
Abstract
Glycogen, a key branched glucose polymer, acts as a vital energy reservoir in mammalian cells, particularly during intense activity or fasting. The glycogen debranching enzyme (GDE) plays a key role in glycogen degradation by removing branches, ensuring efficient glucose release. Dysfunction of GDE leads to the accumulation of limit dextrin and is implicated in the pathogenesis of Glycogen Storage Disease Type III (GSD III). We present the cryo-EM structure of human GDE (hsGDE) at 3.23 Å resolution, providing molecular insights into its substrate selectivity and catalytic mechanism. Our study further investigates the molecular consequences of disease-associated mutations by correlating structural data with enzymatic activities of representative GSD III-causing variants. We discover that these mutations induce GSD III through diverse mechanisms, including significant reductions in enzymatic activity, and disruptions to the glycogen-bound region and overall structural integrity. The elucidation of these pathways not only advances our understanding of hsGDE’s role in substrate recognition and catalysis but also illuminates the molecular pathology of GSD III. Our findings pave the way for the development of targeted therapeutic strategies for this disease.
Similar content being viewed by others


Introduction
Glycogen, an intricate glucose polymer exhibiting a highly branched structure, serves as a crucial energy reservoir in numerous animals, including humans1. Predominantly stored in the liver and muscle cells, glycogen is capable of being promptly mobilized to satisfy the body’s energy demands and to maintain stable blood glucose levels during periods of intense activity or fasting. Furthermore, glycogen has also been detected in other tissues such as the brain2,3, heart, and kidney4,5. In the human body, the breakdown of glycogen primarily occurs within the cytoplasm. Additionally, a minor fraction of glycogen degradation is mediated by α-glucosidase within lysosomes1,6. The glycogen degradation in the cytoplasm involves a synergistic action between two enzymes: glycogen phosphorylase (GP) and glycogen debranching enzyme (GDE). Through their collaborative efforts, glycogen is converted into glucose and glucose-1-phosphate, thereby stabilizing blood glucose levels and providing essential energy.
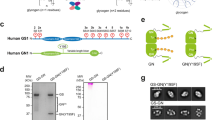
GDE occupies a pivotal role in glycogen degradation. The glucose residues of glycogen are connected through α−1,4 glycosidic bonds, with branch points being linked by α−1,6 glycosidic bonds (Supplementary Fig. 1). GP sequentially removes glycosyl units from the non-reducing terminus of glycogen, generating limit dextrin and glucose-1-phosphate7. Subsequently, GDE, a bifunctional enzyme possessing both 4-alpha-glucanotransferase (GT) and amylo-α−1,6-glucosidase (GC) activities, eliminates the branches of limit dextrin in two steps4,5,8. Initially, the GT activity transfers the maltotriosyl group, linked by α−1,4 glycosidic bonds, from the branch to a proximate non-reducing end of glycogen. Then the GC activity hydrolyzes the α−1,6 glycosidic bonds at branch points, catalyzing the removal of glucose residues and releasing free glucose (Fig. 1a).

a The degradation pathway of glycogen. The glucosyl residues in the main chain are shown in green; the glucosyl residues in the non-reducing end are in brown; the branched point is in salmon and the last three residues of the branched chain are depicted in blue. This figure was created by referring to some elements from the published figure8. b Cartoon of the hsGDE domain organization. hsGDE domain boundaries are drawn to scale. This coloring scheme is used throughout the manuscript unless otherwise indicated. c, d Front view and back view of hsGDE cryo-EM density map (c) and atomic model (d). The color scheme is the same as in (b). All figures are prepared with Chimera (https://www.cgl.ucsf.edu/chimera/) and ChimeraX (https://www.cgl.ucsf.edu/chimerax/).
Dysfunction of hsGDE contributes to the abnormal accumulation of limit dextrin in the human body. This pathological accumulation is the hallmark of GSD III, a complex condition with distinct subtypes determined by specific losses of activities9,10. The majority of GSD III cases, comprising 78%, exhibit a GT/GC activities loss in both liver and muscle, classifying them as the GSD IIIa subtype. Alternatively, 15% of patients exclusively suffer from GT/GC activity loss in the liver, defining the GSD IIIb subtype. Rarely, the loss of a single activity, whether GC or GT activity, leads to the GSD IIIc or GSD IIId subtypes, respectively11. Patients diagnosed with GSD III often present with a range of symptoms, including hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation4,5,12. However, the molecular basis underlying the functional losses exhibited by hsGDE mutants remain elusive. To bridge this knowledge gap and gain a deeper understanding of GSD III, further structural and functional studies of hsGDE are urgently needed.
Furthermore, hsGDE occupies a pivotal position in numerous metabolic processes. It plays a crucial role in suppressing cancer growth13,14, modulating AMP-activated protein kinase (AMPK) activity15, and facilitating the degradation of Polyglucosan Bodies (PBs)16,17, whose intricate molecular mechanisms, however, remain unclear necessitating further exploration.
Despite the fact that the structure of cgGDE (glycogen debranching enzyme from Candida glabrata) has been resolved8 and exhibits a high sequence similarity of 37.5% (Supplementary Fig. 2) with its human counterpart, the existing structural information alone remains insufficient to fully elucidate the mechanisms governing the selection and catalysis of more complex glycogen in mammalian species, as well as the molecular difference underlying GSD III in human. The elucidation of hsGDE atomic structures will propel this inquiry. Therefore, obtaining a high-resolution atomic-level structure of hsGDE is crucial for providing molecular insights into these unresolved questions and advancing our understanding of hsGDE’s multifaceted role in glycogen metabolism, GSD III, and its non-enzymatic activity.
Here, we present the structure of hsGDE at a resolution of 3.23 Å, providing a detailed understanding of its substrate selectivity and catalytic mechanism. Furthermore, we design 21 mutant variants associated with GSD III, identified in patients, to gain deeper insights into this condition. This comprehensive approach enhances our understanding of hsGDE ‘s function in cellular metabolism and disease pathogenesis, paving the way for future therapeutic advancements.
Results
Structure determination of hsGDE
The full-length wild-type hsGDE was engineered with an N-terminal 3×FLAG tag for purification and transiently expressed in HEK293F cells. We isolated hsGDE using affinity chromatography, followed by size exclusion chromatography (Supplementary Fig. 3), preparing it for subsequent negative-stain transmission electron microscopy and cryo-electron microscopy (cryo-EM) analysis. Details of data collection, image processing, and 3D reconstruction are delineated in Supplementary Fig. 4 and Supplementary Table 1. Our research successfully resolved the cryo-EM structure of hsGDE to a gold-standard resolution of ~3.23 Å (Supplementary Fig. 4). The electron density map and the corresponding atomic model of hsGDE are presented in Fig. 1c, d.
The overall structure of hsGDE
The human glycogen debranching enzyme (hsGDE) is composed of four distinct domains: the GT domain, the GC domain, and two middle domains, M1 and M2 (Fig. 1b–d). The GT domain (residues 116 to 947), which includes the A, B, and C subdomains, is conserved in the GH13 family and is responsible for transferring the final three glycosyl residues from branched to main chains, a process known as 4-alpha-glucanotransferase (GT) activity (EC 2.4.1.25) (Fig. 1b). The GC domain, located at the C-terminus (residues 1035 to 1532), belongs to the GH133 family and is characterized by its specific amylo-α−1,6-glucosidase activity (GC activity, EC 3.2.1.33), which hydrolyzes α−1,6-glycosidic bonds to release free glucose. These two main domains are positioned at opposite ends of the molecule with minimal direct interaction, suggesting its unique spatial organization.
Moreover, the M1 and M2 domains serve as connectors between the GT and GC domains, contributing to the overall stability of the protein (Fig. 1c, d). Specifically, the interfaces between the M1/M2 domains and the GT domain bury an extensive area of 4,453 Ų (Interface I), while the interface with the GC domain buries 2,531 Ų (Interface II) (Supplementary Fig. 5a, b). These buried interfaces may be crucial for the structural integrity and functional competence of hsGDE within the cellular milieu. Despite their proximity, the M1 domain and the M2 domain exhibit minimal contact with each other, separated by a cavity that provides a distinctive spatial arrangement. A loop within the A domain, spanning residues 596-617, extends into this cavity, as depicted in Supplementary Fig. 5c. This feature adds a layer of complexity to the structural dynamics of hsGDE.
Before the preparation of human GDE’s structure, the cgGDE one was published8,18. In our study, therefore, we also conducted a detailed structural comparison between the hsGDE and the cgGDE structure (Supplementary Fig. 6). The three-dimensional architecture of hsGDE and cgGDE shows a high degree of similarity (Supplementary Fig. 6a), with a Cα RMSD value of 1.352 Å for all residues, indicating a close match in their overall structure and highlighting the evolutionary importance of these proteins (Supplementary Fig. 6b). However, differences are evident between the two enzymes. Specifically, the increased number of negative residues in cgGDE could potentially lead to electrostatic repulsion with glycogen, hindering its utilization of complex glycogen molecules (Supplementary Fig. 6d), which may correlate with the cgGDE’s poor ability to handle glycogen19. In contrast, the substrate-binding pocket of hsGDE harbors a higher number of positively charged residues. These residues facilitate favorable electrostatic interactions with the negatively charged regions of glycogen, thereby enhancing both the binding affinity and catalytic efficiency of hsGDE (Supplementary Fig. 6c). Moreover, hsGDE exhibits superior stabilization of the transition state compared to cgGDE, as evidenced by the structural comparisons (Supplementary Fig. 6h–m). At the atomic level, hsGDE forms stronger interactions with glycogen (Supplementary Fig. 6p), whereas cgGDE displays weaker interactions (Supplementary Fig. 6n–o). These weaker interactions in cgGDE result in reduced binding affinity and catalytic efficiency. Collectively, these structural and functional differences highlight the species-specific adaptations that have shaped these enzymes. hsGDE appears to be more optimized for glycogen utilization, whereas cgGDE may encounter challenges in processing more complex glycogen structures due to its distinct structural and charge distribution characteristics.
The molecular basis for the glycogen selectivity within the GT domain
To elucidate the substrate selectivity and translocation mechanisms of hsGDE catalysis, we selected maltopentaose as a representative substrate and conducted all-atom molecular dynamics (MD) simulations (Supplementary Fig. 7). Our unbiased MD simulations revealed significant dynamics in the GT domain, as indicated by notably higher root mean square fluctuations (RMSF) (Supplementary Fig. 7a), consistent with the relatively lower cryo-EM density observed in this domain. An ambiguous cryo-EM density in the B domain was observed, suggesting a potential association with the glycan. To confirm whether this blurred density corresponded to the glycan, we further conducted MD simulations of the maltopentaose-bound GDE complex. In these simulations, the conformation of GDE was constrained according to the cryo-EM density map, and maltopentaose underwent a restrain-and-release protocol (initially restrained in the binding pocket and subsequently unrestrained to explore freely, as detailed in the Methods section), aimed at investigating the glycan binding modes. The MD docking complex illustrated extensive interactions between maltopentaose and the GDE binding pocket within GT domain (Fig. 2a), involving residues like W487, D627, and F557 (Fig. 2b and Supplementary Fig. 7e–g).

a Conformational distribution of the substrate derived from three simulations of the complex with restrained GDE conformation. The x-axis, y-axis, and color bar illustrate the center of mass (COM) distance between the substrate and key residues in the binding pocket of the GT domain of GDE, root mean square deviation (RMSD) for the Cα atoms of the substrate relative to the final MD-docking structure, and the number of protein-substrate contacts within a 0.5 nm cutoff. b Contact frequencies of key residues interacting with the substrate, sorted by their contact frequencies. c The hsGDE’s maltopentaose-binding structure achieved by MD. The maltopentaose is in a sphere form and shown in green. d The constriction of the pocket. All loops composing the pocket migrate inward, induced by the binding of maltopentaose. The maltopentaose-binding structure is colored, and the wild-type structure is shown in gray. e, f The surface models show that maltopentaose binds in a polar (e) and hydrophobic (f) chemical environment in hsGDE. g Interactions between maltopentaose and surroundings residues. The dash lines in green indicate hydrogen bonds, and the dashed lines in black mark the CH-π interactions. h Analysis of interaction between maltopentaose and surroundings residues is achieved by the LigPlot+ program. The dash lines in green indicate hydrogen bonds and the red arc mark the hydrophobic interaction. i−k The activity of potential substrate-binding residues. i The glycogen binding affinity. j The GT activity (k) The overall enzymatic activity. All data in figures indicate ratios to the wild-type value with the wild-type indicated at 1.
These residues, exhibiting high contact frequencies with the glycan, likely play crucial roles in the selectivity and catalysis of glycogen branches (Fig. 2b). Comparing the maltopentaose-bound structure with the wild-type enzymes may provide insights into the glycogen selectivity within the GT domain. Our MD simulations of the maltopentaose-bound GDE revealed that maltopentaose binds to a pocket enclosed by the A and B domains, where the inherent flexibility of the B domain facilitates substrate translocation. A closer examination of the binding mode indicates that maltopentaose adopts a vertical orientation within the pocket (Fig. 2c and Supplementary Fig. 8a). The non-reducing end, positioned at subsite +2, engages in specific interactions with residues such as F557 and E579 (Fig. 2g and Supplementary Fig. 8b). This orientation positions the 4-OH group of the glucose at subsite +2 near the catalytic residues, thereby possibly favoring the second transglycosylation step—where transfer to the main chain’s α−1,4 linkage occurs. By comparison, in the cgGDE structure, the hydroxy group at position 6 (6-OH) of glucose at subsite +2 is closest to subsite –1 (Supplementary Fig. 6n–o), a configuration that aligns with the first step of transglycosylation.
Although the substrate-binding residues are largely conserved between hsGDE and cgGDE, the structural distinctions—specifically, the size and chemical environment of the binding pocket—result in divergent binding modes. In hsGDE, the more constrained pocket and its mixed polar/hydrophobic character direct maltopentaose to bind such that the 4-OH is optimally positioned for the second transglycosylation step. This contrasts with the broader pocket of cgGDE, which accommodates a binding mode suitable for the initial transglycosylation reaction.
In hsGDE, key residues such as F557, E579, D627, N628, E629, E667, H412, G413, and P414 are strategically positioned at the pocket’s entrance, further restricting the entry of longer chains, which otherwise would introduce steric hindrance with surrounding residues (Supplementary Fig. 8b). On the other hand, the binding of maltopentaose induces a conformational change, causing the flexible loops (residues 166−176, 410−427, 457−480, 579−588, 666−676) to migrate inward and constrict the pocket (Fig. 2d). This adaptation results in a more confined space that may selectively bind maltopentaose while excluding longer glycogen side chains.
Moreover, the binding of maltopentaose to the ligand-binding pocket is facilitated by a unique chemical environment that is both polar and hydrophobic. This side of the pocket, which is polar in nature, houses maltopentaose and engages in extensive hydrogen bonding and CH-π interactions with surrounding residues (Fig. 2e, g). Upon analyzing the binding interface, crucial residues such as N527, E555, E579, H626, E629, E667, I668, and V670 are identified to participate in these interactions (Fig. 2g, h and Supplementary Fig. 8c). Significantly, a CH-π interaction is observed between the pyranose ring of Glc1 and the side chains of F557 and W461, which is a common interaction pattern in sugar-binding proteins20,21 (Fig. 2g and Supplementary Fig. 8d). In addition to this, the aromatic and hydrophobic residues on another side of the pocket create a hydrophobic environment that complements the nonpolar regions of the pyranose rings (Fig. 2f–h). This distinctive chemical environment within the ligand-binding pocket may represent a unique mechanism by which GDE facilitates the entry of glycogen into the active site of GT. All these results show that the combination of size, shape, and the precise chemical environment within the pocket plays a specialized role in the recognition, binding, and induction of maltopentaose, which is essential for the function of GT activity.
To substantiate our hypothesis, we generated a series of mutants to assess their impact on substrate-binding affinity and GT activity (Fig. 2i–k). Consistent with our predictions, the majority of these mutants exhibited a loss of GT activity, while their substrate-binding affinity remained largely unaffected. This outcome could be attributed to the extensive interactions between glycogen and the protein, suggesting that the single mutations we introduced do not significantly alter the affinity of glycogen for the protein. Consequently, despite the mutations’ large influence on GT activity, the binding affinity remained minimally changed. This suggests that enzyme activity may be regulated by complex mechanisms beyond substrate affinity alone, such as its specific chemical environment. However, we acknowledge that further research is needed to validate these mechanisms.
Catalytic mechanisms of glycogen debranching enzyme domains
As mentioned before, the GC and GT domains belong to the GH133 and GH13 families, which share conserved conformation and catalytic residues. In the following sections, we will individually explore the catalytic mechanisms of the GC and GT domains to gain a deeper understanding of their specific contributions to the enzyme’s overall activity.
The GC domain’s (α/α)6-barrel fold is a common structural signature among GH133 enzymes (Fig. 3a and Supplementary Fig. 9a). In these enzymes, one acidic residue typically acts as a proton donor, while the other serves as a general base catalyst that facilitates the nucleophilic attack by a water molecule. To investigate the conserved catalytic mechanism of hsGDE’s GC domain, we compared it with homologous proteins. However, due to the limited availability of high-resolution structures within the GH133 family, we also included a GH15 family protein, specifically the glucodextranase from Arthrobacter globiformis I4222, in our comparison. GH15 family enzymes are characterized by their typical (α/α)6-barrel fold, which is also present in the GC domain of hsGDE. This conserved folding pattern ensures the spatial conservation of catalytic residues, which is crucial for enzymatic catalysis. Additionally, both this GH15 enzyme and hsGDE are capable of hydrolyzing α−1,6-glycosidic bonds, further justifying its use as a comparison target. In hsGDE, the conserved catalytic residues D1261 and E1502 show significant homology to those in cgGDE8 (D1241 and E1492) and the glucodextranase from Arthrobacter globiformis I4222 (E430 and E628) (Fig. 3b, c). To experimentally confirm the importance of D1261 and E1502 in GC activity, we introduced mutations D1261A and E1502A. The resultant mutant enzymes showed a substantial decrease in GC activity (Fig. 3g), validating their role. As expected, these mutations had little effect on GT activity (Fig. 3h), suggesting that while crucial for GC activity, these residues are not essential for GT activity. Similar mutations in cgGDE8 and Saccharomyces cerevisiae GDE (scGDE)23 also significantly affected GC activity, indicating a conserved function in different enzymes.

a, d The catalytic region from hsGDE GC (a) or GT (d) domains. The catalytic residues are shown in atoms. b, c and e, f Structural comparison of the catalytic region of hsGDE with glucodextranase from Arthrobacter globiformis I42 (b PDB ID:1UG9), Aspergillus oryzae alpha-Amylase (e PDB ID: 2TAA) and cgGDE (c, f PDB ID: 5D06). Glucodextranase, alpha-Amylase, and cgGDE were colored in light purple, pink, and dim gray, respectively. g−i Specific debranching activities of hsGDE and its GT/GC-defective mutants, including GC activity (g), GT activity (h), and Holoenzyme activity (i). All data in the figures indicate ratios to the wild-type value with the wild-type indicated at 1. More details about statistical analysis are shown in Supplementary Fig. 10.
Despite low sequence identity with most GH13 enzymes24, the key catalytic region in hsGDE shows a strikingly similar (β/α)8-barrel structure (Fig. 3d–f and Supplementary Fig. 9b). The catalytic triad in hsGDE, consisting of Asp526, Glu555, and Asp627, closely resembles that of other GH13 enzymes, with Asp526 as the nucleophile, Glu555 as the acid/base, and Asp627 stabilizing the transition state. This arrangement is supported by the structural elements defining the catalytic site (Fig. 3d). To assess the impact of these residues on hsGDE’s enzymatic functions, we created some mutants. As anticipated, these mutants showed a significant loss of debranching activity, confirming the importance of these residues for GT activity (Fig. 3g–i). Consistent with our findings, equivalent mutations in cgGDE8 and scGDE23 also compromised GT activity, highlighting the conserved role of these residues.
Molecular difference underlying pathogenic mutations in glycogen storage disease type III
To better understand the molecular differences associated with GSD III, we analyzed 21 pathogenic mutation sites identified in affected patients. By mapping these mutations onto the enzyme’s structure, we found that all mutations are located in the GT/GC domains except for R864P, suggesting that they may impair enzymatic activity and contribute to the disease (Fig. 4a, b).

a, b Mapping of GSD III-causing mutations in the hsGDE structure. Disease-causing mutations are shown in sphere form with only backbones. c, d GSD III-causing mutations located in the GC (c) or GT (d) active pocket. The catalytic residues are in salmon/blue and GSD III-causing mutations are in purple. e−g Specific activities of wild-type hsGDE and GSD III-causing mutants, including GC activity (e), GT activity (f), and Holoenzyme activity (g). All data in the figures indicate ratios to the wild-type value with the wild-type indicated at 1. More details about statistical analysis are shown in Supplementary Fig. 11.
Notably, one-third cluster in regions critical for enzymatic activity, potentially compromising the functions of the GT or GC domains (Fig. 4c, d). Several mutations, such as D215N, R524H, L620P, H626R, and G655R, are situated in the catalytic core of the GT domain, specifically within the α/β−8 barrel (Fig. 4d). These mutations significantly affect GT activity by altering the conformation of the catalytic region, with minimal impact on GC activity (Fig. 4e–g). Other disease-causing mutations are located near the GC active pocket’s extension loop, close to catalytic residues, including G1087R, R1147G, and G1448R (Fig. 4c). They exert a profound effect on GC activity, nearly abolishing overall enzyme function (Fig. 4e–g).
Some mutations, such as N219D and R494H, are associated with the compromise of glycogen-bound regions. N219 is homologous to N121 from α-amylase in a conserved calcium-binding site, which is crucial for maintaining loop stability and regulating glycogen-binding sub-sites25,26 (Supplementary Fig. 12a). Mutations like N219D and R494H may disrupt the calcium-binding region or key glycogen-bound regions, reducing glycogen affinity and affecting enzymatic activity (Supplementary Fig. 12b, f).
The integrity and stability of hsGDE are essential for its normal activity. Mutations such as G138E and R675W, located at the junction of the B domain and C domain, adversely affect the GT catalytic region, leading to significant impairments in GT activity (Supplementary Fig. 13). Additionally, proline-induced mutations, like A253P, L400P, and A1120P, destabilize proteins by disrupting secondary structures27, affecting enzymatic activity and its glycogen affinity (Supplementary Fig. 14). The C234R mutation, at the intersection of A and B domains, replaces a Cys with a more rigid and bulky Arg to greatly reduce the conformational flexibility and undermine all enzymatic functions (Supplementary Fig. 11). This observation highlights the B domain’s importance for hsGDE activity.
Additionally, specific mutations, such as R343W28, H62628, and D1364H29, identified in distinct patient cohorts, are associated with GSD III. These mutations are hypothesized to precipitate the disease through mechanisms that reduce protein expression and stability (we did not obtain the mutant proteins under the same conditions). Although not examined under identical conditions, these alterations may intensify the disease phenotype, suggesting a complex interplay between genetic mutations and disease severity.
Mutations in non-catalytic domains, such as R864P, may contribute to disease progression and phenotypic heterogeneity through some specific mechanisms, despite minimal effects on hsGDE activity. R864P is located precisely at the junction where the GT domain connects with the M2 domain within the protein’s structure, which may have significant implications for the overall integrity of the protein. Specifically, this mutation can disrupt the stability of the entire protein structure by interfering with the proper folding process. Moreover, it may also predispose the protein to form aggregates (the comparison of their size exclusion chromatography data is included in the Source Data file). Once proteins aggregate and form insoluble clumps within the cell, these insoluble aggregates can severely disrupt the cell’s normal functioning, thereby leading to the manifestation of the disease. In addition to these structural implications, GSD III mutations show allelic heterogeneity across the AGL gene (gene encoding hsGDE), with some mutations prevalent in specific ethnic groups12,30, like R864P in the United States, affecting 10.3% of GSD IIIa cases12. This finding suggests a role for R864P in ethnic heterogeneity and emphasizes the complexity of GSD III, warranting further investigation into the molecular mechanisms of these mutations.
hsGDE exists as a monomer and dimer
An unexpected finding was the discovery of hsGDE existing in both monomeric and dimeric forms, with dimerization facilitated by the GC domain (Fig. 5). Previous studies have demonstrated that glycogen debranching enzymes from Streptomyces venezuelae31 and Sulfolobus solfataricus32 exhibit different assembly patterns under different pH conditions, as well as under the influence of various allosteric effectors, with ATP having the most significant effect. These changes in assembly are associated with alterations in their enzymatic activity. Remarkably, this allosteric effectors/pH-mediated regulatory mechanism appears to be a conserved feature across diverse species and organisms, including porcine muscle33,34, chicken muscle35 and the bovine brain36. In summary, the presence of dimeric hsGDE is reasonable, and its activity appears to be modulated by pH, allosteric effectors, and its own assembly state. To validate this, we manipulated the pH of the solution, and the small molecule added, as well as quantified the percentage of dimers (Supplementary Fig. 15). We confirmed that GDE assembly is pH-dependent and ATP plays a regulatory role (Supplementary Fig. 15). Despite our extensive efforts, we have been unable to achieve a stable dimeric state, thereby precluding the acquisition of a high-resolution EM map. Consequently, our ability to further elucidate the structural mechanisms underlying dimer formation remains limited.

a, b The result of 2D classification exhibits monomer and dimer. a The 2D classification of monomeric state. b The 2D classification of dimeric state. c The electron density map of the GDE dimer. Maps in the picture are rotated by the X-axis. d The structure of dimeric GDE. The color scheme follows the previous one. The structure colored green indicates the interaction between two protomers.
Discussion
For decades, the structure of hsGDE has remained elusive. Our study now presents the high-resolution structure of full-length hsGDE, unraveling a possible catalytic mechanism and revealing molecular differences underlying GSD III. Besides, we also provide a structural insight into the glycogen selectivity and stable binding, which may be achieved by the special pocket shape and the chemical environment. Compared to the previously published structures of cgGDE and its maltopentaose complex8,18, our analysis reveals similar overall architectures but distinct differences in the A domain loop, electronic potential surface, and substrate positioning and orientation (Supplementary Fig. 6), which may correlate with the cgGDE’s poor ability to handle glycogen’s heterogeneous size, polymerization, and branching19. Specifically, the human one has a higher density of positive charges (Supplementary Fig. 6g), and its binding pocket is more constrained (Supplementary Fig. 6g). Additionally, amino acids are arranged in a more organized fashion on both sides of the pocket (Supplementary Fig. 6k–m), which may facilitate the transfer of substrates. We speculate that this may be due to the evolutionary adaptations of the two enzymes in processing glycogen of varying complexities (Supplementary Fig. 2).
Employing the hsGDE structure and MD simulations, we provide a comprehensive analysis of the enzyme’s substrate selectivity and attachment. Our proposed model illustrates how hsGDE selectively recognizes glycogen and efficiently transfers it to the catalytic region within the GT domain (Fig. 6a). The binding pocket’s size, shape, and chemical environment are critical for glycogen recognition and stabilization, facilitated by hydrophobic and charged residues that guide binding and enhance the sliding efficiency, reminiscent of the Maltoporin Membrane Channel mechanism37,38,39. In addition, the B domain also may play a crucial role in substrate binding. This region usually varies in length and irregular structure inserted in the loop between the β3-strand and α3-helix of the barrel40 (Supplementary Fig. 5d). Furthermore, recent studies have found that the B domain has a function in opening the (β/α)8 catalytic barrel outward, exposing the key catalytic amino acid24. The limited electron density of the B domain in our study also demonstrates its high flexibility (Supplementary Fig. 4). This flexibility may allow it to modulate the number of embedded glucose molecules through conformational adjustments (Fig. 6a). This suggests a possible regulatory function for the B domain in controlling the enzyme’s activity and substrate interaction. However, further research is warranted to validate this regulatory function.

a The structural basis of substrate selectivity in hsGDE GT domain. The selectivity for proper number glycosyl units is achieved by the size and the specifical chemical environment of the binding pocket. Lines in green indicate the H-bond, and circles mark the hydrophobic interaction. b Mutations that underlie GSD III trigger the disease through diverse pathways, including but not limited to impairment of catalytic function (magenta), reduction in substrate affinity (blue), disruption of structural integrity (orange), and other pathways (green), thereby manifesting the molecular differences that underlie this condition. Disease-causing mutations are shown in sphere form with only backbone.
Phylogenomic analysis41 reveals that GDE, as a bifunctional enzyme, is conserved across animals and fungi, but not in bacteria, where separate enzymes perform α−1,6-glucosidase and 4-alpha-glucanotransferase functions, contrasting with the intra-molecular translocation of the substrate in eukaryotic GDEs23,42,43. Evidence, in addition, suggests that the GT and GC activities of hsGDE may operate independently, each with distinct substrate specificity8,44 (Fig. 3). The larger size of hsGDE, approximately twice that of glycogen branching enzyme (GBE)45, may be an adaptation to limit dextrin structures46, allowing simultaneous binding to different glycogen branches and concurrent domain function, thus enhancing glycogen degradation.
Without structural information, understanding the impact of GSD III patient mutations has been challenging. Our structures and biochemical assays offer a framework to demystify this complexity (Fig. 6b). Mutations clustering in the GT/GC domain’s crucial catalytic regions significantly impact activity and contribute to disease pathogenesis (Fig. 4).
During our research, we unexpectedly discovered that hsGDE exists as a dimer, a finding not previously reported. Notably, many mammalian homologs, such as those from chickens35, pigs33,34 and cattle36, also exhibit dimerization, with their activity regulated by this process. Given the sequence and structural similarities between these homologs and the human protein, this suggests a conserved dimerization mechanism across species, likely crucial for hsGDE function in specific physiological processes. Additionally, structural studies of two prokaryotic homologs—GlgX31 from Streptomyces venezuelae (dimerization mediated by c-di-GMP) and TreX47 from Sulfolobus solfataricus (oligomerization)—have shown that their dimerization or oligomerization affects protein activity. Although these archaeal proteins differ from mammalian homologs, their findings hint at a potential regulatory mechanism: dimerization influenced by small molecules and impacting activity, which may also apply to human homologs. Experimental evidence suggests that the human protein exists as a dimer, with ATP modulating its dimerization. This supports the idea that ATP, as a key cellular energy molecule, regulates hsGDE dimerization and activity. The ATP-dependent dimeric structure may allow cells to respond to varying energy states or intracellular signals, thereby fine-tuning enzyme activity to ensure proper glycogen metabolism and cellular homeostasis.
In conclusion, our studies elucidate the structure, substrate selectivity, and catalytic mechanism of hsGDE. The expanded understanding of hsGDE’s structural and functional implications opens avenues for therapeutic strategies targeting this enzyme for the treatment of GSD III.
Methods
Protein expression and purification
The full-length open reading frames (ORFs) encoding homo sapiens GDE (hsGDE, UniProt ID: P35573) were amplified from cDNA by PCR. Subsequently, both wild-type hsGDE and its mutants (detailed primers for WT and mutation are provided in Source data) were cloned into modified pCAG vectors containing an N-terminal 3×Flag tag and an HRV 3C protease site. All plasmids were confirmed by sequencing. These plasmids were then transfected to HEK293F cells (provided by Union-Biotech(shanghai)) utilizing Polyethylenimine Linear (PEI, Sigma) as a transfection agent. Transfected cells were cultured at 37 °C for 48 h before harvesting. The cell pellets were frozen in liquid nitrogen and stored at −80 °C until needed.
Cell pellets containing either wild-type hsGDE or its mutants were lysed in a buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton, 1 × Cocktail (Novagen), 1 mM EDTA, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF) at 4 °C for 30 mins. Subsequently, the lysate was clarified by centrifugation at 13,000 × g at 4 °C for 30 mins, and the supernatant was incubated with Anti-DYKDDDDK G1 Affinity Resin (GenScript) for 1.5 h followed by on-column washing at 4 °C in buffer containing 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 2 mM MgCl2, 10 mM ATP. The proteins were eluted out with 200 μg/ml peptide and concentrated for further purification by size exclusion chromatography (SEC) on Superdex 200 columns (GE Healthcare). The freshly purified hsGDE was utilized for cryo-EM sample preparation, while the remaining proteins were promptly frozen in liquid nitrogen and stored at –80 °C for future processing in activity assays. All hsGDE proteins and mutants were confirmed by SDS-PAGE.
GDE activity assays
Purification of glycogen phosphorylase-digested glycogen
Glycogen phosphorylase-digested glycogen was generated following a standardized protocol8,48. Specifically, 1 gram of glycogen from oyster (Sigma) and 200 U muscle glycogen phosphorylase-\(\alpha\) from rabbit (Sigma) were dissolved in 20 ml of digestion buffer containing 200 mM phosphate pH 6.8. This mixture was subjected to overnight dialysis against 2 L digestion buffer at 37 °C. Subsequently, glycogen phosphorylase-\(\alpha\) was precipitated by heating at 100 °C for 30 mins and removed by centrifugating at 13,000 × g for 30 mins. The resulting products were further precipitated by adding double-volume ethanol, followed by collection via centrifugation. The obtained precipitate was then resolved in H2O and dialyzed against 2 L H2O overnight at 4 °C. The product was precipitated once more by adding 2 times ethanol and collected by centrifugation. The precipitate was dried in a 50 °C oven until its weight remained constant three times, indicating complete dryness. That is a common method for quantifying the weight, citing Pharmacopoeia of the People’s Republic of China 2020 Edition (in 4 volumes). It was then weighed and dissolved in water to achieve a final concentration of 40.8 μg/ml.
Overall GDE activity assay
The overall activity of hsGDE was assayed by the glucose produced from glycogen phosphorylase-digested glycogen. 16.7 mM citric acid at pH 6.5, varying concentrations of glycogen phosphorylase-digested glycogen, and 0.3 mM hsGDE were mixed and each experiment was repeated three times. Glucose production was measured using the Glucose Colorimetric Assay Kit (GOD/POD Method) (Solarbio Life Science), and the resulting absorption change at 505 nm was monitored on an Ultraspec 2100 pro spectrophotometer (GE Healthcare). The specific activity in our study is defined using the catalytic constant (Kcat). The Kcat value was calculated using the formula: Kcat = Vmax / [E]. This experiment was repeated three times.
GT activity assay
The GT activity was assayed by monitoring the production of additional oligosaccharides from maltopentaose. The reaction mixture contained 50 mM citric acid pH 6.5, 20 mM maltopentaose, and 10 mM hsGDE. The reaction was allowed to proceed at 37 °C for 12 h and was terminated by heating at 100 °C for 10 mins. After removing the precipitated protein by centrifugation, the supernatant was dried with an SPD121P speed vacuum concentrator (Thermo Scientific) and redissolved in 70% methanol.
The products were analyzed by UPLC-MS/MS. UPLC-MS/MS analysis was performed on a Thermo Vanquish UHPLC system (Thermo Fisher Scientific, United States) coupled to a Q Exactive mass spectrometer (Thermo Scientific, Thermo Fisher Scientific, United States). Chromatographic separation was achieved on an ACQUITY UPLC® BEH Amide column (100 × 2.1 mm, 1.7 μm; Waters) at 50 °C. Ultrapure water with 0.1% (v/v) of Formic Acid (FA) and acetonitrile were employed as mobile phases A and B, respectively, operated at a constant flow rate of 0.3 mL/min throughout separation. The injection volume was 2 μL. The UPLC-MS/MS analysis of each sample was performed three times.
GC activity
The measurement of GC activity was similar to that of the overall activity assay while the substrate was produced from glycogen phosphorylase-digested glycogen and hsGDE R1147G mutant. We purified a substantial amount of hsGDE R1147G mutant, a mutation featured by GC activity null state. Glycogen phosphorylase-digested glycogen (manufactured by us) was mixed with an excess of hsGDE R1147G mutant (1:5 ratio) ensuring the limit dextrin degraded into the substrate for the GC activity completely. The mixture was subjected to overnight dialysis against 2 L digestion buffer at 37 °C. Proteins were removed by heating and centrifugation and then the product in the supernatant was further purified by ethanol and water. Finally, the product was dried at 50 °C until its weight remained constant three times. It was then weighed and dissolved in water.
The reaction mixture comprised 16.7 mM citric acid pH 6.5, an equal amount of substrate, and 0.3 mM hsGDE. The products, glucose, were measured using the Glucose Colorimetric Assay Kit (GOD/POD Method) (Solarbio Life Science) and monitored on an Ultraspec 2100 pro spectrophotometer (GE Healthcare) at 505 nm. This experiment was repeated three times.
Glycogen binding assay
The binding capability of glycogen and hsGDE was assayed via pull-down assay utilizing glycogen immobilized on concanavalin A (ConA) agarose beads. A total of 100 μL ConA agarose beads (Sigma-Aldrich) pre-equilibrated in the binding buffer (67 mM HEPES pH 7.5, 0.2 mM CaCl2, 10 mM MgCl2, 1 mM MnCl2) were incubated with 30 mg of glycogen (Sigma Aldrich) at 4 °C for 1 h. Following five washes with the binding buffer, the prepared beads were incubated with increasing amounts of hsGDE (10, 25, and 50 mg) at 4 °C for 1 h. Subsequently, unbound hsGDE was removed through five washes with the binding buffer, and the quantity of bound hsGDE was assessed by SDS-PAGE analysis with Coomassie blue staining. The experiment was repeated three times. A representative uncropped scan of the SDS-PAGE is presented in Gel bands, which were quantified by ImageJ49 (https://imagej.net/software/imagej/).
Statistical analysis
Each data point in the activity measurements was repeated at least three times. Data are presented as the mean ± SEM. Error bars represent the SEM. Statistical analyses were performed with Ordinary one-way ANOVA in GraphPad Prism 9.0 (GraphPad Software, Boston, Massachusetts USA, www.graphpad.com) to compare differences in mean values.
Cryo-EM sample preparation and data collection
The purified hsGDE sample (~3 mg/ ml) was applied to freshly glow-discharged Quantifoil Cu R1.2/1.3 300 mesh grids. Under controlled conditions of 4 °C temperature and 100% humidity, the grids were blotted for 4 s using an FEI Vitrobot (Mark IV, Thermo Fisher Scientific) and were promptly plunge-frozen in liquid ethane cooled by liquid nitrogen.
The cryo-EM grids were loaded onto an FEI Titan Krios transmission electron microscope operating at 300 kV with a nominal magnification of × 105,000. Utilizing a Gatan K2 Summit direct electron detector and a Gatan GIF Quantum energy filter (slit width: 20 eV) in the super-resolution counting mode, all cryo-EM images were automatically recorded using EPU2 software. The defocus range spanned from 1.0 to 2.0 mm.
Each stack was exposed for 5.6 s in super-resolution mode with an exposure time of 0.175 s per frame, resulting in 32 frames per stack. The total dose for each stack was 50 electrons per Å2. From the complete dataset of 3867 micrographs, 3782 micrographs were selected for further processing.
Image processing
All 32 frames in each stack were aligned and summed using the whole-image motion correction program MotionCor2. Dose-weighted micrographs were used for Contrast Transfer Function (CTF) estimation using Patch-CTF in cryoSPARC50. During manual curation, micrographs exhibiting CTF fitting resolutions worse than 3.5 Å were excluded. Initial particles were picked from all micrographs using the blob picker tool in cryoSPARC. Subsequently, particle extraction was accomplished using a box size of 300 pixels, followed by the generation of 2D averages. Final particle picking was done by template picker, employing templates derived from the aforementioned 2D results.
A total of 1,228,386 particles were extracted from 3,782 micrographs. Following multiple rounds of 2D classification, 457,185 high-quality particles were acquired. Leveraging ab-initio reconstruction, as well as hetero refinement for guided multi-reference 3D classification, a 3.47 Å map was generated from those particles. Following non-uniform refinement and local refinement, the dataset gave rise to a reconstruction at an average resolution of 3.23 Å.
Model building and refinement
The initial structure model for hsGDE was generated by AlphaFold251, subsequently docked into the density map, and manually adjusted and reconstructed by WinCoot52,53. Further refinement of the hsGDE model against the corresponding maps was conducted using PHENIX54 in real space with secondary structure and geometry restraints. Overfitting of the model was monitored by refining the model in one of the two independent maps from the gold-standard refinement approach and testing the refined model against the other map. Validation of the hsGDE structures was performed through examination of the Molprobity scores and statistics of the Ramachandran plots.
Molecular dynamics simulations
Maltopentaose was initially docked into the binding pocket (residues: 400−418, 580−590, and 630−640) of the cryo-EM structure of GDE using AutoDock55 guided by cryo-EM density. The grid box (80 Å × 80 Å × 80 Å) was set up based on the binding pocket (residues: 400−418, 580−590, and 630−640) and all parameters were kept as default. The top docking pose was slightly adjusted by PyMol and subsequently verified and refined through atomistic MD simulations. The simulation systems were set up using CHARMM-GUI, Glycan Reader & Modeler56,57, to prepare the topology and parameter files for glycogen and protein. Specifically, the glucose residue forming an aldehyde open-chain at the reducing end of the maltopentaose pentasaccharide was modified to alpha-D-glucose. The protein termini were neutralized, and the protonation states of specific residues were assigned by PROPKA58. The protein complex was placed in a cubic box having a volume of 16.3 nm3, ensuring a minimum separation of 2.0 nm between periodic images. The system was solvated using the TIP3P water model with 0.15 M NaCl. Van der Waals and short-range electrostatic interactions were cut off at 1.2 nm, while Particle-mesh Ewald (PME)59 algorithm was used to calculate long-range electrostatic interactions with a 1.2 nm real-space cutoff. The Linear Constraint Solver (LINCS) algorithm was used to constrain all bond lengths, with a 2 fs time step used for integration. Periodic boundary conditions (PBC) were applied in all directions. Energy minimization was performed using the steepest descent algorithm to remove steric clashes. During the equilibration step, harmonic positional restraints of 400 kJ mol⁻¹ nm⁻² on the backbone and 40 kJ mol⁻¹ nm⁻² on the side chains were applied to the heavy atoms of the protein to maintain the structural integrity of the complex while allowing the solvent and ions to equilibrate around the system. Additionally, the system was equilibrated in a single step lasting 125 ps using the Nosé-Hoover thermostat. The production steps were performed at a temperature of 303.15 K and a pressure of 1.0 bar, controlled using the v-rescale temperature coupling algorithm60 and the Parrinello–Rahman barostat61, respectively. All simulations were performed with GROMACS62 (version 2024.2) using the CHARMM36m force field63.
The GT domain, particularly the highly dynamic B domain (residues 301-366), shows significant flexibility, which can affect the size and shape of the binding pocket. To ensure that the simulations reflected meaningful biological interactions, we applied positional restraints on select residues to preserve the size and shape of the binding pocket as observed in the cryo-EM structure. This approach balances maintaining a realistic dynamic system and preventing the artificial escape of the substrate due to structural flexibility. In particular, the unbiased MD simulation lasted 200 ns with six repeats, during which the root-mean-square fluctuation (RMSF) values of protein backbone atoms and the center of mass distance between the substrate and key residues (Y173, Y406, W461, F557, W487) were used as reference values for position restraints and flat-bottom restraints, respectively. Positional restraints were selectively applied to protein Cα atoms in four domains, specifically where RMSF values exceeded 0.8 nm and were distant from the active binding site. A total of 34 Cα atoms at residues 115, 155, 161, 204, 219, 230, 241, 252, 287, 302, 317, 325, 428, 438, 621, 635, 777, 782, 865, 903, 911, 930, 943, 1027, 1041, 1049, 1114, 1128, 1214, 1305, 1321, 1382, 1429, and 1444 (Supplementary Fig. 7b) were restrained using a force constant of 1000 kJ mol⁻¹ nm⁻² to preserve the protein’s structural integrity while allowing for natural dynamics. Flat-bottom restraints, using an upper wall harmonic potential relative to the center of mass distance between the substrate and the binding site centered at 0.6 nm with a force constant of 10 kJ mol−1 nm−2, were applied to facilitate the substrate searching its optimal conformations within the binding site. Simulations with both position and distance restraints were performed in three repeats, each lasting 200 ns (Supplementary Fig. 7c). Additionally, these simulations were extended by an extra 500 ns to assess the substrate binding stability, with only positional restraints on GDE to maintain the shape of the binding pocket consistently. Specifically, in repeat 3, the center-of-mass distance between glycogen and the protein increased, resulting in fewer contacts and a less favorable interaction pose. This difference can be attributed to the inherent flexibility of the GT domain, particularly the B domain, and the stochastic nature of MD simulations, which may lead to sampling limitations. Despite the extended simulation time, the conformational space explored by the system may still be limited, resulting in certain runs sampling less favorable binding modes, as seen in repeat 3. The restraints were applied using the Collective Variables module (Colvars) implemented in Gromacs2024.264. Details of the simulations are provided in Table S2.
MD analysis and visualization
The trajectory analysis, including RMSD, RMSF, number of contacts calculations, and block analysis was performed using MDAnalysis65,66 and plotted with the Matplotlib module of Python. The average RMSF values were mapped onto the protein structure using an in-house Python script. The substrate-protein contact frequencies were calculated using GetContacts (https://getcontacts.github.io/). Protein structures were visualized using ChimeraX67 or Chimera68.
Measurement of assembly in different pH conditions
Size-exclusion chromatography on a Superdex 200 columns (GE Healthcare) was used to estimate the molecular mass of native GDE at 4 °C. The column was equilibrated in 50 mM Tris-HCl buffer (pH 8.5, 8.0), 50 mM Hepes buffer (pH 7.5, 7.0), 50 mM sodium phosphate buffer (pH 6.5) or 50 mM sodium acetate buffer (pH 6.0, 5.5). The enzyme (100 μL at 1 mg/ml) was applied to the column and eluted with the equilibration buffer at a flow rate of 0.4 ml/min.
Measurement of assembly in the presence of the ATP
The dimer purification method was similar to the previous one, but the buffer pH (pH 8.5) was different. The enzymes from two peaks (0.025 mg/ml) were preincubated with ATP at 4 °C for an hour. After incubation, the enzyme was subjected to Mass Photometry (REFEYN) to determine the assembly state of GDE.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates of human GDE have been deposited in the PDB under the accession code 8ZEQ. The corresponding EM maps have been deposited in the Electron Microscopy Data Bank under the accession codes EMD-60043 (monomer), EMD-63640 (dimer). Source data is provided with this paper as a Source data file, and the data regarding the molecular dynamics analysis can be accessed on GitHub [https://github.com/yongwangCPH/papers/tree/main/2024/GDE] and Zenodo [https://zenodo.org/records/15525495]. Source data are provided with this paper.
References
Roach, P. J., Depaoli-Roach, A. A., Hurley, T. D. & Tagliabracci, V. S. Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787 (2012).
Swanson, R. A. A thermodynamic function of glycogen in brain and muscle. Prog. Neurobiol. 189, 101787 (2020).
Nadeau, O. W., Fontes, J. D. & Carlson, G. M. The regulation of glycogenolysis in the brain. J. Biol. Chem. 293, 7099–7107 (2018).
Adeva-Andany, M. M., Gonzalez-Lucan, M., Donapetry-Garcia, C., Fernandez-Fernandez, C. & Ameneiros-Rodriguez, E. Glycogen metabolism in humans. BBA Clin. 5, 85–100 (2016).
Kanungo, S., Wells, K., Tribett, T. & El-Gharbawy, A. Glycogen metabolism and glycogen storage disorders. Ann. Transl. Med. 6, 474 (2018).
Raben, N., Plotz, P. & Byrne, B. J. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr. Mol. Med. 2, 145–166 (2002).
Watanabe, Y., Makino, Y. & Omichi, K. Activation of 4-alpha-glucanotransferase activity of porcine liver glycogen debranching enzyme with cyclodextrins. J. Biochem. 140, 135–140 (2006).
Zhai, L., Feng, L., Xia, L., Yin, H. & Xiang, S. Crystal structure of glycogen debranching enzyme and insights into its catalysis and disease-causing mutations. Nat. Commun. 7, 11229 (2016).
Okubo, M. et al. Glycogen storage disease type IIIa: first report of a causative missense mutation (G1448R) of the glycogen debranching enzyme gene found in a homozygous patient. Hum. Mutat. 14, 542–543 (1999).
Hoof, F. & Hers, H. G. The subgroups of typeglycogenosis. Eur. J. Biochem. 2, 265–270 (1967).
Mili, A. et al. Molecular and biochemical characterization of Tunisian patients with glycogen storage disease type III. J. Hum. Genet. 57, 170–175 (2012).
Kishnani, P. S. et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med 12, 446–463 (2010).
Richmond, C. S. et al. Glycogen debranching enzyme (AGL) is a novel regulator of non-small cell lung cancer growth. Oncotarget 9, 16718–16730 (2018).
Guin, S. et al. Role in tumor growth of a glycogen debranching enzyme lost in glycogen storage disease. J. Natl. Cancer Inst. 106, dju062 (2014).
Sakoda, H. et al. Glycogen debranching enzyme association with beta-subunit regulates AMP-activated protein kinase activity. Am. J. Physiol. Endocrinol. Metab. 289, E474–481 (2005).
Worby, C. A., Gentry, M. S. & Dixon, J. E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 283, 4069–4076 (2008).
Cheng, A. et al. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 21, 2399–2409 (2007).
Shen, M., Gong, X. & Xiang, S. Crystal structures of glycogen-debranching enzyme mutants in complex with oligosaccharides. Acta Crystallogr F. Struct. Biol. Commun. 77, 420–426 (2021).
Miao, J. et al. Glycogen metabolism in Candida albicans impacts fitness and virulence during vulvovaginal and invasive candidiasis. mBio 14, e0004623 (2023).
Kiessling, L. L. & Diehl, R. C. CH-pi interactions in glycan recognition. ACS Chem. Biol. 16, 1884–1893 (2021).
Ma, D. et al. Structural basis for sugar perception by Drosophila gustatory receptors. Science 383, eadj2609 (2024).
Mizuno, M. et al. Structural insights into substrate specificity and function of glucodextranase. J. Biol. Chem. 279, 10575–10583 (2004).
Nakayama, A., Yamamoto, K. & Tabata, S. Identification of the catalytic residues of bifunctional glycogen debranching enzyme. J. Biol. Chem. 276, 28824–28828 (2001).
Xi, S. et al. Conserved residues at the family and subfamily levels determine enzyme activity and substrate binding in glycoside hydrolase family 13. Int J. Biol. Macromol. 253, 126980 (2023).
Brzozowski, A. M. & Davies, G. J. Structure of the Aspergillus oryzae alpha-amylase complexed with the inhibitor acarbose at 2.0 A resolution. Biochemistry 36, 10837–10845 (1997).
Boel, E. et al. Calcium binding in alpha-amylases: an X-ray diffraction study at 2.1-A resolution of two enzymes from Aspergillus. Biochemistry 29, 6244–6249 (1990).
Choi, E. J. & Mayo, S. L. Generation and analysis of proline mutants in protein G. Protein Eng. Des. Sel. 19, 285–289 (2006).
Sentner, C. P., Vos, Y. J., Niezen-Koning, K. N., Mol, B. & Smit, G. P. Mutation analysis in glycogen storage disease type iii patients in the netherlands: novel genotype-phenotype relationships and five novel mutations in the AGL Gene. JIMD Rep. 7, 19–26 (2013).
Oh, S. H., Park, H. D., Ki, C. S., Choe, Y. H. & Lee, S. Y. Biochemical and molecular investigation of two Korean patients with glycogen storage disease type III. Clin. Chem. Lab. Med. 46, 1245–1249 (2008).
Lu, C. et al. Spectrum of AGL mutations in Chinese patients with glycogen storage disease type III: identification of 31 novel mutations. J. Hum. Genet. 61, 641–645 (2016).
Schumacher, M. A. et al. Allosteric regulation of glycogen breakdown by the second messenger cyclic di-GMP. Nat. Commun. 13, 5834 (2022).
Park, J. T. et al. Oligomeric and functional properties of a debranching enzyme (TreX) from the archaeonSulfolobus solfataricusP2. Biocatal. Biotransform. 26, 76–85 (2009).
Kyla-Puhju, M., Ruusunen, M. & Puolanne, E. Activity of porcine muscle glycogen debranching enzyme in relation to pH and temperature. Meat Sci. 69, 143–149 (2005).
Makino, Y. & Omichi, K. Purification of glycogen debranching enzyme from porcine brain: evidence for glycogen catabolism in the brain. Biosci. Biotechnol. Biochem. 70, 907–915 (2006).
Ylä-Ajos, M., Ruusunen, M. & Puolanne, E. Glycogen debranching enzyme and some other factors relating to post-mortem pH decrease in poultry muscles. J. Sci. Food Agric. 87, 394–398 (2007).
Narahara, E., Makino, Y. & Omichi, K. Glycogen debranching enzyme in bovine brain. J. Biochem. 130, 465–470 (2001).
Van Gelder, P. et al. Sugar transport through maltoporin of Escherichia coli: role of the greasy slide. J. Bacteriol. 184, 2994–2999 (2002).
Dutzler, R., Schirmer, T., Karplus, M. & Fischer, S. Translocation mechanism of long sugar chains across the maltoporin membrane channel. Structure 10, 1273–1284 (2002).
Denker, K., Orlik, F., Schiffler, B. & Benz, R. Site-directed mutagenesis of the greasy slide aromatic residues within the LamB (maltoporin) channel of Escherichia coli: effect on ion and maltopentaose transport. J. Mol. Biol. 352, 534–550 (2005).
Janecek, S., Svensson, B. & Henrissat, B. Domain evolution in the alpha-amylase family. J. Mol. Evol. 45, 322–331 (1997).
Zmasek, C. M., Godzik, A. Phylogenomic analysis of glycogen branching and debranching enzymatic duo. BMC Evol. Biol. 14, 183 (2014).
Woo, E. J. et al. Structural insight into the bifunctional mechanism of the glycogen-debranching enzyme TreX from the archaeon. J. Biol. Chem. 283, 28641–28648 (2008).
Park, H. S. et al. TreX from ATCC 35092 displays isoamylase and 4-a-glucanotransferase activities. Biosci. Biotech. Biochem. 71, 1348–1352 (2007).
Gillard, B. K. & Nelson, T. E. Amylo-1,6-glucosidase/4-alpha-glucanotransferase: use of reversible substrate model inhibitors to study the binding and active sites of rabbit muscle debranching enzyme. Biochemistry 16, 3978–3987 (1977).
Froese, D. S. et al. Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design. Hum. Mol. Genet. 24, 5667–5676 (2015).
Sullivan, M. A. et al. Nature of α and β particles in glycogen using molecular size distributions. Biomacromolecules 11, 1094–1100 (2010).
Woo, E. J. et al. Structural insight into the bifunctional mechanism of the glycogen-debranching enzyme TreX from the archaeon Sulfolobus solfataricus. J. Biol. Chem. 283, 28641–28648 (2008).
Makino, Y. & Omichi, K. Acceptor specificity of 4-alpha-glucanotransferases of mammalian glycogen debranching enzymes. J. Biochem. 139, 535–541 (2006).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Jones, T. A. Interactive electron-density map interpretation: from INTER to O. Acta Crystallogr. D. Biol. Crystallogr. 60, 2115–2125 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D. Struct. Biol. 74, 531–544 (2018).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Park, S. J. et al. CHARMM-GUI glycan modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 29, 320–331 (2019).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Olsson, M. H., Sondergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: Consistent treatment of internal and surface residues in empirical pka predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. chem. phys. 98, 10089–10092 (1993).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Fiorin, G., Klein, M. L. & Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 111, 3345–3362 (2013).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 (2011).
Gowers, R. et al. MDAnalysis: A python package for the rapid analysis of molecular dynamics simulations. In Proc. 15th Python in Science Conference 98–105 (SciPy, 2016).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank all staff members of the Cryo-EM Center, Southern University of Science and Technology for providing data collection and computation support. We are grateful for the assistance of the SUSTech Core Research Facilities. GT activity data were obtained using equipment maintained by Southern University of Science and Technology IPF Core Facilities. This work was supported by the National Natural Science Foundation of China (32271251 to K.Y.), Guangdong Innovative and Entrepreneurial Research Team Program (2021ZT09Y104 to K.Y., 2021QN02Y429 to K.Y.), Shenzhen Science and Technology Program (No. JCYJ20220530115214033 to K.Y., No. KQTD20210811090115021 to K.Y., and No. 20231120160100001 to K.Y.), Guangdong basic and applied basic research foundation (2025A1515011166 to R.M.). K.Y. and Z.L. are investigators of the SUSTech Institute for Biological Electron Microscopy. Y.W. thanks the Information Technology Center and State Key Lab of CAD&CG, Zhejiang University, for computational support.
Author information
Authors and Affiliations
Contributions
K.Y. designed and supervised the whole project; H.G. (Author 1), H.C. and R.M. prepared the samples; H.G. (Author 1), H.C. and R.M. collected the cryo-EM data; H.G. (Author 1), H.C. processed the cryo-EM data; H.G. (Author 1) built the model. H.G. (Author 1) and H.G. (Author 3) performed the activity assay; Y.C. and Y.W. performed the MD simulations and the analysis; H.G. (Author 1), Y.C., Z.L., Y.W. and K.Y. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks X and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guan, H., Chen, H., Geng, H. et al. Molecular architecture and catalytic mechanism of human glycogen debranching enzyme. Nat Commun 16, 5962 (2025). https://doi.org/10.1038/s41467-025-61077-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61077-6
