
Abstract
Translesion synthesis polymerases efficiently incorporate nucleotides opposite DNA lesions. Pol ι, for example, bypasses minor-groove and exocyclic purine adducts. Conventional X-ray crystallography showed that this enzyme incorporates nucleotides by forming Hoogsteen base pairs with the incoming nucleotide rather than Watson-Crick base pairs. While this revealed the structural basis of nucleotide selection during nucleotide binding, it did not allow the visualization of the process of phosphodiester bond formation or the detection of reaction intermediates that form during nucleotide incorporation. Here, we use a combination of time-lapse crystallography and molecular dynamics simulations to examine the mechanism of pol ι-catalyzed nucleotide incorporation. We show that this enzyme maintains Hoogsteen base pairing with the incoming dNTP during the entire reaction. We also show that pol ι possesses a pyrophosphatase activity that generates two monophosphates within its active site. Our findings provide insights into the features of pol ι’s active site that allow it to translocate along DNA and catalyze processive DNA synthesis.
Similar content being viewed by others


Introduction
DNA lesions are poor templates for replicative DNA polymerases and block normal DNA replication. Translesion synthesis polymerases, by contrast, efficiently incorporate nucleotides opposite certain DNA lesions and allow cells to bypass these replication blocks1,2,3,4,5,6,7,8,9,10. In mammals, there are four Y-family translesion synthesis polymerases: pol η, pol ι, pol κ, and Rev111. Each of these enzymes has one or more cognate lesions, opposite which they incorporate nucleotides with high efficiency4,9,10. Moreover, the structural basis by which these polymerases direct the incorporation of the incoming nucleotide differs. Pol η, for example, selectively binds the dNTP that forms the usual Watson–Crick base pairs with the template nucleotide12,13,14,15. Rev1, by contrast, flips the template G residue out of the DNA double helix and selectively binds dCTP using a conserved arginine side chain in the enzyme’s active site16,17,18.
The cognate lesions of pol ι include minor-groove and exocyclic purine adducts19,20,21,22. Ground state structures of the complex of pol ι with DNA and dNTP substrates have shown that this enzyme directs the incorporation of the incoming nucleotide by rotating the template purine base around the N-glycosyl bond from the usual anti conformation to the syn conformation22,23,24. This leads to the formation of a Hoogsteen base pair between the template nucleotide and the dNTP rather than the usual Watson–Crick base pair. Like most other DNA polymerases, two metal ions are found in the active site of the substrate complex in a manner consistent with a two-metal ion-assisted mechanism of nucleotide incorporation25,26,27.
While conventional X-ray crystallography has revealed the structural basis of incoming nucleotide selection during the nucleotide-binding step, it does not allow one to visualize the process of phosphodiester bond formation or to detect the presence of any reaction intermediates that form during the nucleotide-incorporation reaction. By contrast, time-lapse X-ray crystallography, which allows one to observe the nucleotide-incorporation reaction as it occurs in the protein crystal, has provided important insights into the mechanisms of other DNA polymerases9,28,29,30,31,32,33,34,35,36,37,38. This has led to the discovery of specific active site re-arrangements in either the protein or the DNA substrate that potentially function as fidelity checkpoints in pol β, pol λ, pol μ, and Rev131,32,33,34,35. In addition, this has led to the discovery of a third metal ion associated with product formation in these polymerases27,36,37. Finally, this has also led to the discovery of a pyrophosphatase activity in Rev1 and bacterial pol IV that cleaves the pyrophosphate product to two monophosphates18,38. These examples underscore the importance of studying the structures of DNA polymerases at each step of the nucleotide-incorporation reaction.
Here we have used a combination of time-lapse crystallography and molecular dynamics simulations to examine the mechanism of pol ι-catalyzed nucleotide incorporation. We show that this enzyme maintains Hoogsteen base pairing between the template base and the incoming nucleotide during the entire nucleotide-incorporation reaction. We also show that two important intermediate states occur along this reaction pathway: one bound to a pyrophosphate product and another bound to two monophosphate products. This demonstrates that pol ι possesses a pyrophosphatase activity that converts pyrophosphate to two monophosphates. Finally, during the course of the nucleotide-incorporation reaction, the interactions between pol ι and the DNA are altered, and the newly formed Hoogsteen base pair becomes less stable. These findings provide substantial insights into how pol ι incorporates nucleotides via Hoogsteen base pairing and how it alters its interactions with the DNA in preparation for translocation.
Results
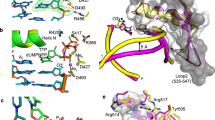
We used time-lapse X-ray crystallography to examine the mechanism of pol ι-catalyzed nucleotide incorporation (Fig. 1A). To do this, we first crystallized the substrate binary complex of pol ι and DNA with a template A nucleotide. We then crystallized the substrate ternary complex of pol ι, DNA, and an incoming dTTP in the presence of CaCl2. The Ca2+ ions allow for incoming nucleotide binding, but do not allow for catalysis. To obtain structural snapshots of pol ι in the act of catalyzing nucleotide incorporation, the crystals of the substrate ternary complex were soaked in a cryoprotectant solution containing MgCl2 for various lengths of time prior to flash freezing in liquid nitrogen. Because the Mg2+ ions rapidly replace the Ca2+ ions and allow for catalysis, these crystals contained complexes of pol ι bound to reaction products.

(A) Scheme of the time-lapse X-ray crystallography approach. (B) Overall structure of the pol ι-DNA binary substrate complex. (C) Close-up of the active site of the pol ι-DNA binary substrate complex with the template base labeled n and the primer-terminal base pair labeled n-1. (D) Close-up of the active site of the pol ι-DNA-dTTP substrate complex with the nascent base pair labeled n, the primer-terminal base pair labeled n-1, and the base on the 5′ side of the template base labeled n + 1 and with a polder map contoured at σ = 3.0 around the two active site calcium ions and the incoming dTTP (green mesh). The conclusion that the electron density shown here corresponds to calcium ions rather than water molecules or sodium ions is supported by observing an increase in R-free when modeling water molecules or sodium ions. (E) Close-up of the active site of the pol ι-DNA-dTTP substrate complex showing the polar contacts between the dTTP, the Ca2+ ions, and select amino acid residues.
Organization of the pol ι active site prior to catalysis
To understand the organization of the pol ι active site bound to the DNA substrate, we solved the structure of the substrate binary complex of pol ι and DNA in the presence of Ca2+ to a resolution of 3.2 Å (Fig. 1B, C and Table S1). The asymmetric unit contains one polymerase iota and the primer strand of the DNA substrate. The primer strand of a symmetry mate serves as the template strand (Fig. S1). The RMSD between the α-carbons of this structure and a previously determined structure of a substrate binary complex using a different DNA sequence and a primer-terminal dideoxy nucleotide is 0.69 Å23. We found that the template A nucleotide (position n) exists in two different conformations around the N-glycosyl bond (Fig. S1). The template nucleotide is in the anti conformation with an occupancy of 50% and in the syn conformation with an occupancy of 50%. The primer terminus is in a C3′ endo conformation. All of the base pairs in the duplex portion of the DNA substrate (positions n-1 to n-7) are in the standard Watson–Crick configuration. Additionally, all three of the nucleotides in the single-stranded region on the 5′ side of the template nucleotide (positions n + 1 to n + 3) are disordered in this structure, indicating that these nucleotides are more flexible in the substrate binary complex than they are in subsequent substrate and product complexes (see below).
We next determined the structure of the substrate ternary complex of pol ι, DNA, and an incoming dTTP in the presence of Ca2+ to a resolution of 2.2 Å (Fig. 1D). The RMSD between the α-carbons of this structure and a previously determined structure of a substrate complex of pol ι that was trapped using a primer-terminal dideoxy nucleotide is 0.64 Å24. We found that template A (position n) is now only in the syn configuration around the N-glycosyl bond and forms a Hoogsteen base pair with the incoming dTTP. The N7 and N6 atoms of the template A hydrogen bond to the N3 and O4 atoms of the dTTP, respectively. Unlike ternary substrate complexes of other polymerases, the primer terminus is in a C3′ endo conformation, aligning with the α-phosphate of the dTTP. The remaining base pairs in the duplex portion of the DNA substrate (positions n-1 to n-7) are in the Watson–Crick configuration. Unlike the substrate binary complex, the single-stranded region of the template strand in the substrate ternary complex is bent by 120° and the aromatic side chain of Tyr-61 stacks with the first unpaired nucleotide on the 5′ side of the template nucleotide (position n + 1) so that this nucleotide is excluded from the enzyme active site (Fig. 1D). The second and third unpaired nucleotides on the template strand (positions n + 2 and n + 3) lack density and are disordered.
Similar to other DNA polymerases, there are two metal ions in the active site of the pol ι substrate ternary complex that were not present in the binary complexes. The first is bound to the α-phosphate of the incoming dTTP, the 3′-oxygen of the primer terminus, and the side chains of Asp-34, Asp-126, and Glu-127 (metal A), and the second is bound to the α-, β-, and γ-phosphates, the side chains of Asp-34 and Asp-126, and the backbone of carbonyl of Leu-35 (metal B) (Fig. 1E). In addition, the side chain of Arg-71 interacts with the β- and γ-phosphates of the incoming dTTP. Together, the side chain of Arg-71 and these metal ions properly position the incoming dTTP for catalysis. Overall, the protein does not significantly change its conformation upon binding the incoming dTTP (Fig. S2).
Characterization of Hoogsteen-directed nucleotide incorporation by pol ι
To obtain structural snapshots of pol ι in the act of catalyzing nucleotide incorporation, the crystals of the substrate ternary complex were transferred to a cryoprotectant solution containing MgCl2 and soaked for various lengths of time prior to flash freezing in liquid nitrogen. Following a 4-min soak, we determined the structure of a complex comprised of pol ι, DNA, and pyrophosphate (PPi) to a resolution of 2.2 Å (Fig. 2A and Table S1). Overall, this structure is similar to that of the substrate ternary complex (Fig. S3). However, small changes in the positions of the 3′-carbon of the primer terminus and the 5′-carbon of the incoming nucleotide indicate that a phosphodiester bond has formed between these atoms. This is reflected in the α-phosphate of the incoming nucleotide moving 1.6 Å toward the primer terminus and the primer-terminal 3′-oxygen moving 0.9 Å toward the incoming nucleotide (Fig. 2B). The newly formed base pair between the template A and the incoming T remains in the Hoogsteen configuration. Both metal ions are retained in the active site of this complex. We do not observe density corresponding to the presence of a third metal ion in the active site of the PPi-bound structure, as was observed previously in the structures of several other polymerases27,36,37. Furthermore, the Arg-71 side chain interacts with both phosphates of the PPi product (Fig. 2A).

(A) Close-up of the active site of the pol ι complex with pyrophosphate bound and with the newly formed base pair labeled n, the primer-terminal base pair labeled n-1, and the base on the 5′ side of the template base labeled n + 1 and with a polder map contoured at σ = 3.0 around the pyrophosphate (green mesh). (B) Overlay of the DNA and dTTP substrates of the pol ι-DNA-dTTP ternary complex (white) and the DNA of the pol ι complex with pyrophosphate bound (yellow). The distances moved by the α-phosphate of the dTTP, the 3′ oxygen of the primer terminus, and the two metal ions is indicated. (C) Close-up of the active sites of the pol ι complex with two monophosphate products bound and with the newly formed base pair labeled n, the primer-terminal base pair labeled n-1, and the base on the 5′ side of the template base labeled n + 1 and with a polder map contoured at σ = 3.0 around the two monophosphates (green mesh). The conclusion that the electron density shown here corresponds to monophosphates rather than water molecules is supported by observing an increase in R-free when modeling water molecules. (D) Graph showing the production of monophosphate by pol ι in the presence of DNA and dTTP. Data are presented as mean values ± SEM. Four technical replicates of each experimental condition were carried out. Source data are provided as a Source Data file. The p values were derived from one-way ANOVA using GraphPad Prism, and **** indicates p < 0.0001.
Following a 6-min soak, we determined the structure of a complex comprised of pol ι, DNA, and two inorganic monophosphate (Pi) products to a resolution of 2.5 Å (Fig. 2C and Table S1). Again, the structure of this complex is similar to that of the substrate ternary complex. The newly formed base pair remains in the Hoogsteen configuration, and both metal ions occupy approximately the same positions as they do in the ternary substrate complex and in the PPi-bound complex. In addition, we do not observe the presence of a third metal ion within the active site of this intermediate structure. The Pi derived from the γ-phosphate of the incoming dTTP has an occupancy of 100% with B-factors of 56, and the Pi derived from the β-phosphate has an occupancy of 50% with B-factors of 50, indicating that this latter Pi dissociates prior to the former one. This is likely because in this structure, the Arg-71 side chain now interacts only with the Pi derived from the γ-phosphate of the incoming dTTP.
The presence of Pi molecules in an intermediate complex shows that pol ι can hydrolyze the PPi product into two Pi molecules in the active site of the enzyme. In this respect, pol ι resembles the Y-family polymerases Rev1 and pol IV in possessing a pyrophosphatase activity18,38. This conclusion is further supported by the observation that the distance between the phosphorus atoms in the two phosphate molecules is 3.8 Å, which is greater than the distance between these two atoms in the pyrophosphate molecule, which is 3.0 Å. To demonstrate that pol ι possesses a pyrophosphatase reaction in solution, we monitored the formation of Pi during nucleotide incorporation as done previously with other polymerases18,38 (Fig. 2D). When pol ι (1 μM) was incubated with DNA (15 μM) and dTTP (100 μM), we observed the production of 27 ± 2 μM Pi. Significantly less Pi was observed in the absence of pol ι (9 ± 1 μM), the absence of DNA (18 ± 1 μM), or the absence of dTTP (1.9 ± 0.5 μM). Approximately half of the Pi produced in this assay by pol ι is DNA-dependent, while about half is DNA-independent. This shows that although pol ι has a less specific pyrophosphatase activity than does Rev1, pol ι does indeed produce Pi during nucleotide incorporation in solution.
Organization of the pol ι active site after catalysis
Following a 20-min soak, we determined the structure of the product binary complex of pol ι and DNA to a resolution of 2.2 Å (Fig. 3A and Table S1). In this complex, the newly incorporated T remains in a Hoogsteen base pair with the template A within the nascent base pair binding pocket. There are no metal ions present in the active site of the product complex. Aside from the formation of a phosphodiester bond between the newly incorporated T (position n) and the adjacent C in the primer strand (position n-1), the overall structure of the protein and DNA has not changed significantly between the substrate and product complexes (Fig. S4).

(A) Close-up of the active site of the pol ι binary product complex formed in crystallo with the newly formed base pair labeled n, the primer-terminal base pair labeled n-1, and the base on the 5′ side of the template base labeled n + 1. (B) Close-up of the active site of the co-crystallized pre-translocation pol ι binary product complex with the newly formed base pair labeled n in the nascent base pair binding pocket and the primer-terminal base pair labeled n-1 in the primer-terminal base pair binding pocket. (C) Close-up of the active site of the co-crystallized post-translocation pol ι binary product complex with the newly formed base pair labeled n in the primer-terminal base pair binding pocket, and the new template base labeled n + 1 is in the nascent base pair binding pocket.
Pol ι is capable of processive nucleotide incorporation, meaning that it can translocate one nucleotide along the DNA ligand following a nucleotide-incorporation event. We did not observe the translocation of pol ι along the DNA in the crystal. Thus, we attempted to determine the structure of the pol ι product complex after the enzyme has translocated by co-crystalizing pol ι with a DNA ligand that is identical to the DNA product. Surprisingly, this structure, which was determined to a resolution of 2.4 Å, was of the pre-translocation product state rather than the expected post-translocation state (Fig. 3B and Table S1). The A-T base pair, which would be equivalent to the newly formed base pair in the prior structure, was located in the nascent base pair binding site rather than the primer-terminal base pair binding pocket (Fig. 3B). This suggests that the register of the DNA substrate within the enzyme active site is determined by the crystal lattice. Importantly, this also shows that constraints imposed by the nascent base pair binding pocket in the active site cause this A-T base pair to adopt a Hoogsteen configuration.
The structure of this co-crystalized pre-translocation product complex is very similar to the structure of the time-lapse product complex formed catalytically in crystallo with an RMSD of alpha-carbons equal to 0.53 Å (Fig. S4). The primary difference between these two structures is the dynamics of Tyr-61 and the single-stranded region of the template strand (positions n + 1, n + 2, and n + 3). In the time-lapse product complex, the side chain of Tyr-61 is positioned between the template nucleotide in the newly formed base pair (position n) and the next available unpaired nucleotide in the template strand (position n + 1). The other unpaired nucleotides on the template strand (positions n + 2 and n + 3) are disordered. By contrast, in the co-crystallized pre-translocation product complex, the Tyr-61 side chain and all three unpaired nucleotides on the template strand (positions n + 1 to n + 3) are disordered. This suggests that the unpaired template nucleotide (position n + 1) is no longer bound to the protein in this product structure. Because the transient release of the single-stranded region of the DNA product is necessary for translocation, this structure most likely reflects an intermediate of the translocation process.
After obtaining the structure of the co-crystallized pre-translocation product complex and recognizing that the register of the DNA is dictated by the crystal lattice, we attempted again to obtain a structure of the co-crystallized post-translocation product complex. To do this, we reduced the length of the duplex region of the DNA product by two base pairs so that the crystal lattice would promote a register of the DNA product more appropriate for the post-translocation complex. We successfully obtained a co-crystallized structure of the post-translocation complex at a resolution of 2.3 Å (Fig. 3C). In this structure, the newly formed base pair (position n) is in the primer-terminal base pair binding pocket and the next unpaired nucleotide (position n + 1) is now in the templating position in the nascent base pair binding pocket. Similar to the co-crystallized pre-translocated structure, the additional two unpaired nucleotides on the template strand (positions n + 2 to n + 3) and the side chain of Tyr-61 are disordered. As observed with the initial binary substrate complex, the new template residue (position n + 1) is in the anti conformation with an occupancy of 15% and in the syn conformation with an occupancy of 85%. The newly formed base pair (position n) is in the Watson–Crick configuration, showing that the movement of this base pair from the nascent base pair binding pocket to the primer-terminal base pair binding pocket is accompanied by a shift from a Hoogsteen configuration to a Watson–Crick configuration.
Role of active site residues in pol ι-catalyzed nucleotide incorporation
We have identified two active site residues that may play an important role in facilitating nucleotide incorporation. First, the side chain of Tyr-61 stacks with the nucleotide in the n + 1 position, helping to exclude it from the active site in the substrate ternary complex. Second, the side chain of Arg-71 interacts with the triphosphate moiety of the incoming dTTP as well as the PPi and Pi products in the substrate ternary complex and intermediate complexes. To gain insights into the roles of these active site residues in catalysis, we examined the nucleotide-incorporation activity of the Y61A and R71A mutant proteins (Fig. 4A). We incubated wild-type or mutant pol ι protein (50 nM) with DNA (100 nM) and dTTP (10 μM), and we found that the Y61A mutant protein has nucleotide incorporation activity similar to that of the wild-type protein. By contrast, the R71A mutant protein has no detectable nucleotide incorporation activity.

(A) Nucleotide-incorporation activity of the wild-type, mutant Y61A, and mutant R71A pol ι proteins. These experiments were repeated four times to ensure reproducibility. The uncropped gel images are provided as a Source Data file. (B) Close-up of the active site of the mutant R71A substrate complex with the newly formed base pair labeled n, the primer-terminal base pair labeled n-1, and the base on the 5′ side of the template base labeled n + 1 and with a polder map contoured at σ = 3.0 showing the weak electron density corresponding to the incoming dTTP (green mesh).
While we were unable to obtain crystals of the Y61A mutant protein that diffracted to a sufficient resolution to obtain a structure, we determined the structure of the R71A mutant protein bound to DNA in the presence of dTTP and Ca2+ to a resolution of 2.5 Å (Fig. 4B). Overall, aside from the unpaired nucleotide on the 5′ side of the template nucleotide (position n + 1), which is in a different position from where it is located in the wild-type ternary substrate complex, the structures of the wild-type and R71A mutant substrate ternary complexes are similar (Fig. S5). Like the binary substrate complex, template A is in the anti conformation with an occupancy of 50% and in the syn conformation with an occupancy of 50%. However, unlike the wild-type substrate ternary complex, we only observe weak density corresponding to the incoming dTTP and have therefore modeled the incoming dTTP with only 40% occupancy. When the dTTP is present, it forms Hoogsteen base pairs with the template A in the syn conformation. This shows that Arg-71 plays an important role in both binding the incoming dTTP and positioning it for metal ion binding and catalysis.
Stability of the Hoogsteen base pair during nucleotide incorporation
The nascent/newly formed base pair was in the Hoogsteen configuration in the X-ray crystal structures of the substrate ternary complex and the various product complexes. In order to understand the relative stability of the Hoogsteen base pair in each of these structures during the nucleotide-incorporation reaction, we carried out MD simulations in triplicate of (i) the substrate ternary complex, (ii) the complex containing PPi product, (iii) the complex containing two Pi products, (iv) the complex containing one Pi product corresponding to the γ-phosphate of the incoming dTTP, and (v) the product binary complex. We measured the glycosidic angle, the Hoogsteen hydrogen bond distances (N7-N3 and N6-O4), the Watson–Crick hydrogen bond distances (N1-N3 and N6-O4), and the C1′-C1′ distance of the nascent/newly formed base pair at position n after every ns of simulation time. As described in the Methods section, we used these values to determine whether the base pair was a Hoogsteen pair, was a Watson–Crick pair, or was fraying.
For the substrate ternary complex, the nascent base pair remained in the Hoogsteen configuration for the entire 1-μs duration in two of the three simulations and for 600 ns in the third simulation before fraying (Fig. 5A, B and Figs. S6–S9). For the complex containing PPi, the newly formed base pair remained in the Hoogsteen configuration for the entire duration in one of the three simulations and for less than 200 ns in the other two simulations before fraying and subsequently forming Watson–Crick base pairs (Fig. 5C, D and Figs. S10–S13). For the remaining complexes (the complex containing two Pi molecules, the complex containing one Pi, and the product binary complex) the newly formed base paired remained in the Hoogsteen configuration for less than 200 ns—often less than 20 ns—in all of the simulations before either fraying or forming Watson–Crick base pairs (Fig. 5E–J and Figs. S14–S25). Overall, these simulations showed that the Hoogsteen base pair at position n is more stable in the substrate ternary complex than it is in any of the product complexes.

(A) Close-up of the active site of the pol ι-DNA-dTTP substrate complex with an overlay of 20 structures of the nascent base pair (shown as sticks) obtained after every 50 ns of simulation time for the first replicate. (B) Graph showing the glycosidic angle of the template nucleotide as a function of time. The shaded region (0 to 90°) represents the typical range for a Hoogsteen base pair. (C, D) Same as above for the pol ι complex bound to the pyrophosphate product. (E, F) Same as above for the pol ι complex bound to two monophosphate products. (G, H) Same as above for the pol ι complex bound to one monophosphate product. (I, J) Same as above for the pol ι binary product complex.
Discussion
One of the most striking features about the mechanism of pol ι is that it incorporates nucleotides using Hoogsteen base pairing between the template purine nucleotide and the incoming dNTP rather than Watson–Crick base pairing22,23,24. When pol ι binds DNA in the absence of an incoming dNTP, the template nucleotide (position n) can adopt either the anti or syn configuration, and the structure of the binary substrate complex (pol ι - DNA) shows the template nucleotide occupying both configurations. When the incoming dNTP binds, steric constraints within the active site imposed by residues from the fingers subdomain (specifically Gln-59, Lys-60, and Tyr-61) prevent the nascent base pair from forming a Watson–Crick base pair. Instead, the template nucleotide rotates from the anti to the syn configuration and forms a Hoogsteen base pair with the incoming dNTP. Using time-lapse crystallography, we showed here that this Hoogsteen base pair persists through the entire nucleotide-incorporation reaction in the crystal. Moreover, when we determined the structure of the co-crystalized pre-translocation product complex, the new base pair in the nascent base pair binding pocket formed a Hoogsteen base pair. In solution, these nucleotides would presumably be in a Watson–Crick base pair. Thus, in the crystal, the active site of pol ι can convert an already formed Watson–Crick base pair to a Hoogsteen base pair by placing it within the nascent base pair binding pocket.
Time-lapse crystallography with other DNA polymerases has shown that some of these enzymes possess a third metal ion (metal C) that is associated with product formation25,27,36. These include X-family polymerases pol β, pol λ, and pol μ, as well as Y-family polymerase pol η. This third metal ion contacts the former α-phosphate of the incoming dNTP, which has become the phosphate group between the newly incorporated nucleotide (position n) and the nucleotide on its 5′ side (position n-1). This metal also contacts the β-phosphate of the former incoming dTTP, which has become a phosphate group in the pyrophosphate product. It is difficult to determine whether this third metal ion binds immediately prior to phosphodiester bond formation and facilitates catalysis or whether it binds immediately after catalysis28. In the case of pol ι, we observe metal A and metal B in the substrate and product complexes with pyrophosphate and with two monophosphates. We do not observe electron density in any of these complexes corresponding to a third metal ion in the position that metal C occupies in other polymerases. In this respect, pol ι resembles another Y-family polymerase, Rev1, for which there is no structural evidence of a third metal ion appearing at this position during the nucleotide-incorporation reaction18.
As we have reported here, pol ι also resembles Rev1 in that both enzymes possess a pyrophosphatase activity18. Although the pol ι pyrophosphatase activity is somewhat less specific than is Rev1’s activity, both of these polymerases are capable of hydrolyzing the pyrophosphate product to two monophosphate molecules. This thermodynamically favors the forward reaction by removing the pyrophosphate product, a task that is often carried out by a separate pyrophosphatase enzyme during normal DNA replication. It should be noted that in the case of Rev1, no pyrophosphate product has been observed, which raised the possibility that instead of Rev1 possessing a genuine pyrophosphatase activity, it could remove the two monophosphates sequentially. For example, it is possible that the γ-phosphate is removed prior to phosphodiester bond formation, forming a dNDP intermediate, and that the β-phosphate is subsequently removed during phosphodiester bond formation. The observation of a pyrophosphate intermediate in pol ι rules out such a mechanism and demonstrates a genuine pyrophosphatase activity. It is also worth noting that Rev1 and pol ι are the only known eukaryotic polymerases that both lack a third metal ion and possess pyrophosphatase activity. It may be the case that the presence of a third metal ion and the possession of pyrophosphatase activity are mutually exclusive.
Given a series of template purines, pol ι is capable of processive DNA synthesis21,39,40. Thus, it can translocate along the DNA substrate one step following each nucleotide incorporation event. Translocation shifts the newly formed base pair from the nascent base pair binding pocket to the primer-terminal base pair binding pocket. Because the base pair in the former binding pocket is a Hoogsteen base pair and the base pair in the latter binding pocket is a Watson–Crick base pair, translocation requires the conversion of the newly formed Hoogsteen base pair into a Watson–Crick base pair. The translocation process remains poorly understood for all polymerases, and it is particularly complicated for pol ι, given the additional need to convert the template nucleotide from the syn to anti configuration. We attempted to obtain a post-translocation structure of pol ι by co-crystallizing it with a product DNA ligand. We expected to see the base pair at position n in the primer-terminal base pair binding pocket, but instead, we obtained a pre-translocation product structure with this base pair in the nascent base pair binding pocket. This is likely due to crystal contacts preventing pol ι translocation in the crystal.
Interestingly, this co-crystallized binary product structure has several differences with the time-lapse binary product structure formed by the reaction in crystallo. For example, both the Tyr-61 side chain and the next available template nucleotide (position n + 1) are disordered in the co-crystallized pre-translocation product complex but are ordered in the time-lapse product complex. Based on this, we suggest that the structure of the co-crystallized binary product complex is an intermediate in the translocation process immediately prior to moving one step along the DNA. In this intermediate, the enzyme has lost contact with the next available template nucleotide (position n + 1), and the Tyr-61 side chain has moved away from the active site. Together, these changes could assist in moving this nucleotide into the nascent base pair binding pocket as the polymerase moves ahead one step along the DNA.
We were ultimately able to obtain a structure of the co-crystallized post-translocation product complex by shortening the length of the DNA product, thereby allowing the crystal contacts to position the DNA in a post-translocation register. The newly formed base pair (position n) moved from the nascent base pair binding pocket to the primer-terminal base pair binding pocket. Similarly, the next unpaired template residue (position n + 1) moved into the nascent base pair binding pocket. These co-crystallized pre-translocation and post-translocation product structures showed that the movement of the newly formed base pair (position n) indeed shifts from a Hoogsteen to a Watson–Crick configuration upon translocation.
Because the translocation process requires the shift of the newly formed base pair (position n) from a Hoogsteen pair to a Watson–Crick pair, we examined the relative stability of the Hoogsteen pair at this position at each step along the reaction pathway without any constraints imposed by crystal packing. To do this, we carried out a series of MD simulations with five models derived from the X-ray crystal structures determined here. We found that the Hoogsteen base pair is relatively more stable in the substrate ternary complex than it is in the other complexes. It becomes less stable upon phosphodiester bond formation and even less stable upon the cleavage or release of the pyrophosphate product. We suggest that the cleavage or release of the pyrophosphate impacts the stability of the newly formed base pair in the Hoogsteen configuration, because the pyrophosphate product interacts with the fingers subdomain via Arg-71. Because the fingers subdomain constrains the active site to stabilize the Hoogsteen configuration, disrupting this interaction could release steric constraints enough to allow the newly formed base pair to fray. Further support for this notion comes from the MD simulations, which show that the fingers subdomain is quite mobile and can occupy a range of different positions relative to the DNA substrate and remainder of the protein (Fig. S26). For example, in both the substrate ternary complex and the complex containing pyrophosphate, the α-carbons in the fingers subdomain move as much as 21 Å during the course of the simulation. After the cleavage of the pyrophosphate and the release of one or both of the phosphates, the fingers become more mobile with the α-carbons moving as much as 32 Å during the simulation. This additional flexibility causes the fingers subdomain to move away from the newly formed base pair, which would allow this base pair to fray more readily. Ultimately, the fraying of the newly formed base pair within the nascent base pair binding pocket, combined with the alterations in the pol ι-DNA interactions discussed above, would facilitate the translocation of pol ι one nucleotide along the DNA. This would place the newly formed base pair in the primer-terminal base pair binding pocket and flip the next available nucleotide (position n + 1) into the nascent base pair binding pocket to allow for another cycle of nucleotide incorporation.
Methods
Protein overexpression and purification
The catalytic core of pol ι (residues 1–418) was over-expressed in BL21 (DE3) cells harboring plasmid pKW800, which was derived from pET11a. This plasmid produced pol ι as an N-terminally labeled glutathione-S-transferase (GST) fusion protein with a PreScission cleavage site. Cells were grown in LB media to an OD600 of 0.6 before induction with 0.1 mM IPTG and growth at 18 °C overnight. Cells were lysed using an EmulsiFlex (Avestin) in the presence of 1 mM PMSF, Complete EDTA-free Protease Inhibitor Cocktail (Roche), and DNase. After centrifuging at 10,000×g for an hour, the clarified crude extract was incubated with glutathione beads (Cytiva) for 2 h. The beads were washed with 25 column volumes of buffer containing 50 mM Tris pH 7.4, 300 mM NaCl, 5 mM DTT, 5 mM EDTA, and 10% glycerol. The beads were resuspended in the same buffer with PreScission protease (Cytiva) at 4 °C overnight. The cleaved pol ι protein was extracted from the beads with 5 column volumes of buffer. The protein was then loaded on an SP Sepharose column (GE Healthcare) and eluted using a salt gradient of 50 mM to 2 M NaCl. The eluted protein was further purified using a HiLoad Superdex 200 size-exclusion column (GE Healthcare) in buffer containing 50 mM Tris pH 7.4, 250 mM NaCl, 5 mM DTT, 5 mM EDTA, and 10% glycerol before concentrating. The Y61A and R71A mutant pol ι proteins were produced in BL21 (DE3) cells harboring plasmids pKW801 and pKW802, respectively, and were purified using the same approach.
DNA substrates
All DNA oligodeoxynucleotides were ordered from IDT. For time-lapse x-ray crystallography, a single oligodeoxynucleotide (5′-TCAAGGGTCCTAGGACCC) was self-annealed by heating to 95 °C and slowly cooling over several hours to 4 °C in 10 mM TrisCl pH 8.0 and 50 mM NaCl. For the co-crystallization pre-translocation complex with product DNA, the same procedure was used with a longer oligodeoxynucleotide (5′-TCAAGGGTCCTAGGACCCT). For the co-crystallization post-translocation complex with product DNA, the same procedure was used with a shorter oligodeoxynucleotide (5′-TCAAGGGTCCGGACCCT). For nucleotide-incorporation assays, the primer strand (5′-GCCTCGCCTAGGACCC), which was 5′-32P-end-labeled with polynucleotide kinase and α-32P-ATP, was annealed to the template strand (5′- TCGAGGGTCCTAGGCGAGGC) as described above. For phosphate-production assays, the primer strand (5′-CTGCAGCTGATGCAGCGTCAT) was annealed to the template strand (5′-CATAAAATGACGCTGCATCAGCTGCAG) as described above.
Crystallization
Pol ι (20 mg/ml), the DNA substrate (500 μM), and dTTP (1 mM, New England Biolabs, catalog number N0446S) were co-crystallized in the presence of 5 mM CaCl2. The best diffracting crystals were obtained using the hanging drop method with a mother liquor containing 100 mM HEPES pH 7, 5 mM EDTA, and 1.5 to 1.8 M ammonium sulfate. To obtain time-lapse structures, crystals were soaked in a solution containing 50 mM MgCl2 and 10% glycerol for various time intervals before flash freezing in liquid nitrogen. Crystals were subsequently used for data collection at 100 K at the 4.2.2 synchrotron beamline at the Advanced Light Source in the Lawrence Berkeley National Laboratory. Crystals for the R71A mutant protein were used for data collection at 100 K on a Rigaku MicroMax-007 HF rotating anode diffractometer equipped with a Dectris Pilatus3R 200 K with a wavelength of 1.54 Å. Crystals for the co-crystalized post-translocation product complex were used for data collection at the CHESS Beamline 7B2.
Structural determination
Initial models were generated in Phenix by molecular replacement using a previously solved pol ι substrate complex (PDB: 3H4B) as a reference structure23. Subsequent refinement was carried out using Phenix, and model building was carried out using WinCoot41,42. Structures were visualized and analyzed using PyMol (Schrödinger).
Polymerase activity assay
The DNA primer strand was 32P 5′-end labeled using polynucleotide kinase and 32P-γ-ATP (PerkinElmer), and the labeled primer and unlabeled template strands were annealed by heating to 95 °C and slowly cooling over several hours to room temperature19,43,44. Experiments were performed with 50 nM pol ι, 100 nM DNA, and 10 µM dTTP in 50 mM TrisCl pH 7.5, 5 mM MgCl2, and 10% glycerol. Reactions were quenched after 0 to 10 min in 90% formamide and loaded onto a 15% polyacrylamide gel with 8 M urea. Gels were dried and then imaged with a Typhoon 7000 PhosphorImager (Molecular Dynamics).
Phosphate-production assay
Experiments were performed in quadruplicate with 1 µM pol ι, 15 µM DNA, and 100 µM dTTP in 50 mM TrisCl pH 7.5, 300 mM NaCl, 5 mM MgCl2, 5 mM EDTA, and 10% glycerol at 25 °C. After an hour of reaction time, reactions were quenched with 100 mM EDTA. Control experiments were performed lacking pol ι, lacking DNA, lacking dTTP, and every combination of these. To determine the concentration of Pi produced, the quenched reactions were incubated for 30 min with Malachite green (Sigma Aldrich, catalog number MAK307). The absorbance was measured at 620 nM, and the concentration of Pi was determined using a standard curve generated with purified Pi. The baseline concentration of Pi determined from the buffer-only reactions was subtracted from all experiments and controls. The DNA substrate was a 21-mer primer strand (5′-CTGCAGCTGATGCAGCGTCAT) annealed to a 27-mer template strand (5′-CATAAAATGACGCTGCATCAGCTGCAG). No data were excluded from these experiments, and no randomization or blinding were done.
Molecular dynamics simulations
MD simulations were performed with explicit water solvent for five pol ι-DNA complexes: the substrate complex bound to dTTP, the complex bound to pyrophosphate, the complex bound to two monophosphates, the complex bound to one monophosphate, and the binary product complex. Starting models were prepared using PyMol and WinCoot. Input files were generated using CHARMM-GUI for the GROMACS software suite using the CHARMM36m forcefield with TIP3P water45,46,47,48,49,50,51,52,53,54,55,56,57. MD simulations were run on the University of Iowa’s Argon High-Performance Computing system.
Starting models were derived from the X-ray crystal structures determined here as follows. First, any disordered nucleotides (n + 1 to n + 3) from the single-stranded region of the template strand were built, and the double-stranded region of the DNA was truncated to only include DNA bound to the polymerase (n-1 to n-7). Second, disordered regions of pol ι were built as loops using WinCoot. Hydrogen atoms were added, the system was neutralized by adding K+ and Cl- ions, and the resulting system was solvated in a rectangular water box with a 10 Å distance between the system and the edge of the box. The prepared system was then minimized for 5 ps and equilibrated for 125 ps under NVT conditions. The equilibrated systems were then used to run a 1-μs simulation under constant NPT conditions. Each simulation contained ~100,000 atoms, was run in triplicate with pseudo-randomly generated seeds. The exact commands used to run the simulations and the parameter files (.mdp) used are included in the Supplemental Methods section.
The simulations were analyzed using VMD and the GROMACS distance and angle commands49,58. After each ns of simulation time, we determined whether the base pair at position n was a Hoogsteen pair, was a Watson–Crick pair, or was fraying. It was a Hoogsteen pair if the following four criteria were satisfied: (i) the glycosidic angle was between 0 and 90°, (ii) the N7-N3 and N6-O4 distances were between 2 and 4 Å, (iii) the N1-N3 distance was greater than 4 Å, and (iv) the C1′-C1′ distance was between 8 and 10 Å. It was a Watson–Crick pair if the following four criteria were satisfied: (i) the glycosidic angle was between −90 and −180°, (ii) the N1-N3 and N6-O4 distances were between 2 and 4 Å, (iii) the N7-N3 distance was greater than 4 Å, and (iv) the C1′-C1′ distance was between 10 and 12 Å. It was fraying in all other cases.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The structure factors and atomic coordinates for all of the X-ray crystal structures are available in the Protein Data Base under the following accession numbers: 9DQT (binary substrate complex), 9DDR (ternary substrate complex), 9DQU (pyrophosphate product complex), 9DR7 (phosphate product complex), 9DR9 (binary product complex), 9DRB (co-crystallized pre-translocation product complex), 9DRC (R71A mutant ternary substrate complex), and 9NJH (co-crystallized post-translocation product complex). The previously published reference structure used for molecular replacement is available in the Protein Data Base under the following accession number: 3H4B. Source data are provided with this paper.
References
Friedberg, E. C., Wagner, R. & Radman, M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science 296, 1627–1630 (2002).
Prakash, S. & Prakash, L. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 16, 1872–1883 (2002).
Lehmann, A. R. Replication of damaged DNA. Cell Cycle 2, 299–301 (2003).
Waters, L. S. et al. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154 (2009).
Washington, M. T., Carlson, K. D., Freudenthal, B. D. & Pryor, J. M. Variations on a theme: eukaryotic Y-family DNA polymerases. Biochim. Biophys. Acta1804, 1113–1123 (2010).
Pryor, J. M., Dieckman, L. M., Boehm, E. M. & Washington, M. T. in Nucleic Acid Polymerases (eds Murakami, K. S. & Trakselis, M. A.) Ch. 4 (Springer, 2014).
Powers, K. T. & Washington, M. T. Eukaryotic translesion synthesis: choosing the right tool for the job. DNA Repair 71, 127–134 (2018).
Marians, K. J. Lesion bypass and the reactivation of stalled replication forks. Annu. Rev. Biochem. 87, 217–238 (2018).
Ling, J. A., Frevert, Z. & Washington, M. T. Recent advances in understanding the structures of translesion synthesis DNA polymerases. Genes 13, 915 (2022).
Prakash, S., Johnson, R. E. & Prakash, L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74, 317–353 (2005).
Burgers, P. M. J. et al. Eukaryotic DNA polymerases: proposal for a revised nomenclature. J. Biol. Chem. 276, 43487–43490 (2001).
Carlson, K. D. & Washington, M. T. Mechanism of efficient and accurate nucleotide incorporation opposite 7,8-dihydro-8-oxoguanine by Saccharomyces Cerevisiae DNA polymerase η. Mol. Cell. Biol. 25, 2169–2176 (2005).
Haracska, L., Yu, S. L., Johnson, R. E., Prakash, L. & Prakash, S. Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase η. Nat. Genet. 25, 458–461 (2000).
Johnson, R. E., Prakash, S. & Prakash, L. Efficient Bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 283, 1001–1004 (1999).
Washington, M. T., Johnson, R. E., Prakash, S. & Prakash, L. Accuracy of thymine–thymine dimer bypass by Saccharomyces Cerevisiae DNA polymerase η. Proc. Natl Acad. Sci. USA 97, 3094–3099 (2000).
Pryor, J. M. & Washington, M. T. Pre-steady state kinetic studies show that an abasic site is a cognate lesion for the yeast Rev1 protein. DNA Repair 10, 1138–1144 (2011).
Haracska, L. et al. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 15, 945–954 (2001).
Weaver, T. M. et al. Visualizing Rewv1 catalyze protein-template DNA synthesis. Proc. Natl Acad. Sci. USA 117, 25494–25504 (2020).
Washington, M. T. et al. Efficient and error-free replication past a minor-groove DNA adduct by the sequential action of human DNA polymerases ι and κ. Mol. Cell. Biol. 24, 5687–5693 (2004).
Pence, M. G. et al. Lesion bypass of N 2-ethylguanine by human DNA polymerase i. J. Biol. Chem. 284, 1732–1740 (2009).
Johnson, R. E., Washington, M. T., Haracska, L., Prakash, S. & Prakash, L. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406, 1015–1019 (2000).
Nair, D. T., Johnson, R. E., Prakash, S., Prakash, L. & Aggarwal, A. K. Replication by human DNA polymerase-ι occurs by Hoogsteen base-pairing. Nature 430, 377–380 (2004).
Jain, R. et al. Replication across template T/U by human DNA polymerase-ι. Structure 17, 974–980 (2009).
Nair, D. T., Johnson, R. E., Prakash, L., Prakash, S. & Aggarwal, A. K. An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase-ι active site. Structure 14, 749–755 (2006).
Perera, L., Freudenthal, B. D., Beard, W. A., Pedersen, L. G. & Wilson, S. H. Revealing the role of the product metal in DNA polymerase β catalysis. Nucleic Acids Res. 45, 2736–2745 (2017).
Beese, L. S. & Steitz, T. A. Structural basis for the 3′-5′ exonuclease activity of Escherichia Coli DNA polymerase I: a two metal ion mechanism. EMBO J. 10, 25–33 (1991).
Wang, J. & Konigsberg, W. H. Two-metal-ion catalysis: inhibition of DNA polymerase activity by a third divalent metal ion. Front. Mol. Biosci. 9, 824794 (2022).
Weaver, T. M., Washington, M. T. & Freudenthal, B. D. New insights into DNA polymerase mechanisms provided by time-lapse crystallography. Curr. Opin. Struct. Biol. 77, 102465 (2022).
Nakamura, T., Zhao, Y., Yamagata, Y., Hua, Y. J. & Yang, W. Watching DNA polymerase η make a phosphodiester bond. Nature 487, 196–201 (2012).
Freudenthal, B. D., Beard, W. A. & Wilson, S. H. Watching a DNA polymerase in action. Cell Cycle 13, 691–692 (2014).
Freudenthal, B. D., Beard, W. A., Shock, D. D. & Wilson, S. H. Observing a DNA polymerase choose right from wrong. Cell 154, 157 (2013).
Jamsen, J. A., Shock, D. D. & Wilson, S. H. Watching right and wrong nucleotide insertion captures hidden polymerase fidelity checkpoints. Nat. Commun. 13, 3193 (2022).
Weaver, T. M. et al. Mechanism of nucleotide discrimination by the translesion synthesis polymerase Rev1. Nat. Commun. 13, 2876 (2022).
Chang, C., Lee Luo, C. & Gao, Y. In crystallo observation of three metal ion promoted DNA polymerase misincorporation. Nat. Commun. 13, 2346 (2022).
Jamsen, J. A., Sassa, A., Shock, D. D., Beard, W. A. & Wilson, S. H. Watching a double strand break repair polymerase insert a pro-mutagenic oxidized nucleotide. Nat. Commun. 12, 2059 (2021).
Gao, Y. & Yang, W. Capture of a third Mg2+ Is essential for catalyzing DNA synthesis. Science 352, 1334–1337 (2016).
Stevens, D. R. & Hammes-Schiffer, S. Exploring the role of the third active site metal ion in DNA polymerase η with QM/MM free energy simulations. J. Am. Chem. Soc. 140, 8965–8969 (2018).
Kottur, J. & Nair, D. T. Pyrophosphate hydrolysis is an intrinsic and critical step of the DNA synthesis reaction. Nucleic Acids Res. 46, 5875–5885 (2018).
Makarova, A. V. et al. Inaccurate DNA synthesis in cell extracts of yeast producing active human DNA polymerase iota. PLoS ONE 6, e16612 (2011).
McIntyre, J. Polymerase iota - an odd sibling among Y family polymerases. DNA Repair 86, 102753 (2020).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in phenix. Acta Crystallogr. Sect. Struct. Biol. 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Washington, M. T., Johnson, R. E., Prakash, L. & Prakash, S. The mechanism of nucleotide incorporation by human DNA polymerase η differs from that of the yeast enzyme. Mol. Cell. Biol. 23, 8316–8322 (2003).
Washington, M. T., Johnson, R. E., Prakash, S. & Prakash, L. Fidelity and processivity of Saccharomyces Cerevisiae DNA polymerase eta. J. Biol. Chem. 274, 36835–36838 (1999).
Brooks, B. R. et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Bauer, P., Hess, B. & Lindahl, E. GROMACS 2022.3 source code. Zenodo https://doi.org/10.5281/zenodo.7037338 (2022).
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Lindahl, E., Hess, B. & van der Spoel, D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol. Model. Annu. 7, 306–317 (2001).
Páll, S., Abraham, M. J., Kutzner, C., Hess, B. & Lindahl, E. in Solving Software Challenges for Exascale (eds Markidis, S. & Laure, E.) Springer International Publishing, 2015).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
This work was supported by award R35 GM148186 to M.T.W. and by award R35-GM128562 to B.D.F. from the Nation Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health. We thank Lynne Dieckman, Christine Kondratick, Justin Ling, and Tyler Woodward for discussions. We thank Michael Schnieders and Adrian Elcock for their assistance in setting up the simulations.
Author information
Authors and Affiliations
Contributions
Z.F.: Conceptualization, methodology, validation, formal analysis, investigation, and writing. D.T.R. and M.S.G.: Conceptualization and methodology. S.M.J. and B.J.R.: Methodology. B.D.F.: Conceptualization, writing, and funding acquisition. M.T.W.: Conceptualization, writing, supervision, project administration, and funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Frevert, Z., Reusch, D.T., Gildenberg, M.S. et al. Visualizing DNA polymerase ι catalyze Hoogsteen-directed DNA synthesis. Nat Commun 16, 5979 (2025). https://doi.org/10.1038/s41467-025-61245-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61245-8
