
Abstract
Deep learning methods have revolutionised our ability to predict protein structures, allowing us a glimpse into the entire protein universe. As a result, our understanding of how protein structure drives function is now lagging behind our ability to determine and predict protein structure. Here, we describe how topology, the branch of mathematics concerned with qualitative properties of spatial structures, provides a lens through which we can identify fundamental organising features across the known protein universe. We identify topological determinants that capture global features of the protein universe, such as domain architecture and binding sites. Additionally, our analysis identifies highly specific properties, so-called topological generators, that can be used to provide deeper insights into protein structure-function and evolutionary relationships. We present a practical methodology for mapping the topology of the known protein universe at scale. We then use our approach to determine structural, functional and disease consequences of mutations. Our approach reveals and helps to explain differences in properties of proteins in mesophiles and thermophiles, and the likely structural and functional consequences of polymorphisms in a protein. For eukaryotes we find striking differences between protein topologies in multi-cellular and single-celled organisms.
Similar content being viewed by others


Introduction
Proteins are the executors of cellular function and the main building blocks of cellular structures. Each protein within the protein universe, defined as a collection of all proteins from all organisms, has a three-dimensional (3D) shape (or an ensemble of 3D shapes for a subset of proteins that contain regions of intrinsic disorder), usually referred to as protein structure. One of the main principles of protein science is that the shape of the protein determines its function; therefore, determination of the protein structure has been a core interest in molecular biology over the last seven decades. Collectively, structural biology, structural bioinformatics, and, more recently, deep learning approaches have jointly amassed vast amounts of exquisitely detailed data1,2,3,4. AlphaFold2, together with a growing suite of similar tools, represents one of the latest breakthroughs in this area. AlphaFold2 is a deep learning model for predicting protein structures that has outperformed accuracy and volume of other protein structure predictions methods. Currently, the AlphaFold2 database contains models for more than 214 million unique proteins across all kingdoms of life, thus likely covering almost the entire protein universe. Direct analyses of this many structures has thus far been impossible. Capitalising on the vast computational advances in sequence alignments (e.g., MMseqs25), it is now possible to use sequence information as a guiding principle for the analysis of the AlphaFold2 database. This allows, for example, sequence-based clustering of structures6, or sequence-based clustering followed by structural alignment of a subset of AlphaFold2 structures7. The combination of structural bioinformatics and deep learning has been recognised with the 2024 Nobel Prize in Chemistry.
However, the majority of commonly used tools for analysing protein structures and extracting comprehensive and overarching principles governing protein structure and function have been developed to handle much smaller datasets (with notable exceptions, c.f.8), and to our knowledge, no tool has yet been applied in a structural analysis of the entire AlphaFold2 database. This highlights the need for developing new methods that can be applied for such analyses to unravel the organising principles of the protein universe.
Protein structures can be described in terms of topology, a powerful framework for understanding connectivity and arrangement of secondary structural elements (e.g., α-helices, β-strands and β-sheets) within a protein9. Mapping of these secondary structural elements and their relationships provides a reductionist view of complex 3D structures of the proteins, and represents a powerful strategy for identifying recurring motifs, spatial arrangements and functional regions within proteins from different organisms and/or protein families10,11. Therefore, analysis of protein topological features is a cornerstone of protein science that is often used to understand protein structure-function relationships, deduce evolutionary relationships, and engineer proteins with novel functions.
In mathematics, topology is the field that focuses on qualitative features of spatial structures. Qualitative features of spatial structures include: connectedness, and holes or voids12. Topology considers any two structures to be identical if they can be turned into one another by stretching, twisting, bending the structures, but not cutting or gluing them. The advantage of the topological perspective is that it allows identification of features that are not strongly dependent on the (spatial or temporal) scale at which data are interrogated. In terms of proteins, two proteins that bind the same ligand through similar interactions and similar pockets can be regarded as topologically equivalent, irrespective of their size or detailed global tertiary structure. Therefore, using mathematical topology formalisms to analyse protein structures could enable the detection of hidden (or latent) structures in complex multidimensional data13.
This might seem vague and unhelpfully general, but the topological perspective has proven advantageous in many settings. Two examples come from physics, and, more recently and pertinently in the present context, topological data analysis (TDA). In physics, Morse theory and Floer homology give exquisite structures to the laws of quantum field theory and cosmology14,15,16. Recently, TDA emerged as a new approach in mathematical topology (i.e., topology)17,18. The essence of TDA lies in analysing the shape of data using algebraic concepts. The most effective approach to do that is persistent homology (PH)19,20, a computational tool that transforms scattered points into a sequence of revealing shapes, to identify the system’s features that persist across different scales. When applied to spatial objects (e.g., protein structures), this corresponds to analysing how the system’s shape evolves, as its data points become increasingly more spatially extended, overlap and create changing patterns. Thus, PH tracks topological features as they appear and vanish over the course of this spatial filtration, and uses persistence, the measure of how long the feature exists, to distinguish robust signal from noise, as the longer a feature persists, the more reliably it captures a feature of the data13,20. The collection of these features, together with their persistence values, are used as descriptors of the underlying system and have proven extremely effective for clustering, parameter inference, and pattern detection in natural and physical systems21,22,23,24.
Here, we develop, optimise, and implement a PH-based TDA method to analyse all 214 million structures predicted by AlphaFold22,25. We use this approach to statistically derive organising principles, topology-function relationships, and to obtain a topological “tour guide” to the vast AlphaFold2 resource. In this manner, we address the key need for the field and present a systematic strategy for analysing the currently largest protein structure dataset in a way that yields insights into structure-function relationships and protein evolution at an unprecedented scale.
Results
Developing a pipeline for topological analysis of protein universe reveals its topological richness
A recent advance in PH is the ability to efficiently determine “homology generators”26 and to analyse them systematically27. Topology generators pinpoint the specific aspects and regions in the data that are responsible for the creation of topological features. At the level of a single protein, topology generators may reveal groups of highly interacting amino acids that form higher-order structural features, e.g., specific conformations28, or entanglement in knotted proteins29. Here, we extended this methodology to analyse more than 214 million protein structures available in the AlphaFold2 database. In order to be able to handle the unprecedentedly large set of topology generators, we developed computational processes for bulk persistent homology calculation and to improve memory requirements. The subsequent analysis of the topological output follows the approach developed in27,29 using the pipeline in Fig. 1B and Supplemental Fig. 3. As can be seen, in Step 1, we model each protein structure via the α-carbon atoms to generate the point cloud representation of the structures. The point cloud representation has the advantage of reducing the complex 3D shape into a single point in the (x, y, z) coordinate space for each given residue. The point cloud is used as an input for PH pipeline to compute persistent diagrams and topology generators that provide information about persistence (signal strength or relative relevance/contribution) of each topological feature (in dimensions 1 and 2, i.e., loops and voids) and interpretation of abstract topological information as local features of the data, respectively (Step 2, Fig. 1B). Thus, the output of this step are topological features, together with their persistence, with each amino acid having the potential to contribute to several, distinct topological features, with different persistence values (Step 3). To understand how important a single region is in affecting the topology of the protein, we compute the point-wise “topological influence score” (TIF), which provides a ranking of amino acids based on the persistence of their connections (Step 4). TIFs are computed as normalised centrality values on the network of topology generators27, and the TIF values are higher for residues colocated in significant topology generators, see also Supplementary Section 1. Collectively, these steps required circa 10,560 CPU hours, performed on Oracle Cloud Compute (see “Methods”). These computations yielded more than 9.85 terabytes of topological data and mapped the topology of the currently known protein universe, which we have made freely available online (see ”Methods”).

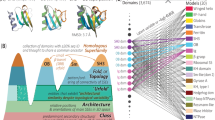
The 214M AlphaFold2 protein structures, organised by species and plotted as a tree of life (A). Their topological analysis (sketched in the purple box, B) reveals intricacy and variety of topological features and a remarkable complexity across the evolutionary tree, which we represent using a circle packing plot. The area of each circle is proportional to the number of proteins grouped in it (C). Circles saturation represents average topological richness, which is also shown numerically for domains, kingdoms, phyla, and selected species of interest. The average richness is approximated here by normalising by group-averaged protein size. The colour scale has the upper bound set by the 95% quantile to ignore outliers and emphasise differences, and has domain averages indicated by black lines (D). Boundaries of circles are coloured by topological variance (E). Zooming into humans, each protein is shown as a dot, with colour saturation proportional to its topological richness. Haemoglobin (G) is plotted showing its most persistent one-dimensional (left, a loop) and two-dimensional (right, a void) topological features (H), and below with amino-acids coloured by their topological influence score (I), with a white-blue-purple scale (F).
To examine the resulting data in the broadest and most general context, we use it to construct the topological tree of life (Fig. 1A). The tree is visualised as a circle packing plot, with the area of the circle corresponding to the number of AlphaFold2 predicted structures available for each species. Next, we connect and rank each genus and related them one genus at the time, so that the area of higher ranks is approximately representative of the number of structures (Fig. 1C). The three domains of life, bacteria, archaea, and eukaryotes, are all well represented in the topological tree of life, and include organisms with vastly varying proteome sizes. Furthermore, we are able to map the topological richness for each protein, each organism, and across domains (areas of low richness in light colours, areas of high richness in dark colours, (Fig. 1D). Topological richness is the measure of how many unique, persistent topological features each protein has, averaged across all proteins and normalised by number of residues. We observe that, comparatively speaking, bacterial and archaeal proteins exhibit lower topological richness, whereas eukaryotes exhibit several areas of heightened richness, especially within the mammalian class.
A couple of notable highlights among species include Acinonyx jubatus (cheetahs) and Pipra filicauda (wire-tailed manakin), while humans are outliers among other mammals in terms of their relatively low richness value. It might seem surprising that humans show a lower richness than other species in their class. However, similarly to the case of gene count, which were found to be unexpectedly low for humans, this arguably reflects that topology is just one among many ways to assess complexity. In this specific case, human complexity at the protein level is equally or more likely to arise from intricate layered regulation and developmental programmes30, alternative splicing, and the complexity of the protein-protein interaction network31.
The curation of topological properties for millions of proteins provides a unique opportunity to quantify protein properties emerging at the scale of the known protein universe. For entire domains of life a pattern emerges differentiating eukaryota from bacteria and archaea (Fig. 2). By focusing on topologically rich proteins, we observe a slight shift in the distributions for each domain of life (see Fig. 2A). Eukaryotic proteins appear to have more intricate structures, while the predicted archaeal structures contain more topology with lower intricacy. Topological richness is defined with a high threshold for counts of “loops”, which means most protein structures have richness scoring equal or very close to zero, and are excluded from the density plot (see Table 1 for protein richness counts).

Topological richness for high-quality proteins exhibits a slight shift for proteins from each domain with non-zero richness (A). Shown on a log-scale, see Table 1 for the numbers of excluded zero-valued entries. Number of topological representatives – either “loops” or “voids” – normalised by protein length to indicate the average residue’s topological feature membership (B). Size of the single largest loop and void in terms of the number of simplices normalised by protein length (C). For loops, this roughly corresponds to the fraction of a protein contained in its largest loop. Equal weight is given to each species in calculations of all subfigure bin frequencies. Each density plot is a scaled histogram, scaled such that the total area sums to 1. Low-quality structures were filtered out with the threshold mean pLDDT > 70. We find that proteins from eukaryotes have different topological tendencies and typically exhibit higher topological complexity than proteins from the other two domains of life. Source data are provided as a Source Data file in Source_data/source_data_Figure2ABC.tsv.
By comparing the average number of loops and voids an amino acid in a given protein belongs to, we find that eukaryota contain many protein examples with large membership (Fig. 2B). For bacteria and archaea, by contrast, we find a more even, flatter distribution. We next consider the sizes of the largest loops and voids in each protein (see Fig. 2C). Again, the eukaryota stand out. We estimate the size of the largest loop as the simplex count divided by the number of residues. For loops, this translates into the fraction of a protein sequence that is contained in the largest loop. Eukaryotic proteins appear to have less of their structure contained in a single loop or void, and tend to show increased topological complexity with multiple loops/voids. For bacteria and archaea, the distributions over topological features are, by contrast, relatively uniform.
A yet more striking observation are the pronounced peaks in the eukaryotic distributions, whereas the distributions of bacteria and archaea are generally uniform. At this scale of analysis, a uniform distribution appears more intuitive, as millions of proteins from distinct species are aggregated. The sharp peaks for eukaryota are unexpected, and may allude to a very specific level of protein complexity favourable to achieve the intricate regulations within multicellular lifeforms. To explore this further, the eukaryotic data was divided into two sub-groups for further analysis: one group, representing multicellular organisms (approximately), was formed by taking all proteins from the Metazoa and Embryophyta (informally, animals and land plants); the remaining eukaryotic proteins were assigned to the other group. The pronounced peaks in Fig. 2 are confined to the first grouping corresponding (approximately) to the multicellular organisms (see Supplementary Fig. 17). We should note that possible biases in the dataset (e.g., those caused by an over-studied protein family) might be a factor contributing to such peaks. However, because we assign equal weights to the species contributing to the distribution, we safeguard against highly studied eukaryotic model organisms skewing the results.
The results in Fig. 2 are normalised by species, meaning that each species is weighted equally. If this had not been the case, model organisms would greatly skew the figures in their favour due to the massive efforts by the research community in sequencing their genomes. We find that eukaryotic model organisms have lower complexity when compared to the species-wise distributions, which can also be seen from Table 2, where we list individual model organisms. For some model organisms, this observations makes perfect sense: they may have specifically been selected as model organisms for their simplicity, and the protein topology could reflect this. Interestingly Homo sapiens is an outlier among the model organisms and the distribution has more pronounced peaks.
We mapped the topological variance (Fig. 1E), which can be taken as a measure of the evolutionary robustness of topological characteristics. The topological variance is computed as the variance of the number of 1-dimensional topological features in a given circle, normalised by the number of proteins in the circle. The variance is shown in the figure as the outline colour of discs, using a black-yellow colour code. Similarly to richness, topological variance is higher for eukaryotes than for Bacteria and Archaea. This is particularly evident in insects, especially at the species level, and suggests an increased diversity in their topological features. On the other hand, when variance is consistently low across ranks (as for Bacteria), this could be interpreted as topological complexity levels being preserved through evolution.
Lastly, we mapped TIFs onto all proteins, which provides insights at the residue level (Fig. 1F and I). In each protein structure, TIF values quantify how topologically important individual residues are; this, in turn, leads us to identify structurally significant regions, and potential locations for candidate damaging mutations, as we show in Section Topological analysis detects protein regions enriched for disease-associated mutations.
We can zoom in on individual proteins such as human haemoglobin subunit alpha (Fig. 1G), where our analysis identified its most persistent loop and void (Fig. 1H), and how they influence the topology as measured via TIFs (Fig. 1I). Taken together, our method offers a powerful, flexible, and timely tool for analysing topology of the protein universe. Applying our pipeline to the whole AlphaFold2, a database with close to a quarter of a billion protein structures, reveals both the intricacies and variety of topological features across the tree of life.
Topological analysis of the protein universe enables nuanced protein structure analysis
We have recently shown that PH can be analysed using network theory, which reveals further relationships between topological features27. To extract further insights from our topological map of the protein universe, we interpret protein topology via networks, where edges are defined by loops (dimension 1) and voids (dimension 2). In this framework, intensity and overlaps of these connections induce a grouping of amino acids into units, which we call “topological clusters” (Fig. 3). This approach allows us to capture global structural properties of the protein universe that detect characteristics that are beyond conventional protein structure analysis strategies.

Topological clusters in dimension 1 often provide a refinement of protein domains. As an example, we see protein kinase, coloured by its two CATH domains (A) and topological clusters (B). C Clustering homogeneity can be used to check if a topological cluster contains only residues belonging to a single domain, with 1 corresponding to a perfect sub-partition, and 0 to each cluster containing all the same labels. The bar plot shows the distribution of homogeneity scores for a set of 38,171 non-redundant AlphaFold2 predictions with identified CATH domains. D Two-dimensional topological cluster boundary points are enriched for binding sites from the Mechanism and Catalytic Site Atlas (M-CSA) dataset. The bar chart shows the distribution of distances (in number of residues) from cluster boundaries to residues of either binding sites or other residues. E The topological analysis is robust to small perturbations. The bar plot shows the distribution of correlation coefficients between the topological influence scores for AlphaFold2 predicted structures and their experimentally solved counterparts. The high values show that topological features tend to correspond and to interest the same residues. F Topological clusters for horse apomyoglobin (UniProt accession P68082) differentiated by colour. Folding events based on experimental evidence is indicated by transparency, where fully opaque sections are formed. Source data are provided as a Source Data file in Source_data/source_data_Figure3C-S8.csv, Source_data/source_data_Figure3D.csv, and Source_data/source_data_Figure3E.csv.
For example, we observe that topological clusters of dimension 1 (loops) are closely associated with protein domains relating to semi-independent units of folding, as classified by the CATH Protein Structure Classification Database32. We illustrate this by examining more closely the relationship between CATH domains (Fig. 3A) and topological clusters (Fig. 3B) of a protein kinase (UniProt33 ID Q4DF08) as a representative example. We note that in this case, as well as many others, topological clusters of dimension 1 capture the essence of CATH domain classification; more specifically, we note that here a single CATH domain is partitioned into multiple topological clusters: the topological analysis refines on the resolution provided by domain assignments. We used the homogeneity score to quantify whether the topological clusters of dimension 1 provide an exact subdivision of CATH domains (score = 1), or whether the two partitions are completely unrelated (score = 0). We analysed 38,171 AlphaFold2 structural predictions, representing different protein families, domains, and organisms (Supplementary Table 2), which correspond to all non-redundant, high-confidence AlphaFold2 predictions, containing at least two distinct identified CATH domains25,32 (see also “Methods”). As seen in Fig. 3C (see also Supplemental Fig. 8, showing the same computation, but including redundant structures), the vast majority of topological clusters belong to a single domain; thus, the topological analysis refines on the resolution provided by domain assignments, revealing that many domains are formed by distinct topological features. This may have important implications for evolutionary analysis as well as protein engineering efforts, given that work in these areas often uses protein domains as the basic unit for analysis. Our results suggest that, for the majority of proteins, mathematical topology is consistent with, and sometimes refines into more nuanced features, known protein domains catalogued in CATH and similar databases.
As CATH domains relate to folding, we may further speculate that the 1D topological clusters can identify individual folding units. Except for counting connected components with 0-dimensional topology, the simplest features are found by dimension 1 topology (loops), which effectively captures qualitative spatial features of a shape or structure12,13,20. In the case of proteins, previous work has shown that loops can subtly capture geometric substructures, including entanglement and other non-trivial spatial features27,29. Our results align with this perspective: we observe that 1-dimensional loops are intricately interwoven within CATH domains, while loops traversing separate domains are obfuscated by the clustering. Experimental folding intermediates are difficult to obtain, but evidence exists for partial folds of apomyoglobin forming within micro- and milliseconds34,35,36. In this case, the 1-dimensional topological analysis captures the initial folding core (the blue cluster in Fig. 3F). While this is encouraging preliminary evidence that the topological perspective can augment the analysis of protein folding, as noted elsewhere37, AlphaFold does not provide the structural ensembles necessary to shed light on protein folding dynamics.
Unlike dimension 1 topological features (loops) that could inform on substructures, dimension 2 (voids) may be associated with binding sites. To examine if this is the case, we investigate the distances between the clusters of voids and the binding sites as defined in the Mechanism and Catalytic Site Atlas (M-CSA) dataset38. Our analysis includes 866 AlphaFold2 predicted protein structures (862 predicted with high confidence), representing a broad range of enzyme families and other proteins known to engage ligands (Supplementary Table 2). These structures were obtained by mapping to UniProt all 1033 RCSB Protein Data Bank (RCSB PDB)39 entries of experimental structures having M-CSA annotated sites, and then by selecting those corresponding to high-confidence AlphaFold2 predictions. We mapped the distance in terms of number of residues between the void boundary and the binding site, and we find that some 70% of binding sites are either immediately at the boundary of a void or one amino acid away (Fig. 3D, see also Supplemental Fig. 7, showing the same computation, but including low-confidence predictions). Again, this makes sense from a structural perspective, as binding sites must correspond to areas of accessibility and flexibility, and our topological analysis allows the detection of such sites across 214 million predicted structures. Thus, mapping voids has the potential to identify cryptic and/or unknown binding sites within the protein universe.
Despite its remarkable accuracy, AlphaFold2 predictions can sometimes fail to fully capture the structural complexity of certain proteins37,40,41. One of PH’s most powerful features is its robustness: input data differing by small to moderate perturbations will have similar topological fingerprints42. Thus, we can reasonably assume that topological analyses will be agnostic to possible misinterpretation of local 3D conformations. To assess this, we compare the output of our pipeline for experimental structures catalogued in RCSB PDB with their AlphaFold2 counterpart; results are shown in Fig. 3E. We quantify the discrepancy between topological features in the experimental and predicted datasets by looking at TIFs in dimension 1 and 2; as shown by the bar-plot, per residue values are highly correlated in both cases, ensuring that the topological analysis is transferable from simulations to experiments.
Taken together, these results demonstrate the value of topology in identifying features of protein structural organisation.
Topological comparison of thermophilic and mesophilic proteins
How thermophilic proteins achieve stability while maintaining functionality remains heavily debated in protein science, structural, and evolutionary biology. Factors such as differences in hydrophobicity, secondary structure, ion-pairing, hydrogen bonds, and numbers and sizes of cavities have been proposed as key determinants of thermophilic protein stability and function43. However, the lack of statistical power and the need to correct meticulously for potentially confounding factors has impeded analyses. Given the wealth of structural information generated by AlphaFold2 and the robust nature of topology, we hypothesise that we can detect topological differences between – even structurally very similar – thermophilic and mesophilic proteins, and that these differences may provide insights into how thermophilic proteins maintain their structure and function. Finding such differences is especially challenging as, across different organisms, specific enzymes often present highly similar, almost super-imposable structures. This is the case for Glucose-6-phosphate 1-dehydrogenase, shown in Fig. 4A in E. coli (mesophile) and M. thermoacetica (thermophile).

Highly persistent topological features in dimension 2 identify voids in protein structures. Voids in thermophilic organisms are in general, smaller in volume than in their mesophilic counterparts, as exemplified here by Glucose-6-phosphate 1-dehydrogenase (A). The observation is made consistently across enzymes from 10 different EC numbers, shown here in a stacked horizontal bar chart (B). Thus, the area of each bin corresponds to fractions of voids within a certain size range. Molecules with approximately representative sizes are illustrated for each of the four bins. Error bars shown in grey around bin boundaries (dotted lines) are calculated as the standard deviation from sampling 1000 voids. Furthermore, no compensating influence of amino acid frequencies around voids has been detected to reduce the significance of the results. This is illustrated for each amino acid (sorted from lowest to highest AA volume) by TIF distributions, which indicate their occupancy around voids (C). Dashed lines show averages. The ATP and polypeptide icons were produced using BioRender. Source data are provided as a Source Data file in Source_data/source_data_Figure4C-S16.tsv.
To address this, we select 10 different Enzyme Commission (EC) numbers based on their relevance to biotechnology (see Supplemental Table 2 and Supplemental Figs. 9, 10 and 12 for details). The selected enzymes covered 30 thermophilic and 8 mesophilic organisms, for a total of 1656 high-confidence AlphaFold2 predictions. We compare topological features of dimension 2 - i.e., voids - in mesophiles (blue) and thermophiles (red) (Fig. 4B). For this analysis, we choose to focus on voids because we are interested in understanding whether more compact topological features could be associated with high-temperature preferences.
In addition, we focus on comparing orthologous proteins with matching amino acid sequence length to minimise potential compounding effects of variable protein sequence length, substrate/binding partner properties, and function. We observe that voids in predicted protein structures from thermophilic organisms are smaller and more compact than their mesophilic equivalents (Fig. 4B). This difference is statistically significant according to a one-sided Mann–Whitney U test of void volumes after excluding noise by filtering out persistence < 1 A˚ngstrom (p = 2.789 × 10−6 and U = 7782508. n = 1937 and 8603 voids for thermo- and mesophiles, respectively.) We conduct an additional test to control for the effect of EC numbers, where random samples of equal size (1000) are taken from each EC number for meso- and thermophiles. This test also indicates a significance difference (p = 8.372 × 10−14 and U = 46624045. n = 10000 voids for both thermo- and mesophiles). See Supplemental Figs. 11 and 12 for visual comparisons.
We next consider whether the differences in voids may be explained or diminished by compensating differences in amino acid volumes in the voids. We compare the amino acid constituents of voids in terms of TIF from two-dimensional homology, as this amino acid–wise importance measure secondarily indicates the abundance of a given amino acid in the backbone adjacent to voids (Fig. 4C). While the shapes of these empirical distributions for each amino acid are significantly different for meso- and thermophiles (see Supplementary Table 3), we can only detect insignificant differences in terms of the association between TIFs and AA volumes (see Supplementary Fig. 16). The Pearson’s correlation coefficient in both cases is 0.155, indicating a weak tendency for larger amino acids at voids in general, which is unsurprising and consistent with our interpretation. Correlation tests yield p-values < 2.2 × 10−16. (For mesophiles: t-statistic = 117.04, n = 558297, degrees of freedom = 558295, and 95% confidence interval from 0.152 to 0.157. For thermophiles: t-statistic = 52.445, n = 112136, degrees of freedom = 112134, and 95% confidence interval from 0.149 to 0.160.) To test if there are compensating effects from AA occupancies around voids, we use a simple linear regression displayed as trend lines in the Supplementary Fig. Just as for the correlations, the 95% confidence intervals of estimated regression slopes overlap, thus, we cannot detect a compensating effect of AA volumes on TIFs. By contrast, the median AA volume is slightly higher for thermophiles (140 vs. 138.4). The difference is significant according to a Mann–Whitney U test (p-value < 2.2 × 10−16, U = 3.038 ⋅ 1010). Similarly, the estimated regression slope is marginally larger for thermophiles (9.112 × 10−4 vs. 8.936 × 10−4). This indicates AA volume distribution is not compensating for the difference in void volumes, but may have a slight influence on compacting thermophilic proteins further.
In light of these results, we suggest that the topological differences between thermophiles and mesophiles may reflect the different thermodynamic pressures experienced by the different organisms in their respective habitats, where binding pockets with larger voids may neither be able to provide the correct specificity of binding at higher temperatures, nor adequate thermodynamic stability.
Topological analysis detects protein regions enriched for disease-associated mutations
Because protein function depends on protein structure and sequence, we examine whether topological analysis can detect protein regions that are enriched in damaging, disease-associated mutations. To test this, we use a dataset of disease-causing and neutral variants that contains experimental structures of a few hundred wild-type and mutated proteins44,45. This dataset was previously analysed to establish the link between damaging mutations and their effect on structures44. As above, we restrict our analysis to structures predicted with high confidence. For each of the proteins analysed, we want to identify residues that are structurally important, and thus, more likely to accommodate mutations leading to structural damage, and in turn, to the occurrence of disease-associated polymorphisms. TIF values provide a measure of the topological significance of each residue; a natural question is whether a high 1- or 2-dimensional TIF directly estimates the influence on structural stability. Overall, we find that mutations that give rise to structural variants, those that give rise to disease, and those that give rise to both disease and structural effects, are more likely to be co-located with topology generators than non-disease causing variants, or polymorphic sites that have no known structural role. Figure 5A and B show the 3D structures of human ACE2 (top) and HBB (bottom), coloured by their per-residue two-dimensional TIFs. On the right-hand side, we see the distribution of 2-dimensional TIFs on residues whose substitution induce polymorphisms that are predicted to be structurally damaging and associated with disease, or neutral45. In these examples, the pattern discussed above is clearly visible. A similar result is observed in other individual proteins (see e.g., Human Adenylosuccinate lyase, Fig. 5C, and CTFR, Supplemental Fig. 15), in the whole dataset considered (Fig. 5B, and Supplemental Fig. 14), and for 1-dimensional features alike (Supplemental Fig. 13).

A The 3D structures of human ACE2 (top) and HBB (bottom), coloured by their per-residue two-dimensional TIFs. Structural analysis of missense variants for these genes predicts a number of them to be damaging44,45. Our topological analysis shows that amino acid substitutions causing structural damage are more likely to happen where the TIF is high, as shown by the violin plots on the right-hand site. This pattern is maintained across a dataset of disease-associated missense variants (B). As a further example, (C) shows human Adenylosuccinate lyase, with its missense variants highlighted on the 3D structure, and a plot of its 2-dimensional TIFs on the bottom. The n damaging and neutral variants for ACE2 are 49 and 200, for HBB are 81 and 396, and overall: 1418 and 6600, respectively. All box plots are shown with a box from the first to third quantile, median as a solid line, mean as a dashed line, and a line from the minimum to maximum value, excluding outliers. Source data are provided as a Source Data file in Source_data/source_data_Figure5A_ACE2.csv, Source_data/source_data_Figure5A_HBB.csv, and Source_data/source_data_Figure5B-S13-S14.csv.
Discussion
In this work, we demonstrate that topology can serve as an interpretative tool for the wealth of data contained in AlphaFold2. Our pipeline provides a topological analysis of all 214 million predicted protein structures in a time- and cost-effective manner. Topological information extracts novel and global insights into the features and properties of the protein universe. We illustrate these insights in several use case scenarios, including: using topology to analyse large-scale structural features, such as domains and binding sites; to identify differences between thermophiles and mesophiles; and examine effects of disease-causing mutations. To make this topological perspective accessible to the broader research community, we provide access to all one and two-dimensional persistence diagrams, topological features, and TIFs (per residue) via an online resource of approximately 20 TB.
Overall, this analysis shows how topology allows us to make sense of the vast amount of protein structural data. Importantly, our analysis was done using solely structural data on positions of Cα provided by AlphaFold2 (and the PDB for validation) without additional biological information, including sequence information. Thus, in the future, incorporating additional information, such as the biophysical and biochemical properties of amino acids and their three-dimensional arrangements, may capture additional factors that influence protein function. Already, our work highlights that topology adds an additional set of features for function prediction and an additional dimension to the biophysical analysis of protein structure. Although topology may not be enough to fully understand (or design) protein function, we are confident that topology offers a natural and direct route for making sense of the wealth of data in AlphaFold2 and that the topological information generated here will aid the functional and evolutionary analysis of the molecular machinery of life.
An intriguing direction for future research would be the integration of secondary structure information into the topological analysis, as other authors have developed persistent homology–based approaches incorporating secondary structure or even atomistic details46,47. While secondary structure annotation has become standard for solved structures, given a sequence, it remains a matter of prediction48. Thus, the potential unreliability of AlphaFold2 predictions presents a challenge for additional preprocessing of the raw structural data.
Methods
Persistent homology
Persistent homology13,20 is a method in computational topology for analysing the shape of data via topological features. Persistent homology is built on the concepts of simplicial complexes and simplicial homology12. Intuitively, a simplicial complex is a space constructed by gluing together simplices (i.e., points, line segments, triangles, and their higher-dimensional counterparts), for a formal definition, see e.g., [12, Ch.2]. Let \(PC=\{{p}_{1},\ldots,{p}_{n}\}\subset {{\mathbb{R}}}^{n}\) be a point cloud, i.e., a set of scattered points in the Euclidean space \({{\mathbb{R}}}^{n}\); the shape of PC can be described by constructing a simplicial complex PCε that approximates the connectivity of the points pi at a given spatial scale ε. Common choices of such a simplicial complex are:
-
The Vietoris-Rips complex [13, Ch.III.2] PCε = VRε(PC); this is constructed by adding a k-simplex \([{v}_{{i}_{0}},{v}_{{i}_{1}},\cdots \,,{v}_{{i}_{k}}]\) if the distance between all pairs of points in \(\{{v}_{{i}_{0}},{v}_{{i}_{1}},\cdots \,,{v}_{{i}_{k}}\}\) is less than ε.
-
The Ĉech complex [13, Ch.III.2] PCε = Cε(PC); this is constructed as the nerve complex [12, Ch.3] of the union of balls of radius ε centred in PC.
-
The Alpha complex [13, Ch.III.4] PCε = Aε(PC); this is similar to the Ĉech complex, but has a canonical geometric realisation, and it is a sub-complex of both the Delanauy complex and the Ĉech complex.
Note that for each of these choices, \(P{C}_{{\varepsilon }_{1}}\subset P{C}_{{\varepsilon }_{2}}\) whenever ε1 < ε2. More information on these complexes, their differences, and their properties can be found, e.g., in ref. 13; see also Supplemental Fig. 1 for one example.
The qualitative features of PCε can be analysed by computing its k-dimensional simplicial homology \({{{\rm{H}}}}_{k}(P{C}_{\varepsilon };{{\mathbb{F}}}_{2})\), where \({{\mathbb{F}}}_{2}\) is the field with two coefficients. For each choice of dimension k, \({{{\rm{H}}}}_{k}(P{C}_{\varepsilon };{{\mathbb{F}}}_{2})\) is a vector space, and its rank corresponds to the number of k-dimensional topological feature (called homology classes) of PCε. The 0-dimensional homology \({{{\rm{H}}}}_{0}(P{C}_{\varepsilon };{{\mathbb{F}}}_{2})\) counts the “connected components” (i.e., separate pieces) that form PCε, while 1 and 2-dimensional homologies \({{{\rm{H}}}}_{1}(P{C}_{\varepsilon };{{\mathbb{F}}}_{2})\) and \({{{\rm{H}}}}_{2}(P{C}_{\varepsilon };{{\mathbb{F}}}_{2})\) count loops and voids, respectively. For a formal definition of simplicial homology, see e.g., ref. 12.
Persistent homology studies the shape of the initial data PC at different spatial resolutions, by looking at the simplicial complexes PCε for increasing values of ε > 0, see Supplemental Fig. 1. This results in a nested sequence of simplicial complexes
which in turn yields a sequence of vector spaces and maps between them
called the k-dimensional filtered homology of PC.
We are interested in looking at how topological features evolve in this sequence of simplicial complexes and homology spaces. Thanks to the Structure Theorem [19, Thm 2.1], we can summarise the information contained in each sequence \({{{\rm{H}}}}_{k}(P{C}_{{\varepsilon }_{0}};{{\mathbb{F}}}_{2})\to {{{\rm{H}}}}_{k}(P{C}_{{\varepsilon }_{1}};{{\mathbb{F}}}_{2})\cdots \to {{{\rm{H}}}}_{k}(P{C}_{{\varepsilon }_{N}};{{\mathbb{F}}}_{2})\) as a “persistent diagram” PD. This is a finite collection of points PD = {(bi, di)}, where bi and di are the birth and death scales of the ith k-dimensional feature. The “persistence” of each feature is given by the difference d − b, which gives a measure of its significance.
For each homology class, it is possible to compute a “representative” or “generator”, that is, a specific set of simplices creating the corresponding homology feature12. Homology generators provide an interpretation of the abstract topological information as local, structural features of the data27,28,29,49.
Topological analysis of protein structures
The topological analysis of the protein universe follows the methodology developed in refs. 27,29, see Supplemental Fig. 3 for a schematic representation.
Step 1. We model each protein structure as the point cloud given by its α-carbon atoms, i.e., by the set PC = {p1, …, pn}, where each pi = (xi, yi, zi) is the triple of the predicted xyz-coordinates of its ith residue.
Step 2. We then feed the point cloud PC = {p1, …, pn} to the persistent homology pipeline, and compute its filtered homology in dimension 1 and 2:
From these, we compute the persistent diagrams in dimensions 1 and 2.
Step 3. We compute a representative cycle for each homology class. Note that these correspond to loops and voids appearing in the sequence of simplicial complexes \(P{C}_{{\varepsilon }_{0}}\hookrightarrow P{C}_{{\varepsilon }_{1}}\hookrightarrow \cdots \hookrightarrow P{C}_{{\varepsilon }_{N}}\).
Step 4. We compute the 1 and 2-dimensional point-wise topological influence score (TIF) of residues in PC. This is achieved by first computing centrality values centrality(res) for each residue, as in ref. 27 and using spectral methods developed in ref. 50. Then, centrality scores are normalised over all the residues in the protein to obtain values in [0, 1]:
TIFs provide a ranking of residues based on how often they contribute to topological features (i.e., how often they appear in generators) and how persistent these features are.
Software
Persistent diagrams and generators are computed using the Julia software Ripserer.jl26. Specifically, we use the Alpha filtration to construct the nested simplicial complexes, and the involutive algorithm26,51 to compute homology and representatives.
TIFs are computed using the hyperTDA method developed in ref. 27. Specifically, for each protein structure and dimension considered, we construct the hypergraph having as vertices the residues, and having a (weighted) hyperedge for each generator. Then, we compute node centrality using the software from refs. 50,52, using the max centrality flavour. More details are contained in the hyperTDA paper27 and the corresponding GitHub repository.
Similarly, topological clusters are computed as graph-communities, as explained in ref. 27 and using Python’s Louvain module53.
How we handled computations
Large-scale computations were performed on Oracle Cloud Compute. All computations were performed on a single instance with 160 CPU cores and 1 TB memory. The compute shape is named BM.Standard.A1.160 which is Arm-based Ampere A1 compute (Ampere Altra processor). A 32 TB block storage volume was attached for storage of AlphaFold2’s predicted structures as well as general storage, and a separate 32 TB volume for the outputs of our topological analyses. The former was mounted at the project root, and the latter at data/alphafold/PH/.
AlphaFold2 structures were downloaded as sharded proteomes according to their bulk download instructions.
Benchmarking was performed on a single large structure (accession “A0A009DWL0”) to assess the computational viability and reduce time, cost, and environmental impact (Supplemental Fig. 4). Only homology dimension one was computed. Julia methods were run multiple times before the recorded run, to remove the impact of compilation. Note that some bars appear to have zero height, since methods in compiled languages such as C++ have significantly lower memory consumption than Julia and Python methods. Considerations to computational cost were also important in terms of the memory usage, as the cluster becomes unstable when the 1 TB is exceeded (See Supplementary Fig. 4). Tools that were benchmarked:
Eirene.jl the initial method used in previous works due to its ability to compute representative cycles.
Eirene.jl mod a modified version of Eirene.jl, which was made in an attempt to tailor it to this specific project, however, this barely improved time at the cost of increased memory consumption.
giotto-ph a method written in C++ and Python which takes advantage of CPU parallelisation. It was not considered further, as it does not compute representatives.
Gudhi a toolkit with numerous Python modules, however no module was found for computing representative cycles.
Ripser A popular method written in C++54. There is experimental support for computing representative cycles (in a separate branch).
Ripser.py builds on top of Ripser with computations of representative cocyles. As it is built on Ripser it might be possible to also get representative cycles, however, it was not trivial.
Ripser.py sparse an approximate sparse filtration with a sparse distance matrix tested to reduce computational time.
Ripser++ The only GPU method tested46. Clearly this is a big advantage, however, it was not possible to compute representative cycles.
Ripserer.jl a Julia implementation of Ripser26.
Ripserer.jl alpha by default, Ripserer.jl (and all other listed tools) uses Vietoris-Rips filtration (Supplemental Fig. 1). Alpha filtration was tested here, which can be much more efficient on low-dimensional point clouds.
Computational time for Eirene.jl and Ripserer.jl with Alpha filtration were estimated simply by multiplying the computational time observed in Supplemental Fig. 4 by 214M and dividing by 160 (Supplementary Fig. 4). The runs are assumed to be completely parallel since multiple identical calls to Ripserer.jl will be performed, each given a single core. It is a rough estimate since the average number of residues is around 333, however, computational time does not scale linearly with residue count; larger point clouds take up a disproportionate amount of the total time. The estimated time was more than 16 times longer than the actual. This is partly explained by the large point cloud used for benchmarking, essentially making it a worst-case estimate, and partly explained by a few other optimisations:
TAR iteration Instead of extracting and reading files in the sharded proteomes, it was found to be much more efficient to stream the content of the TAR archives directly using TarIterators.jl (with a minor tweak).
CIF parsing Instead of reading the CIF files with a standard CIF reader, they were instead streamed line-by-line, only reading a required subset of the file contents.
Centrality on sparse H The hypergraph centrality code was rewritten and tailored to this project’s specific use-case, particularly with a sparse representation of the hypergraph H, paired with an efficient implementation of the sparse encoding itself.
The output of the topological analyses was written to compressed JSONs matching the structure of the shared proteomes, and later repackaged into HDF5 files to organise by UniProt accessions33, to allow for partial read/write and in order to add additional protein metadata.
The topological tree of life
The taxonomy tree is visualised in Fig. 1 of the main text with a circle packing plot, is generated by constructing circles for each species with area proportional to its number of AlphaFold2 structures (including any entries annotated with its subspecies and other lower ranks). Circles associated with child nodes of genuses are then circle packed, one genus at a time. This process is repeated for each rank, going up, which means that the area of higher ranks is only approximately representative of their number of structures.
The lightness of circles indicates the topological richness of the proteins belonging to a taxonomy ID. The richness is defined as the persistence of the 1-dimensional topological features, restricted to those having persistence ≥ 10, divided by the number of residues in the protein and averaged across proteins.
Each edge in the taxonomy tree is represented visually as an outline around the circles. The outlines are sized according to the taxonomy rank, with slightly thinner outlines for lower ranks. The outlines are coloured in a black-to-yellow palette, indicating the variance of the number of 1-dimensional topological features in each protein, normalised by the number of proteins in the circle. Differences in the outlines are made clearer by a log-transform, specifically log10 of one minus the correlation. To check whether the variance was influenced by the number of residues in each protein, we further normalised by this quantity. The output of this latter computation has a 0.925 correlation coefficient with the non-normalised one, showing thus high consistency.
Circles are packed within each container circle with the R library packcircles55 and visualised with ggplot256. Data is from the tables TreeNode and TreeEdge from the Postgres database, as well as the table AF for the zoomed example for Haemoglobin.
Mediaflux
We share the output of our topological analysis on Mediaflux. Here, the data is organised into three folders: compressed JSON files, HDF5 files, and a Postgres database. The entire dataset can be downloaded with the following links: JSON (~ 10 TB), HDF5 (~ 9 TB), and Postgres database (~ 210 GB). The links will not immediately start downloads but rather prompt for installing a helper utility “Mediaflux Data Mover” which will then aid in the download process.
See Supplemental Fig. 5 for an overview of the data structure. Some data containers are left blank for simplicity.
JSON
Protein structures predicted by AlphaFold2 and topological data is stored in GZip compressed JSONs. The organisation is similar to the proteome sharing provided by AlphaFold2. In addition, sharded proteomes are placed in folders according to the first three numbers of the taxonomy id. The JSONs contain integers and floats (floating-point values). Numbers are either provided as a scalar, in a list or lists of lists. Newline in the figure indicates the highest grouping level for JSON values.
n number of residues (scalar).
x, y, zα-carbon coordinates in Å (list of floats).
cent1, cent2 TIFs for dimensions 1 and 2 (list of floats).
bars1, bars2 Birth and death filtration times for each topological feature in dimensions 1 and 2 (list of floats).
reps1, reps2 Representative cycles for dimensions 1 and 2. Stored as a list of lists of integers. Each representative cycle is a set of either 1- or 2-simplices, provided as node indices (1-indexed).
For each proteome, we also include the topological clusters computed as graph-communities, as explained in ref. 27 and using Python’s Louvain module53. The result is written to a compressed JSON with one entry per accession, containing community indexes for each residue.
HDF5
The data is also provided in Hierarchical Data Format version 5 organised by UniProt accession. Proteins are placed together in HDF5 files based on the first five characters of their accessions. Each protein is found as an HDF5 group, which contains HDF5 attributes and HDF5 datasets. Here, each dataset is always a table of unnamed columns, stored as a numerical matrix.
AA One-letter amino acid sequence encoded as an ASCII string.
n Number of residues.
tax, taxv Taxonomy ID and sharding index used by AlphaFold2.
Cas Values for each node, i.e., α-carbons. The columns are the x, y, z coordinates in Å, pLDDT score (AlphaFold2 confidence score), and TIFs in dimensions 1 and 2.
bars1, bars2 Birth and persistence (death − birth) filtration times for each topological feature in dimensions 1 and 2.
reps1, reps2 Representative cycles for dimensions 1 and 2. The first column is an index for the feature, starting at 1. The remaining columns are node indexes for members of a simplex (one simplex per row).
Remark
(Decompression step needed to access files). The HDF5 files are uncompressed except for the datasets reps1 and reps2, which require a ZStd plugin (Zstandard) for access. For example, in Python import h5py, zstandard and in Julia, using HDF5, H5Zzstd will suffice to read the compressed datasets.
Postgres
Protein metadata is collected in a Postgres database (see Supplementary Fig. 6 and Supplementary Table 1).
AF The main table, which contains summary statistics computed on the topological analysis results for each protein.
JSON Path to JSON file for a given UniProt accession.
Tax Taxonomy ID associated with a vast amount of identifiers (NCBI taxonomy FTP server).
TaxTree Taxonomy ids at the species level or lower, with species parent indicated. Species as child nodes are also included with themselves as parents. This table (in combination with Tax) is used for connecting any relevant accession to a species.
TaxParent All direct and indirect child nodes for a subset of taxonomy ranks (Domain, Kingdom, Phylum, Class, Order, Family, Genus, and Species).
TreeNode Taxonomy tree nodes with summary statistics for the same subset of taxonomy ranks as in TaxParent.
TreeEdge Taxonomy tree edges and summary statistics between the nodes from TreeNode.
Figure 1 in the main text is build from the tables TaxNode and TaxEdge, after further data processing (see data/alphafold/vis/ in the code repository).
Datasets
The datasets discussed in the results are summarised in Supplementary Table 2. In each of these datasets, we removed structures with low-confidence AlphaFold2 predictions. AlphaFold2 produces a per-residue confidence score (pLDDT)2, which assigns a value between 0 and 100 to each residue in a structure; values below 70 are considered low. Here, to select proteins with an overall good prediction, we average the pLDDTs over all the residues in a structure and discard those scoring an average below 70. The remaining ones are considered high-confidence predictions and are kept in the dataset.
Comparison with experimental structures (RCSB)
To compare between the topological analysis performed on AlphaFold2 predictions and on experimental structures, we considered all the 2712 UniProt entries with full structure available on PDB. These UniProt accessions correspond in total to 28,309 different experimentally solved protein chains. Out of the 2712 AlphaFold2 predictions, only 2637 have a high-confidence score. For each of these structures, we considered the 1 and 2-dimensional TIFs and computed the correlation coefficient between the resulting vector for each predicted structure and its experimental counterparts.
Complete lists of the structures considered in each dataset, and the correlation coefficients, are available for download, see Data Availability. This folder contains:
-
a file uniprot2PDB_fullstructures.json, containing a mapping between UniProt accessions and PDB entries.
-
a file centrality_correlation.csv, containing, for each UniProt id, the correlation coefficient between its 1 and 2-dimensional topological influence vectors and the experimental counterparts.
Correlation coefficients were computed using numpy’s corrcoef function.
M-CSA dataset
To analyse the relation between 2-dimensional topological clusters and binding sites, we looked at the Mechanism and Catalytic Site Atlas (M-CSA)38, a database of enzyme reaction mechanisms, which provides catalytic residues of hundreds of enzymes. We downloaded all 1033 PDB entries of experimental structures with annotated sites, and performed the topological analysis on the corresponding structures, see Supplementary Fig. 7 for the result of our analysis.
To reproduce the result on AlphaFold2 predicted structures, we then mapped each PDB entry to the corresponding UniProt accession, when found. This left us with a total of 866 different proteins, 862 of which are predicted by AlphaFold2 with a high confidence score.
A complete list of the structures considered in each dataset, and code to reproduce the results, are available for download, see Data Availability.
This folder contains:
-
a file CSA_site.tsv, containing PDB entries and residue numbers of binding sites.
-
a file CSA_AF.csv, containing mapping between PDB and UniProt accessions, as well as the confidence score of the AlphaFold2 predictions.
-
files communities.json and communities-experimental.json, containing the partition of each structure into 2-dimensional topological clusters. The organisation of these JSON files is as described in Section JSON.
-
notebooks Results.ipynb and Results_experimental.ipynb to compute boundary points between 2-dimensional topological clusters and to reproduce the results.
CATH
To investigate the relation between topological features and protein domains, we looked at all the 73,749 AlphaFold2 predictions containing at least two distinct identified CATH domains32. These structures, and the corresponding domain mapping, were recently identified in ref. 25. We then excluded low-confidence predictions (leaving 62,861 proteins) and reduce the dataset to a list of 38,171 non-redundant structures. This last step was achieved using the software CD-HIT57 and a threshold of 70% sequence similarity.
To quantify the agreement between the partition induced by CATH domains and by 1-dimensional topological clusters, we computed the homogeneity score using the homogeneity_score function in Python’s sklearn package. The homogeneity score is a value between 0 and 1; a clustering satisfies homogeneity (and thus has homogeneity 1) if all of its clusters (in our case, 1-dimensional topological clusters) contain only data points which are members of a single class (in our case, a single CATH domain). For completeness, Supplemental Fig. 8 shows the results for the 62,861 high-confidence AlphaFold2 predictions, including redundant ones.
A complete list of the considered structures, the corresponding homogeneity scores, their partition into 1-dimensional topological clusters and CATH domains are available for download, see Data Availability. This folder contains:
-
a hom_scores_red.csv, with UniProt entries, homogeneity score, confidence score of the prediction, and whether they are non-redundant or not.
-
a file domain_vectors.json with the partition into CATH domains.
-
a file communities_all.json with 1-dimensional topological clusters.
Thermophiles and mesophiles
To investigate structural differences between enzymes in thermophilic and mesophilic organisms, we selected 10 different Enzyme Commission (EC) numbers based on their biotech relevance, a total of 30 thermophilic and 8 mesophilic organisms, and we listed all UniProt entries with these characteristics. In total, we considered 1815 different protein structures, that became 1656 after excluding low-confidence predictions. On this latter dataset, we were interested in analysing the distribution of volumes of significant 2-dimensional features (i.e., with high persistence). The distribution of features with persistence < 1 turned out to be almost identical across EC numbers and thermal characteristics, see Supplemental Fig. 9. For this reason, we restricted our attention to topological features with persistence ≥ 1, that show more variation, see Supplemental Fig. 10. The volume of each feature was computed using scipy ConvexHull function, as the volume of the convex-hull of residues in the generator. Our results show that mesophile organisms have on average larger voids in their enzymes, and that this patter is robust. In Supplementary Fig. 11, error bands are given by sampling 1000 different voids in thermophiles and mesophiles, respectively, and then looking at the standard deviation.
A natural question is whether this pattern is maintained for single EC numbers. Volume, number, and persistence of voids are all strongly influenced by the size and length of the protein. Since the distribution of lengths in individual EC numbers is different for thermophiles and mesophiles, to analyse EC numbers, we first selected thermophilic and mesophilic proteins in the same range of length. For a given EC number, this is achieved by randomly selecting a mesophilic enzyme for each thermophilic one, with a difference in length of at most 5 residues. The result of this analysis are shown in Supplemental Fig. 12.
A complete list of the structures considered, and code to reproduce the results, are available for download, see Data Availability. This folder contains:
-
a file thermozymes-acc-unjag-summ.tsv, containing accessions and taxonomy information of the structures considered
-
a file summary.csv, containing confidence scores of the structure considered.
-
a file thermo_all.csv containing the volumes and persistence values of the 2-dimensional topological features.
-
a file SEQ.csv containing TIFs for different amino acids
-
a file Samples.csv, containing the distribution of volumes for the sampled dataset.
-
a notebook Results.ipynb containing code to reproduce the sampling used for the result in Supplementary Fig. 12.
Mutations
To check if our analysis is effective in the detection of protein regions that are enriched for damaging mutations, we looked at the datasets of disease-causing and neutral variants studied in the paper44, where the authors consider a few hundreds experimental structures and their disease-associated missense variants, and link damaging mutations to structurally damaging changes in their mutant structures.
As usual, we restrict our analysis to structures with high-confidence predictions. Results in the manuscript show the distribution of 2-dimensional TIFs for residues accommodating neutral mutations that do not cause structural damage, and disease-associated mutations that modify the structure. Supplemental Fig. 13A shows the distributions for the full set of labels, and Supplemental Fig. 13B shows the same result for the control dataset used in ref. 44. As shown in Supplemental Fig. 14, the pattern is maintained for 1-dimensional TIFs, although the differences are weaker.
The data for the ACE2 and HBB examples shown in the manuscript is taken from the Missense3D database45, which catalogues amino-acid substitutions that are predicted to be structurally damaging44,45. A third example we analysed is CFTR, the results are shown in Supplemental Fig. 15.
A complete list of the structures considered is available for download, see Data Availability. This folder contains:
-
files mutations.csv, mutations_control.csv, containing UniProt accessions of the proteins considered and the list of mutations with labels and TIFs;
-
files ace2_cent.csv, cftr_cent.csv,hbb_cent.csv, containing the list of mutations with labels and TIFs for the examples shown;
-
a file thermo_all.csv containing the volumes and persistence values of the 2-dimensional topological features;
-
a notebook Results.ipynb containing code to visualise the results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. Due to the large size of the datasets, all raw data have been deposited in MediaFlux and are also available upon request. The datasets analysed for the current study are available for direct download from MediaFlux via: https://bit.ly/protTDA(1.31 GB). Topology outputs can be bulk downloaded from MediaFlux: JSON files via https://bit.ly/protTDAjson(~ 10 TB), and HDF5 files via https://bit.ly/protTDAhdf5(~ 9 TB). The Postgres database containing protein and taxonomy data is available via https://bit.ly/protTDApostgres(13.4 GB). Install Postgres 15.2, then run pg_restore -U opc -d protTDA Postgres. Source data are provided in this paper.
Code availability
The code used to develop the model, perform the analyses and generate results in this study is publicly available and has been deposited in the protTDA repository at https://github.com/degnbol/protTDA, under GPL-3.0 license license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://zenodo.org/records/1512915958.
References
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583 (2021).
Varadi, M. et al. Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123 (2023).
Steinegger, M. & Söding, J. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026 (2017).
Durairaj, J. et al. Uncovering new families and folds in the natural protein universe. Nature 622, 646–653 (2023).
Barrio-Hernandez, I. et al. Clustering predicted structures at the scale of the known protein universe. Nature 622, 637–645 (2023).
van Kempen, M. et al. Missense3d-db web catalogue: an atom-based analysis and repository of 4m human protein-coding genetic variants. Nat. Biotechnol. 43, 243 (2023).
Richardson, J. S. β-sheet topology and the relatedness of proteins. Nature 268, 495 (1977).
Minami, S., Sawada, K. & Chikenji, G. How a spatial arrangement of secondary structure elements is dispersed in the universe of protein folds. PLOS ONE 9, 1 (2014).
Cummings, C. G. & Hamilton, A. D. Disrupting protein–protein interactions with non-peptidic, small molecule α-helix mimetics. Curr. Opin. Chem. Biol. 14, 341 (2010).
Hatcher, A. Algebraic Topology (Cambridge Univ. Press, Cambridge, 2000).
Edelsbrunner, H., Harer, J. Computational Topology: An Introduction (American Mathematical Soc., 2010).
Seiberg, N. & Witten, E. Electric-magnetic duality, monopole condensation, and confinement in n= 2 supersymmetric Yang-Mills theory. Nucl. Phys. B 426, 19 (1994).
Atiyah, M. Progress in Mathematics 133. The Floer memorial volume (Hofer, H. Taubes, C. H., Weinstein, A. & Zehnder, E.) 105–108 (Birkhäuser (Basel), 1995).
Donaldson, S. K. Floer Homology Groups in Yang-Mills Theory. (Cambridge University Press, 2002).
Edelsbrunner, Letscher, Zomorodian. Topological persistence and simplification. Discrete Comput. Geom. 28, 511 (2002).
Zomorodian, A. Topological data analysis. Adv. Appl. Comput. Topol. 70, 1 (2012).
Zomorodian, A. & Carlsson, G. Computing persistent homology. Discrete Comput. Geom. 33, 249 (2005).
Ghrist, R. Barcodes: the persistent topology of data. Bull. Am. Math. Soc. 45, 61 (2008).
Saggar, M. et al. Towards a new approach to reveal dynamical organization of the brain using topological data analysis. Nat. Commun. 9, 1399 (2018).
Sørensen, S. S., Biscio, C. A., Bauchy, M., Fajstrup, L. & Smedskjaer, M. M. Revealing hidden medium-range order in amorphous materials using topological data analysis. Sci. Adv. 6, eabc2320 (2020).
Vipond, O. et al. Multiparameter persistent homology landscapes identify immune cell spatial patterns in tumors. Proc. Natl. Acad. Sci. USA 118, e2102166118 (2021).
Thorne, T., Kirk, P. D. & Harrington, H. A. Topological approximate Bayesian computation for parameter inference of an angiogenesis model. Bioinformatics 38, 2529 (2022).
Bordin, N. et al. Alphafold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun. Biol. 6, 160 (2023).
Čufar, M. Ripserer.jl: flexible and efficient persistent homology computation in Julia. J. Open Source Softw. 5, 2614 (2020).
Barbensi, A. et al. Hypergraphs for multiscale cycles in structured data. Preprint at https://doi.org/10.48550/arXiv.2210.07545 (2022).
Kovacev-Nikolic, V., Bubenik, P., Nikolić, D. & Heo, G. Using persistent homology and dynamical distances to analyze protein binding. Stat. Appl. Genet. Mol. Biol. 15, 19 (2016).
Benjamin, K. et al. Homology of homologous knotted proteins. J. R. Soc. Interface 20, 20220727 (2023).
Ewing, B. & Green, P. Analysis of expressed sequence tags indicates 35,000 human genes. Nat. Genet. 25, 232 (2000).
Stumpf, M. P. H. et al. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 105, 6959–6964 (2008).
Sillitoe, I. et al. Cath: increased structural coverage of functional space. Nucl. Acids Res. 49, D266 (2021).
The UniProt Consortium. Uniprot: the universal protein knowledgebase in 2023. Nucl. Acids Res. 51, D523 (2023).
Ballew, R. M., Sabelko, J. & Gruebele, M. Observation of distinct nanosecond and microsecond protein folding events. Nat. Struct. Biol. 3, 923 (1996).
Gruebele, M. The fast protein folding problem. Annu. Rev. Phys. Chem. 50, 485 (1999).
Jennings, P. A. & Wright, P. E. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science 262, 892 (1993).
Nussinov, R., Zhang, M., Liu, Y. & Jang, H. Alphafold, artificial intelligence (ai), and allostery. J. Phys. Chem. B 126, 6372–6383 (2022).
Ribeiro, A. J. M. et al. Mechanism and catalytic site atlas (m-csa): a database of enzyme reaction mechanisms and active sites. Nucl. Acids Res. 46, D618 (2018).
Berman, H. M. et al. The protein data bank. Nucl. Acids Res. 28, 235 (2000).
Chakravarty, D. & Porter, L. L. Alphafold2 fails to predict protein fold switching. Protein Sci. 31, e4353 (2022).
Dabrowski-Tumanski, P. A. Stasiak Alphafold blindness to topological barriers affects its ability to correctly predict proteins’ topology. Molecules 7, 7462 (2023).
Cohen-Steiner, D., Edelsbrunner, H. & Harer, J. Stability of persistence diagrams. Discrete Comput. Geom. 37, 103 (2007).
Szilágyi, A. & Závodszky, P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure 8, 493–504 (2000).
Ittisoponpisan, S. et al. Can predicted protein 3d structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 431, 2197 (2019).
Khanna, T., Hanna, G., Sternberg, M. J. & David, A. Missense3d-db web catalogue: an atom-based analysis and repository of 4m human protein-coding genetic variants. Hum. Genet. 140, 805 (2021).
Zhang, S, Xiao, M, & Wang, H. GPU-Accelerated Computation of Vietoris-Rips Persistence Barcodes. In 36th International Symposium on Computational Geometry (SoCG 2020). Leibniz International Proceedings in Informatics (LIPIcs), Vol 164, 70:1-70:17 (Schloss Dagstuhl – Leibniz-Zentrum für Informatik, 2020)
Bramer, D. & Wei, Guo-Wei Atom-specific persistent homology and its application to protein flexibility analysis. Comput. Math. Biophys. 8.1, 1–35 (2020).
Drozdetskiy, A., Cole, C., Procter, J. & Barton, G. J. JPred4: a protein secondary structure prediction server. Nucl. Acids Res. 43, W389–W394 (2015).
Emmett, K., Schweinhart, B., & Rabadan, R. Multiscale topology of chromatin folding. BICT’15: Proceedings of the 9th EAI International Conference on Bio-inspired Information and Communications Technologies, 177–180 (2016).
Tudisco, F. & Higham, D. J. Node and edge nonlinear eigenvector centrality for hypergraphs. Commun. Phys. 4, 1–10 (2021).
Čufar, M. & Virk, Ž. Fast computation of persistent homology representatives with involuted persistent homology. Found. Data Sci. 5, 466–479 (2023).
Sofía Urbieta, M. et al. Thermophiles in the genomic era: Biodiversity, science, and applications. Biotechnol. Adv. 33, 633–647 (2015).
Aynaud, T. Python-louvain x.y: Louvain algorithm for community detection. https://github.com/taynaud/python-louvain (2020).
Bauer, U. Ripser: efficient computation of vietoris–rips persistence barcodes. J. Appl. Comput. Topol. 5, 391–423 (2021).
Wang, W., Wang, H., Dai, G., & Wang, H. Visualization of large hierarchical data by circle packing. In Proceedings of the SIGCHI Conference on Human Factors in Computing Systems. (2006).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York, (2016).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Madsen, C.D. degnbol/protTDA https://zenodo.org/records/15129159 (2024).
Acknowledgements
CDM gratefully acknowledges funding through a University of Melbourne PhD studentship. A.B. gratefully acknowledges funding through a MACSYS Centre Development initiative from the School of Mathematics & Statistics, the Faculty of Science and the Deputy Vice-Chancellor Research, University of Melbourne. S.Y.Z. gratefully acknowledge funding through a University of Melbourne PhD studentship. M.P.H.S. is funded through the University of Melbourne DRM initiative, through an ARC Laureate Fellowship (FL220100005) and acknowledges financial support from the Volkswagen Foundation through a “Life?” programme grant. L.H. is funded through the University of Melbourne DRM initiative. A.D. gratefully acknowledges funding through Wellcome Trust grant 218242/Z/19/Z. D.E.V.P. received funding from an Oracle for Research Grant. The authors wish to thank Ellen Leffler for helpful discussions.
Author information
Authors and Affiliations
Contributions
C.D.M., A.B., D.E.V.P. and M.P.H.S. designed the research; C.D.M. and A.B. wrote the software; C.D.M., A.B., S.Y.Z., LH, AD, D.E.V.P. and M.P.H.S. analysed the data; all the authors were involved in writing the manuscript, and all authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks David Gleich and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Madsen, C.D., Barbensi, A., Zhang, S.Y. et al. The topological properties of the protein universe. Nat Commun 16, 7503 (2025). https://doi.org/10.1038/s41467-025-61108-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61108-2
