
Abstract
PMEL amyloids serve as essential scaffolds for melanin deposition in melanosomes, playing a crucial role in pigmentation. Despite their importance, the high-resolution structure of PMEL amyloids has remained unresolved. Using cryo-electron microscopy, we determine near-atomic resolution structures of wild-type PMEL amyloid core, revealing two distinct polymorphic forms with structural features. We further investigate the pathogenic G175S mutation associated with pigment dispersion syndrome (PDS). Structural analysis reveales that G175S introduces an additional hydrogen bond, stabilizing an alternative fibril conformation. In vitro, the G175S mutant exhibits a fourfold increase in polymerization efficiency compared to the wild type. In cells, G175S expression resultes in a twofold increase in intracellular amyloid content and a ~70% increase in extracellular amyloids, without altering melanosome morphology or number. These results indicate that the G175S mutation enhances amyloidogenesis within melanosomes, elevating amyloid load and potentially contributing to PDS pathophysiology. This study provides molecular insights into PMEL amyloid formation, highlighting its structural diversity and dysregulation in pigmentation disorders.
Similar content being viewed by others


Introduction
Amyloids are protein aggregates traditionally associated with neurodegenerative diseases, but they can also play essential roles in normal physiological processes1,2,3,4,5,6. One example is PMEL (Pmel17/gp100), a pigment-cell-specific protein that forms amyloid fibrils in melanosomes to scaffold melanin deposition7,8,9,10,11,12. These fibrils underscore the dual nature of amyloids as both pathological and functional entities.
PMEL amyloidogenesis occurs within specialized organelles called melanosomes, which progress through four distinct stages (I–IV) of maturation (reviewed in ref. 13). During the transition from stage I to stage II, PMEL fibrils form and assemble laterally into sheets, a process essential for the structural organization of melanosomes. While Seiji et al. (1961) first described fibrillar morphology using thin-section electron microscopy14, the three-dimensional sheet-like architecture of PMEL fibrils was revealed by electron tomography in Hurbain et al. (2008)15. These fibrillar sheets are crucial for the transition to stage III, where melanin deposition begins. The structural organization of these fibrils is integral to their function, yet their high-resolution structure has remained unresolved.
PMEL was first demonstrated to be an amyloid-forming protein by Fowler et al. (2006), who established the presence of cross-β amyloid fibrils in melanosomes5. However, the precise region of PMEL that forms the core of these amyloids has been the subject of significant debate. Several in vitro studies, primarily involving recombinant, unglycosylated fragments, have proposed that the repeat (RPT) domain of PMEL constitutes the amyloid core, based on its polymerization under artificial conditions16,17,18,19,20,21,22,23,24,25,26,27. Graham et al. (2019) demonstrated that the O-glycosylation of the RPT domain, rather than its peptide backbone, is essential for organizing the lateral sheet architecture of PMEL fibrils by facilitating their proper spacing and alignment within stage II melanosomes28. Leonhardt et al. (2013) further demonstrated that deletion of the RPT domain disrupted sheet organization, supporting its structural role in the supramolecular architecture of fibrils rather than in amyloid core formation29.
In contrast, multiple lines of biochemical and cellular evidence consistently point to the CAF domain as the physiological amyloid core. Watt et al. (2009) demonstrated that the CAF domain is required for fibrillogenesis in vitro, as its removal abolished amyloid formation30. This was supported in vivo by Hee et al. (2017), who showed that mutations in the CAF domain disrupted PMEL fibril formation in cells31. Leonhardt et al. (2013) also found that deletion of the CAF domain abolished the formation of fibrillar structures in melanocytic cells, though the study did not confirm whether these were amyloid or organized into sheets29. Together, these studies reinforce the view that the CAF domain forms the structural core of PMEL amyloids in vivo, while the RPT domain, via its O-glycosylation, contributes to higher-order sheet organization. This distinction is critical for resolving ongoing controversy over PMEL domain functions in amyloidogenesis and pigmentation.
Mutations in the PMEL gene, such as Gly175Ser (G175S), are associated with pigment dispersion syndrome (PDS), a condition in which pigment granules are released into the anterior chamber of the eye. This aberrant pigment release can elevate intraocular pressure, increasing the risk of pigmentary glaucoma (PG), a sight-threatening complication that affects 15–20% of PDS patients32,33. Although the G175S mutation has been proposed to alter PMEL amyloid formation, its precise molecular and structural effects remain unclear34.
In this work, we present near-atomic resolution structures of native PMEL amyloid cores from both wild-type and G175S mutant forms. We reveal fibril polymorphism and show that the G175S mutation introduces an additional hydrogen bond that stabilizes an alternative amyloid conformation.
Results
Cryo-EM Structure of Native PMEL Amyloids Reveals Two Polymorphs
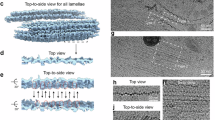
Native PMEL amyloids were isolated from the human melanoma cell line HMV-II through deglycosylation, protease digestion, and sonication. Cryo-electron microscopy (cryo-EM) and 2D classification revealed two distinct fibrillar forms: thick and thin filaments, corresponding to two-protofilament and single-protofilament structures, respectively (Fig. 1a and Fig. S1). High-resolution structures were obtained from two-protofilament fibrils as shown in Fig. 1b. However, our attempts to resolve the single-protofilament structures were unsuccessful, possibly due to their inherent instability or disruption during the extraction and processing stages. This suggests that the single-protofilament forms may represent immature or structurally less stable amyloid species, which are challenging to analyze at high resolution under the conditions applied in this study.

a Cryo-EM images of native PMEL amyloids extracted from the human melanoma cell line, showing both thick (two-protofilament) and thin (single-protofilament) fibrils (Fig. S1, squares). For each condition, the imaging experiment was independently repeated three times using separately prepared grids, yielding consistent results. b Side views of the reconstructed 3D maps of the two-protofilament fibrils, illustrating the overall helical structure of the amyloids.
Two polymorphic forms of two-protofilament fibrils, termed Polymorph 1 and Polymorph 2, were identified in the native amyloids (Figs. 1b and 2). Polymorph 1 adopts a two-start helical architecture, characterized by a helical twist of 177.7° and a helical rise of 2.34 Å. In contrast, Polymorph 2 forms a one-start helical architecture with C2 symmetry, featuring a helical twist of −4.1° and a helical rise of 4.67 Å. Despite similar β-sheet configurations, the main chain morphology between the two polymorphs is markedly different (Figs. 3a–c and 4a). Notably, Polymorph 2 encompasses a central cavity, which is absent in Polymorph 1, thereby rendering Polymorph 1 as a more densely packed structure.

Side and cross-sectional views of cryo-EM maps and atomic models of wild-type PMEL polymorph 1 (top), polymorph 2 (middle), and the G175S mutant (bottom), each composed of two interacting protofilaments. Left panels show filament side views aligned to the fibril axis, with maps in gray and models colored by chain (green and cyan). Right panels show cross-sectional views highlighting inter-protofilament interfaces. Key residues with bulky side chains and/or interfacial contacts are labeled. Red dashed curves indicate the inter-protofilament interface, which is formed predominantly by hydrophobic packing rather than polar contacts. No hydrogen bonds were detected between protofilaments based on ChimeraX hydrogen bond analysis. Prominent nonpolar residues—such as Phe149, Tyr151, Leu163, Pro166, and Gly169—contribute to shape complementarity and tight packing across the interface. Gray: reconstructed maps; carbon: green/cyan; oxygen: red; nitrogen: blue; sulfur: yellow. Scale bars = 1 nm.

a Ribbon diagrams of native PMEL amyloid fibrils, illustrating the four-layered structure of β-strands in the fibril core. b Amino acid sequence of the N-terminal portion of the CAF domain, with the positions of β-strands (indicated by arrows) predicted using ModelAngelo. c Close-up views of the parallel cross-β sheets, highlighting the structural differences between wild-type and G175S mutant fibrils. Insets (left) show cross-sectional views of the fibril core to indicate the positions of the β-sheets (boxes). In the G175S mutant, Thr155 (box, left) separates β1-1 and β1-2, resulting in the division of the β-sheet. Note the alignment of aromatic residues along the fibril axis, emphasizing the positioning of these bulky residues in both wild-type and G175S mutant fibrils. Panel (c) highlights intra-protofilament interactions along the fibril axis, which are maintained primarily through backbone hydrogen bonding between stacked β-strands. Carbon: green; oxygen: red; nitrogen: blue; sulfur: yellow.

a Packing schemes of one cross-sectional layer of the fibrils, illustrating the structural arrangement of residues in wild-type and G175S mutant fibrils. Asterisks (*) indicate the inner cavities present in Polymorph 2 and G175S. Blue and red circles and arrowheads highlight the positions of Tyr159 and Gly/Ser175, respectively. Numbers of key residues are labeled. Residues are color-coded by chemical properties: hydrophobic (white), polar (green), basic (blue), glycine (magenta), and proline (purple). b Close-up views of the G175S fibril highlighting the additional hydrogen bond formed between Tyr159 and Ser175. Insets show the cross-sectional orientation of the fibril and the corresponding regions displayed in the top view (top) and side view (bottom).
Each polymorph comprises two protofilaments that pack against each other along a distinct interface. ChimeraX hydrogen bond analysis35 revealed no hydrogen bonds at the protofilament interface in any of the structures. Instead, the protofilaments interact via extensive hydrophobic contacts between bulky nonpolar side chains, such as Phe149, Tyr151, Leu163, Pro166, and Gly169. These residues are laterally positioned at the interface and form a tightly packed hydrophobic surface, which likely stabilizes the fibril architecture through shape complementarity and van der Waals interactions (Fig. 2, red dashed lines). This contrasts with the vertical stacking of β-strands within each protofilament—illustrated in Fig. 3c—which is stabilized by canonical backbone hydrogen bonds along the fibril axis.
Cryo-EM maps revealed that the side chains of aromatic residues, including tyrosine (Tyr), phenylalanine (Phe), and tryptophan (Trp), are aligned along the fibril surface. However, these residues do not form π-π stacking interactions, as their rings are not directly aligned (Fig. 3c).
Structural alterations in G175S mutant amyloids
To investigate the effects of the G175S mutation on PMEL amyloids, we expressed G175S PMEL in HMV-II melanoma cells in a PMEL-knockout (KO) background and isolated the resulting amyloids (Figs. 1–4, G175S). Similar to the wild-type, cryo-EM revealed thick and thin filaments, with high-resolution reconstructions obtained exclusively from the thick filaments (Fig. S2, 2-start). The G175S amyloids exhibited a two-start helical architecture with a helical twist of 178.4° and a helical rise of 2.35 Å, similar to Polymorph 1 in wild-type amyloids. However, the β-sheet configuration and packing within the G175S fibrils were markedly altered.
In G175S amyloids, the first β-sheet (β1) is divided into three distinct segments (β1-1, β1-2, and β1-3), in contrast to the single continuous β1 observed in wild-type amyloids. β2 and β3 maintain similar configurations to those in the wild-type, but the overall packing density is reduced due to the division of β1 (Fig. 3a–c). A notable structural feature in G175S amyloids is the formation of an additional hydrogen bond between Ser175 and Tyr159, which may contribute to the fragmented β1 configuration and potentially enhance fibril stability (Fig. 4b).
In Vitro Polymerization of Wild-Type and G175S PMEL CAF Domains
To further investigate the biochemical properties of PMEL amyloids, we expressed and purified the CAF domain (residues 148–223) of wild-type and G175S PMEL in E. coli. The purified proteins were subjected to in vitro polymerization assays, and the resulting amyloids were analyzed using cryo-EM and thioflavin T (ThT) fluorescence.
Cryo-EM analysis revealed that the wild-type CAF domain polymerized into fibrils structurally identical to Polymorph 1 of native PMEL amyloids, confirming that the in vitro polymerization recapitulates the native fibril structure. Similarly, the G175S CAF domain polymerized into fibrils indistinguishable from the G175S native amyloids, further validating that the extraction process from melanoma cells did not disrupt the amyloid structure (Fig. 5a–c, Fig. S3, 4). Notably, no one-start helical fibrils were observed in the in vitro polymerized samples, suggesting that thin fibrils represent immature intermediates or are disassembled during the extraction process.

a Cryo-EM images of PMEL CAF domain fibrils polymerized in vitro, showing thick fibrils corresponding to two-protofilament structures. Wild-type fibrils are shorter than G175S ones. For each condition, the experiment was independently repeated three times, with fibrils imaged from separately prepared reactions, and yielded similar results. b Side views of the reconstructed 3D maps of the in vitro polymerized fibrils, highlighting the overall helical structure. c Side (top) and cross-sectional (bottom) views of the reconstructed maps (gray), superimposed with models (green and cyan) of the CAF domain. Key residues are labeled as in Fig. 2. Scale bars = 1 nm.
G175S mutation enhances amyloid formation in vitro
Next, we performed ThT fluorescence assays to measure the amyloid-forming efficiency of wild-type and G175S CAF domains in vitro5 (Fig. 6a, b). The G175S mutant polymerized approximately four times more efficient than the wild-type, as shown by the rapid increase in ThT signal. Additionally, both G175S and wild-type polymerization curves plateaued, demonstrating that the final yield of polymerized amyloids is approximately four times greater for the G175S mutant than for the wild-type. The increased amyloid-forming efficiency of the G175S mutant is consistent with its structural alterations, particularly the possible stabilizing effect of the additional Ser175-Tyr159 hydrogen bond.

a Negative-stain EM images of CAF domain fibrils polymerized in vitro. Wild-type fibrils form thick bundles by 24–48 h, whereas G175S fibrils show earlier bundling at 14 h and further thickening by 24 h. b Thioflavin T (ThT) fluorescence assay measuring polymerized amyloids over time. Data represent means with individual data points from five independent polymerization reactions (N = 5), each using separately prepared protein. G175S fibrils showed greater fluorescence than wild type. A two-sided Mann-Whitney U test confirmed a significant difference (U = 0, Z = –2.6548, p = 0.007937, standardized effect size r = 0.79, 95% CI of acceptance region for U = [3,22]). c ThT fluorescence of intracellular amyloids from detergent-insoluble lysates after 96 h of doxycycline (Dox) induction. Data from six biological replicates (N = 6), each an independently induced and harvested well. G175S-expressing cells showed a twofold increase over wild type. Fluorescence was normalized using propidium iodide signals to account for differences in cell density and lysis efficiency. One-way ANOVA detected a significant effect across groups (F(4,25) = 42.46, p = 8.49 × 10⁻¹¹, η² = 0.87, 95% CI for F = [0, 2.76]). Post-hoc Tukey HSD revealed a significant increase in G175S Dox(+) over WT Dox(+) (mean difference = 312.1, p = 2.14 × 10⁻⁵, 95% CI = [161.5, 462.7]) (asterisk). d ThT fluorescence assay of extracellular amyloids in supernatants normalized to 1 × 10⁷ cells per replicate. Data from eight biological replicates (N = 8). WT/G175S Dox (–) represents knockout cells; WT/G175S Dox (+) indicates amyloid levels after 0.2 µg/ml Dox induction for 96 h. G175S Dox (+) samples showed a ~ 70% increase over WT Dox (+). One-way ANOVA revealed a significant group effect (F(4,35) = 57.22, p = 7.22 × 10−15, η² = 0.87, 95% CI for F = [0, 2.64]). Tukey’s HSD post-hoc test showed that G175S Dox(+) had significantly higher extracellular amyloid than WT Dox(+) (mean difference = 73.50, p = 3.36 × 10⁻⁵, 95% CI = [35.05, 111.95]) (asterisk).
While negative stain EM images (Fig. 6a) showed fewer discrete fibrils at 48 h compared to earlier time points, this reflects a natural progression in PMEL fibril assembly rather than a decrease in total amyloid. Over time, fibrils undergo lateral bundling into thicker aggregates or sheets, leading to fewer visible individual filaments despite an overall increase in amyloid mass. This phenomenon is consistent with the quantitative ThT fluorescence measurements (Fig. 6b), which track total amyloid content and show continued accumulation over time. In G175S samples, fibrils exhibit more extensive bundling than in wild type, consistent with enhanced polymerization efficiency. It is also important to note that uneven adhesion of fibrils to carbon film during sample preparation contributes to local variation in fibril density in the EM images and limits direct visual quantification. Accordingly, the EM images are intended to illustrate morphological progression (i.e., bundling and thickness) rather than the absolute number of fibrils.
G175S mutation increases intracellular and extracellular amyloids
To evaluate the impact of the G175S mutation on amyloidogenesis within the cells, we quantified intracellular and extracellular amyloids produced by MNT1 cells expressing wild-type or G175S PMEL using ThT fluorescence assays (Fig. 6c, d). Here, extracellular amyloids refer to PMEL aggregates recovered from the culture medium, reflecting material released into the extracellular space. Cells expressing the G175S mutant PMEL exhibited approximately twofold higher levels of intracellular amyloids compared to wild-type PMEL-expressing cells, consistent with enhanced amyloidogenic efficiency. Similarly, extracellular amyloids were significantly elevated in G175S-expressing cells, showing approximately 70% higher levels than wild-type. Cells lacking PMEL expression showed negligible ThT fluorescence for both intracellular and extracellular fractions, confirming that the observed signals were specifically attributable to PMEL amyloids.
To rule out the possibility that the elevated extracellular amyloids in G175S-expressing cells were due to increased cell death, we performed a cell viability assay (Fig. S5a). No significant differences in viability were observed between wild-type and G175S-expressing cells, indicating that the extracellular PMEL amyloids are not the result of passive leakage from dying cells. Rather, these data suggest that amyloids are actively released into the medium—possibly via secretory pathways such as exosomes or melanosome extrusion10,36,37,38. Importantly, the increase in extracellular ThT signal closely paralleled the increase in intracellular amyloids, suggesting that G175S does not preferentially enhance secretion or release, but instead promotes overall amyloid accumulation both within and outside the cell. These findings are consistent with the enhanced polymerization observed in vitro and further support the conclusion that the G175S mutation enhances amyloid formation and accumulation within melanosomes.
G175S mutation does not affect melanosomal structure in situ
To investigate whether the G175S mutation affects melanosome architecture, we employed cryo-focused ion beam scanning electron microscopy (cryo-FIB-SEM) and cryo-electron tomography to analyze melanosomes in MNT1 cells expressing wild-type or G175S PMEL (Fig. 7a, Fig. S6, Videos S1, 2). Stage III melanosomes, characterized by pigmented fibrillar lamellae, were the primary focus of this analysis15,28. Tomographic slices revealed no discernible differences in the overall structural organization of melanosomes between wild-type and G175S mutant cells. Both cell types displayed lamellar structures, which were reconstructed into three-dimensional models to further examine the spatial arrangement of the fibrillar networks.

a Tomographic slices (top panels) and 3D models (bottom panels) of stage III melanosomes from MNT1 cells expressing either WT or G175S PMEL, with Dox induction. Green represents the melanosomal membrane, and cyan indicates the fibrillar lamellae within the melanosome. Scale bars, 100 nm. b Quantification of melanosomal structural features from 12 melanosomes per condition (N = 12), derived from three independent biological replicates. Parameters measured include lamella number, thickness, cross-sectional area, length, curvature, and inter-lamellar distance. No significant differences were observed between WT and G175S. Two-sided Mann-Whitney U test results were: – Lamella number: U = 73.5, Z = 0.0588, p = 0.9531, standardized effect size r = 0.012, 95% CI = [38.65, 105.35]. – Lamella thickness: U = 63, Z = -0.482, p = 0.6297, r = 0.1, 95% CI = [38, 106]. – Cross-sectional area: U = 52, Z = -1.113, p = 0.266, r = 0.23, 95% CI = [38, 106]. – Lamella length: U = 65, Z = -0.369, p = 0.7125, r = 0.077, 95% CI = [38, 106]. – Lamella curvature: U = 74, Z = 0.0849, p = 0.9323, r = 0.018, 95% CI = [38, 106]. – Inter-lamellar distance: U = 57, Z = -0.825, p = 0.4095, r = 0.17, 95% CI = [38, 106]. Individual data points are shown with horizontal bars indicating mean values. These results indicate that G175S expression does not significantly alter the ultrastructure of stage III melanosomes compared to wild type.
The ultrastructure visualized by cryo-electron tomography was highly consistent with the lamellar architecture previously described in osmium-stained thin-section electron microscopy and tomographic reconstructions15, confirming that PMEL fibrils are laterally arranged into sheets within stage III melanosomes. The use of cryo-ET allowed visualization of these structures in a near-native, unstained state, providing complementary confirmation of prior findings.
Quantitative analysis demonstrated no significant differences in the number of lamellae per melanosome, the thickness of the lamellae, the length of the lamellae, or their average curvature between wild-type and G175S melanosomes (Fig. 7b). Additionally, the interlamellar distance and the cross-sectional area of melanosomes were consistent across the two groups. These results suggest that the G175S mutation does not significantly alter the gross morphological properties of melanosomes or their lamellar organization.
G175S Alters Melanosomal Stage Distribution
To determine whether the G175S mutation affects melanosome maturation, we quantified melanosome density and analyzed the distribution of melanosome stages (II, III, IV) using ultrathin-section electron microscopy of MNT1 cells expressing wild-type or G175S PMEL (Fig. 8a, b, and Fig. S7)39,40. Melanosome density, calculated as the number of melanosomes per square micrometer of cytoplasm, showed no significant difference between wild-type and G175S mutant cells, indicating that the mutation does not alter melanosome abundance (Fig. 8c, left panel).

a Representative images of melanosomes in stages I–IV. Stage I: multi-vesicular bodies. Stage II: vesicles containing fibrillar lamellae without melanin deposition. Stage III: vesicles containing fibrillar lamellae with melanin deposition. Stage IV: vesicles with fully melanized, electron-dense granules. b Representative ultrathin-section electron microscopy images of MNT1 cells expressing wild-type (WT) or G175S PMEL with Dox induction. Melanosomes throughout the cytoplasm are annotated with red numerals to indicate stages I–IV. c Quantification of melanosomes. Left: Melanosome density (number per square micrometer of cytoplasm). Right: Percentage of melanosomes in stages II, III, and IV. Horizontal bars represent mean values, and dots represent individual data points (N = 39 cells for each condition). No significant difference in melanosome density was observed between groups. A two-sided Mann-Whitney U test using the normal approximation with tie correction yielded U = 766.5, Z = 0.055, p = 0.9562, standardized effect size r = 0.0062, 95% CI of acceptance region for U = [564.44, 956.56]. A two-sided Wilcoxon signed-rank test revealed a significant increase in stage III melanosomes in G175S cells compared to WT cells (W⁺ = 690, p = 2.575 × 10⁻⁷, r = 0.84, 95% CI for W⁺ = [236, 505], single asterisk). Additionally, the proportion of stage II melanosomes was significantly decreased in G175S cells compared to WT (W⁺ = 35, p = 1.568 × 10−8, r = 0.91, with 95% CI for W⁺ = [250, 530], double asterisk), while no significant differences were observed in the proportion of stage IV melanosomes (W⁺ = 324, Z = –0.91, p = 0.3607, r = 0.15, with 95% CI for W⁺ = [0, 780]). Stage I melanosomes were not included in the quantification due to their low abundance and difficulty in identification.
Next, we assessed the proportion of melanosomes in stages II, III, and IV. The G175S mutation significantly increased the proportion of stage III melanosomes compared to wild-type, as determined by a Wilcoxon signed-rank test (Fig. 8c, single asterisk). Additionally, the proportion of stage II melanosomes was significantly decreased in G175S cells compared to WT (Fig. 8c, double asterisk), while no significant differences were observed in the proportion of stage IV melanosomes.
To assess whether the G175S mutation affects melanin production, we measured intracellular melanin content at 96 h after Dox induction. No significant differences in melanin content were observed between cells expressing wild-type or G175S PMEL (Fig. S5b), suggesting that the increase in stage III melanosomes in G175S cells does not result from altered melanin synthesis. These results collectively indicate that the G175S mutation modulates melanosomal stage distribution without affecting intracellular melanin levels.
Discussion
In this study, we present the near-atomic resolution structures of PMEL amyloids, revealing two distinct polymorphic forms and the structural alterations induced by the G175S mutation. PMEL amyloids play a critical role in melanosomes by providing a scaffold for melanin deposition, which is essential for pigmentation. While PMEL amyloid formation has been studied extensively for its physiological relevance, their high-resolution structure has remained elusive until now9,12,16,31. Our cryo-EM analysis addresses this gap, offering crucial insights into PMEL amyloid architecture and the structural consequences of the G175S mutation associated with PDS.
Polymorphism and Structural Insights into PMEL Amyloids
Our analysis of native PMEL amyloids revealed two polymorphic forms, Polymorph 1 and Polymorph 2, both adopting a two-protofilament helical architecture. Despite their similar β-sheet configurations, the main chain morphology differs significantly. Polymorph 2 exhibits an inner cavity, absent in Polymorph 1, resulting in a more loosely packed structure. Such polymorphism highlights the structural plasticity of PMEL amyloids, which may facilitate interactions with melanin precursors or melanosomal proteins under varying physiological conditions. This flexibility is a common feature of functional amyloids and may be critical for the regulation of melanosomal architecture and pigmentation41,42,43.
Factors such as post-translational modifications or variations in the cellular environment may influence the formation of these polymorphs. The observation of multiple fibril forms within a single cell type suggests greater structural heterogeneity in PMEL amyloids than previously recognized. Future studies are needed to determine how polymorphism affects interactions with melanin precursors or melanosomal proteins under diverse physiological conditions.
Our cryo-EM structures of wild-type PMEL fibrils (Polymorphs 1 and 2) and the G175S mutant fibril reveal critical roles for specific residues within the 148–183 region in stabilizing the amyloid core. Trp160 consistently forms the inner core of the β-strand in all three fibril structures, underscoring its essential role in maintaining structural integrity. Tyr151 and Trp153, although not engaged in π–π stacking, exhibit aligned aromatic rings along the fibril length, contributing to β-sheet stabilization. These findings align with previous studies showing that mutation of these residues disrupts the formation of PMEL fibril-like structures in cells, underscoring their importance in fibril assembly29,31.
Our cryo-EM structures reveal extra densities within the inner cavities of wild-type polymorph 2 and the G175S mutant fibrils (Fig. S8). These densities, present in both native and in vitro polymerized structures, are unlikely to originate from melanin pigments, particularly in melanin-free in vitro samples. While these observations suggest potential pigment-binding sites, further studies are required to confirm their functional relevance.
Our cryo-EM structures resolve the N-terminal half of the PMEL CAF domain (residues 148–183) in the fibril core, while additional densities distal to Thr183 were observed but could not be reliably resolved due to noise. This suggests that the C-terminal portion of the CAF domain may be more flexible relative to the structured N-terminal part. The flexibility of this region could contribute to its inability to adopt a consistent conformation suitable for cryo-EM analysis.
Structural comparison with other amyloids
The PMEL amyloids described in this study exhibit an unusually high density of hydrophobic and bulky residues, such as Trp and Tyr, on their exterior surface. This is distinct from the hydrophilic or charged external surfaces often seen in pathological amyloids like Aβ fibrils, AL, ATTR, SOD1 amyloids44,45,46,47,48,49,50. While these pathological amyloids utilize external hydrophilic interactions to promote aggregation and toxicity, PMEL amyloids may rely on the glycosylated RPT domain to shield their hydrophobic surfaces and prevent non-specific aggregation.
Structural implications of the G175S mutation
The G175S mutation in PMEL, strongly associated with PDS, introduces an additional hydrogen bond between Ser175 and Tyr159, altering the β-sheet configuration. The mutation divides the first β-sheet (β1) into three shorter segments while preserving the configuration of β2 and β3. These changes enhance amyloid polymerization, as demonstrated by the four-fold increase in polymerization efficiency in vitro. Furthermore, cells expressing G175S PMEL exhibited increased levels of both intracellular and extracellular amyloids compared to wild-type, suggesting more efficient amyloidogenesis.
Despite these biochemical changes, cryo-electron tomography revealed no significant differences in melanosomal architecture between wild-type and G175S cells. This suggests that while the G175S mutation enhances amyloidogenesis, the overall melanosomal structure remains intact. The enhanced amyloidogenic capacity likely enables a greater proportion of PMEL proteins to convert into amyloids, contributing to increased amyloid content without disrupting melanosome morphology.
Impact on melanosome maturation
Quantification of melanosomal stages revealed a significant increase in stage III melanosomes and a corresponding decrease in stage II melanosomes in G175S mutant cells, with no significant changes in stage IV or overall melanosome density. These findings suggest that the G175S mutation enhances amyloid formation efficiency, potentially providing a scaffold for melanin deposition earlier than in wild-type cells. The increased amyloid content in G175S melanosomes likely enhances the efficiency of melanin binding, facilitating the observed shift toward stage III melanosomes. However, no differences in intracellular melanin content were detected (Fig. S5b), indicating that amyloidogenic efficiency, rather than melanin synthesis, drives these changes.
Functional Implications and Pathophysiology of PDS
PDS is characterized by the release of melanin granules into the anterior chamber of the eye, leading to increased intraocular pressure and an elevated risk of PG. Our findings suggest that the G175S mutation enhances the amyloidogenic efficiency of PMEL, resulting in a higher amyloid content within melanosomes. This structural alteration likely contributes to the rigidity and altered biophysical properties of melanosomes, as evidenced by the requirement for 4 M urea treatment to disintegrate G175S melanosomes during amyloid extraction (Fig. S9b).
These changes in melanosome properties may increase their susceptibility to mechanical stress within the eye, facilitating pigment dispersion under intraocular forces. While our study does not directly demonstrate a link between G175S-induced melanosome rigidity and pigment dispersion, the observed structural and biochemical changes provide a plausible explanation for the increased extracellular amyloids and their potential contribution to PDS pathophysiology. Further investigations into the mechanical properties of G175S melanosomes and their behavior under physiological conditions could shed additional light on this mechanism.
Roles of the CAF, RPT, and PKD domains
Our analysis focused on the CAF domain as the core structural element of PMEL amyloids. Although several in vitro studies using unglycosylated recombinant fragments have proposed that the RPT domain can form amyloid fibrils under artificial conditions16, in vivo evidence suggests a different role. In particular, Graham et al. (2019) demonstrated that it is not the RPT polypeptide itself, but its O-glycosylation, that is essential for the lateral sheet organization of PMEL fibrils within stage II melanosomes. These findings support a model in which the RPT domain, while not forming the amyloid core, contributes to fibril architecture by spacing and aligning fibrils through its glycan moieties.
A recent review by Buchanan et al. (2023) asserted a field-wide consensus that the amyloidogenic region responsible for the characteristic PMEL fibrillar matrix lies within the RPT domain51. While this view reflects findings from in vitro studies using unglycosylated RPT fragments, it does not incorporate substantial in vivo evidence pointing to the CAF domain as the physiological amyloid core. Our structural data, along with prior cellular and biochemical studies, support the CAF domain as the primary component of PMEL fibrils. Moreover, as shown by Graham et al. (2019)28, the RPT domain contributes to fibril architecture not through its peptide sequence but via O-glycosylation, which mediates lateral sheet organization. These observations call for a more nuanced view of PMEL domain function and underscore the importance of integrating structural and cell-based evidence when defining amyloidogenic regions.
While our cryo-EM structures did not reveal RPT-derived structures in the fibril core, we cannot exclude the possibility that our extraction methods, which involved deglycosylation and protease digestion, may have selectively degraded RPT-derived structures or preferentially preserved CAF-derived fibrils.
In addition to the CAF and RPT domains, the PKD (polycystic kidney disease-1 repeat) domain has also been implicated in PMEL amyloid formation. The PKD domain (residues 235–297) was originally defined by Theos et al. (2005) as part of the Mα fragment52, and was first shown to be present in PMEL fibrils in 2001 using domain-specific antibodies7,8. Subsequent studies demonstrated that both the PKD and CAF domains are required for amyloid formation in vivo53,54. In vitro, the PKD domain has also been shown to be amyloidogenic30. Biochemical evidence suggests that it may be transiently incorporated into native PMEL fibrils during early stages of maturation, particularly as part of the MαC fragment55, although it is likely removed or remodeled during later processing steps. In our cryo-EM structures, we did not observe density corresponding to the PKD domain, which may reflect its absence in the final stable amyloid core or degradation during sample preparation involving protease digestion and sonication. This highlights the value of complementary in situ approaches to further define the structural contributions of the CAF, RPT, and PKD domains to PMEL amyloid architecture.
Our cryo-EM analysis reveals that the CAF domain alone can form fibrils resembling those observed in vivo. This suggests that the CAF domain is the primary driver of amyloid core formation. However, the roles of other PMEL domains, such as the N-terminal fragment (NTF), PKD, and RPT domains, cannot be disregarded. Previous studies have shown that the NTF domain is essential for CAF aggregation in vivo, while the PKD and RPT domains are implicated in stabilizing and organizing PMEL fibrils30,55. These domains likely contribute to fibril formation and assembly in the cellular context, where additional factors such as post-translational modifications and environmental conditions may modulate fibril morphology and function. Further studies are needed to elucidate how these domains interact with the CAF domain to regulate PMEL amyloidogenesis.
Taken together, our findings support a model in which the CAF domain serves as the core structural scaffold of PMEL amyloids, while the RPT and PKD domains contribute peripheral or regulatory roles modulated by glycosylation, proteolytic processing, and intracellular context.
Challenges in in situ structural analysis
Although cryo-electron tomography (cryo-ET) has been successfully used to study amyloids in their native cellular contexts, PMEL amyloids pose challenges. Their entangled lamellar organization within melanosomes complicates the isolation and visualization of individual fibrils (Fig. S9a). Furthermore, our 2D classification revealed higher-order assemblies that lack helical twists, suggesting a more complex packing arrangement. These findings indicate that in situ structural analysis of PMEL amyloids is technically challenging due to their densely packed and entangled nature.
Future directions
Our study provides a foundation for understanding the molecular mechanisms underlying PMEL amyloid formation and its dysregulation in PDS. Future research could focus on exploring the effects of other PMEL mutations, elucidating the role of RPT domain glycosylation, and identifying potential therapeutic strategies to mitigate the pathological impact of enhanced amyloidogenesis in PDS.
In conclusion, our cryo-EM analysis reveals the structural basis of PMEL amyloid polymorphism and the significant impact of the G175S mutation on amyloidogenesis and melanosome maturation. These findings provide insights into the functional and pathological roles of PMEL amyloids and their broader implications in pigmentation biology and disease.
Methods
Cell culture
Human melanoma cell lines HMV-II (TKG-0318) and MNT-1 (CRL-3450) were obtained from the Cell Resource Center for Biomedical Research at Tohoku University and the American Type Culture Collection, respectively. HMV-11 cells were maintained in F12 medium (Fujifilm, Osaka, Japan) supplemented with 10% fetal bovine serum (FBS). MNT-1 cells were maintained in DMEM medium (Fujifilm) supplemented with 20% FBS, 10% AIM-V (ThermoFisher Scientific, Waltham, MA), 0.1 mM MEM non-essential amino acids (Fujifilm). Cell lines were authenticated by analysis of short tandem repeat profiling (BEX, Tokyo, Japan).
For isolating native PMEL fibrils, we used the HMV-II human melanoma cell line. MNT1 cells, while also suitable for PMEL expression, were not used for fibril isolation because their melanin granules are too tightly packed to be efficiently disintegrated during extraction. Conversely, MNT1 cells were used for the rest of the experiments. HMV-II cells, although useful for fibril isolation, become unstable after extended culture and multiple passages, leading to a gradual decrease in their capacity to produce melanin granules. Thus, MNT1 cells were more suitable for experiments requiring stable, long-term granule production.
PMEL gene knockout and transfection
First, we constructed two lentiviral transfer plasmids: pCW57.1-X330-gRNA/SpCas9-mCeru-PuroR and pCW57.1-PMEL-DsRed-HygR-GFP-rTetR (Fig. S10a).
Construction of pCW57.1-X330-gRNA/SpCas9-mCeru-PuroR
The expression cassette backbone for Cas9 and the gRNA scaffold was derived from the pX330_sgRNA/hSpCas9 plasmid (Addgene number 172832)56. The original gRNA scaffold sequences were replaced with those targeting the PMEL gene.
The sequences of the sgRNA target sites are listed below, with PAM sites underlined:
-
AAGTGACTGTCTACCATCGC CGG
-
CGTGTCCCAGTTGCGGGCCT TGG
-
TCCATCCAAGGCCCGCAACT GGG
-
CTCCATCCAAGGCCCGCAAC TGG
Following the SpCas9 sequence, we inserted mCerulean3 (synthesized by ThermoFisher Scientific), a T2A sequence, and the puromycin-resistance gene (derived from pCW57.1, Addgene number 99283)57. This plasmid enables knockout of the PMEL gene and selection of clones via mCerulean fluorescence and puromycin resistance. The entire cassette, from gRNA to SpCas9 and mCerulean-PuroR, was inserted into pCW57.1 to convert it into a lentiviral transfer plasmid.
Construction of pCW57.1-PMEL-DsRed-HygR-GFP-rTetR: The backbone for this plasmid is the Tet-on plasmid pCW57.158. We inserted the PMEL gene with or without G175S mutation, along with DsRed (excised from the tdTomato sequence, derived from pCDH-EF1-Luc2-P2A-tdTomato, Addgene number 72486, a gift from Kazuhiro Oka), under the tight TRE promoter. In addition to introducing the G175S mutation, the PAM sites in the PMEL expression plasmid were mutated to prevent cleavage by Cas9. Following the hPGK promoter, we inserted the hygromycin-resistance gene (derived from pCEP4-AD8gp160, Addgene number 123260)59, a T2A sequence, the EGFP, another T2A sequence, and the rTetR gene. This plasmid enables doxycycline (Dox)-dependent expression of PMEL G175S and allows clone selection via hygromycin resistance and EGFP fluorescence.
Virus production
We transfected Lenti-X 293 T cells (Takara Bio, Shiga, Japan) with the transfer plasmids, alongside the packaging plasmid pCMV-dR8.2 delta-vpr (Addgene number 8455),60 and the envelope plasmid pCMV-VSV-G (Addgene number 8454), using the Avalanche-Everyday transfection reagent (EZ Biosystems, College Park, MD). Twenty-four hours post-transfection, the cells were washed three times with PBS and incubated in fresh medium. Virus-containing medium was collected at 48-, 72-, and 96-hours post-transfection. Cell debris was removed by centrifugation at 1000 × g, followed by filtration through a 0.45 µm membrane (Millipore). The lentivirus-containing supernatant was concentrated using polyethylene glycol precipitation61.
Generation of PMEL-expressing cell lines in a PMEL background
To generate PMEL knockout cell lines, HMV-II and MNT1 cells were incubated with the concentrated lentiviral particles carrying pCW57.1-X330-gRNA/SpCas9-mCeru-PuroR for 24 h. Cells were then selected using 2 µg/ml puromycin. PMEL gene knockout was confirmed via western blotting using a PMEL-specific antibody (1/2,000 dilution, E-7, Santa Cruz Biotechnology, Dallas, TX) (Fig. S10b). PMEL-knockout cells were subsequently transduced with the concentrated lentivirus carrying pCW57.1-PMEL-DsRed-HygR-GFP-rTetR and selected using 200 µg/ml hygromycin. Expression levels of PMEL were adjusted to match wild-type levels by optimizing Dox concentration (Fig. S10b). Primer sequences are summarized in Supplementary Data 1.
We initially attempted to generate PMEL(G175S)-expressing cells by directly modifying the genomic sequence of the native PMEL gene using CRISPR-Cas9. However, no clones successfully proliferated following drug selection. This outcome is likely due to the additional stress imposed on single isolated cells during the proliferation stage, which is inherently a challenging process. While doxycycline-induced expression of G175S PMEL protein did not result in cell death, its expression may impose a level of cellular stress that interferes with the proliferation of isolated single cells under these conditions. Consequently, we employed the Tet-on system to control the expression of PMEL(G175S) in a PMEL-knockout background, allowing for Dox-inducible expression and avoiding the issues associated with constitutive expression of the mutant protein.
Extraction of PMEL amyloids
PMEL amyloid fibrils were isolated following a modified version of the previously described method31. Melanoma cells were resuspended in PBS supplemented with 2.5 µg/ml cytochalasin D (Fujifilm) and 10 µM nocodazole (Fujifilm) and incubated at 37 °C for 30 min. The cells were then resuspended in 10 mM Tris-HCl (pH 7.4) containing a protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) and incubated on ice for 10 min. The cells were disrupted by Dounce homogenization, followed by centrifugation at 800 ×g for 10 min at 4 °C to remove cell debris. The membrane fraction was collected by ultracentrifugation at 100,000 × g for 60 min at 4 °C, and the resulting pellets were rinsed twice with PBS. Rinsed membranes were lysed in 2% Triton X-100 in PBS for 1 h at 4 °C and then layered onto a discontinuous sucrose gradient consisting of 30%, 45%, and 55% sucrose in 50 mM Tris-HCl (pH 7.4), 200 mM NaCl, and 1 mM EDTA. The samples were centrifuged for 2 h at 100,000 × g at 4 °C. The 55% sucrose fraction was diluted 10-fold and ultracentrifuged at 100,000 ×g for 2 h at 4 °C. The resulting pellet was rinsed twice with PBS and resuspended in 50 mM Tris-HCl (pH 7.4) containing 5 mM CaCl₂ and disrupted by sonication for 10 s using a Q125 sonicator (Qsonica, Newtown, CT). For the G175S specimen, the pellet was incubated in 4 M Urea in 50 mM Tris-HCl (pH 7.4) for 10 min at room temperature to loosen the structure before sonication (Fig. S9b). Deglycosylation was carried out by adding 10 µl of O-glycosidase (New England Biolabs, Ipswich, MA) and 10 µl of neuraminidase (New England Biolabs) to the sample, followed by overnight incubation at 37 °C. The digested product was centrifuged at 20,000 × g for 10 min at 4 °C, and the pellet was resuspended in 50 mM Tris-HCl (pH 8.0) containing 5 mM CaCl₂ and 1 mM DTT. The pellet was then digested with 10 µg of trypsin for 1 h at 37 °C16,62. After centrifugation at 20,000 × g for 10 min at 4 °C, the pellet was resuspended in 50 mM Tris-HCl (pH 8.0) containing 5 mM CaCl₂ and 1 mM DTT, followed by digestion with 20 µg of Arg-C endopeptidase overnight at 37 °C. The digested product was centrifuged at 20,000 × g for 10 min at 4 °C, then disrupted by sonication for 20 s. After ultracentrifugation at 100,000 × g for 2 h at 4 °C, the pellet was sonicated for an additional 20 s. The final specimen was centrifuged at 20,000 × g for 10 min at 4 °C and prepared for cryo-EM analysis (Fig. S9).
In vitro polymerization of PMEL CAF domain
The CAF domain (residues 148–223) of human PMEL was subcloned into the pET24a plasmid (Merck Millipore, Darmstadt, Germany) and transformed into E. coli BL21 (DE3) (New England Biolabs). Transformed bacteria were cultured in LB medium at 37 °C until the optical density (OD600) reached 0.8. Expression of the CAF domain was induced by the addition of 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG), followed by overnight incubation at 18 °C. The cells were harvested and disrupted by sonication. Inclusion bodies were collected and sequentially washed with 2 M, 3 M, 4 M, 6 M, and 8 M urea. The resulting pellets were solubilized in 6 M guanidine hydrochloride (GuHCl), and insoluble debris were removed by centrifugation at 20,000 × g for 10 min at 30 °C. For G175S inclusion bodies, extensive sonication without cooling (up to 40 °C) was required to achieve solubilization. The clarified supernatant was loaded onto Ni-NTA resin (Nacalai Tesque) pre-equilibrated with 50 mM Tris-HCl (pH 8.0), 8 M urea, and 20 mM imidazole. The resin was washed with the equilibration buffer, and bound proteins were eluted with 50 mM Tris-HCl (pH 8.0), 8 M urea, and 300 mM imidazole. The eluted protein was then diluted with 150 mM sodium acetate (pH 4.4) to reach a final concentration of 0.3 mg/ml, and incubated with vigorous shaking (200 rpm) at 37 °C for 24 ~ 30 h for wild-type protein. Initially, we incubated the G175S mutant protein at 37 °C; however, the rapid growth of amyloids at this temperature led to the formation of thick, bundled rods that were unsuitable for analysis. To obtain analyzable fibrils, we incubated the G175S protein at 18 °C for 30 h. For the wild-type protein, we incubated at 18 °C for 66 h, resulting in fibrils that exhibited the same structure as those incubated at 37 °C.
Cryo-electron microscopy of PMEL amyloids
Samples were resuspended in 150 mM sodium acetate (pH 4.4) at a final concentration of 0.3 mg/ml. A 2 µl aliquot of the sample was applied to each side of freshly glow-discharged Ultra Au foil R1.2/1.3 300 mesh grids (Quantifoil Micro Tools GmbH, Großlöbichau, Germany). The grids were blotted from both sides for 3 s at 12 °C under 100% humidity and subsequently plunge-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific).
Images were recorded on a CRYO ARM 300 II (JEOL, Tokyo, Japan) at the University of Tokyo, operated at 300 keV. An Omega filter with a slit width of 20 eV and a Gatan K3 direct electron detector in correlated-double sampling (CDS) mode were used for imaging. The nominal magnification was set to 60,000×, yielding a physical pixel size of 0.8784 Å/pixel. Movies were acquired using the SerialEM software, with a target defocus range of 0.8–1.5 µm. Each movie was recorded for 5.52 s with a total electron dose of 50 e⁻/Ų, divided into 50 frames.
Data processing
Image processing was carried out using CryoSPARC v4.5.3 and Relion v3.1 and v563,64. Raw movie frames were motion-corrected, and the contrast transfer function (CTF) was estimated using the patch-CTF estimation method. Fibrils were automatically picked using the filament tracer tool, and segments were extracted with a box size of 300 pixels and an inter-box distance set to 10% of the box size. Several rounds of 2D classification were performed to exclude bad classes and those that lacked discernible helical twists.
For the native specimen, we identified both 1-start and 2-start helical fibrils (Fig. S1, squares), but only the 2-start helical fibrils were processed further. The 1-start helical particles were discarded due to insufficient resolution. In the native wild-type specimen, two distinct polymorphs were identified and processed separately. After selection, the final segments were re-extracted with a box size of 280 (native fibrils) or 300 (in vitro fibrils) pixels and reconstructed using helical refinement.
Initial references were generated from 2D class averages with a box size of 600 pixels, using the relion_helix_inimodel2D subroutine65. Further refinement included local and global CTF refinement, reference-based motion correction, and 3D classification. Final maps were generated from 3D classes with the highest resolution using helical refinement. The handedness of the fibrils was determined by generating mirror structures of the 3D maps. Models were built using ModelAngelo66 for both the original and mirrored structures. Only the left-handed structure produced reasonable and consistent models, confirming the fibril handedness.
For the in vitro polymerized specimen, additional refinement steps were performed, including Relion CTF refinement and Bayesian polishing after 3D classification. Resolution estimates were determined using the Fourier shell correlation (FSC) at a threshold of 0.143. Image processing workflows were summarized in Figs. S1–4.
Initial models were generated using Relion v5.0 ModelAngelo and refined using ChimeraX and ISOLDE tool35,67,68. The refined models were validated using Phenix (v.1.19.2-4158)69 (Table 1). Image rendering was conducted using Chimera X and MaskChains tool70. We used pyem tools for parameter conversion from CryoSPARC to Relion71.
Preparation of specimen for cryo-FIB-SEM
A formvar-coated gold grid stabilized with an evaporated carbon film (FCF100-Au-EC, Electron Microscopy Sciences, Hatfield, PA) was prepared by coating with a 0.1% gelatin solution (Nacalai Tesque) for 1 h at 37 °C. The grid was then washed with culture medium to remove excess gelatin. MNT1 cells were applied to the prepared grid surface and cultured for 96 h in the presence of 0.2 µg/mL Dox to induce expression of PMEL. Hoechst-33258 was added to the medium at a final concentration of 1 µg/mL and incubated with the cells for 10 min to stain nuclei.
Following staining, grids were transferred into a TBS buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl) supplemented with 0.1% BSA, 5 mM CaCl₂, 5 mM MgCl₂, and 9% propylene glycol72. The grids were incubated in this buffer for 5 min at room temperature to enhance cryoprotection. After incubation, grids were blotted from both sides for 25 s at 25 °C in a controlled 100% humidity environment using a Vitrobot Mark IV (Thermo Fisher Scientific). Finally, the grids were plunge-frozen in liquid ethane cooled by liquid nitrogen.
Cryo-FIB-SEM lamella preparation
We used an Aquilos cryo-focused ion beam scanning electron microscope (ThermoFisher Scientific) for preparing lamellae73 (Fig. S11a). The preparation was performed in a stepwise manner, beginning with a lamella thickness of 3 μm, followed by successive milling to reduce the thickness to 2 μm and finally to 0.88 μm. These steps utilized gallium ion beam milling currents of 1 nA, 0.5 nA, and 0.3 nA, respectively. For the final polishing step, milling currents between 13 and 50 pA were applied to reach a final lamella thickness of less than 300 nm. After achieving the desired thickness, the lamellae were coated with an additional thin layer of inorganic platinum by sputter coating at 30 mA for 3 s to enhance conductivity and protect the sample during subsequent imaging.
Cryo-electron tomography data acquisition and processing
Tomographic tilt series were recorded on a Talos Arctica transmission electron microscope (Thermo Fisher Scientific) operating at 200 keV at the University of Tokyo (Fig. S11b). The microscope was equipped with a Gatan Quantum-LS Energy Filter set to a slit width of 30 eV and a Gatan K2 BioQuantum direct electron detector in electron counting mode. Imaging was performed at a nominal magnification of 49,000×, resulting in a physical pixel size of 2.2 Å/pixel. Movies were acquired using Thermo Fisher’s Tomography software, with a target defocus set between 4 and 6 µm. Tilt series were collected over an angular range from -62° to +42°, with a 4.0° increment between images and an initial tilt angle of -10°. Each movie was recorded for 2.0 s, with a per-frame dose of 2.0 electrons/Ų, subdivided into 15 frames to capture high-resolution details. The total accumulated dose for one tilt series was 54 electrons/Ų. This dose was carefully optimized to balance the need for sufficient image contrast and resolution while minimizing electron radiation damage to the carbohydrate-rich structures present in the sample74,75. Raw tilt series were subjected to motion correction using the Alignframes program in IMOD76. Reconstruction of the corrected tilt series was performed with AreTomo277 using a simultaneous iterative reconstruction technique (SIRT) method. Tomograms were denoised using Topaz Denoise3D78. 3D modeling of the tomograms was accomplished using the Drawing tools and Interpolator tool in IMOD.
Lamella thickness was measured in the XY plane using 3dmod. The lamellar structure was visualized in Zap View, and the boundaries of the lamella were identified in the XY plane of the tomogram. The Distance Tool in 3dmod was used to measure the width (thickness) of the lamellae in nanometers. Calibration was performed using the tomogram’s pixel size.
The length of lamellae was measured using Fiji. A midline of each lamella was traced in a 2D slice using the Freehand Tool. The length of the traced midline was calculated using the Analyze > Measure function in Fiji and recorded in µm.
The curvature of lamellae was measured using the Kappa Curvature plugin in Fiji. A midline of the lamella was traced in a 2D slice and the curvature was calculated at multiple points along the traced midline, and the average curvature across the entire lamella was reported in µm-1.
The inter-lamellar distance was measured in 3dmod. The Distance Tool in Zap View was used to determine the distance between the midlines of adjacent lamellae within the same melanosome. Multiple measurements were taken for each melanosome to account for variability in spacing, and the average distance was reported in nanometers.
The cross-sectional area of melanosomes was measured from 2D slices extracted from tomograms in Fiji. The melanosome boundary was manually traced in a single Z-slice using the Polygon ROI Tool. The area was calculated using the Analyze > Measure function in Fiji and converted into nm2 based on the pixel calibration.
Thioflavin T assay
For in vitro polymerized fibrils, wild-type and G175S mutant CAF domains (30 µM) were polymerized at 37 °C with shaking at 200 rpm. At each time point, 5 µl of 1 mM ThT solution was added to 100 µl of the reaction mixture. Fluorescence was measured using a SpectraMax GeminiEM plate reader (Molecular Devices, San Jose, CA) with an excitation wavelength of 444 nm and an emission wavelength of 480 nm.
For extracellular amyloid quantification, PMEL-knockout MNT-1 cells, transformed with Tet-on-inducible wild-type or G175S PMEL expression plasmids, were cultured in the presence or absence of 0.2 µg/ml Dox for 96 h. The culture supernatant from 10 cm dishes was recovered and centrifuged at 800 × g for 5 min to remove cell debris. The clarified supernatant was further centrifuged at 20,000 × g for 15 min at 4 °C, and the resulting pellets containing melanin granules were resuspended in PBS. After two washes with PBS, the pellets were demembranated by incubating in PBS containing 1% Triton X-100 for 1 h at 4 °C. The demembranated melanin granules were then washed twice with PBS and resuspended in 100 µl PBS after disintegration. For ThT fluorescence measurement, 10 µl of the granule suspension was added to 200 µl of 50 µM ThT solution in PBS. Fluorescence was measured as described above using the SpectraMax GeminiEM plate reader. To normalize the ThT fluorescence for extracellular amyloid content, the cell number of each culture was determined using a hemocytometer. The fluorescence values were adjusted to reflect extracellular amyloids from 1 × 107 cells per data point.
To quantify the intracellular amyloid content, PMEL-knockout MNT-1 cells with Dox-inducible expression of wild-type or G175S mutant PMEL were cultured in 6-well plates. Dox (0.2 µg/mL) was added to the medium to induce PMEL expression at time 0 h. Cells were collected at 96 h after induction with six replicates (N = 6) for each condition. Cells were harvested by scraping into 1 mL ice-cold lysis buffer per well (TBS plus 1% Triton X-100, and protease inhibitor cocktail) and homogenized using a homogenization pestle. The lysate was centrifuged at 15,000 × g for 10 min at 4 °C to separate detergent-soluble (supernatant) and detergent-insoluble (pellet) fractions. The detergent-insoluble pellet was resuspended in 500 µL TBS and washed twice by repeating centrifugation and resuspension. After the final wash, the pellet was disintegrated and resuspended in 100 µL of ThT assay buffer (10 mM Tris-NaOH pH 8.0, 50 µM ThT, 1 µg/mL propidium iodide (PI)). For each measurement, 10 µL of the detergent-insoluble fraction was mixed with 90 µL ThT solution in a 96-well black plate. ThT fluorescence was measured using a SpectraMax GeminiEM plate reader with excitation at 444 nm and emission at 480 nm. PI fluorescence was measured as an internal control for cell count with excitation at 535 nm and emission at 615 nm. ThT fluorescence values were standardized using PI fluorescence readings to account for differences in cell density and lysis efficiency. PI fluorescence was assumed to reflect genomic DNA content proportional to the number of cells. Uninduced cells (no Dox) served as negative controls to account for background fluorescence. All measurements were performed with six biological replicates per condition. Normalized ThT fluorescence values were compared between wild-type and G175S PMEL-expressing cells over time to determine differences in intracellular amyloid content.
Cell viability assay
To assess the effect of wild-type or G175S PMEL expression on cell viability, we utilized PMEL-knockout MNT-1 cells transduced with Dox-inducible constructs for wild-type or G175S PMEL expression. Cells were seeded in 6-well plates and induced by adding 0.2 µg/mL Dox to the culture medium. Cell viability was determined by sequential staining first with Propidium iodide (PI, 1 µg/mL; Fujifilm) and followed by Hoechst 33342 (1 µg/mL). Propidium iodide was used to label dead cells, as its strong nuclear staining distinguishes it from the cytoplasmic localization of the dsRed-PMEL reporter. Hoechst 33342 was used to stain the nuclei of all cells, providing the total cell count. Imaging and quantification were performed using a fluorescence microscope. Comparisons were made between Dox-treated and untreated cells, as well as between wild-type and G175S PMEL-expressing cells at each time point. Data were collected from six times biological replicates to ensure statistical robustness.
Intracellular melanin content assay
To quantify intracellular melanin content, PMEL-knockout MNT1 cells with Dox-inducible expression of wild-type or G175S mutant PMEL were cultured in 6-well plates. Dox (0.2 µg/mL) was added to the medium to induce PMEL expression at time 0 h. Cells were collected at 96 h after induction, with six replicates for each condition. Cells were harvested by trypsinization and split into two equal portions for melanin content measurement and DNA quantification. For melanin measurement, one portion was centrifuged at 500 × g for 5 min at room temperature to obtain a cell pellet. Melanin Extraction The pellet was resuspended in 1 mL of 1 N NaOH containing 10% DMSO. The mixture was heated at 80 °C for 1 h to dissolve melanin79. After heating, the optical density (OD) of the solution was measured at 405 nm. The second portion of the cells was lysed as described in the intracellular amyloid content assay, and genomic DNA content was quantified using PI fluorescence (excitation 535 nm, emission 615 nm). The OD405 values were normalized to genomic DNA content to account for variability in cell number. Uninduced cells (no Dox) were used as negative controls to account for background melanin levels. All measurements were performed with six biological replicates per condition and time point. Normalized melanin content was compared between wild-type and G175S PMEL-expressing cells over time to assess differences in intracellular melanin accumulation.
Ultrathin section electron microscopy of melanosomes
After inducing wild-type or G175S mutant PMEL expression for 96 h, MNT1 cells were collected and fixed with 4% paraformaldehyde and 0.1% glutaraldehyde for 1 h at 4 °C. The samples were post-fixed with 1% osmium tetroxide, followed by staining with 1% uranyl acetate. Dehydration was performed using a graded ethanol series and acetone, and the samples were embedded in Quetol 812 resin (Nissin EM, Tokyo, Japan). Ultrathin sections of 60 nm were prepared using an ULTRACUT microtome (Reichert Leica) and mounted onto Formvar-coated copper grids. Electron microscopy images were acquired using a JEM-2100F microscope (JEOL, Tokyo, Japan) equipped with an F216 CMOS camera (TVIPS GmbH, Gauting, Germany), operated at 200 keV, at the University of Yamanashi. Melanosomes were identified and counted, and their areas were measured using Fiji software80.
Statistical analysis
For statistical comparison of amyloid yield between the wild-type and G175S mutant CAF domains, two analyses were conducted. First, we used the Mann-Whitney U test due to the non-normal distribution of the data. The test was performed as a two-tailed analysis with an exact p-value calculation. The results showed that the G175S mutant yielded significantly higher levels of polymerized amyloids compared to the wild-type (p = 0.0079). The test statistic, Z = -2.6548, was outside the 95% confidence interval ([-1.96, 1.96]), indicating a statistically significant difference between the two groups. The observed standardized effect size was 0.79, suggesting a large magnitude of difference in amyloid yield between the wild-type and G175S groups.
For the quantification of intracellular amyloids, statistical analysis was performed using one-way analysis of variance (ANOVA) to test for overall differences between groups (native MNT1, WT Dox (-), WT Dox (+), G175S Dox (-), and G175S Dox (+), N = 6). A significant difference was found between the groups (p = 8.4903e-11). To further analyze pairwise differences, Tukey’s HSD (Honestly Significant Difference) post-hoc test was performed. Tukey’s HSD revealed that the difference between WT Dox (+) and G175S Dox (+) was statistically significant (p = 2.144e-4).
For the quantification of extracellular amyloids, statistical analysis was performed using one-way analysis of variance (ANOVA) to test for overall differences between groups (native MNT1, WT Dox (-), WT Dox (+), G175S Dox (-), and G175S Dox (+), N = 8). A significant difference was found between the groups (p = 7.216e-15). To further analyze pairwise differences, Tukey’s HSD (Honestly Significant Difference) post-hoc test was performed. Tukey’s HSD revealed that the difference between WT Dox (+) and G175S Dox (+) was statistically significant (p = 3.36e-5), and there was a highly significant difference between G175S Dox (-) and G175S Dox (+) (p = 6.635e-12).
For the quantification of melanosome stages, the proportions of stage II, III, and IV melanosomes were compared between wild-type PMEL and G175S mutant PMEL expressing cells using the Wilcoxon signed-rank test, as the proportions of the three stages are dependent (their sum equals 100%). Significant differences were observed in the proportion of stage III melanosomes (p = 2.575e-7, effect size r = 0.8358) and stage II melanosomes (p = 1.568e-8, effect size r = -0.9054). However, no significant difference was detected for stage IV melanosomes (p = 0.3607, effect size r = -0.1464).
All the statistical analyses were conducted using an online calculator (https://www.statskingdom.com/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes: EMD-61782 (native wild-type PMEL fibrils, polymorph 1); EMD-61783 (native wild-type PMEL fibrils, polymorph 2); EMD-61784 (native G175S PMEL fibrils); EMD-61785(in vitro polymerized wild-type CAF domain fibrils); and EMD-61786 (in vitro polymerized G175S CAF domain fibrils). The atomic coordinates have been deposited in the Protein Data Bank (PDB) under accession codes: 9JST (native wild-type PMEL fibrils, polymorph 1); 9JSU (native wild-type PMEL fibrils, polymorph 2); 9JSV (native G175S PMEL fibrils); 9JSW (in vitro wild-type CAF domain fibrils); and 9JSX (in vitro G175S CAF domain fibrils). The source data underlying Figs. 6b–d, 7b, 8c, Supplementary Fig. 5a, b, and 10b are provided as a Source Data file. Source data are provided with this paper.
References
Maury, C. P. The emerging concept of functional amyloid. J. Intern. Med. 265, 329–334 (2009).
Jackson, M. P. & Hewitt, E. W. Why are Functional Amyloids Non-Toxic in Humans?. Biomolecules 7, 71 (2017).
Chuang, E., Hori, A. M., Hesketh, C. D. & Shorter, J. Amyloid assembly and disassembly. J. Cell. Sci. 131, jcs189928 (2018).
Chiti, F. & Dobson, C. M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006).
Fowler, D. M. et al. Functional amyloid formation within mammalian tissue. PLoS. Biol. 4, e6 (2006).
Fowler, D. M. & Kelly, J. W. Functional amyloidogenesis and cytotoxicity-insights into biology and pathology. PLoS. Biol. 10, e1001459 (2012).
Raposo, G., Tenza, D., Murphy, D. M., Berson, J. F. & Marks, M. S. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell. Biol. 152, 809–824 (2001).
Berson, J. F., Harper, D. C., Tenza, D., Raposo, G. & Marks, M. S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell. 12, 3451–3464 (2001).
Berson, J. F. et al. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell. Biol. 161, 521–533 (2003).
Watt, B., van, Niel, G., Raposo, G. & Marks, M. S. PMEL: a pigment cell-specific model for functional amyloid formation. Pigment. Cell. Melanoma Res. 26, 300–315 (2013).
Bissig, C., Rochin, L. & van, Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 17, 1438 (2016).
Hellström, A. R. et al. Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS. Genet. 7, e1002285 (2011).
Le, L., Sirés-Campos, J., Raposo, G., Delevoye, C. & Marks, M. S. Melanosome Biogenesis in the Pigmentation of Mammalian Skin. Integr. Comp. Biol. 61, 1517–1545 (2021).
Seiji, M., Fitzpatrick, T. B. & Birbeck, M. S. The melanosome: a distinctive subcellular particle of mammalian melanocytes and the site of melanogenesis. J. Invest. Dermatol. 36, 243–252 (1961).
Hurbain, I. et al. Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc. Natl Acad. Sci. Usa. 105, 19726–19731 (2008).
Dean, D. N. & Lee, J. C. Modulating functional amyloid formation via alternative splicing of the premelanosomal protein PMEL17. J. Biol. Chem. 295, 7544–7553 (2020).
Louros, N. N. & Iconomidou, V. A. Identification of an amyloid fibril forming segment of human Pmel17 repeat domain (RPT domain). Biopolymers 106, 133–139 (2016).
Chrystal, P. W. et al. Functional Domains and Evolutionary History of the PMEL and GPNMB Family Proteins. Molecules 26, 3529 (2021).
McGlinchey, R. P. et al. Repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J. Biol. Chem. 286, 8385–8393 (2011).
Pfefferkorn, C. M., McGlinchey, R. P. & Lee, J. C. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl Acad. Sci. Usa. 107, 21447–21452 (2010).
McGlinchey, R. P., Yap, T. L. & Lee, J. C. The yin and yang of amyloid: insights from alpha-synuclein and repeat domain of Pmel17. Phys. Chem. Chem. Phys. 13, 20066–20075 (2011).
Jiang, Z. & Lee, J. C. Lysophospholipid-containing membranes modulate the fibril formation of the repeat domain of a human functional amyloid, pmel17. J. Mol. Biol. 426, 4074–4086 (2014).
Dean, D. N. & Lee, J. C. Purification and characterization of an amyloidogenic repeat domain from the functional amyloid Pmel17. Protein Expr. Purif. 187, 105944 (2021).
McGlinchey, R. P. & Lee, J. C. Why Study Functional Amyloids? Lessons from the Repeat Domain of Pmel17. J. Mol. Biol. 430, 3696–3706 (2018).
Pedersen, J. N. et al. Lysophospholipids induce fibrillation of the repeat domain of Pmel17 through intermediate core-shell structures. Biochim. Biophys. Acta Proteins Proteom. 1867, 519–528 (2019).
Dean, D. N. & Lee, J. C. pH-Dependent fibril maturation of a Pmel17 repeat domain isoform revealed by tryptophan fluorescence. Biochim. Biophys. Acta Proteins Proteom. 1867, 961–969 (2019).
Dean, D. N. & Lee, J. C. Defining an amyloid link Between Parkinson’s disease and melanoma. Proc. Natl Acad. Sci. Usa. 117, 22671–22673 (2020).
Graham, M., Tzika, A. C., Mitchell, S. M., Liu, X. & Leonhardt, R. M. Repeat domain-associated O-glycans govern PMEL fibrillar sheet architecture. Sci. Rep. 9, 6101 (2019).
Leonhardt, R. M., Vigneron, N., Hee, J. S., Graham, M. & Cresswell, P. Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol. Biol. Cell. 24, 964–981 (2013).
Watt, B. et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 284, 35543–35555 (2009).
Hee, J. S., Mitchell, S. M., Liu, X. & Leonhardt, R. M. Melanosomal formation of PMEL core amyloid is driven by aromatic residues. Sci. Rep. 7, 44064 (2017).
Niyadurupola, N. & Broadway, D. C. Pigment dispersion syndrome and pigmentary glaucoma-a major review. Clin. Exp. Ophthalmol. 36, 868–882 (2008).
Tandon, A. et al. The Heritability of Pigment Dispersion Syndrome and Pigmentary Glaucoma. Am. J. Ophthalmol. 202, 55–61 (2019).
Lahola-Chomiak, A. A. et al. Non-Synonymous variants in premelanosome protein (PMEL) cause ocular pigment dispersion and pigmentary glaucoma. Hum. Mol. Genet. 28, 1298–1311 (2019).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Rochin, L. et al. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl Acad. Sci. Usa. 110, 10658–10663 (2013).
Perez-Gonzalez, R., Gauthier, S. A., Kumar, A. & Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 287, 43108–43115 (2012).
Ando, H. et al. Melanosomes are transferred from melanocytes to keratinocytes through the processes of packaging, release, uptake, and dispersion. J. Invest. Dermatol. 132, 1222–1229 (2012).
Wasmeier, C., Hume, A. N., Bolasco, G. & Seabra, M. C. Melanosomes at a glance. J. Cell. Sci. 121, 3995–3999 (2008).
Cichorek, M., Wachulska, M., Stasiewicz, A. & Tymińka, A. Skin melanocytes: biology and development. Postepy. Dermatol. Alergol. 30, 30–41 (2013).
Wetzel, R., Shivaprasad, S. & Williams, A. D. Plasticity of amyloid fibrils. Biochemistry 46, 1–10 (2007).
El, Mammeri, N., Duan, P., Dregni, A. J. & Hong, M. Amyloid fibril structures of tau: Conformational plasticity of the second microtubule-binding repeat. Sci. Adv. 9, eadh4731 (2023).
Guerrero-Ferreira, R. et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife 8, e48907 (2019).
Fernandez, A. et al. Cryo-EM structures of amyloid-beta and tau filaments in Down syndrome. Nat. Struct. Mol. Biol. 31, 903–909 (2024).
Yang, Y. et al. Cryo-EM structures of amyloid-beta 42 filaments from human brains. Science 375, 167–172 (2022).
Wang, L. Q. et al. Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion. Nat. Commun. 13, 3491 (2022).
Schmidt, M. et al. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 10, 5008 (2019).
Steinebrei, M. et al. Cryo-EM structure of an ATTRwt amyloid fibril from systemic non-hereditary transthyretin amyloidosis. Nat. Commun. 13, 6398 (2022).
Radamaker, L. et al. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 10, 1103 (2019).
Kollmer, M. et al. Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 10, 4760 (2019).
Buchanan, J. A., Varghese, N. R., Johnston, C. L. & Sunde, M. Functional Amyloids: Where Supramolecular Amyloid Assembly Controls Biological Activity or Generates New Functionality. J. Mol. Biol. 435, 167919 (2023).
Theos, A. C., Truschel, S. T., Raposo, G. & Marks, M. S. The Silver locus product Pmel17/gp100/Silv/ME20: controversial in name and in function. Pigment. Cell. Res. 18, 322–336 (2005).
Hoashi, T. et al. The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J. Biol. Chem. 281, 21198–21208 (2006).
Theos, A. C. et al. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell. 10, 343–354 (2006).
Harper, D. C. et al. Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J. Biol. Chem. 283, 2307–2322 (2008).
Zhong, H. et al. High-fidelity, efficient, and reversible labeling of endogenous proteins using CRISPR-based designer exon insertion. Elife 10, e64911 (2021).
Shadle, S. C. et al. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS. Genet. 13, e1006658 (2017).
Das, A. T., Tenenbaum, L. & Berkhout, B. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene. Ther. 16, 156–167 (2016).
Heredia, J. D., Park, J., Choi, H., Gill, K. S. & Procko, E. Conformational Engineering of HIV-1 Env Based on Mutational Tolerance in the CD4 and PG16 Bound States. J. Virol. 93, e00219 (2019).
Stewart, S. A. et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 (2003).
Kutner, R. H., Zhang, X. Y. & Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4, 495–505 (2009).
Schönfelder, J. et al. Protease resistance of ex vivo amyloid fibrils implies the proteolytic selection of disease-associated fibril morphologies. Amyloid 28, 243–251 (2021).
Burt, A. et al. An image processing pipeline for electron cryo-tomography in RELION-5. Febs. Open. Bio. 14, 1788–1804 (2024).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Elife 11, e83724 (2022).
Scheres, S. H. W. Amyloid structure determination in RELION-3.1. Acta Crystallogr. D. Struct. Biol. 76, 94–101 (2020).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Struct. Biol. 74, 519–530 (2018).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Hoq, M. R. et al. Cryo-EM structures of cotton wool plaques’ amyloid beta and of tau filaments in dominantly inherited Alzheimer disease. Acta Neuropathol. 148, 20 (2024).
Asarnow, D., Palovcak, E. & Cheng, Y. UCSF pyem v0.5. Zenodo https://doi.org/10.5281/zenodo.3576630 (2019).
Osei-Bempong, C., Ghareeb, A. E., Lako, M., Figueiredo, F. C. & Armitage, W. J. Defining the optimal cryoprotectant and concentration for cryopreservation of limbal stem cells. Cryobiology 84, 98–102 (2018).
Wagner, F. R. et al. Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nat. Protoc. 15, 2041–2070 (2020).
Yanagisawa, H., Kita, Y., Oda, T. & Kikkawa, M. Cryo-EM elucidates the uroplakin complex structure within liquid-crystalline lipids in the porcine urothelial membrane. Commun. Biol. 6, 1018 (2023).
Oda, T., Yanagisawa, H., Shinmori, H., Ogawa, Y. & Kawamura, T. Cryo-electron tomography of Birbeck granules reveals the molecular mechanism of langerin lattice formation. Elife 11, e79990 (2022).
Mastronarde, D. N. & Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol. 197, 102–113 (2017).
Zheng, S. et al. AreTomo: An integrated software package for automated marker-free, motion-corrected cryo-electron tomographic alignment and reconstruction. J. Struct. Biol. X. 6, 100068 (2022).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Chung, S., Lim, G. J. & Lee, J. Y. Quantitative analysis of melanin content in a three-dimensional melanoma cell culture. Sci. Rep. 9, 780 (2019).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
We thank Natsuko Maruyama (University of Yamanashi) and Eri Komatsu (Application Engineer, Thermo Fisher Scientific) for technical assistance. This research is partially supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from Japan Agency for Medical Research and Development (AMED) under Grant Number JP24ama121002. This work was supported by the Takeda Science Foundation (to T.O.), the Japan Society for the Promotion of Science (KAKENHI Grant numbers 21H02654, 24H02285 (to T.O.), 21H04762, 21H04762, and 21H05248 (to M.K.)), and a Research Grant from the Human Frontier Science Program (Grant number RGP006/2023 to T.O.) (https://doi.org/10.52044/HFSP.RGP0062023.pc.gr.168592).
Author information
Authors and Affiliations
Contributions
T.O. conceived and designed the experiments. H.Y. conducted cryo-EM data collection and analysis. H.A. and H. M. conducted cryo-FIB-SEM and cryo-electron tomography. H.A. and T. W. established the protocol for cryo-FIB-SEM milling operation. T.O. prepared samples and analyzed the tomography and cell biology/biochemistry data. T.O. and M.K. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Michael Marks, Ralf Leonhardt and Guillaume van Niel for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yanagisawa, H., Arai, H., Wang, T. et al. Cryo-EM of wild-type and mutant PMEL amyloid cores reveals structural mechanism of pigment dispersion syndrome. Nat Commun 16, 5411 (2025). https://doi.org/10.1038/s41467-025-61233-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61233-y
This article is cited by
-
Atomic structure and in situ visualization of native PMEL lamellae in melanosomes
Nature Communications (2025)
