
Abstract
Human immunoglobulin G (IgG) antibodies are a major class of biotherapeutics and undergo N-linked glycosylation in their Fc domain, which is critical for immune functions and therapeutic activity. Hence, technologies for producing authentically glycosylated IgGs are in high demand. Previous attempts to engineer Escherichia coli for this purpose have met limited success due in part to the lack of oligosaccharyltransferase (OST) enzymes that can install N-glycans at the conserved N297 site in the Fc region. Here, we identify a single-subunit OST from Desulfovibrio marinus with relaxed substrate specificity that catalyzes glycosylation of native Fc acceptor sites. By chemoenzymatic remodeling the attached bacterial glycans to homogeneous, asialo complex-type G2 N-glycans, the E. coli-derived Fc binds human FcγRIIIa/CD16a, a key receptor for antibody-dependent cellular cytotoxicity (ADCC). Overall, the discovery of D. marinus OST provides previously unavailable biocatalytic capabilities and sets the stage for using E. coli to produce fully human antibodies.
Similar content being viewed by others

Introduction
Protein glycosylation is an important post-translational modification that occurs in all domains of life1. It is estimated that over half of all naturally occurring proteins in eukaryotes are glycoproteins2,3,4, with an even greater proportion among therapeutic proteins5. Of the different types of protein glycosylation, asparagine-linked (N-linked) glycosylation is the most common4,6. The central reaction in the pathway is catalyzed by the oligosaccharyltransferase (OST), which transfers a preassembled oligosaccharide from a lipid-linked oligosaccharide (LLO) donor to an asparagine residue within a consensus acceptor site or sequon (typically N-X-S/T where X ≠ P) in a newly synthesized protein7.
While N-linked glycosylation in eukaryotes, archaea, and bacteria share many mechanistic features, some notable differences have been observed, especially with respect to the OSTs that are central to these systems1,8,9. For example, most eukaryotic OSTs are hetero-octameric complexes comprised of multiple non-catalytic subunits and a catalytic subunit, STT310,11,12,13. In contrast, archaea and bacteria possess single-subunit OSTs (ssOSTs) that are homologous to STT311,14,15. Another difference among the various OSTs is their distinct but overlapping acceptor sequon preferences. The prototypical bacterial ssOST, namely PglB from Campylobacter jejuni (CjPglB), recognizes a more stringent D/E-X-1-N-X+1-S/T (X-1,+1 ≠ P) sequon compared to the N-X-S/T sequon recognized by eukaryotic and archaeal OSTs16. However, this requirement for an acidic residue in the –2 position of the sequon, known as the minus two rule, is not universally followed by all bacterial ssOSTs. Indeed, several PglB homologs from the Desulfobacterota (formerly Deltaproteobacteria) phylum including D. alaskensis G20 (formerly D. desulfuricans G20) PglB (DaPglB), D. gigas DSM 1382 PglB (DgPglB), and D. vulgaris Hildenborough PglB (DvPglB) exhibit sequon specificities that are relaxed compared to CjPglB and overlap with those of eukaryotic and archaeal OSTs17.
To date, these and other functional details about bacterial ssOSTs come from studies where the C. jejuni protein glycosylation machinery has been functionally reconstituted in laboratory strains of Escherichia coli, a feat that was first demonstrated >20 years ago18. Since that time, many groups have leveraged CjPglB and its homologs for performing N-linked glycosylation of diverse protein substrates. Included among these substrates are fragments of human immunoglobulin (IgG) such as CH2 or CH2-CH3 (hereafter fragment crystallizable (Fc) domain), which hold promise in the treatment of autoimmune disorders19,20. However, the use of engineered E. coli for producing glycosylated Fc domains has been limited to the attachment of non-human glycan structures at mutated acceptor sequons17,21,22,23,24. While some progress has been made to overcome these shortcomings, the overall poor glycosylation efficiency of Fc domains in E. coli (<5%) remains an unsolved problem that has discouraged efforts to develop this user-friendly host for biosynthesis of Fc domains, as well as their parental IgG counterparts, with relevant glycosylation.
Here, we sought to discover ssOSTs capable of N-glycosylation of the authentic QYNST sequon in human Fc fragments and full-length IgGs expressed in E. coli. We hypothesized that uncharacterized PglBs with broader substrate recognition and higher glycosylation efficiency might exist in the genomes of other Desulfobacterota. To test this hypothesis, a collection of 19 PglB homologs was generated by genome mining of Desulfovibrio spp. and screened in E. coli for the ability to glycosylate canonical and non-canonical acceptor sequons in periplasmically expressed acceptor proteins. This screening campaign led to the discovery of a PglB homolog from D. marinus strain DSM 18311 (DmPglB) that could efficiently glycosylate eukaryotic-type N-X-T motifs in different model acceptor proteins regardless of the residue at the –2 position. We further show that the relaxed sequon specificity of DmPglB enabled glycosylation of authentic QYNST sequons in the context of both a human hinge-Fc fragment and a full-length chimeric IgG composed of murine antigen-binding regions (Fv) and human constant domains. Glycosylation by DmPglB reached ~30–50% for hinge-Fc and 10–14% for IgG, which was significantly higher than the efficiencies of <2% and 0%, respectively, achieved with the next-best bacterial ssOST, DgPglB. As a result of this improved efficiency, it was possible to chemoenzymatically remodel the bacterial monoantennary N-glycan on E. coli-derived hinge-Fc with a nonfucosylated and fully galactosylated N-glycan called G2, a transformation that was not previously possible due to the very low glycosylation efficiency of this protein. Importantly, the glycoengineered hinge-Fc bearing homogeneous G2 glycans displayed strong affinity for a human Fc gamma receptor (FcγR), specifically FcγRIIIa. Collectively, these results deepen our understanding of substrate selection by bacterial ssOSTs and pave the way for using glycoengineered E. coli to customize the glycan-sensitive properties (e.g., anti-inflammatory activity, effector function, FcγR signaling, etc.) of IgGs and their fragments.
Results
Bioprospecting Desulfobacterota genomes for interesting ssOST candidates
The current armamentarium of characterized bacterial ssOSTs is insufficient for glycoprotein engineering applications that endeavor to recapitulate human-type glycosylation of biotherapeutic proteins22,24,25. Therefore, we sought to identify previously uncharacterized PglB homologs from Desulfovibrio spp. that have relaxed sequon specificity and catalyze glycosylation of diverse sequons with higher efficiency than previously discovered enzymes. To this end, we curated a collection of 19 candidate OSTs with similarity to DaPglB and DgPglB (Fig. 1a). We chose DaPglB and DgPglB as the query sequences because these OSTs previously exhibited the most efficient glycosylation of non-canonical sequences (e.g., AQNAT)17 and thus do not conform to the –2 rule that has been established for CjPglB16. For context, DaPglB and DgPglB share 30% Sequence identity with each other and only ~15–20% with the prototypic bacterial ssOSTs, CjPglB and C. lari PglB (ClPglB). In fact, the catalytic region of Desulfovibrio PglBs, which contains the signature WWDXG motif that is essential for function and thought to play a key role in catalysis26, is more similar to the catalytic region of eukaryotic and archaeal OSTs than to the same region of CjPglB17,27. Here, a total of 10 DgPglB homologs were selected, with DmPglB and D. indonesiensis DSM 15121 PglB (DiPglB) exhibiting the highest identity (42% and 47%, respectively) and D. desulfuricans DSM 642 PglB exhibiting the lowest (30%) identity. A further 9 PglB homologs with homology to DaPglB were selected, with Desulfovibrio sp. A2 PglB and D. litoralis DSM 11393 PglB exhibiting the highest (38%) and lowest (30%) identity, respectively.

a Phylogenetic tree of the PglB homologs evaluated in this study. The curated list of enzymes was generated from a BLAST search using DaPglB and DgPglB as the query sequences. CjPglB and ClPglB were added for comparison. The tree was generated by the neighbor-joining method from multiple sequence alignment using Molecular Evolutionary Genetics Analysis version 11 (MEGA11) software65. b Immunoblot analysis of periplasmic fractions from CLM24 cells transformed with plasmid pMW07-pglΔBCDEF encoding genes for biosynthesis of a modified C. jejuni heptasaccharide glycan (GalNAc5(Glc)GlcNAc), plasmid pBS-scFv13-R4DQNAT encoding the scFv13-R4DQNAT acceptor protein, and a derivative of plasmid pMLBAD encoding one of the PglB homologs as indicated. The first two lanes in left and right panels of (a) and (c) were loaded with the same positive and negative control samples (marked with asterisk). Blots were probed with polyhistidine epitope tag-specific antibody (anti-His) to detect the C-terminal 6x-His tag on the acceptor protein (top panel) and hR6 serum specific for the C. jejuni heptasaccharide glycan (bottom panel). Molecular weight (MW) markers are indicated on the left. The g0 and g1 arrows indicate un- and monoglycosylated acceptor proteins, respectively. Blots are representative of biological replicates (n = 3). Schematic created with BioRender. DeLisa, M. (2025) https://BioRender.com/wesqljn. c Glycosylation efficiency was determined by densitometric analysis, with data reported as mean ± SD. Red bars correspond to positive and negative controls generated with CjPglB and CjPglBmut, respectively; blue bars correspond to samples generated with Desulfovibrio PglBs. Statistical significance was determined by unpaired two-tailed Student’s t-test with calculated p-values represented as: *p < 0.05; ***p < 0.001; ns, not significant. Actual p-values relative to CjPglB are (from left-to-right): p = 0.541, p = 0.680, p = 0.0001, p = 0.0112, p = 0.0002, and p = 0.9586. Source data are provided as a Source Data file.
A subset of Desulfovibrio OSTs exhibit efficient glycosylation activity
To functionally evaluate the curated list of Desulfobacterota OSTs, we employed an ectopic trans-complementation assay17. The assay is based on E. coli strain CLM24, which lacks native glycosylation but is rendered glycosylation competent by transformation with one plasmid encoding enzymes for N-glycan biosynthesis, a second plasmid encoding a candidate PglB homolog, and a third plasmid encoding a glycoprotein target bearing either an engineered or natural N-glycan acceptor site. Using this assay, candidate PglB homologs were provided in trans and tested for their ability to promote glycosylation activity in E. coli.
To minimize microheterogeneity so that modified acceptor proteins were homogeneously glycosylated, we used plasmid pMW07-pglΔBCDEF that was previously shown to yield glycoproteins that were predominantly glycosylated (>98%) with GalNAc5(Glc)GlcNAc, a mimic of the C. jejuni N-glycan but with reducing-end GlcNAc replacing bacillosamine28. This reducing-end GlcNAc could be further advantageous as a substrate for PglB enzymes from Desulfovibrio spp. given that at least one glycoprotein from D. gigas, the 16-heme cytochrome HmcA, involves the formation of a GlcNAc-asparagine linkage at N261 of HmcA29. Moreover, this linkage also occurs in eukaryotic N-glycoproteins and can be remodeled to create a eukaryotic complex-type glycan via a two-step enzymatic trimming/transglycosylation process22. Codon-optimized versions of each Desulfovibrio PglB homolog were expressed from plasmid pMLBAD. For the acceptor protein, anti-β-galactosidase single-chain Fv antibody clone 13-R4 (scFv13-R4) fused with an N-terminal co-translational Sec export signal and a C-terminal DQNAT glycosylation tag24 was expressed from plasmid pBS-scFv13-R4DQNAT. We chose scFv13-R4DQNAT as a model acceptor protein because it is well expressed in the E. coli periplasm and can be efficiently glycosylated by diverse PglB homologs17,24,30. It should be noted that DQNAT is an optimal sequon for CjPglB31 and has been widely used as a tag for studying PglB-mediated glycosylation in E. coli21.
Glycosylation of the periplasmic scFv13-R4DQNAT protein was evaluated by immunoblot analysis with a polyhistidine epitope tag-specific antibody (anti-His) or C. jejuni heptasaccharide-specific serum (hR6)23. As expected, positive control cells complemented with wild-type (wt) CjPglB produced two proteins that were detected with the anti-His antibody, which corresponded to the un- (g0) and monoglycosylated (g1) forms of scFv13-R4DQNAT (Fig. 1b). Subsequent detection of the higher molecular weight g1 band with hR6 serum specific for the C. jejuni glycan confirmed glycosylation of this protein by wt CjPglB. In contrast, negative control cells complemented with a CjPglB mutant rendered inactive by two active-site mutations, D54N and E316Q (hereafter CjPglBmut), produced only the g0 form of scFv13-R4DQNAT with no detectable signal from the hR6 serum (Fig. 1b), confirming lack of glycosylation in these cells. Of the 22 Desulfobacterota PglB homologs tested here (19 newly curated and 3 that were tested previously, namely DaPglB, DgPglB and DvPglB17), a total of four (DgPglB, DiPglB, DmPglB, and D. gilichinskyi PglB (DgilPglB)) were functionally expressed based on their ability to promote clearly detectable glycosylation of the canonical DQNAT motif as determined by immunoblot analysis with the anti-His antibody and hR6 serum (Fig. 1b). Of these, DgPglB, DiPglB and DmPglB showed the highest glycosylation efficiency (all >89% based on densitometry analysis), rivaling that observed for CjPglB (91%) (Fig. 1c). These three highly efficient OSTs also produced an additional slower migrating band in the anti-His and hR6 blots, corresponding to a diglycosylated (g2) form of scFv13-R4DQNAT. This g2 form likely resulted from the glycosylation of a native 75RDNAT79 motif in scFv13-R4 that was previously observed to be glycosylated by Desulfovibrio PglB homologs such as DgPglB having relaxed sequon specificity17. Two additional enzymes, D. bastini PglB (DbPglB) and D. ferrireducens PglB (DfPglB), showed weak glycosylation that appeared following longer exposure of the same blots (Supplementary Fig. 1).
Desulfovibrio OSTs that efficiently glycosylate non-canonical sequons
To determine whether any of the Desulfovibrio PglB homologs also recognized sequons with a non-acidic amino acid in the –2 position, we tested glycosylation of the acceptor protein scFv13-R4AQNAT, which carries an AQNAT motif at its C-terminus. AQNAT is considered a non-canonical sequon because it is not glycosylated by CjPglB16. Hence, the ability to glycosylate AQNAT and other related sequons in which D/E residues are absent from the –2 position serves as a measuring stick for relaxed substrate specificity17,23,27,30. To eliminate any potential confounding results related to additional sequons, we additionally used an scFv13-R4 variant in which two putative internal glycosylation sites (32FSNYS36 and 75RDNAT79) were mutated by introducing N34L and N77L substitutions. These mutations were previously shown to eliminate the g2 form of this protein arising from glycosylation at position N77 (N34 was not observed to be glycosylated)17.
Of the six Desulfobacterota PglB homologs that showed activity towards scFv13-R4DQNAT above, all but DbPglB could glycosylate the scFv13-R4(N34L/N77L)AQNAT construct based on immunoblot analysis with anti-His antibody and hR6 serum (Fig. 2a). In contrast, CjPglB was unable to glycosylate the AQNAT motif, as expected given its preference for D/E in the –2 position16. The remaining OSTs failed to show any measurable activity, which together with their lack of activity above, suggests that they either prefer sequons besides (D/A)QNAT or were non-functional in our trans-complementation assay for other reasons, such as poor expression or incompatibility with the GalNAc5(Glc)GlcNAc glycan and/or C-terminal location of the sequon. Importantly, DgPglB, DiPglB and DmPglB were again among the most active OSTs in terms of glycosylation efficiency, with DmPglB reaching 90% (Fig. 2b). Interestingly, DfPglB and DgilPglB showed significantly stronger glycosylation of scFv13-R4(N34L/N77L)AQNAT versus scFv13-R4DQNAT, suggesting that these enzymes may possess a bias for sequons with non-acidic residues in the –2 position. It should be noted that while DaPglB was previously observed to glycosylate scFv13-R4(N34L/N77L)AQNAT 17, we were unable to detect any activity for this OST with the AQNAT sequon under the conditions tested here.

a Immunoblot analysis of periplasmic fractions from CLM24 cells transformed with the following plasmids: pMW07-pglΔBCDEF for making GalNAc5(Glc)GlcNAc; a derivative of pMLBAD encoding one of the PglB homologs as indicated; and pBS-scFv13-R4AQNAT encoding the scFv13-R4(N34L/N77L) acceptor protein with AQNAT sequon. Blots were probed with anti-His antibody (top panel) and hR6 serum (bottom panel). Molecular weight (MW) markers are indicated on the left. The g0 and g1 arrows indicate un- and monoglycosylated acceptor proteins, respectively. Blots are representative of biological replicates (n = 3). b Glycosylation efficiency was determined by densitometric analysis, with data reported as mean ± SD. Red bars correspond to positive and negative controls generated by CjPglB with scFv13-R4DQNAT or scFv13-R4(N34L/N77L)AQNAT as acceptors, respectively; blue bars correspond to samples generated with Desulfovibrio PglBs. (c, d) Same as (a, b) but with plasmid pBS-scFv13-R4QYNST encoding scFv13-R4(N34L/N77L) with QYNST sequon. Red bars correspond to positive and negative controls generated by CjPglB with scFv13-R4DQNAT or scFv13-R4(N34L/N77L)QYNST as acceptors, respectively; blue bars correspond to samples generated with Desulfovibrio PglBs. The first two lanes in left and right panels of (a) and (c) were loaded with the same positive and negative control samples (marked with asterisk). Schematics created with BioRender. DeLisa, M. (2025) https://BioRender.com/wesqljn. Statistical significance was determined by unpaired two-tailed Student’s t-test with calculated p-values represented as: *, p < 0.05; ***, p < 0.001; ns, not significant. Actual p-values relative to CjPglB (DQNAT) in panel (b) are (from left-to-right): p = 0.1072, p = 0.3838, p = 0.0008, p = 0.2436, and p = 0.2462. Actual p-values relative to CjPglB (DQNAT) in panel (d) are (from left-to-right): p = 0.6619, p = 0.0144, p = 0.0363 (DgPglB vs. DmPglB), p = 0.0017, and p = 0.0027. Source data are provided as a Source Data file.
To further investigate the ability of PglB homologs from Desulfovibrio to recognize non-canonical sequences, we tested glycosylation of the acceptor protein scFv13-R4(N34L/N77L)QYNST, which carries a QYNST motif at its C-terminus. We chose QYNST because IgG antibodies, one of the most abundant glycoproteins in human serum, are invariably decorated with N-glycans at a highly conserved QYNST motif in their Fc region. Whereas scFv13-R4(N34L/N77L)QYNST was not glycosylated by CjPglB, consistent with its restricted sequon specificity16, four Desulfovibrio ssOSTs – DgPglB, DmPglB, DiPglB, and DgilPglB – exhibited glycosylation of the non-canonical QYNST sequon as revealed by immunoblotting (Fig. 2c and Supplementary Fig. 1) and mass spectrometry (MS) analysis (Supplementary Fig. 2a; shown for DmPglB). Of these, DmPglB displayed the highest glycosylation efficiency (95% based on densitometry; 92% based on MS) (Fig. 2d and Supplementary 2b), making it the only OST that could glycosylate all three sequons with ~90% efficiency or higher.
OST from D. marinus exhibits highly relaxed sequon specificity
During these experiments, we observed autoglycosylation of DmPglB (Supplementary Fig. 3a), indicating that DmPglB itself is a glycoprotein, just like CjPglB and ClPglB15,32. Specifically, MS analysis identified two sequons clustered at the extreme C-terminus of DmPglB that were autoglycosylated, namely 751EANGT755 and 756AANAT760 (Supplementary Figs. 3b and 4), with the latter site providing further evidence of the relaxed sequon specificity for DmPglB. Considering this relaxed acceptor-site specificity, we decided to investigate DmPglB’s amino acid preferences at the –2 position of the sequon more systematically. This analysis took advantage of a set of plasmids encoding scFv13-R4 variants in which the –2 position of the C-terminal acceptor motif was varied to include all 20 amino acids30. Like DaPglB and DgPglB17, DmPglB exhibited greatly relaxed acceptor-site specificity in this assay (Fig. 3a and b). However, unlike the more variable relaxation observed for DaPglB and DgPglB previously, with certain sequons becoming strongly glycosylated and others only weakly or not at all (Supplementary Fig. 5; shown for DgPglB), DmPglB exhibited non-preferential and highly efficient glycosylation (77–100%) of all 20 sequons (Fig. 3b and Supplementary Fig. 6). At this point, we also constructed a catalytically inactive DmPglB variant (hereafter DmPglBmut) by mutating two residues, D55N and E363Q, in the catalytic pocket. Sequence alignment and structural modeling indicated that these two residues corresponded to D56 and E319 in ClPglB or D54N and E316Q in CjPglB (Supplementary Fig. 7), which are essential for catalytic activity15,30. As expected, DmPglBmut was unable to glycosylate scFv13-R4DQNAT (Fig. 3a), confirming the DmPglB-dependent nature of the glycosylation results above.

a Immunoblot analysis of periplasmic fractions from CLM24 cells transformed with the following plasmids: pMW07-pglΔBCDEF for making GalNAc5(Glc)GlcNAc; pMLBAD encoding DmPglB, DmPglBmut, CjPglB or CjPglBmut; and pBS-scFv13-R4XQNAT encoding the scFv13-R4 with each of the 20 amino acids in the –2 position of the C-terminal sequon as indicated. Blots were probed with anti-His antibody (top panel) and hR6 serum (bottom panel). Molecular weight (MW) markers are indicated on the left. The g0 and g1 arrows indicate un- and monoglycosylated acceptor proteins, respectively. Blots are representative of biological replicates (n = 3). Schematic created with BioRender. DeLisa, M. (2025) https://BioRender.com/wesqljn. b Heatmap analysis of the relative –2 amino acid preference of CjPglB, DgPglB, and DmPglB. Relative preferences (weaker = white; stronger = dark cyan) were determined based on densitometry analysis of the glycosylation efficiency for each acceptor protein in the anti-His immunoblot (see Supplementary Figs. 5, 6 for efficiency data). c Sequence logo showing experimentally determined acceptor-site specificity of DmPglB using glycoSNAP-based library screening of YebF(N24L)-Im7XXNXT. Source data are provided as a Source Data file.
To analyze acceptor-site specificity of the DmPglB enzyme in a more unbiased manner, we utilized a previously established genetic screen called glycoSNAP (glycosylation of secreted N-linked acceptor proteins)30. GlycoSNAP is a high-throughput colony blotting assay based on glycosylation and extracellular secretion of a reporter protein composed of E. coli YebF, a small (10 kDa in its mature form) extracellularly secreted protein33, or YebF fusion proteins modified with an acceptor sequon28,30. To eliminate potentially confounding internal glycosylation in the YebF protein itself, we used an N24L mutant of YebF that was not glycosylated by any relaxed OST homologs17,30. The compatibility of one such reporter fusion, YebF(N24L)-Im728, with DmPglB was first evaluated in the context of a C-terminal DQNAT sequon, with clear extracellular accumulation of glycosylated YebF(N24L)-Im7DQNAT detected for cells co-expressing wild-type DmPglB (Supplementary Fig. 8a). In contrast, there was no evidence for glycosylation of the YebF(N24L)-Im7DQNAT construct that had been secreted by cells co-expressing DmPglBmut. Encouraged by this result, we next used glycoSNAP to screen a combinatorial library of acceptor-site sequences for glycosylation by DmPglB. A combinatorial library of ~1.1 × 105 YebF(N24L)-Im7XXNXT variants was generated by randomizing the amino acids in the –2, –1, and +1 positions of the C-terminal acceptor sequon by PCR amplification using NNK degenerate primers. The resulting library was screened by glycoSNAP replica plating to identify clones that produced glycosylated YebF(N24L)-Im7 in culture supernatants (Supplementary Fig. 8b). A total of 65 positive hits were recovered (Supplementary Fig. 8c, d) and used to generate a consensus motif representing sequons that were preferentially glycosylated by DmPglB (Fig. 3c). Overall, DmPglB exhibited highly relaxed specificity at all three variable sequon positions with only a slight preference for threonine at the –1 position and alanine or serine at the +1 position. The –2 and –1 positions showed the most variability with nearly all 20 amino acids represented at each site (Supplementary Fig. 8d). Importantly, these results were in good agreement with the findings above in which DmPglB indiscriminately glycosylated every XQNAT sequon with high efficiency.
Glycosylation reaction kinetics of bacterial OSTs
To compare the rates and Michaelis–Menten constants of DmPglB relative to the prototypic CjPglB ssOST, we employed a fluorescently labeled peptide with either a DQNAT or QYNST glycosylation sequon and solvent-extracted LLOs bearing the GalNAc5(Glc)GlcNAc glycan to track the glycosylation reaction using in-gel fluorescence34. The glycosylation of these peptides was determined by examining the increase of molecular weight corresponding to the addition of the ~1 kDa heptasaccharide using tricine-SDS-PAGE gels. Following purification of CjPglB and DmPglB, each was added to an in vitro glycosylation reaction with one of the fluorescently tagged peptide substrates along with the GalNAc5(Glc)GlcNAc LLOs as glycan donor. The glycosylated products were separated from the unmodified substrate by gel electrophoresis, and the educt/product ratio was determined by measuring the in-gel fluorescence intensities of both educt and product bands as a function of time and peptide concentration (Supplementary Fig. 9a, b).
To determine turnover rates, time course analysis was performed, and the initial turnover rates were determined for the linear range of the reaction (Supplementary Fig. 9c). Importantly, the turnover rates confirmed that the DQNAT-containing peptide was an active substrate for both CjPglB and DmPglB, whereas the QYNST-containing peptide was only an active substrate for DmPglB (Table 1), consistent with our in vivo findings. Although kcat for DmPglB with the QYNST sequon was ~30–40% lower than the turnover rates measured for each enzyme with DQNAT, it was still on par with the kcat value reported previously for CjPglB using a DANYT-containing peptide and synthetic LLO35. Next, Michaelis–Menten kinetics were determined for both enzymes using increasing concentrations of peptide substrate (Supplementary Fig. 9d). From this analysis, we determined Km values of 10.7 ± 0.98 μM for CjPglB with the DQNAT sequon and 4.30 ± 0.95 and 5.16 ± 1.13 for DmPglB with the DQNAT and QYNST sequons, respectively (Table 1). Importantly, these values were on par with Km values reported previously for CjPglB using synthetic LLO substrates35 and quantifiably confirmed the relaxed acceptor site specificity of the DmPglB enzyme.
Structural features of OSTs that correlate with relaxed specificity
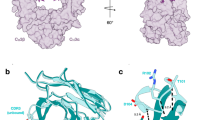
To better understand the observed functional differences for DmPglB relative to other OSTs, we generated a structural model of DmPglB using the AlphaFold2 protein structure prediction algorithm implemented with ColabFold36,37. Comparing the predicted structure of DmPglB with the solved structure of ClPglB15 revealed clear variations in the structures of the catalytic pockets. Based on our electrostatic surface calculations38, it is apparent that the entrance to the peptide-binding cavity that hosts the –2 position of the acceptor sequon is positively charged in ClPglB but neutral in DmPglB (Fig. 4a). This difference in surface charge results from residues in the vicinity of the arginine at position 331 in ClPglB(R375 in DmPglB), which is strongly conserved in bacterial ssOSTs (Fig. 4b) and provides a salt bridge to the aspartic acid in a bound DQNATF substrate peptide in the ClPglB crystal structure15. Specifically, in the case of ClPglB, R331 is surrounded by primarily hydrophobic residues (I323, V327, and L374) that cluster to form a positively charged patch in this region of the protein (Fig. 4a and c). Conversely, the same region in DmPglB is significantly more neutral due to the occurrence of negatively charged and neutral amino acids (L367, E371, D374 and T418) that surround R375, providing a possible explanation for the more relaxed substrate specificity of this enzyme. Another visible difference is the peptide-binding cavity in DmPglB, which is more spacious and lined with more negatively charged residues than the cavity in ClPglB. It is worth noting that structural models of eukaryotic STT3s, which themselves do not require an acidic residue in the –2 position of the sequon, exhibited features akin to DmPglB including an even more voluminous peptide-binding cavity with a similarly neutral entrance and a highly negatively charged lining (Fig. 4a).

a Electrostatic potential of various OST peptide-binding pockets modeled with either DQNAT (top) or QYNST (bottom) acceptor peptides (yellow). Electrostatic surfaces were generated based on calculations using the adaptive Poisson-Boltzmann solver (APBS)38. b Sequence alignments of conserved, short motifs in eukaryotic STT3s (human and plant STT3A and STT3B, protozoan Leishmania major STT3D and Trypanosoma brucei TbSTTA) and bacterial ssOSTs (ClPglB, CjPglB, DgPglB, DmPglB, DiPglB). Alignments shown were made using Clustal Omega web server multiple alignment editor64. Conserved residues are shaded gray while notable residues that deviate between eukaryotic and bacterial sequences are shaded yellow. c Structural model of QYNST peptide (yellow) in the peptide-binding pocket of the same OSTs in (a). Depicted in green are amino acids at the entrance to the peptide-binding cavity that cluster to create a positively charged patch in ClPglB but are neutral in all other OSTs. The SVXE/SVIE/TIXE motifs are depicted in gold.
Multiple sequence alignment revealed that the Desulfovibrio PglBs possessed all the short, conserved motifs that have been previously documented for OSTs across all kingdoms, albeit with subtle deviations from the Campylobacter and eukaryotic OSTs including WWDWG instead of WWDYG, DGGR instead of DGGK, and NL instead of DK/MI (Fig. 4b and Supplementary Fig. 10). A more dramatic difference was observed for the SVXE/TIXE motif, which occurs in the fifth external loop (EL5) and is involved in recognizing sequons at the main-chain level with the glutamic acid serving as a coordination switch that responds to ligand binding39. It has been widely reported that the conserved SVXE motif is unique to eukaryotes whereas the conserved TIXE motif is confined to archaeal and eubacterial OSTs. To our surprise, all Desulfovibrio PglBs including DgPglB, DmPglB and DiPglB possessed SVIE/SIIE motifs that were more like the eukaryotic SVXE motif than the canonical bacterial TIXE motifs found in ClPglB and CjPglB (Fig. 4b and Supplementary Fig. 10). Moreover, in eukaryotic and Desulfovibrio OSTs we observed a highly conserved glutamine located two residues downstream of this motif, with the Desulfovibrio PglB homologs also possessing a highly conserved glutamine immediately upstream of the motif.
Glycosylation of native acceptor site in human Fc domain by DmPglB
Encouraged by the ability of DmPglB to recognize minimal N-X-T motifs, we next evaluated the extent to which it could glycosylate the native QYNST site found in the Fc region of an IgG antibody. To this end, we created a pTrc99S-based plasmid that encoded the native Fc region and hinge derived from human IgG1 (hereafter hinge-Fc). For the N-glycan, we utilized the same pMW07-pglΔBCDEF plasmid from above as well as a derivative, plasmid pMW07-pglΔBICDEF, that produces GalNAc5GlcNAc without the branching glucose. We added this latter glycan because it facilitates enzymatic removal of GalNAc5 to reveal a GlcNAc primer that can be used for chemoenzymatic glycan remodeling22. For the PglB homologs, we evaluated DmPglB alongside both CjPglB and DgPglB, with the latter two enzymes having been shown previously to glycosylate Fc domains but with very low efficiency17,21,22,23,24. Each of the PglB homologs were expressed from pMLBAD as above.
In agreement with previous work17, CjPglB was unable to glycosylate the native QYNST sequon in the hinge-Fc with either of the tested N-glycan structures as revealed by non-reducing immunoblot analysis using an anti-IgG antibody and hR6 serum for detection (Fig. 5a). In the case of DgPglB, there was also no glycosylation detected with the GalNAc5GlcNAc glycan and only weak glycosylation (<2%) with the GalNAc5(Glc)GlcNAc glycan (Fig. 5a, b), consistent with earlier observations in which DgPglB only glycosylated a small fraction (<5%) of Fc molecules17. In stark contrast, DmPglB glycosylated the hinge-Fc regardless of the N-glycan used, in agreement with the highly relaxed acceptor-site specificity observed above for this ssOST. Densitometric analysis revealed that DmPglB glycosylated the hinge-Fc with an efficiency of 52% when using GalNAc₅(Glc)GlcNAc and 30% with GalNAc₅GlcNAc, which reflected a statistically significant increase over the very low glycosylation efficiency achieved with DgPglB (Fig. 5b).

a Non-reducing immunoblot analysis of protein A-purified proteins from whole-cell lysate of CLM24 cells transformed with the following plasmids: pMW07-pglΔBCDEF for making GalNAc5(Glc)GlcNAc (left) or pMW07-pglΔBICDEF for making GalNAc5GlcNAc (right); pMLBAD encoding CjPglB, DgPglB, DmPglB, or DmPglBmut; and pTrc99S-spDsbA-hinge-Fc encoding hinge-Fc derived from human IgG1. Blots were probed with anti-human IgG (anti-IgG) to detect human Fc (top panel) and hR6 serum (bottom panel). Molecular weight (MW) markers are indicated on the left. The g0, g1, and g2 arrows indicate un-, mono-, and diglycosylated Fc proteins, respectively. Blots are representative of biological replicates (n = 3). b Glycosylation efficiency was determined as above with data reported as the mean ± SD. Statistical significance was determined by unpaired two-tailed Student’s t-test with calculated p-values represented as: **p < 0.01; ***p < 001; ****p < 0.0001. Actual p-values in top panel are (from left-to-right): p < 0.0001 and p = 0.0002. Actual p-values in bottom panel are (from left-to-right): p = 0.0017 and p = 0.0017. c Same as in (a) but with JUDE-1 cells transformed with plasmid pMAZ360-YMF10-IgG encoding a full-length chimeric IgG1 specific for PA along with plasmids for glycan biosynthesis and ssOST as indicated. Asterisks denote band shifts due to glycosylation of HC-LC dimer. Schematics created with BioRender. DeLisa, M. (2025) https://BioRender.com/wesqljn. Source data are provided as a Source Data file.
Importantly, this activity was completely absent in cells carrying the DmPglBmut variant, confirming the OST-dependent nature of the glycosylation. Moreover, the observation of doubly and singly glycosylated hinge-Fc indicated that a mixture of fully and hemi-glycosylated products, respectively, were generated under the conditions tested, with roughly equal quantities of both based on the comparable g2 and g1 band intensities in the anti-glycan blot. To unequivocally prove glycosylation of the native QYNST sequon in hinge-Fc by DmPglB, LC-MS/MS analysis of the glycosylation products was performed under reduced and protease-digested conditions. The MS/MS spectrum of a tryptic peptide (99EEQYNSTYR107) containing the known glycosylation sequon conclusively revealed the presence of a HexNAc6Hex1 structure, consistent with the GalNAc5(Glc)GlcNAc glycan (Supplementary Fig. 11a).
We next investigated whether DmPglB could glycosylate a full-length IgG1 antibody, namely YMF10, which is a chimeric IgG clone (murine VH and VL regions and human constant regions) with high affinity and specificity for Bacillus anthracis protective antigen (PA)40. YMF10 was chosen because it can be expressed in the E. coli periplasm at high levels, and its heavy chain (HC) and light chain (LC) can be properly assembled into a functional full-length IgG. To ensure efficient IgG expression, we used JUDE-1 E. coli cells carrying plasmid pMAZ360-YMF10-IgG40. These cells were further transformed with plasmid pMLBAD encoding a PglB homolog and either pMW07-pglΔBCDEF or pMW07-pglΔBICDEF encoding the N-glycan biosynthesis genes.
Non-reducing immunoblot analysis revealed formation of fully assembled heterotetrameric YMF10 as well as other intermediate products for each of the strain/plasmid combinations tested (Fig. 5c), in line with expression patterns observed previously40. Importantly, only cells carrying DmPglB were capable of YMF10 glycosylation as evidenced by detection of HC-linked glycans with hR6 serum, whereas no glycosylation was observed for cells carrying either CjPglB or DgPglB (Fig. 5c). Although all products containing at least one HC were detected by hR6 serum, the fully assembled IgG tetramer was one of the major glycoforms along with the HC-HC and HC-LC dimers based on relative band intensities. While it was difficult to see a band shift in the anti-IgG blot indicative of glycosylation of the full-length protein due to poor resolution at higher molecular weights (>100 kDa), a band shift was observed for the half antibody product (HC-LC dimer) at ~70 kDa. As expected, there was no detectable glycosylation activity when the catalytically inactive mutant DmPglBmut was substituted for wt DmPglB. Further confirmation of IgG glycosylation was obtained by LC-MS/MS analysis of reduced and digested IgG-containing samples. Specifically, the MS/MS spectrum confirmed glycosylation of a tryptic peptide (293EEQYNSTYR301) containing the known glycosylation sequon and modified with HexNAc6Hex1 or HexNAc6, consistent with the GalNAc5(Glc)GlcNAc and GalNAc5GlcNAc glycans, respectively (Supplementary Fig. 11b, c). From the LC-MS/MS analysis, the glycosylation efficiency of YMF10 was estimated to range from 10–14% with these two N-glycans (Supplementary Table 1).
Remodeling of E. coli-derived hinge-Fc with eukaryotic N-glycans
Upon confirming the ability of DmPglB to glycosylate the authentic QYNST sequon in human hinge-Fc, we sought to transform the installed GalNAc5GlcNAc glycan into a more biomedically relevant glycoform (Fig. 6a). To this end, we adapted a previously described glycan remodeling strategy that used engineered E. coli to produce glycoproteins bearing GalNAc5GlcNAc glycans, which were subsequently trimmed and chemoenzymatically remodeled in vitro by an enzymatic transglycosylation reaction. Using this method, it was possible to install eukaryotic N-glycans including an asialo afucosylated complex-type biantennary glycan (Gal2GlcNAc2Man3GlcNAc2; G2) onto a model bacterial acceptor protein22. However, this method could not be extended to a human CH2 domain because of the low glycosylation efficiency (<5%) achieved with CjPglB, even after the introduction of a preferred DFNST sequon in place of QYNST in this protein. Here, we hypothesized that transglycosylation by this strategy would be possible with our glycosylated hinge-Fc protein due to the much higher glycosylation efficiency (~30–50%) achieved with DmPglB. To test this hypothesis, we first subjected the protein A-purified hinge-Fc bearing GalNAc5GlcNAc to enzymatic trimming with exo-α-N-acetylgalactosaminidase, with GalNAc removal being continuously monitored by LC-ESI-MS (Supplementary Fig. 12a) and confirmed by immunoblot analysis (Fig. 6b). The resulting hinge-Fc bearing only a GlcNAc stump was then subjected to transglycosylation catalyzed by the glycosynthase mutant, EndoS2-D184M41, with preassembled G2-oxazoline as donor substrate22 in a reaction that was again monitored by LC-ESI-MS (Supplementary Fig. 12b) and confirmed by immunoblot analysis (Fig. 6b). This sequence of steps produced a hinge-Fc protein bearing the G2 glycoform (G2-hinge-Fc).

a Schematic representation of the chemoenzymatic reaction for trimming and remodeling hinge-Fc glycans. b Immunoblot analysis of the four E. coli-derived glycoforms (from left to right): aglycosylated hinge-Fc, glycosylated GalNAc5GlcNAc-hinge-Fc, GlcNAc-hinge-Fc, and G2-hinge-Fc. Blot was probed with anti-human IgG (anti-IgG) to detect human Fc. Molecular weight (MW) markers are indicated on the left. The g0, g1, and g2 arrows indicate un-, mono-, and diglycosylated Fc proteins, respectively. Blot is representative of biological replicates (n = 3). c ELISA analysis of same constructs in (b) with FcγRIIIA-V158 as immobilized antigen. Data are average of biological replicates (n = 3) ± SD. Schematics created with BioRender. DeLisa, M. (2025) https://BioRender.com/wesqljn.
To evaluate the functional consequences of installing eukaryotic glycans onto the E. coli-derived hinge-Fc, we investigated the binding affinity between different hinge-Fc glycoforms and a human Fc gamma receptor (FcγR). Specifically, we chose the clinically relevant FcγRIIIa-V158 allotype42 because it is the high-affinity allele and interactions between this receptor and different IgG subclasses have been extensively studied43,44. It is also worth noting that glycosylated hinge-Fc antibodies including those containing terminal galactose residues, such as G2, exhibit affinity for FcγRIIIa45. In total, we examined four E. coli-derived glycoprotein forms: aglycosylated hinge-Fc, glycosylated GalNAc5GlcNAc-hinge-Fc, GlcNAc-hinge-Fc, and G2-hinge-Fc. Among these glycoforms, G2-hinge-Fc displayed the highest binding affinity for FcγRIIIA-V158 as determined by enzyme-linked immunosorbent assay (ELISA), with a half-maximal effective concentration (EC50) of 28.5 ± 3.2 nM (Fig. 6c). In contrast, little to no binding above background was observed for the trimmed GlcNAc-hinge-Fc, untrimmed hinge-Fc bearing the GalNAc5GlcNAc glycan, and aglycosylated hinge-Fc, confirming the importance of the human N-glycan structure on FcγRIIIA-V158 binding. By way of comparison, we measured an EC50 of 5.4 ± 0.6 nM for commercial trastuzumab (Fig. 6c), consistent with the EC50 value of 1.4 nM for FcγRIIIA-V158 that was measured for another IgG product, rituximab, following glycan remodeling to acquire the G2 glycan46. The weaker FcγRIIIA affinity of our G2-hinge-Fc relative to these full-length IgGs may be due to differences in their glycosylation levels and/or the absence of Fab domains in hinge-Fc that stabilize IgG-FcγRIIIA interactions47. Regardless, our results provide proof-of-concept for chemoenzymatic conversion of E. coli-derived IgG-Fc glycans into glycoforms that preserve important Fc effector functions.
Discussion
The engineered expression of glycosylated antibodies in E. coli depends on OSTs that can install N-linked glycans within the QYNST sequon of the IgG CH2 domain. To this end, we identified a previously uncharacterized ssOST, DmPglB, that was able to glycosylate minimal N-X-S/T sequons with high efficiency and without preference for the residues in the −2, −1 or +1 positions. In fact, the breadth of sequons recognized by DmPglB and the efficiency with which they were modified was unmatched by any of the ~50 bacterial ssOSTs that have been tested here and elsewhere16,17,23,27,30. Importantly, DmPglB promoted glycosylation of the native QYNST motif in a human hinge-Fc fragment and a full-length, chimeric IgG antibody, with efficiencies that ranged from ~30–52% and ~10–14%, respectively, which were significantly higher than any of the efficiencies reported previously for PglB-mediated Fc glycosylation in E. coli17,21,22,23,24. Although the installed glycans were bacterial-type structures, we side-stepped this limitation by in vitro chemoenzymatic transformation of bacterial GalNAc5GlcNAc into complex-type G2, a glycan that is known to enhance ADCC activity in vitro and anticancer efficacy in vivo48. The complete conversion to G2 on hinge-Fc observed here was significantly more efficient than the roughly 50% conversion achieved with a model bacterial glycoprotein22. This difference was presumably due to the use of a more efficient glycosynthase mutant, EndoS2-D184M, that potently remodels antibodies with complex-type glycans including G241. Importantly, the remodeled G2-hinge-Fc engaged FcγRIIIa while the hinge-Fc bearing the bacterial glycan did not, demonstrating the potential of our strategy for creating antibodies with native effector functions.
While the precise sequence determinants responsible for the unique substrate specificity of DmPglB remain to be experimentally determined, we hypothesize that acceptor substrate selection is governed in part by the EL5 loop including the SVXE/TIXE motif and neighboring residues. This hypothesis is supported by our structural models that showed the SVXE/TIXE motifs of bacterial and eukaryotic OSTs in close proximity to the acceptor peptide. This positioning is consistent with recently determined crystal structures of archaeal and bacterial ssOSTs, namely AglB from Archaeoglobus fulgidus (AfAglB) and ClPglB, respectively, with bound substrate peptide, which revealed that the TIXE motif lies side-by-side in an anti-parallel β-sheet configuration with the sequon and forms two interchain hydrogen bonds with the +1 and +3 residues of the sequon39,49. Interestingly, whereas ClPglB and CjPglB each possess a canonical bacterial TIXE motif and follow the minus two rule, the DgPglB, DiPglB, and DmPglB enzymes possess eukaryotic-like SVIE motifs. We speculate that this motif in Desulfovibrio ssOSTs may contribute to their more eukaryotic-like sequon requirements relative to Campylobacter ssOSTs. However, the fact that archaeal OSTs also possess a TIXE motif and yet do not require an acidic residue in the –2 position of the sequon indicates that this motif alone is insufficient to explain the differences in sequon preference among these OSTs.
We speculate that additional residues in the vicinity of the SVXE/TIXE motif might also be important in determining acceptor substrate preferences. In support of this notion, alanine scanning mutagenesis of the EL5 loop of AfAglB confirmed that the TIXE motif as well five adjacent downstream residues that are positioned near the –2 position of the acceptor peptide are essential for glycosylation activity39. These residues are in the immediate vicinity of the highly conserved arginine that, in ClPglB, forms a stabilizing salt bridge with the aspartic acid in the –2 position of the sequon15. This residue appears to be a key regulator of sequon selection based on mutagenesis studies in which substitution of the analogous arginine in CjPglB or DgPglB with residues such as leucine or asparagine was sufficient to reprogram the −2 preferences of each enzyme17,30. Another key feature in sequon selection may be the electrostatic charge of this region of the enzyme, which forms the peptide-binding cavity and is more neutral in DmPglB and eukaryotic OSTs but positively charged in ClPglB. A more spacious peptide-binding cavity in DmPglB may also contribute to its ability to accommodate sequons having bulkier sidechains such as the aromatic residue at −1 of QYNST.
It has long been known that the E. coli periplasm can support the proper assembly of antibody HC and LC50. However, while E. coli-derived antibodies bind strongly to their cognate antigens and the neonatal Fc receptor (FcRn), they show no significant binding to complement component 1q (C1q) or FcγRs due to lack of glycosylation50,51. This deficiency can be overcome by introducing specific mutations to the IgG Fc domain that confer FcγR binding52,53,54, but all aglycosylated IgG mutants isolated so far exhibit selective binding to a single FcγR, which is in contrast to glycosylated IgGs derived from mammalian cells that bind all FcγRs. Hence, there remains great interest in combining Fc or IgG expression with protein glycosylation in E. coli. Unfortunately, previous attempts to glycosylate Fc fragments in E. coli have largely been limited to attachment of bacterial N-glycans17,21,22,23, which are insufficient to confer Fcγ receptor binding as we showed here. While it is possible to attach eukaryotic N-glycans to the Fc domain using CjPglB in E. coli, this approach was met with inefficient glycosylation (~1%)24. Our combined strategy overcomes the deficiencies of these previous works in two important ways. First, the use of DmPglB greatly increases the efficiency of Fc glycosylation including at the authentic QYNST sequon and second, the chemoenzymatic remodeling strategy introduces eukaryotic complex-type glycans that permit the full spectrum of Fc effector functions that have until now been inaccessible to E. coli-derived IgGs. Although further improvements in glycosylation efficiency and yield will be required to rival IgG expression in mammalian host cell lines, our discovery of DmPglB provides a potent N-glycosylation catalyst to the bacterial glycoprotein engineering toolbox and creates an important foundation on which the production and glycoengineering of IgG antibodies and antibody fragments can be more deeply investigated and optimized in the future.
Methods
Bacterial strains, growth conditions, and plasmids
E. coli strain DH5α was employed for all cloning and library construction. E. coli strain CLM2455 was utilized for all in vivo glycosylation studies except for full-length IgG expression and glycosylation, which used E. coli strain JUDE-140. E. coli strain BL21(DE3) was used to generate acceptor proteins for in vitro glycosylation experiments. Cultures were grown overnight and subsequently subcultured at 37 °C in Luria-Bertani (LB) broth, supplemented with antibiotics as required at the following concentrations: 20 μg/ml chloramphenicol (Cm), 80 μg/ml spectinomycin (Spec), 100 μg/ml ampicillin (Amp), and 100 μg/mL trimethoprim (Tmp). When the optical density at 600 nm (OD600) reached ~1.4, 0.1 mM of isopropyl-β-D-thiogalactoside (IPTG) and 0.2% (w/v) L-arabinose inducers were added. Induction was carried out at 30 °C for 18 h. For expression and glycosylation of full-length IgGs, cultures were grown overnight and subsequently subcultured at 37 °C in terrific broth (TB) supplemented with the necessary antibiotics. When the OD600 reached ~1.4, 0.3 mM of IPTG and 0.2% (w/v) L-arabinose inducers were added. Induction was carried out at 30 °C for 12 h.
Plasmids for expressing different bacterial OSTs were constructed similarly to pMAF10 56 that encodes CjPglB. Specifically, each of the 24 bacterial OST genes were separately cloned into the EcoRI site of plasmid pMLBAD56. Template DNA for bacterial OSTs was codon optimized and obtained as double-stranded DNA fragments known as gBlocks (Integrated DNA Technologies). Plasmid pMAF10-CmPglBmut was constructed previously by performing site-directed mutagenesis on CjPglB in pMAF10 to introduce two mutations, D54N and E316Q, that abolish catalytic activity30. Plasmid pMAF10-DmPglBmut was constructed in a similar fashion by introducing analogous mutations, namely D55N and E363Q, to DmPglB in plasmid pMAF10-DmPglB. For purification of DmPglB, plasmid pSF-DmPglB-10xHis was created by replacing the gene encoding CjPglB in plasmid pSF-CjPglB17 with the gene encoding DmPglB along with an additional 10xHis sequence using Gibson assembly. For heterologous biosynthesis of the GalNAc5(Glc)GlcNAc glycan, we generated plasmid pMW07-pglΔBCDEF by deleting the pglCDEF genes coding for enzymes involved in bacillosamine biosynthesis from the pgl locus in plasmid pMW07-pglΔB30 using Gibson assembly cloning. For biosynthesis of the linear GalNAc5GlcNAc glycan, we generated plasmid pMW07-pglΔBICDEF by additionally deleting the gene coding for the enzyme responsible for transferring the branching glucose (pglI). The gene deletions were confirmed by Oxford nanopore whole plasmid sequencing at Plasmidsaurus. For acceptor protein expression, previously constructed plasmids pBS-scFv13-R4DQNAT, pBS-scFv13-R4XQNAT, and pBS-scFv13-R4AQNAT-GKG-His6 were used17,30. Plasmid pBS-scFv13-R4QYNST-GKG-His6 was created by replacing the AQNAT motif in pBS-scFv13-R4AQNAT-GKG-His6 with QYNST. Plasmid pTrc99S-YebF-Im7DQNAT constructed in previous studies28 was used as template to create pTrc99S-YebF-Im7XXNXT using degenerate primers with NNK bases (N = A, C, T or G; K = G or T) at the −2, −1 and +1 positions of the glycosylation sequon. The resulting plasmid DNA library was used to transform DH5α cells as discussed below. Plasmid pTrc99S-spDsbA-hinge-Fc was created by adding the hinge sequence EPKSCDKTHTCPPCP between the E. coli DsbA signal peptide and the human IgG1 Fc domain in pTrc-spDsbA-Fc21. Plasmid pMAZ360-YMF10-IgG40 was provided as a generous gift from Prof. George Georgiou (University of Texas, Austin). Sequences of all DNA primers and gBlocks (IDT) used in plasmid construction are provided in the Source Data file. All PCRs were performed using Phusion high-fidelity polymerase (New England Biolabs), and the PCR products were gel-purified from the product mixtures to eliminate nonspecific PCR products. The resulting PCR products were assembled using Gibson Assembly Master Mix (New England Biolabs). After transformation of DH5α cells, all plasmids were isolated using a QIAprep Spin Miniprep Kit (Qiagen) and confirmed by Sanger DNA sequencing at the Genomics Facility of the Cornell Biotechnology Resource Center.
GlycoSNAP assay
Screening of the pTrc99S-YebF-Im7XXNXT library was performed using the glycoSNAP assay17,28,30. Briefly, E. coli strain CLM24 carrying plasmid pMW07-pglΔBCDEF and pMLBAD encoding the DmPglB OST was transformed with the pTrc99S-YebF-Im7XXNXT library plasmids, yielding a cell library of ~1.1 × 105 members. The resulting transformants were grown on 150 mm LB-agar plates containing 20 μg/mL Cm, 100 μg/mL Tmp, and 80 μg/mL Spec overnight at 37 °C. The second day, nitrocellulose transfer membranes were cut to fit 150-mm plates and prewet with sterile phosphate-buffered saline (PBS) before placement onto LB-agar plates containing 20 μg/mL Cm, 100 μg/mL Tmp, 80 μg/mL Spec, 0.1 mM IPTG, and 0.2% (w/v) L-arabinose. Library transformants were replicated onto a nitrocellulose transfer membrane (BioRad, 0.45 µm), which were then placed colony-side-up on a second nitrocellulose transfer membrane and incubated at 30 °C for 18 h. The nitrocellulose transfer membranes were washed in Tris-buffered saline (TBS) for 10 min, blocked in 5% bovine serum albumin for 30 min, and probed for 1 h with fluorescein-labeled soybean agglutinin (SBA, Vector Laboratories, Cat # FL-1011, 1:500 dilution) and Alexa Fluor 647 (AF647)-conjugated anti-His antibody (R&D Systems, Cat # IC0501R, 1:2000 dilution) following the manufacturer’s instructions. All positive hits were re-streaked onto fresh LB-agar plates containing 20 μg/mL Cm, 100 μg/mL Tmp, and 80 μg/mL Spec and grown overnight at 37 °C. Individual colonies were grown in liquid culture to confirm glycosylation of periplasmic fractions, and the sequence of the glycosylation tag was confirmed by Sanger DNA sequencing.
Protein isolation
To analyze the products of in vivo glycosylation, periplasmic extracts were derived from E. coli cultures as follows. Following induction, cells were harvested by centrifugation at 6000 × g for 2 min, after which the pellets were resuspended in an amount of 0.4 M arginine such that OD600 values were normalized to 10. Following incubation at 4 °C for 1 h, the samples were centrifuged at 16,360 × g for 1 min and the supernatant containing periplasmic extracts was collected. For purification of proteins containing a polyhistidine (6x-His) tag, cells were harvested after induction by centrifugation at 7600 × g at 4 °C for 25 min and the pellets were resuspended in desalting buffer (50 mM NaH2PO4 and 300 mM NaCl) followed by cell lysis using a Emulsiflex C5 homogenizer (Avestin) at 16,000–18,000 psi. The resulting lysate was centrifugated at 7600 × g at 4 °C for 25 min. The imidazole concentration of the resulting supernatant was adjusted to 10 mM by addition of desalting buffer containing 1 M imidazole. The supernatant was incubated at 4 °C for 1 h with HisPur Ni-NTA resin (ThermoFisher), after which the samples were applied twice to a gravity flow column at room temperature. The column was washed using desalting buffer containing 10 mM imidazole and proteins were eluted in 2 mL of desalting buffer containing 300 mM imidazole. The eluted proteins were desalted using Zeba Spin Desalting Columns (ThermoFisher) and stored at 4 °C.
For protein A purification, harvested cells were resuspended in equilibration buffer (100 mM Na2HPO4, 136 mM NaCl, pH 8), followed by cell lysis using a Emulsiflex C5 homogenizer (Avestin) at 16,000–18,000 psi. The resulting lysate was centrifugated at 7600 × g at 4 °C for 25 min. The supernatant was mixed with the equilibration buffer in a 1:1 ratio by mass, after which the samples were applied to a gravity flow column which contained MabSelect SuRe protein A resin (Cytiva). The column was washed using equilibration buffer. Proteins were eluted using 1 mL of elution buffer (165 mM glycine, pH 2.2). The eluted proteins were collected in a tube containing 100 μL of neutralizing buffer. The eluted fractions were subject to buffer exchange with PBS twice using a 10 K MWCO protein concentrator (ThermoFisher). During buffer exchange, samples were centrifugated at 3900 × g at 4 °C for 20 min.
For purification of CjPglB and DmPglB from E. coli, a single colony of BL21DE3 carrying plasmid pSN1857 or pSF-DmPglB-10xHis, respectively, was grown overnight at 37 °C in 20 mL of LB supplemented with Amp. Overnight cells were subcultured into 1 L of TB supplemented with Amp and grown until the OD600 reached a value of ~0.8. The incubation temperature was adjusted to 16 °C, after which protein expression was induced by the addition of L-arabinose to a final concentration of 0.02% (w/v). Protein expression was allowed to proceed for 16 h at 16 °C. Cells were harvested by centrifugation, resuspended in 10 mL Buffer A (50 mM HEPES, 250 mM NaCl, pH 7.4) per gram of pellet and then lysed using a homogenizer (Avestin C5 EmulsiFlex). The lysate was centrifuged to remove cell debris, and the supernatant was ultracentrifuged (99,000 ×g; Beckman 70Ti rotor) for 2 h at 4 °C. The resulting pellet containing the membrane fraction was partially resuspended in 25 mL Buffer B (50 mM HEPES, 250 mM NaCl, and 1% (w/v%) n-dodecyl-β-D-maltoside (DDM), pH 7.4). The suspension was incubated at room temperature rotating for 1 h and then ultracentrifuged (99,000 × g; Beckman 70Ti rotor) for 1 h at 4 °C. The supernatant containing DDM-solubilized DmPglB was mixed with 0.8 mL of HisPur Ni-NTA resin (ThermoFisher) equilibrated with Buffer B supplemented with protease inhibitor cocktail and incubated rotating for 24 h at 4 ⁰C. After incubation, the material was transferred to a gravity column, washed with Buffer C (50 mM HEPES, 250 mM NaCl, 15 mM imidazole and 1% (w/v) DDM, pH 7.4), and eluted using Buffer D (50 mM HEPES, 250 mM NaCl, 250 mM imidazole and 1% (w/v) DDM, pH 7.4). Purified proteins were stored at a final concentration of 3 mg/mL in a modified OST storage buffer (50 mM HEPES, 250 mM NaCl, 33% (v/v) glycerol, 1% (w/v) DDM, pH 7.5) at −20 °C.
For size exclusion chromatography (SEC), purified CjPglB or DmPglB proteins were concentrated to ~600 μL using Pierce Protein Concentrator, PES, 10 K MWCO (5−20 mL, ThermoFisher). The resulting protein was filter sterilized and further purified using Superdex 200 SEC column (Cytiva) with a buffer containing 50 mM HEPES, 250 mM NaCl, 1% DDM, pH 7.4. The peak fractions were collected and analyzed using Coomassie Brilliant Blue R-250 Staining Solution (Bio-Rad). All fractions containing PglB were concentrated using Pierce Protein Concentrator, PES, 10 K MWCO (5 mL, ThermoFisher).
Immunoblotting
Protein samples (either periplasmic fractions or purified proteins) were solubilized in 10% β-mercaptoethanol (BME) in 4x lithium dodecyl sulfate (LDS) sample buffer and resolved on Bolt Bis-Tris Plus gels (ThermoFisher). The samples were later transferred to immobilon PVDF transfer membranes and blocked with 5% milk (w/v) or 5% bovine serum albumin (w/v) in tris-buffered saline supplemented with 0.1% (w/v) Tween 20 (TBST). The following antibodies were used for immunoblotting: polyhistidine (6x-His) tag-specific polyclonal antibody (Abcam, Cat # ab1187; 1:5000 dilution); F(ab’)2-goat anti-human IgG (H + L) secondary antibody conjugated to horseradish peroxidase (HRP) (ThermoFisher, Cat # A24464; 1:5000 dilution), C. jejuni heptasaccharide glycan-specific antiserum hR6 (kind gift of Marcus Aebi, ETH Zürich; 1:1000 dilution)23, and donkey anti-rabbit IgG conjugated to HRP (Cat # ab7083; 1:5000 dilution). After probing with primary and second antibodies, the membranes were washed three times with TBST for 10 min and subsequently visualized using a ChemiDocTM MP Imaging System (Bio-Rad) and processed using Bio-Rad Image Lab software version 6.1. Glycosylation efficiency was determined by performing densitometry analysis of protein bands in anti-His immunoblots using ImageJ software version 1.37 v (https://imagej.net)58. Briefly, bands corresponding to g0 in each lane were grouped as a row or a horizontal lane and quantified using the gel analysis function in ImageJ. The bands corresponding to g1 were analyzed identically. The resulting intensity data for g0 and g1 was used to calculate percent glycosylated expressed according to the following ratio: g1/[g0 + g1]. Efficiency data was calculated from immunoblots corresponding to three biological replicates, with all data were reported as the mean ± SD. Statistical significance was determined by paired Student’s t-tests (*p < 0.05, **p < 0.01; ***p < 0.001; ****p < 0.0001) using Prism 10 for MacOS version 10.4.1.
Glycoproteomic tandem MS analysis
Glycoproteomic tandem MS analysis was performed on a total of 5 protein samples corresponding to: scFv13-R4(N34L/N77L)DQNAT, DmPglB, GalNAc5(Glc)GlcNAc-hinge-Fc, GalNAc5(Glc)GlcNAc-IgG, and GalNAc5GlcNAc-IgGm. A single biological and technical replicate (n = 1) was performed for each. Fetuin from fetal bovine serum (MilliporeSigma), a common standard used for glycoproteomics MS, was analyzed alongside each of these samples and served as positive control. Purified proteins were reduced by heating in 25 mM DL-dithiothreitol (DTT) at 50 °C for 45 min, then cooled down to room temperature, immediately alkylated by incubating with 90 mM iodoacetamide (IAA) at room temperature in dark for 20 min. Samples were loaded on the top of 10-kDa molecular weight cut-off (MWCO) filters (MilliporeSigma) and desalted by passing through 800 µL of 50 mM ammonium bicarbonate (Ambic). Proteins were recovered from the filters and reconstituted as 1 µg/µL solution in 50 mM Ambic. Sequencing grade trypsin (Promega) was added to samples at a 1:20 ratio and digestion was performed at 37 °C overnight. Trypsin activity was terminated by heating at 100 °C for 5 min. Cooled samples were reconstituted in LC-MS grade 0.1% formic acid (FA) as 0.1 µg/µL solution, passed through 0.2 µm filters (Pall). Follow-up digestion with wild-type α-lytic protease (WaLP, Cell Signaling Technology) was performed on the scFv13-R4(N34L/N77L)QYNST and DmPglB samples at 1:20 ratio at 37 °C overnight, aiming to produce easily identified peptide/glycopeptide lengths.
LC-MS/MS was carried out on an Orbitrap Eclipse Tribrid mass spectrometer equipped with a Nanospray Flex Ion Source and coupled to a Dionex UltiMate 3000 RSLCnano low-flow liquid chromatography system (ThermoFisher). Samples were trap-loaded on a 2 µm pore size 75 µm × 150 mm Acclaim PepMap 100 C18 nanoLC column. The column was equilibrated at 0.300 µL/min flowrate with 96% Buffer A (0.1% FA) and 4% Buffer B (80% acetonitrile (ACN) with 0.1% FA). A 60 min gradient in which Buffer B ramped from 4% to 62.5% was used for peptide separation. Precursors were scanned in Orbitrap at 120,000 resolution and fragments were detected in Orbitrap at 30,000 resolution59. The maximum injection time for precursors was 100 ms and for fragments was 200 ms. To scrutinize the expected glycan attachment at the anticipated sequon on antibody Fc domains, a higher collision energy dissociation (HCD) product triggered collision induced dissociation (CID) (HCDpdCID) MS/MS fragmentation cycle in 3 s frame was used. Precursors were activated by HCD energy of 28, then CID was triggered at 40. A stepped collision energy HCD product-triggered electron transfer dissociation with assisted HCD (EThcD) (stepped HCDpdEThcD) MS/MS program was used to localize multiple N-glycans on the C-terminus of DmPglB. The activation HCD energy was stepped as 20/30/40 and the assist HCD energy for ETD was 25. Common glycan oxonium ions were used as mass triggers.
LC-MS/MS data were searched in Byonic software (Protein Metrics). Peptide-spectrum matches (PSM) were annotated by Byonic considering expected glycans and common post-translational modifications (PTMs). PSMs were manually inspected by Byonic viewer (various versions, see below) and Freestyle software version 1.8 SP1 (ThermoFisher). Relative abundance of glycosylated and aglycosylated peptides was determined based on area under the curve (AUC) of deconvoluted extracted-ion chromatogram (XIC) peaks of precursors processed in Freestyle version 1.8 SP1 using the protein Averagine model60. XIC mass tolerance was determined per data acquisition quality. Accurate precursor masses and retention times were used as additional identification bases, when the fragments of either glycosylated peptide or aglycosylated peptide in a pair, but not both, were suppressed in LC-MS/MS acquisition61.
Peptides of each sample were annotated using different versions of Byonic software as follows: Byonic version 5.2.5 for scFv13-R4(N34L/N77L)QYNST, Byonic version 5.3.5 for DmPglB, Byonic version 5.0.3 for GalNAc5(Glc)GlcNAc-hinge-Fc, Byonic version 4.0.12 for GalNAc5(Glc)GlcNAc-IgG, and Byonic version 5.0.3 for GalNAc5GlcNAc-IgG. All AUC was integrated using Freestyle version 1.8 SP1 and no statistical test was used. Database searches for each sample were performed using each of these specific Byonic versions in conjunction with FASTA sequence files corresponding to each of the protein targets with fully reversed decoys. The precursor mass tolerance was set at 5 ppm, while the fragment mass tolerance was set at 20 ppm. Expected glycan composition HexNAc(6) or HexNAc(6)Hex(1) based on the specific glycosylation pathway was registered in N-glycan list. Protein list output was set with a cutoff at 1% false discovery rate (FDR) or 20 reverse sequences, whichever came last. Carbamidomethylation on cysteine was considered as fixed modification. Oxidation on methionine and deamidation on asparagine and glutamine were considered as variable modifications. For hinge-Fc and full-length IgG analysis, only fully specific trypsin-cleaved peptides with up to 2 mis-cleavages were considered (C-terminal KR). The sequentially digested scFv13-R4(N34L/N77L)DQNAT and DmPglB proteins required consideration of up to 4 mis-cleavages and WaLP specificity (C-terminal KRASTV). A semi-specific tryptic digested 48 amino acid-long peptide sequence was selected for locating multiple N-glycans on the C-terminus of DmPglB. Based on the Byonic software documentation, Byonic scores above 100 typically indicate good absolute PSM quality. Delta Mod Score, which is the score drop of fitting one PSM to the second-best localization of PTM, should be at least 10.0 to back a confident identification62. The score cut-off was taken into account when the glycopeptide data was manually curated63. There was no minimum number of unique peptides for protein identification in Byonic.
In vitro glycosylation
For in vitro glycosylation of DmPglB, 500 μL of in vitro glycosylation buffer (10 mM HEPES, pH 7.5, 10 mM MnCl2, and 0.1% (w/v) DDM) containing 50 μg of purified DmPglB and 50 μL of solvent extracted LLOs were incubated at 30 °C for 16 h. Organic solvent extraction of LLOs bearing the GalNAc5(Glc)GlcNAc glycan from the membrane of E. coli cells was performed as follows. A single colony of CLM24 carrying the plasmid pMW07-pglΔBICDEF was inoculated in LB supplemented with Cm and grown overnight at 37 °C. Overnight cells were then subcultured into 1 L of TB supplemented with Cm and grown until the OD600 reached ~0.8. The incubation temperature was adjusted to 30 °C and expression induced with 0.2% (w/v) L-arabinose. After 16 h, cells were harvested by centrifugation, resuspended in 50 mL MeOH, and dried overnight. The next day, dried cell material was scraped into a 50-mL conical tube and pulverized. The pulverized material was then thoroughly mixed with 12 mL of 2:1 mixture of chloroform:methanol, sonicated in a water bath for 10 min, centrifuged at 1756 ×g and 4 °C for 10 min, and the supernatant discarded. This step was then repeated two more times. Subsequently, 20 mL of water was thoroughly mixed with the pellet, sonicated in a water bath for 10 min, centrifuged at 3900 × g and 4 °C for 10 min, and the supernatant discarded. The pellet was vortexed with 18 mL of a 10:10:3 mixture of chloroform:methanol:water and sonicated in a water bath to homogeneity. 8 mL of methanol was subsequently added, the mixture was vortexed, and then centrifuged at 3900 × g and 4 °C for 10 min. The supernatant was decanted and retained while the pellet discarded. Then, 8 mL of chloroform and 2 mL of water were added to the supernatant, mixed, and centrifuged at 3900 × g and 4 °C for 10 min. The aqueous supernatant was aspirated and discarded, while the organic bottom layer containing the LLO was dried overnight. The next day, dried material was resuspended in cell-free glycosylation buffer (10 mM HEPES, pH 7.5, and 0.1% (w/v) DDM) and stored at −20 °C.
In vitro glycosylation was also performed using fluorescently labeled acceptor peptides. For turnover rate measurements, each reaction was prepared in a total volume of 80 µL containing: 8 µL of in vitro glycosylation buffer (500 mM HEPES, 1% (w/v) DDM), 1.6 µL of 1 M MnCl2, 0.18 μM of purified PglB, 16 µL of solvent-extracted LLOs bearing the GalNAc₅(Glc)GlcNAc structure, 0.5 µM of fluorescently labeled acceptor peptide TAMRA-GSDQNATF-NH₂ or TAMRA-GQYNSTAF-NH₂ (GenScript) and 32 µL of ddH2O. Reactions were incubated in a water bath at 30 °C, with samples collected at different time points. Reactions were stopped by boiling the sample at 90 °C for 5 min. For Michaelis–Menten kinetics, reactions were performed in a total volume of 10 µL containing: 1 µL of in vitro glycosylation buffer, 0.2 µL of 1 M MnCl2, 0.18 μM of purified PglB, 2 µL of solvent-extracted LLOs bearing the GalNAc₅(Glc)GlcNAc structure, varying concentrations of fluorescently labeled acceptor peptide (ranging from 0.25 to 30 µM), and ddH2O as needed. The reactions were incubated for 18 h at 30 °C and stopped by boiling the sample at 90 °C for 5 min.
In-gel fluorescence detection
Samples were diluted 1:6 with Novex Tricine SDS Running Buffer (1x). Each sample was then mixed with dye that was produced in-house and boiled at 80 °C for 2 min. The dye consisted of 200 mM Tris-Cl (pH 6.8), 8% (w/v) sodium dodecyl sulfate (SDS; electrophoresis grade), and 40% (v/v) glycerol. For Michaelis–Menten kinetics, the samples were normalized to a final concentration of 0.25 µM. A total of 8 µL of each sample was loaded onto Novex 16% Tricine Mini Protein Gels (1.0 mm thickness). The Spectra™ Multicolor Low Range Protein Ladder was used as the molecular weight marker. The gel was run at 70 V for 2.5 h at 4 °C and subsequently imaged using a ChemiDoc MP Imaging System (Bio-Rad) and processed using Bio-Rad Image Lab version 6.1 software. DyLight 550 was used to visualize the fluorescently labeled peptides, while the Spectra ladder was visualized using Cy5.5.
Chemoenzymatic glycan remodeling
A total of 400 U of exo-α-N-acetylgalactosaminidase (New England Biolabs, Cat # P0734S) was added to a solution of GalNAc5GlcNAc-hinge-Fc dimer (200 µg) in 100 µL GlycoBuffer 1 (50 mM NaOAc, 5 mM CaCl2, pH 5.5) and the reaction mixture was incubated at room temperature. Reaction progress was monitored by LC-ESI-MS using an Exactive Plus Orbitrap Mass Spectrometer (Thermo Scientific) equipped with an Agilent Poroshell 300SB C8 column (5 μm, 1.0 × 75 mm) and was found to be complete after just 2 h. The sample was then buffer exchanged to 100 mM Tris pH 7 buffer using an Amicon® Ultra 0.5 mL 10 K Centrifugal Filter (Millipore) and concentrated to 2 mg/mL. To this solution was added G2-oxazoline (320 µg, 30 mol eq), followed by 1 µg of EndoS2-D184M to a final concentration of 0.4% (w/w) relative to the hinge-Fc. The sample was incubated at 30 °C, and the reaction monitored by LC-ESI-MS. After 30 min, the reaction was complete, and the G2-hinge-Fc product was purified using a 1 mL Protein A HP column (Cytiva) following standard procedures46. The final product was buffer exchanged to PBS by centrifugal filtration and stored at −80 °C until later use.
ELISA
For binding assays between IgG-Fc domain and Fcγ receptor, FcγRIIIA V158 (10 μg/mL; Sino Biological) in PBS buffer (pH 7.4) was coated onto a high-binding 96-well plate (VWR) overnight at 4 °C. After washing with PBST (PBS, 0.1% Tween 20) the plate was blocked overnight at 4 °C with 200 μL of 5% milk (w/v) in PBST. The plate was washed three times and 100-μL serial dilutions of sample were added to each well. The concentrations of each glycosylated and aglycosylated sample ranged from 0.08 to 10 μg/mL (fivefold serial dilutions). All IgG-Fc glycoforms were purified proteins except for commercial trastuzumab (HY-P9907, MedChem Express). The plate was placed on a shaker and incubated for 1 h at 37 °C. After incubation, the plate was washed three times and incubated for 1 h with 100 μL of F(ab’)2-goat anti-human IgG (H + L) antibody conjugated to HRP (ThermoFisher, Cat # A24464; 1:5000 dilution). After three washes, 100 μL of 3,3’,5,5’ tetramethylbenzidine (TMB) ELISA substrate (ThermoFisher) were added to each well for signal development. The reaction was stopped upon addition of 100 μL of 2 M sulfuric acid. The absorbance of samples was measured at 450 nm using a SpectraMax 190 microplate reader (Molecular Devices) and the data was analyzed using GraphPad Prism 10 software (version 10.4.1) by nonlinear regression analysis.
Sequence alignments and structural models
Sequences were aligned using the Clustal Omega web server64. The structure of ClPglB with bound DQNAT substrate peptide was derived from Protein Data Bank (PDB) entry 5OGL15. Structures for all other OSTs without peptides in the substrate-binding pocket of the enzyme were generated with the AlphaFold2 (AF2) protein structure prediction algorithm implemented with ColabFold36,37. ColabFold was downloaded and installed from the github repository [https://github.com/YoshitakaMo/localcolabfold]. All structures were generated with standard settings, 8 recycles and relaxed with Amber. To obtain structures of OSTs with DQNAT peptide in the substrate-binding pocket of the enzyme, we manually aligned the model OST structures from ColabFold-AF2 to the crystal structure of ClPglB complexed with DQNAT peptide (PDB entry 5OGL)15 in PyMol followed by Monte Carlo minimization (MCM) relaxation with Rosetta. To model the QYNST peptide in the substrate-binding pocket of each OST, we replaced the DQNAT peptide with QYNST peptide in the PyMol-aligned OST-peptide complexes (or in 5OGL directly in the case of ClPglB) and relaxed the QYNST peptide in the peptide-binding pocket by MCM relaxation with Rosetta. For both DQNAT- and QYNST-bound OST complexes, twenty-five structures were generated using the Rosetta relax function with default parameters for each enzyme-peptide complex and the structure with the lowest total score was selected. Electrostatic surfaces were generated based on electrostatics calculations using the APBS plugin in PyMOL, which combines standard focusing techniques and the Bank-Holst algorithm into a parallel focusing method for the solution of the Poisson-Boltzmann equation (PBE) for nanoscale systems38.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Unless otherwise noted, all data supporting the results of this study can be found in the article, supplementary information file, and source data files. Source Data are provided with this paper. Crystal structure data for PglB from C. lari strain RM2100 used in this study are available in the PDB under accession code PDB 5OGL. All protein MS data generated in this study are available in the proteomeXchange.org repository under accession code PXD064379. All OST structural models generated in this study are available in ModelArchive.org under accession code ma-ost-pep-comp [https://www.modelarchive.org/doi/10.5452/ma-ost-pep-comp]. Source data are provided with this paper.
References
Abu-Qarn, M., Eichler, J. & Sharon, N. Not just for Eukarya anymore: protein glycosylation in bacteria and archaea. Curr. Opin. Struct. Biol. 18, 544–550 (2008).
Apweiler, R., Hermjakob, H. & Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 (1999).
Stanley, P., Moremen, K. W., Lewis, N. E., Taniguchi, N. & Aebi, M. In Essentials of Glycobiology, Edn. 4th. (eds. Varki, A. et al.) 103−116 (Cold Spring Harbor (NY), 2022).
Khoury, G. A., Baliban, R. C. & Floudas, C. A. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 1, 90 (2011).
Seeberger, P.H., Freedberg, D.I. & Cummings, R.D. In Essentials of Glycobiology, 4th Edn. (eds. Varki, A. et al.) 771−784 (Cold Spring Harbor (NY), 2022).
Walsh, C. T., Garneau-Tsodikova, S. & Gatto, G. J. Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem. Int Ed. Engl. 44, 7342–7372 (2005).
Shrimal, S., Cherepanova, N. A. & Gilmore, R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin Cell Dev. Biol. 41, 71–78 (2015).
Weerapana, E. & Imperiali, B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology 16, 91R–101R (2006).
Dell, A., Galadari, A., Sastre, F. & Hitchen, P. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010, 148178 (2010).
Kelleher, D. J. & Gilmore, R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R (2006).
Mohanty, S., Chaudhary, B. P. & Zoetewey, D. Structural insight into the mechanism of N-linked glycosylation by oligosaccharyltransferase. Biomolecules 10, 624 (2020).
Ramirez, A. S., Kowal, J. & Locher, K. P. Cryo-electron microscopy structures of human oligosaccharyltransferase complexes OST-A and OST-B. Science 366, 1372–1375 (2019).
Wild, R. et al. Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science 359, 545–550 (2018).
Matsumoto, S. et al. Crystal structures of an archaeal oligosaccharyltransferase provide insights into the catalytic cycle of N-linked protein glycosylation. Proc. Natl. Acad. Sci. USA 110, 17868–17873 (2013).
Lizak, C., Gerber, S., Numao, S., Aebi, M. & Locher, K. P. X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 (2011).
Kowarik, M. et al. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 25, 1957–1966 (2006).
Ollis, A. A. et al. Substitute sweeteners: diverse bacterial oligosaccharyltransferases with unique N-glycosylation site preferences. Sci. Rep. 5, 15237 (2015).
Wacker, M. et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793 (2002).
Anthony, R. M. et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 320, 373–376 (2008).
Debre, M. et al. Infusion of Fc gamma fragments for treatment of children with acute immune thrombocytopenic purpura. Lancet 342, 945–949 (1993).
Fisher, A. C. et al. Production of secretory and extracellular N-linked glycoproteins in Escherichia coli. Appl. Environ. Microbiol. 77, 871–881 (2011).
Schwarz, F. et al. A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat. Chem. Biol. 6, 264–266 (2010).
Schwarz, F. et al. Relaxed acceptor site specificity of bacterial oligosaccharyltransferase in vivo. Glycobiology 21, 45–54 (2011).
Valderrama-Rincon, J. D. et al. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat. Chem. Biol. 8, 434–436 (2012).
Glasscock, C. J. et al. A flow cytometric approach to engineering Escherichia coli for improved eukaryotic protein glycosylation. Metab. Eng. 47, 488–495 (2018).
Yan, Q. & Lennarz, W. J. Studies on the function of oligosaccharyl transferase subunits. Stt3p is directly involved in the glycosylation process. J. Biol. Chem. 277, 47692–47700 (2002).
Ielmini, M. V. & Feldman, M. F. Desulfovibrio desulfuricans PglB homolog possesses oligosaccharyltransferase activity with relaxed glycan specificity and distinct protein acceptor sequence requirements. Glycobiology 21, 734–742 (2011).
Li, M. et al. Shotgun scanning glycomutagenesis: a simple and efficient strategy for constructing and characterizing neoglycoproteins. Proc. Natl. Acad. Sci. USA 118, e2107440118 (2021).
Santos-Silva, T. et al. Crystal structure of the 16 heme cytochrome from Desulfovibrio gigas: a glycosylated protein in a sulphate-reducing bacterium. J. Mol. Biol. 370, 659–673 (2007).
Ollis, A. A., Zhang, S., Fisher, A. C. & DeLisa, M. P. Engineered oligosaccharyltransferases with greatly relaxed acceptor-site specificity. Nat. Chem. Biol. 10, 816–822 (2014).
Chen, M. M., Glover, K. J. & Imperiali, B. From peptide to protein: comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry 46, 5579–5585 (2007).
Bokhari, H., Maryam, A., Shahid, R. & Siddiqi, A. R. Oligosaccharyltransferase PglB of Campylobacter jejuni is a glycoprotein. World J. Microbiol. Biotechnol. 36, 9 (2019).
Zhang, G., Brokx, S. & Weiner, J. H. Extracellular accumulation of recombinant proteins fused to the carrier protein YebF in Escherichia coli. Nat. Biotechnol. 24, 100–104 (2006).
Gerber, S. et al. Mechanism of bacterial oligosaccharyltransferase: in vitro quantification of sequon binding and catalysis. J. Biol. Chem. 288, 8849–8861 (2013).
Liu, F. et al. Rationally designed short polyisoprenol-linked PglB substrates for engineered polypeptide and protein N-glycosylation. J. Am. Chem. Soc. 136, 566–569 (2014).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041 (2001).
Taguchi, Y. et al. The structure of an archaeal oligosaccharyltransferase provides insight into the strict exclusion of proline from the N-glycosylation sequon. Commun. Biol. 4, 941 (2021).
Mazor, Y., Van Blarcom, T., Mabry, R., Iverson, B. L. & Georgiou, G. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli. Nat. Biotechnol. 25, 563–565 (2007).
Li, T., Tong, X., Yang, Q., Giddens, J. P. & Wang, L. X. Glycosynthase mutants of endoglycosidase S2 show potent transglycosylation activity and remarkably relaxed substrate specificity for antibody glycosylation remodeling. J. Biol. Chem. 291, 16508–16518 (2016).
Ravetch, J. V. & Perussia, B. Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J. Exp. Med 170, 481–497 (1989).
Bruhns, P. et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725 (2009).
de Taeye, S. W. et al. FcgammaR binding and ADCC activity of human IgG allotypes. Front Immunol. 11, 740 (2020).
Wei, Y. et al. Glycoengineering of human IgG1-Fc through combined yeast expression and in vitro chemoenzymatic glycosylation. Biochemistry 47, 10294–10304 (2008).
Li, T. et al. Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. USA 114, 3485–3490 (2017).
Kurogochi, M. et al. Glycoengineered monoclonal antibodies with homogeneous glycan (M3, G0, G2, and A2) using a chemoenzymatic approach have different affinities for FcgammaRIIIa and variable antibody-dependent cellular cytotoxicity activities. PLoS ONE 10, e0132848 (2015).
Niwa, R. et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 64, 2127–2133 (2004).
Napiorkowska, M. et al. Molecular basis of lipid-linked oligosaccharide recognition and processing by bacterial oligosaccharyltransferase. Nat. Struct. Mol. Biol. 24, 1100–1106 (2017).
Simmons, L. C. et al. Expression of full-length immunoglobulins in Escherichia coli: rapid and efficient production of aglycosylated antibodies. J. Immunol. Methods 263, 133–147 (2002).
Rashid, M. H. Full-length recombinant antibodies from Escherichia coli: production, characterization, effector function (Fc) engineering, and clinical evaluation. MAbs 14, 2111748 (2022).
Jung, S. T. et al. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcgammaRIIa affinity and selectivity. ACS Chem. Biol. 8, 368–375 (2013).
Jung, S. T. et al. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc. Natl. Acad. Sci. USA 107, 604–609 (2010).
Kang, T. H. et al. An engineered human Fc variant with exquisite selectivity for FcgammaRIIIa(V158) reveals that ligation of FcgammaRIIIa mediates potent antibody dependent cellular phagocytosis with GM-CSF-differentiated macrophages. Front Immunol. 10, 562 (2019).
Feldman, M. F. et al. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. USA 102, 3016–3021 (2005).
Lefebre, M. D. & Valvano, M. A. Construction and evaluation of plasmid vectors optimized for constitutive and regulated gene expression in Burkholderia cepacia complex isolates. Appl. Environ. Microbiol. 68, 5956–5964 (2002).
Kowarik, M. et al. N-linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science 314, 1148–1150 (2006).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Shajahan, A., Supekar, N. T., Gleinich, A. S. & Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 30, 981–988 (2020).
Senko, M. W., Beu, S. C. & McLafferty, F. W. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 6, 229–233 (1995).
Klein, J. & Zaia, J. Relative retention time estimation improves N-glycopeptide identifications by LC-MS/MS. J. Proteome Res. 19, 2113–2121 (2020).
Bern, M., Kil, Y. J. & Becker, C. Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinforma. 40, 13.20.11–13.20.14 (2012).
Chalk, R. et al. Identification, mapping and relative quantitation of SARS-CoV-2 Spike glycopeptides by mass-retention time fingerprinting. Commun. Biol. 4, 934 (2021).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Acknowledgements
We thank Anne Ollis (Merck) for design and construction of plasmid pTrc99S-spDsbA-hinge-Fc, Judith Merritt (Glycobia, Inc.) for providing plasmid pMW07-pgl∆B, George Georgiou (University of Texas, Austin) for providing JUDE-1 E. coli cells and plasmid pMAZ360-YMF10-IgG, and Markus Aebi (ETH Zürich) for providing antiserum used in this work. This work was supported by the Defense Advanced Research Projects Agency (DARPA contract W911NF-23-2-0039 to M.C.J. and M.P.D.), the Defense Threat Reduction Agency (grants HDTRA1-15-10052 and HDTRA1-20-10004 to M.P.D. and M.C.J.), the National Science Foundation (grants CBET-1605242 to M.P.D., CBET-1936823 and MCB-1413563 to M.P.D. and M.C.J., and DMR-1933525 to P.A.), and the National Institutes of Health (grants R01GM127578 to M.P.D. and J.J.G., R01GM080374 to L.-X.W., and R24GM137782 to P.A.). S.W.H. was supported by a training grant from the National Institutes of Health NIBIB (T32EB023860). E.J.B was supported by an NIH/NIGMS Chemical Biology Interface Training Grant (T32GM138826) and an NSF Graduate Research Fellowship (DGE-2139899).
Author information
Authors and Affiliations
Contributions
B.S.: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization. T.C.D.: methodology, validation, formal analysis, investigation, writing – review & editing. S.P.M.: methodology, software, formal analysis, investigation related to structural analysis. M.N.T.: methodology and investigation related to bioprospecting. S.W.H.: methodology and investigation related to glycoSNAP. E.J.B. methodology and investigation related to autoglycosylation. D.N.O.: methodology and investigation related to chemoenzymatic glycan remodeling. A.P.: investigation related to glycosylation analysis. X.Y. methodology, formal analysis, investigation related to protein MS analysis. S.G.: investigation related to enzyme kinetics. C.A.A.: supervision. P.A.: methodology, formal analysis, supervision. J.J.G.: supervision. M.C.J.: conceptualization, supervision, funding acquisition. L.-X.W.: conceptualization, supervision. M.P.D.: conceptualization, methodology, formal analysis, writing – original draft, writing – review & editing, visualization, supervision, project administration, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
M.P.D. and M.C.J. have financial interests in Gauntlet, Inc. and Resilience, Inc. M.P.D. also has financial interests in June Bio APS, Glycobia Inc., UbiquiTx Inc., and Versatope Therapeutics Inc. M.P.D.’s and M.C.J.’s interests are reviewed and managed by Cornell University and Stanford University, respectively, in accordance with their conflict-of-interest policies. M.P.D. and M.C.J. declare no additional competing interests. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Smita Mohanty, Brendan Wren, Di Wu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sotomayor, B., Donahue, T.C., Mahajan, S.P. et al. Discovery of a single-subunit oligosaccharyltransferase that enables glycosylation of full-length IgG antibodies in bacteria. Nat Commun 16, 6152 (2025). https://doi.org/10.1038/s41467-025-61440-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61440-7
This article is cited by
-
Antibody glycosylation in neuroimmune diseases
Journal of Translational Medicine (2025)
