecimen Research and Development (CBRD)/Department of Pathology, and the North American SUDEP Registry (NASR) at NYU Comprehensive Epilepsy Center (CEC). Control cases (n = 20) were age 66 ± 13 (SD) at time of death, sex-balanced, and ≤A1, B1, and C1 on ABC AD neuropathology staging. LOAD and DS cases were matched by pathology. LOAD cases (n = 20) were age 82 ± 6, sex-balanced, and A3, B3, and C3 or Braak VI. DS cases were age 60 ± 5, 7 F/13 M, and equivalent to A3, B3, and C3 score or Braak V-VI. FFPE tissue blocks containing hippocampus and surrounding entorhinal and temporal cortex from n = 20 cases per group were used for laser-capture microdissection (LCM) and proteomic analysis. Detailed information on the LCM proteomics cohort is provided in ref. 28. For IHC, a subset of FFPE brain tissues containing the superior frontal gyrus (Brodmann area 8) from control, DS, and LOAD (n = 6 per group) was used for parvalbumin staining. IHC tissue samples were sourced from the same brain banks as the proteomics cohort, except IDIBAPS and the University of Pennsylvania. Data on ADAD were taken from a prior study in the Dominantly Inherited Alzheimer Network (DIAN) as described in Johnson et al21. Briefly, individuals at 50% risk of carrying an autosomal-dominant Alzheimer’s disease mutation in one of three genes (APP, PSEN1, PSEN2) were enrolled in the DIAN observational study (i.e., mutation carriers and non-carriers from the same family). DIAN participants are assessed at baseline and at subsequent follow-up visits that occur every one to three years. Assessments include collection of body fluids (CSF, blood), clinical testing (CDR), neuropsychological testing, and imaging modalities (MRI, PET with Pittsburgh Compound B (PiB-PET), and 18F-FDG) as previously described82,83,84,85,86. Data on ADAD included a total of 230 controls (mutation non-carriers) and 355 mutation carriers across preclinical and clinical disease stages.
Ethics
All procedures in this study were approved by the Sant Pau Ethics Committee (IIBSP-NGF-2018-36 and IIBSP-DOW-2014-30), following the standards for medical research in humans recommended by the Declaration of Helsinki. All participants or their legally authorized representative gave written informed consent before enrollment. We included all adults with Down syndrome that had CSF samples available. The DIAN study is supervised by the Institutional Review Board at Washington University in St. Louis, USA, and all study procedures were approved by the Human Research Protection Office and the Institutional Review Board at Washington University or the respective participating sites.
Sample collection and measurement of CSF Alzheimer´s disease biomarkers
Briefly, CSF samples were collected in 10 mL polypropylene tubes (Sarstedt, #62.610.018), centrifuged, aliquoted and stored at −80 °C within 2 h81. CSF measures of Aβ1-40, Aβ1–42, pTau181, and tTau were obtained on the automated Lumipulse G600II platform (Fujirebio) as previously described87. NfL was measured using the ultrasensitive equipment Simoa SR-X (Quanterix, Billerica, MA, USA) using commercially available kits (NF-light, Quanterix)43. CSF pTau18188 and pTau231 were measured by in-house Simoa assays at the University of Gothenburg89. CSF pTau217 levels were analyzed with the commercial ALZpath pTau217 assay for Simoa HD-X, as previously described90. CSF YLK-40 levels were measured by YKL-40 human chitinase-3-like 1 immunoassay, DC3L10, R&D Systems, USA (ELISA) using a dilution of 1:100 and by YKL-40 MicroVue, Quidel, San Diego (USA).
CSF digestion and tandem mass Tag (TMT) peptide labeling
Samples of 50 µl CSF were reduced and alkylated. Thirteen µl of 24.2 mM TCEP in 5% sodium deoxycholate (DOC), 0.5 M triethylammonium bicarbonate (TEAB) was added to each sample, followed by incubation for 1 h at 55 °C. Next, 3.2 µl of fresh 400 mM iodoacetamide solution was added to the samples and incubated for 30 min at room temperature in the dark. Samples were digested by adding 2.6 µg of trypsin (Promega Sequencing grade modified trypsin Ref V5115) and incubated overnight at 37 °C in an oven. The next day, samples were labeled using TMT 16-plex Label Reagent (Thermo Fisher, Ref#: A44520; Lot#: XB341490)/TMT pro-134C, TMT pro-135N (Thermo Fisher, Ref: #A52046; Lot: #XA338615). The TMT reagents were equilibrated to room temperature before 10 µl was added to each sample, and then incubated for 1 h at room temperature while shaking. The labeling process was quenched by adding 6 µl 5% v/v hydroxylamine solution (50% hydroxylamine is diluted 1:10 with H2O) and incubating for 30 min. All samples were mixed in a 15-ml Falcon tube and acidified by adding 168.5 µl (10% of the vol.) of 0.5 M HCl, and subsequently, 4.15 mL 0.1% TFA was added to reduce the ACN to <3%. Samples were then further acidified to precipitate DOC, which was then removed by centrifugation at 4000×g for 15 min at 4 °C. The supernatant was desalted by solid-phase extraction (Waters Sep-Pak C18, WAT023501). The column was washed twice with 1 ml 0.1% TFA in 80% acetonitrile and equilibrated with 2 ml 0.1% TFA. The sample was then applied to the column, washed twice with 1 ml 0.1% TFA, and eluted with 1 ml 0.1% TFA in 80% acetonitrile. The collected eluate was split into four aliquots, lyophilized via vacuum centrifugation, before being stored at −80 °C until further use.
Off-line fractionation
The TMT-sets were fractionated by basic reversed-phase chromatography using a Dionex Ultimate 3000 UPLC system (Thermo Fischer Scientific) equipped with a reversed-phase XBridge BEH C18 column (3.5 μm, 3.0 × 150 mm, Waters Corporation). Peptides were eluted with a stepped gradient from 3 to 55% solvent B over 35 min, followed by an increase to 100% B at a flow of 400 µL/min. Solvent A was 10 mM ammonium formate at pH 10, and solvent B was 90% acetonitrile (ACN), 10% solvent A. The 36 primary fractions were combined to 12 final fractions which were evaporated and reconstituted in 3% ACN, 0.1% trifluoroacetic acid for LC-MS analysis.
LC-MS analysis
The fractions were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer equipped with a FAIMS-Pro ion mobility system and interfaced with an Easy-nLC1200 liquid chromatography system (all Thermo Fisher Scientific). Peptides were trapped on an Acclaim Pepmap 100 C18 trap column (100 μm × 2 cm, particle size 5 µm, Thermo Fisher Scientific) and separated on an in-house packed analytical column (38 cm × 75 μm, particle size 3 μm, Reprosil-Pur C18, Dr. Maisch) using a stepped gradient from 6 to 35% acetonitrile in 0.2% formic acid over 77 min at a flow of 300 nL/min. FAIMS-Pro was alternating between compensation voltages (CVs) of −50 and −70, with the same data-dependent settings for both CVs. The precursor ion mass spectra were acquired at a resolution of 120,000 and an m/z range of 375–1375. Using a cycle time of 1.5 s, the most abundant precursors with charges 2–7 were isolated with an m/z window of 0.7 and fragmented by collision-induced dissociation (CID) at 35%. Fragment spectra were recorded in the ion trap at a Rapid scan rate. The ten most abundant MS2 fragment ions were isolated using multi-notch isolation for further MS3 fractionation. MS3 fractionation was performed using higher-energy collision dissociation (HCD) at 55%, and the MS3 spectra were recorded in the Orbitrap at 50,000 resolution and an m/z range of 100–500.
Proteomic data analysis
Data analysis was performed using Proteome Discoverer (Version 2.4, Thermo Fisher Scientific) and Mascot (Version 2.5.1) as a search engine. The data were matched against the swissprot Homo sapiens database (May 2022, 20,377 entries) allowing one missed cleavage. Precursor mass tolerance was set to 5 ppm, and fragment mass tolerance was set to 0.6 Da. Cysteine carbamidomethylation and TMTpro were set as fixed modifications, while methionine oxidation was set as a variable modification. Percolator was used for PSM validation at an FDR of 1%. TMTpro reporter ions peak integration was set to 3 mmu and only unique peptides were considered for protein quantification.
Protein abundance data processing
The Proteome Discoverer v2.4 results were exported as tab-separated values (TSV) using Proteome Discoverer v2.5. The raw TMT reporter ion counts from this file were loaded in Microsoft R Open v4.0.2, and human protein isoforms identified with medium confidence or better were kept. We used TAMPOR—a median polish batch correction method—to remove batch effects91. Only the global internal standard (GIS) sample in each batch was used for TAMPOR batch correction. The open-source TAMPOR R function is available from https://www.github.com/edammer/TAMPOR/. There were 1122 proteins output with less than 50% missing values across the 512 samples, including GIS. Samples were reduced to 365 first-draw CSF samples for 365 unique individuals analyzed by TMT-MS (Table 1). Sample connectivity outliers at least three standard deviations (SD) below the mean connectivity were checked using the WGCNA R package v1.69, both before and after reduction of sample count. Ten outliers were found before sample set reduction, and nine entirely different outliers were found after reduction to 365 first-measure samples. Therefore, no consistent outliers were identified or removed in the dataset. Quality control (QC) of batch correction using the variancePartition package in R identified a number of proteins that retained batch covariance, which may have been due to internal standard variations across batches, as the last batch GIS channel contained only ~50% of the GIS peptides of other batches’ GIS TMT channel. Therefore, bootstrap regression for covariance specific to any of the 29 TMT batches was performed on log2(abundance) output of TAMPOR in R, protecting the diagnosis group in the regression model. After regression of batch effects, variancePartition demonstrated that nearly all proteins had variance explained by batch of less than 5 percent. Additional QC was performed using limma package-based MDS plotting to segregate samples using only blood marker protein quantification (blood markers used were HBA1/HBA2, HBB, HBG1, and HP); no separation of samples into high- or low-blood clusters was found, confirming consistent lumbar punctures had resulted in CSF free of variable blood contamination. After controlling for missingness less than 50% in the first 365 measure samples, 1116 protein isoforms across 365 samples with log2(abundance) in the QC-passed matrix were used for subsequent analyses. SRM data for selected proteins in the DS cohort were generated as previously described in the ADAD DIAN cohort, using the same peptides, transitions, and analysis software for measurement21.
Bayesian modeling
Our analytical approach utilized a Bayesian generalized linear model (GLM) with an identity link function to assess the association between a particular measure and diagnoses over the estimated years to onset (EYO). In this phase of our analysis, we concentrated on 877 measures with less than 20% missing values across the cohort. These measures included ELISA assays for existing CSF biomarkers, proteins measured by TMT-MS, and CSF network module eigenproteins. We examined two diagnostic categories: individuals with DS and controls. The methodology used was similar to the one employed in our previous study on ADAD21. The Bayesian framework’s core advantage is its capability in generating posterior distribution samples that replicate the population distribution of the measure at EYO. This enabled us to quantify the variation of the observed measure compared to the population distribution in both diagnostic groups. For the implementation of the Bayesian GLM, we employed the R package rstanarm, which leverages the Hamiltonian Monte Carlo (HMC) algorithm. HMC is an advanced and dependable Markov Chain Monte Carlo (MCMC) method that enhanced the robustness of our analysis92.
Before delving into the details of our model, it’s important to outline the technique utilized for capturing the non-linear relationships between the measures and EYO. We applied a restricted cubic spline transformation to EYO, setting knots at the 10th, 50th, and 90th percentiles, as depicted in Formula 1. This method breaks down EYO into its linear and cubic components, which guarantees a seamless and consistent model fit at each designated percentile. These components were then used to replace the original EYO for the Bayesian GLM fitting. The use of restricted cubic splines is not only substantiated by prior research validating its capability to model data non-linearities, but also through visual assessments that confirm its concordance with the actual observed data in this study21,22.
The primary goal of our analysis was to examine the patterns of measures in individuals with DS compared to a control group, specifically looking at how the differences between these two diagnostic groups evolved over time. Model 1, which is detailed in Formula 2, was developed for this purpose. Within this model, we included several independent covariates with fixed effects: the diagnosis, a linear term for EYO, a cubic term for EYO, and interaction terms between the diagnosis and both the linear and cubic EYO terms. For the regression coefficients, we used the default weak informative normal priors with a mean of 0 and a variance parameter set to \(2.5\times \frac{{S}_{y}}{{S}_{x}}\), where \({S}_{y}\) represents the standard deviation of the outcome measure and \({S}_{x}\) represents the standard deviation of the independent covariate. Similarly, the intercept’s prior was also the default prior that is a weak informative normal distribution with a mean of 0 and a variance parameter of \(2.5\times {S}_{y}\), promoting an approach that is more objective and data-driven93. The MCMC simulations were conducted by initializing eight chains across four cores. Each chain ran for 10,000 iterations, discarding the first 5000 as a warmup. To reduce the data, every tenth iteration post-warmup was selected. We ensured the reliability of the remaining 4000 post-warmup samples by rigorously monitoring the convergence of the parameters. Bayesian two-sided credible intervals for continuous outcomes were estimated for both the DS group and the control group, as well as for the differences between these groups, at increments of 0.5 EYO units ranging from −32 to 24. Additionally, we computed the empirical p value to evaluate the likelihood of an observed difference under the null hypothesis, \({H}_{M{{\mathrm{1,0}}}}\), where M1 stands for Model 1.
A secondary objective of our study was to assess whether APOE genotype and sex—both important risk factors for AD—influenced these measures. For coding purposes in this study, individuals having one or two copies of the APOE ε4 allele were considered APOE ε4 carriers, individuals having one or two copies of the APOE ε2 allele were considered APOE ε2 carriers, and those without any APOE ε4 or ε2 alleles were defined as non-carriers for the respective ε4 or ε2 analysis.
In Model 2, which builds upon Model 1, we introduced the main effect of APOE ε4 or ε2 status along with its interaction with both the linear and cubic EYO terms. These additional terms are detailed in Formula 3. Three hypotheses were tested under this model. The first null hypothesis, \({H}_{M{{\mathrm{2.1,0}}}}\), examined whether there was a significant amount of evidence to support that significant differences exist between DS individuals and controls by EYO, accounting for the main and interaction effects of APOE ε4 or ε2 status over time. The second null hypothesis, \({H}_{M{{\mathrm{2.2,0}}}}\), evaluated whether there was significant evidence of differences between APOE ε4 carriers and non-carriers or between APOE ε2 carriers and non-carriers across EYO. The analyses for ε4 and ε2 status were conducted separately, each accounting for the main and interaction effects of diagnosis over time. The final hypothesis, \({H}_{M{{\mathrm{2.3,0}}}}\), assessed whether there was a significant amount evidence to support the difference between the control group—who are also APOE ε4 or ε2 non-carriers—and the DS individuals who are also APOE ε4 or ε2 carriers.
The methodology for Model 3 was the same as that of Model 2, which was also built upon Model 1 but with sex replacing APOE status (Formula 4). In Model 3, females were set as the reference group, analogous to APOE ε4 or ε2 non-carriers in Model 2. The interpretation of Model 3 and its hypothesis also mirrors that of Model 2.
Formula 1:
Formula 2:
Nul hypothesis, \({H}_{0}\), in Model 1 (M1):
Formula 3:
Null hypothesis, \({H}_{0}\), in Model 2 (M2):
Formula 4:
Null hypothesis, \({H}_{0}\), in Model 3 (M3):
Differential expression analysis
Differential expression and plotting of results were performed using an in-house open-source set of R functions available via https://www.github.com/edammer/parANOVA/. For every protein in a pairwise comparison, a one-way ANOVA F statistic is calculated, and the probability that a larger F statistic would occur by chance is reported as p, which for a two-group comparison is equivalent to an unpaired two-tailed equal variance t-test p value. These p values were further corrected to FDR within each set of 1116 p values using p.adjust R function with the Benjamini–Hochberg method. Log2 fold change, as the difference of log2(abundance) group means, is also provided with the statistics in Supplementary Data 5 for each of ten pairwise group comparisons. Volcano plots were generated using the parANOVA source function plotVolc().
Protein co-expression network analysis
The 1116 protein × 365 participant log2(abundance) matrix was tested for scale-free topology as evidenced by output of the pickSoftThreshold function in the WGCNA v1.69 package, which determined a power series over which a maximum R2 is approached with diminishing slope or rate of increase in R2 (y) versus power (x). The power occurring in the range of diminished slope has lowered sample connectivity and represents an adjacency matrix where spurious correlations (e.g., due to noise) are minimized. Based on the output of this function, a scale-free power of 9 was chosen, at which median connectivity was 13.5 and R2 equaled 0.89. The blockwiseModules function was employed with this power to cluster modules according to a dissimilarity metric derived from the square adjacency matrix of 1116 × 1116 pairwise correlations calculated using midweight bicorrelation. Parameters used were deepSplit = 4; minModuleSize = 4; mergeCutHeight = 0.07; TOMDenom = “mean”; networkType = “signed”; pamStage = TRUE; pamRespectsDendro = TRUE; reassignThreshold = 0.05, and a maxBlockSize greater than 1116, ensuring a single dendrogram calculation in one block.
Following initial module assignment of the 1116 proteins, resulting in 29 modules, a kME table was calculated, assigning every protein in the network a bicor correlation to each of the module eigenproteins, which were in turn calculated as the first principal component of variance of all proteins assigned to that module. An iterative post hoc kME table cleanup algorithm was employed whereby proteins with module assignments inconsistent with a minimum intramodular kME (>0.30), or having a higher kME to a module eigenprotein for a module to which they are not assigned, respectively, result in demotion to gray (proteins not in a module) or conditional reassignment to the module with which the protein is best correlated (highest kME), as previously described17. The condition for reassignment required that the difference in kMEmax and kMEintramodule as assigned in any iteration of the algorithm be greater than 0.10. The module reassignment proceeds iteratively until no more exceptions to the above rules are found, or a thirtieth iteration is reached. The final kME table after the post hoc cleanup procedure is provided in Supplementary Data 5.
Ontology enrichment
We used our open-source R function GOparallel (https://www.github.com/edammer/GOparallel/) to find ontologies (among those in the following six categories: [1] biological processes, [2] molecular functions, [3] cellular components, [4] wiki pathways, [5] reactome, or [6] the Broad M2 molecular signatures database) which were significantly enriched in co-expression module gene product lists. Briefly, this function leverages the R piano package to perform a one-tailed Fisher’s exact test, which we modified to output signed z score for either enrichment or depletion, as well as p value and Benjamini–Hochberg FDR for the enrichment significance. Human protein ontology annotations were obtained from the Bader lab94.
Cell type marker enrichment analyses
We used a previously published list of five brain cell type specific protein markers17 to determine significant overlap of gene products in each module using an in-house open-source R function (available from https://github.com/edammer/CellTypeFET). The function geneListFET calculates Fisher’s exact p value for overlap with each established list, given the background of all gene products in the input (here, N = 1122 total proteins).
Synthetic eigenprotein analysis
We performed calculations of first principal components of template CSF network module hubs with minimum kME of 0.70 and minimum two hubs in brain laser-capture microdissection proteomics data as described in ref. 28, using log2(abundances). Using these parameters, the calculations were otherwise performed as previously reported17 using the WGCNA moduleEigengenes function.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections (8 µm) from the frontal cortex were collected on glass slides. Sections from each cohort were deparaffinized and rehydrated through a series of xylene and ethanol washes. Antigen retrieval was performed using a 7-min treatment with 88% formic acid, followed by heat-induced citrate buffer treatment (10 mM sodium citrate, 0.05% Tween-20, pH 6). Primary antibodies against parvalbumin (1:150, Thermo Scientific, cat. PA5-47693), MAP2 (1:200, BD Biosciences, cat. 556320), and Aβ (D54D2, 1:1000, Cell Signaling, cat. 8243S) were incubated overnight at 4 °C. Alexa Fluor 488, 568, and 647 (Thermo Scientific) secondary antibodies were used for detection. Whole slide scans were acquired at 10× magnification using a Leica Aperio Versa 8 microscope. Ten regions of interest from each case were analyzed in ImageJ 1.54 f. Images for the parvalbumin and MAP2 channels were converted to 8-bit, and a Median filter with a radius of 1 was applied. The “Enhance Contrast” function was applied with a pixel saturation of 0.1% and the “Normalize” option enabled. Thresholding for both channels was performed using the “Otsu” method. The MAP2 and parvalbumin areas were calculated using the “Analyze Particles” function, with a minimum particle size threshold of 50. Parvalbumin area was normalized to the MAP2 area, and the parvalbumin percentage area was used for statistical analysis.
Other statistics and graphic visualization
iGraph layouts of module member proteins organized by their intramodular kME (bicor, correlation to the first principal component of a module) were generated using the netOps buildIgraphs function (https://www.github.com/edammer/netOps/). Nodes are connected by the strongest correlation-based edges as calculated by topology overlap, which is a derived matrix after calculation of the bicor adjacency matrix in the WGCNA framework. The circular network module-trait correlation and enrichment significance heatmap was generated using statistics calculated by the WGCNA bicor function, one-way ANOVA for two groups, or a Fisher’s exact test for enrichment of brain cell type markers in the gene product lists for each network module as described above. Visualization of the heatmap was performed using the R circlize package v0.4.1095. Scatterplots were plotted using the WGCNA verboseScatterplot function. Midweight bicorrelation rho and associated Student’s p values were calculated using the WGCNA bicorAndPvalue function. The investigators were blinded during data acquisition but not outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium with the dataset identifier PXD064699. Data output after batch correction and regression are available at https://www.synapse.org/DownSynAD. Extended Data, including plots of DS versus control for all significant proteins, proteins influenced by APOE ε4, APOE ε2, and sex, co-expression network plots, network module ontologies, and brain synthetic eigenproteins, are available at https://doi.org/10.6084/m9.figshare.29265053.
Code availability
Custom code is available at https://www.github.com/edammer.
References
Chen, L. et al. Global, regional, and national burden and trends of Down syndrome from 1990 to 2019. Front. Genet. 13, 908482 (2022).
Hithersay, R. et al. Association of dementia with mortality among adults with Down syndrome older than 35 years. JAMA Neurol. 76, 152–160 (2019).
McCarron, M. et al. A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 61, 843–852 (2017).
Iulita, M. F. et al. Association of Alzheimer disease with life expectancy in people with Down syndrome. JAMA Netw. Open 5, e2212910 (2022).
Fortea, J. et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet 395, 1988–1997 (2020).
Fortea, J. et al. Alzheimer’s disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol. 20, 930–942 (2021).
Glenner, G. G. & Wong, C. W. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135 (1984).
Chartier-Harlin, M. C. et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844–846 (1991).
Goate, A. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 (1991).
Fagan, A. M. et al. Comparison of CSF biomarkers in Down syndrome and autosomal dominant Alzheimer’s disease: a cross-sectional study. Lancet Neurol. 20, 615–626 (2021).
Iulita, M. F. et al. An inflammatory and trophic disconnect biomarker profile revealed in Down syndrome plasma: relation to cognitive decline and longitudinal evaluation. Alzheimers Dement. 12, 1132–1148 (2016).
Montoliu-Gaya, L., Strydom, A., Blennow, K., Zetterberg, H. & Ashton, N. J. Blood biomarkers for Alzheimer’s disease in Down syndrome. J. Clin. Med. 10, 3639 (2021).
de Gonzalo-Calvo, D. et al. Evaluation of biochemical and hematological parameters in adults with Down syndrome. Sci. Rep. 10, 13755 (2020).
Dammer, E. B. et al. Multi-platform proteomic analysis of Alzheimer’s disease cerebrospinal fluid and plasma reveals network biomarkers associated with proteostasis and the matrisome. Alzheimers Res. Ther. 14, 174 (2022).
Haque, R. et al. A protein panel in cerebrospinal fluid for diagnostic and predictive assessment of Alzheimer’s disease. Sci. Transl. Med. 15, eadg4122 (2023).
Higginbotham, L. et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci. Adv. 6, eaaz9360 (2020).
Johnson, E. C. B. et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225 (2022).
Johnson, E. C. B. et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780 (2020).
Zhou, M. et al. Targeted mass spectrometry to quantify brain-derived cerebrospinal fluid biomarkers in Alzheimer’s disease. Clin. Proteom. 17, 19 (2020).
Johnson, E. C. B. et al. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 13, 52 (2018).
Johnson, E. C. B. et al. Cerebrospinal fluid proteomics define the natural history of autosomal dominant Alzheimer’s disease. Nat. Med. 29, 1979–1988 (2023).
Preische, O. et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 25, 277–283 (2019).
Schultz, S. A. et al. Serum neurofilament light chain levels are associated with white matter integrity in autosomal dominant Alzheimer’s disease. Neurobiol. Dis. 142, 104960 (2020).
Rosas, H. D. et al. Association of plasma neurofilament light chain with microstructural white matter changes in Down syndrome. Alzheimers Dement. 16, e70023 (2024).
Morcillo-Nieto, A. O. et al. Characterization of white matter hyperintensities in Down syndrome. Alzheimers Dement. 20, 6527–6541 (2024).
Connolly, K. et al. Potential role of chitinase-3-like protein 1 (CHI3L1/YKL-40) in neurodegeneration and Alzheimer’s disease. Alzheimers Dement. 19, 9–24 (2023).
Marazuela, P. et al. MFG-E8 (LACTADHERIN): a novel marker associated with cerebral amyloid angiopathy. Acta Neuropathol. Commun. 9, 154 (2021).
Marta-Ariza, M. et al. Comparison of the amyloid plaque proteome in Down syndrome, early-onset Alzheimer’s disease, and late-onset Alzheimer’s disease. Acta Neuropathol. 149, 9 (2025).
Sohal, V. S., Zhang, F., Yizhar, O. & Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702 (2009).
Duits, F. H. et al. Matrix metalloproteinases in Alzheimer’s disease and concurrent cerebral microbleeds. J. Alzheimer’s. Dis. 48, 711–720 (2015).
Jakel, L. et al. Disturbed balance in the expression of MMP9 and TIMP3 in cerebral amyloid angiopathy-related intracerebral haemorrhage. Acta Neuropathol. Commun. 8, 99 (2020).
Manousopoulou, A. et al. Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 43, 492–504 (2017).
Carmona-Iragui, M., Videla, L., Lleo, A. & Fortea, J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: the complex triangle of brain amyloidosis. Dev. Neurobiol. 79, 716–737 (2019).
Davidson, Y. S., Robinson, A., Prasher, V. P. & Mann, D. M. A. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol. Commun. 6, 56 (2018).
Head, E. et al. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol. Commun. 5, 93 (2017).
Rovelet-Lecrux, A. et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26 (2006).
Hondius, D. C. et al. Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. Commun. 6, 46 (2018).
Rafii, M. S. & Fortea, J. Down Syndrome in a new era for Alzheimer disease. JAMA 330, 2157–2158 (2023).
Aguilar, L. F. & Head, E. 2. Cerebrovascular neuropathology in the brains of people with Down syndrome. Alzheimer’s. Dement. 19, e075657 (2023).
Alruwaili, M. et al. Pathogenic role of fibrinogen in the neuropathology of multiple sclerosis: a tale of sorrows and fears. Neurochem. Res. 48, 3255–3269 (2023).
Davalos, D. et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3, 1227 (2012).
Quiroz, Y. T. et al. Plasma neurofilament light chain in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional and longitudinal cohort study. Lancet Neurol. 19, 513–521 (2020).
Carmona-Iragui, M. et al. Diagnostic and prognostic performance and longitudinal changes in plasma neurofilament light chain concentrations in adults with Down syndrome: a cohort study. Lancet Neurol. 20, 605–614 (2021).
Powell, D. et al. Frontal white matter integrity in adults with Down syndrome with and without dementia. Neurobiol. Aging 35, 1562–1569 (2014).
Bazydlo, A. et al. White matter microstructure associations with episodic memory in adults with Down syndrome: a tract-based spatial statistics study. J. Neurodev. Disord. 13, 17 (2021).
Miyoshi, E. et al. Spatial and single-nucleus transcriptomic analysis of genetic and sporadic forms of Alzheimer’s disease. Nat. Genet. 56, 2704–2717 (2024).
Azrak, O. et al. Early white matter microstructure alterations in infants with Down syndrome. Preprint at medRxiv 2025.2002.2026.25322913 (2025).
Bejanin, A. et al. Association of apolipoprotein E varepsilon4 allele with clinical and multimodal biomarker changes of Alzheimer disease in adults with Down syndrome. JAMA Neurol. 78, 937–947 (2021).
Boerwinkle, A. H. et al. Comparison of amyloid burden in individuals with Down syndrome versus autosomal dominant Alzheimer’s disease: a cross-sectional study. Lancet Neurol. 22, 55–65 (2023).
Lai, F. et al. Sex differences in risk of Alzheimer’s disease in adults with Down syndrome. Alzheimers Dement. 12, e12084 (2020).
Iulita, M. F. et al. Association of biological sex with clinical outcomes and biomarkers of Alzheimer’s disease in adults with Down syndrome. Brain Commun. 5, fcad074 (2023).
Beam, C. R. et al. Differences between women and men in incidence rates of dementia and Alzheimer’s disease. J. Alzheimer’s. Dis. 64, 1077–1083 (2018).
Fortea, J. et al. APOE4 homozygosity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 30, 2093 (2024).
Nilsson, J. et al. Cerebrospinal fluid biomarker panel for synaptic dysfunction in a broad spectrum of neurodegenerative diseases. Brain 147, 2414–2427 (2024).
Belbin, O. et al. Cerebrospinal fluid profile of NPTX2 supports role of Alzheimer’s disease-related inhibitory circuit dysfunction in adults with Down syndrome. Mol. Neurodegener. 15, 46 (2020).
Gansa, W. et al. Dysregulation of the immune system in a natural history study of 1299 individuals with Down syndrome. J. Clin. Immunol. 44, 130 (2024).
Wilcock, D. M. et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 36, 2468–2474 (2015).
Zhang, Y. et al. Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: a meta-analysis. Oncotarget 8, 84489–84496 (2017).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Shi, Q. et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9, eaaf6295 (2017).
Batista, A. F., Khan, K. A., Papavergi, M. T. & Lemere, C. A. The importance of complement-mediated immune signaling in Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 25, 817 (2024).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Karch, C. M. & Goate, A. M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51 (2015).
Ferrer-Raventós, P. et al. Amyloid precursor protein βCTF accumulates in synapses in sporadic and genetic forms of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 49, e12879 (2023).
Jiang, Y. et al. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl Acad. Sci. USA 107, 1630–1635 (2010).
Jiang, Y. et al. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol. Aging 39, 90–98 (2016).
Lee, J. H. et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Abeta in neurons, yielding senile plaques. Nat. Neurosci. 25, 688–701 (2022).
Cataldo, A. M. et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286 (2000).
Botte, A. et al. Ultrastructural and dynamic studies of the endosomal compartment in Down syndrome. Acta Neuropathol. Commun. 8, 89 (2020).
Buser, D. P. & Spang, A. Protein sorting from endosomes to the TGN. Front. Cell Dev. Biol. 11, 1140605 (2023).
Kobayashi, K. et al. Cerebral cortical calbindin D28K and parvalbumin neurones in Down’s syndrome. Neurosci. Lett. 113, 17–22 (1990).
Vossel, K. A. et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 80, 858–870 (2016).
Verret, L. et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 (2012).
Sanchez, P. E. et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl Acad. Sci. USA 109, E2895–E2903 (2012).
Mohs, R. et al. The HOPE4MCI study: a randomized double-blind assessment of AGB101 for the treatment of MCI due to AD. Alzheimers Dement. 10, e12446 (2024).
Bakker, A., Rani, N., Mohs, R. & Gallagher, M. The HOPE4MCI study: AGB101 treatment slows progression of entorhinal cortex atrophy in APOE epsilon4 non-carriers with mild cognitive impairment due to Alzheimer’s disease. Alzheimers Dement. 10, e70004 (2024).
Martorell, A. J. et al. Multi-sensory gamma stimulation ameliorates Alzheimer’s-associated pathology and improves cognition. Cell 177, 256–271 e222 (2019).
He, Q. et al. A feasibility trial of gamma sensory flicker for patients with prodromal Alzheimer’s disease. Alzheimers Dement. 7, e12178 (2021).
Englund, H. et al. Increase in beta-amyloid levels in cerebrospinal fluid of children with Down syndrome. Dement. Geriatr. Cogn. Disord. 24, 369–374 (2007).
Alcolea, D. et al. The Sant Pau Initiative on Neurodegeneration (SPIN) cohort: a data set for biomarker discovery and validation in neurodegenerative disorders. Alzheimers Dement. 5, 597–609 (2019).
Lleo, A. et al. Phosphorylated tau181 in plasma as a potential biomarker for Alzheimer’s disease in adults with Down syndrome. Nat. Commun. 12, 4304 (2021).
Bateman, R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804 (2012).
Gordon, B. A. et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 17, 241–250 (2018).
McKay, N. S. et al. Positron emission tomography and magnetic resonance imaging methods and datasets within the Dominantly Inherited Alzheimer Network (DIAN). Nat. neurosci. 26, 1449–1460 (2023).
Morris, J. C. The clinical dementia rating (CDR): current version and scoring rules. Neurology 43, 2412–2414 (1993).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975).
Fortea, J. et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet Neurol. 17, 860–869 (2018).
Karikari, T. K. et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 19, 422–433 (2020).
Ashton, N. J. et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 141, 709–724 (2021).
Ashton, N. J. et al. Diagnostic accuracy of a plasma phosphorylated tau 217 immunoassay for Alzheimer disease pathology. JAMA Neurol. 81, 255–263 (2024).
Dammer, E. B., Seyfried, N. T. & Johnson, E. C. B. Batch correction and harmonization of -omics datasets with a tunable median polish of ratio. Front. Syst. Biol. 3, 1092341 (2023).
Muth, C., Oravecz, Z. & Gabry, J. User-friendly Bayesian regression modeling: a tutorial with rstanarm and shinystan. Quant. Methods Psychol. 14, 99–119 (2018).
Gabry, J. & Goodrich, B. Prior distributions for rstanarm models. (2020).
Reimand, J. et al. Pathway enrichment analysis and visualization of omics data using g:profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 14, 482–517 (2019).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
Acknowledgements
We thank all the participants and investigators involved in these consortia for their tireless efforts and invaluable contributions to the field. This study was supported by K08AG068604 and R01AG089497 (E.C.B.J.); the Goizueta Alzheimer’s Disease Research Center (P30AG066511); the Jerome Lejeune Foundation (GRT-2023B – 2303), Åhlen-Stiftelsen (2021-203061) and Alzheimerfonden (AF-968621) (L.M.G.); the Fondo de Investigaciones Sanitario, Carlos III Health Institute INT21/00073, PI20/01473, and PI23/01786 (J.F.) and the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas Program 1, partly jointly funded by Fondo Europeo de Desarrollo Regional, Unión Europea, Una Manera de Hacer Europa; R01 AG056850, R21 AG056974, R01 AG061566, R01 AG081394, and R61AG066543 (J.F.), P30 AG066512 (T.W.), R01 AG087280 (T.W.), the Department de Salut de la Generalitat de Catalunya, Pla Estratègic de Recerca i Innovació en Salut (SLT006/17/00119) (J.F.), Fundación Tatiana Pérez de Guzmán el Bueno (IIBSP-DOW-2020-151) (J.F.), and Horizon 2020–Research and Innovation Framework Program from the European Union (H2020-SC1-BHC-2018-2020) (J.F.). Proteomic analysis was performed at the Proteomics Core Facility, Sahlgrenska Academy, Gothenburg University, with financial support from SciLifeLab and BioMS.
Author information
Authors and Affiliations
Contributions
L.M.-G., D.A., M.M.-A., T.W., H.Z., J.F., N.T.S., A.I.L., and E.C.B.J. designed the experiments; L.M.-G., M.S., J.L.-R., J.Fuchs, M.M.-A., T.W., C.M.W., L.P., and D.M.D. carried out experiments; S.B., E.B.D., M.M.-A., T.W., and E.C.B.J. analyzed data; N.J.A., O.B., J.N., I.B., L.V., K.B., B.B., B.R.R., M.C.-I., J.G., and A.L. provided advice on the interpretation of data and manuscript review; E.C.B.J., L.M.-G., M.M.-A., J.F., E.B.D., and S.B. wrote the manuscript with input from coauthors.
Corresponding authors
Ethics declarations
Competing interests
E.C.B.J. has served on an advisory board for Eli Lilly and has received royalties from EmTheraPro (outside submitted work). J.F. reported receiving personal fees for service on the advisory boards, adjudication committees or speaker honoraria from AC Immune, Adamed, Alzheon, Biogen, Eisai, Esteve, Fujirebio, Ionis, Laboratorios Carnot, Life Molecular Imaging, Lilly, Lundbeck, Perha, Roche and outside the submitted work. O.B., D.A., A.L., and J.F. report holding a patent for markers of synaptopathy in neurodegenerative disease (WO2019175379, applicant Fundació Institut de Recerca de L’hospital de la Santa Creu, Sant Pau, “Markers of synaptopathy in neurodegenerative diseases”, EP19709749.6, priority date 16/3/2018, patent pending, inventors O Belbin, A Lleó, A Bayés, J Fortea, and D Alcolea, licensed to ADX NeuroSciences, N.V. EPI8382175.0 to develop antibodies and immunoassays for Calsyntenin-1, GluR2, GluR4, Neuroligin-2, neurexin-2a, neurexin-3a, syntaxin-1b, thy-1, tenascin-r, and vamp-2). N.T.S. and A.I.L. are founders of EmTheraPro (outside submitted work). H.Z. has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, Cognito Therapeutics, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). K.B. has served as a consultant, on advisory boards, or at data monitoring committees for Abcam, Axon, BioArctic, Biogen, and JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Ono Pharma, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this paper. T.W. has served as a consultant or on data monitoring committees for Acumen, Biogen, Grifols, Lilly and ProMIS Neurosciences. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Montoliu-Gaya, L., Bian, S., Dammer, E.B. et al. Proteomic analysis of Down syndrome cerebrospinal fluid compared to late-onset and autosomal dominant Alzheimer´s disease. Nat Commun 16, 6003 (2025). https://doi.org/10.1038/s41467-025-61054-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61054-z
