
Abstract
Outer membrane proteins (OMPs) define the surface biology of Gram-negative bacteria, with roles in adhesion, transport, catalysis and signalling. Specifically, porin beta-barrels are common diffusion channels, predominantly monomeric/trimeric in nature. Here we show that the major OMP of the bacterial predator Bdellovibrio bacteriovorus, PopA, differs from this architecture, forming a pentameric porin-like superstructure. Our X-ray and cryo-EM structures reveal a bowl-shape composite outer β-wall, which houses a central chamber that encloses a section of the lipid bilayer. We demonstrate that PopA, reported to insert into prey inner membrane, causes defects when directed into Escherichia coli membranes. We discover widespread PopA homologues, including likely tetramers and hexamers, that retain the lipid chamber; a similar chamber is formed by an unrelated smaller closed-barrel family, implicating this as a general feature. Our work thus defines oligomeric OMP superfamilies, whose deviation from prior structures requires us to revisit existing membrane-interaction motifs and folding models.
Similar content being viewed by others

Introduction
Outer membrane proteins (OMPs) have major roles in bacterial and organelle biology, with classical beta-barrel structures following accepted common rules and folding pathways1. Proteins in the outer membrane (OM) are dominantly composed of β-strands, and those in the inner membrane (IM) α-helical; one anomaly to this preference comes from rare inter-membrane transfer events, such as the translocation of the Neisseria OMP/porin PorB into mitochondria during pathogenesis2 or the transfer of an OMP from the bacterial predator Bdellovibrio bacteriovorus into the IM of the prey cell3,4. PorB has been characterized, adopting a consensus porin fold2; porins are chiefly monomeric or trimeric barrels of low substrate specificity5. B. bacteriovorus is a model predatory bacterium, whose ability to enter and kill a wide range of other Gram-negative bacteria is of interest to the study of staged lifecycles and bacterial cell physiology, but also of potential use in several applied contexts6. Prey membrane function and manipulation is key to Bdellovibrio predation, factoring during cell entry and exit, and potentially nutrient transfer7. Bdellovibrio synthesizes an unusual neutral lipid A8, but retains identifiable OM Bam, Lpt and Mla transport machineries9. Two independent studies reported the transfer of a Bdellovibrio OMP into the inner membrane of prey3,4 – later determined to be encoded by the bd0427 gene10. Ficoll-separated OM fractions revealed that Bd0427 is the major OMP of HD100, and its homologues are likewise the major component in other predatory strains11.
Here we discover that Bd0427 defines a distinct class of oligomeric porin-like proteins, differing from the hundreds of characterised Gram-negative trimeric barrels by adopting a pentameric state. Our x-ray and cryo-EM structures reveal a plugged lumen and a central chamber that forms from a “folding-down” of the barrel wall and is thus able to trap a lipid monolayer. We use molecular dynamics simulations to visualize interactions with the membrane and demonstrate that recombinant expression in E. coli prey leads to IM defects. Structure-based searches identify homologues in both predatory and non-predatory species, and establish Bd0427 (which we rename PopA) as the exemplar of a distinct class of membrane proteins.
Results
PopA adopts a pentameric assembly distinct from other OMPs
We were able to produce and refold Bd0427 (aa 21-353) using dropwise dilution of 8 M urea-solubilized inclusion material into a buffer containing either β-octylglucoside (βOG) or dodecylmaltoside (DDM) detergents. Refolding, as judged by heat-modifiability (Supplementary Fig. 1), allowed purification and generation of several crystal forms; herein we focus on the best-diffracting C2221 form (2.8 Å resolution, statistics provided in Supplementary Table 1). Representative electron density from several key regions of the fold is provided in Supplementary Fig. 2. There are five Bd0427 copies in the asymmetric unit, forming an unprecedented pentameric OMP; from hereon in, we rename Bd0427 as PopA (pentameric outer membrane protein A).
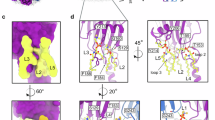
PopA forms a 16-stranded antiparallel β-barrel, with the lumen plugged by an alpha-helical domain, as shown in Fig. 1a,b,c. Distinct from a regular cylinder of uniform height, strands 1–4 and 14–16 are shorter, creating a “flattened edge” 20 Å high on the inward side, considerably shorter than the 55 Å outer rim. PopA retains some features present in “textbook” OMPs, with a hydrophobic band of residues ~25 Å wide for interaction with the bilayer, and identifiable aromatic girdles (Fig. 1d) known to delineate the membrane interface5. PopA has weak structural similarity to several experimentally-determined proteins, including FadL12 (3.3 Å rmsd, Cα) and OmpT13 (3.6 Å rmsd). Like FadL, PopA has a plug and disrupted hydrogen-bonding between barrel strands (but at a different relative location, Supplementary Fig. 3) and has two more β-strands in its fold. PopA also differs from trimeric porins by using seams at its edges to form a continuous outer rim (Fig. 1g). Hence, the PopA fold is divergent from previously characterised OMPs.

A Ribbon diagram of the PopA pentamer. B Two views highlighting the wall-reinforcing alpha helix of loop 2 (pale yellow), plug (pink) and oligomerisation loop 8 (green, forms centre of structure shown in panel A). C Electrostatic representation of PopA pentamer faces (extracellular left, periplasmic right). D Aromatic girdle residues (blue sticks), and inferred membrane spacing. E Lysine ring (purple sticks) at extracellular face. F Hydrophobic span (white) of PopA transmembrane β-strands. G Monomers make a continuous outer sheet wall via seams (backbone shown in stickform) H, I Residue sidechain detail for loop 8 and central chamber. J Oligomer comparison (overlay, centre) of PopA (blue, left) to trimeric anion transporter (1E5414, red, right), demonstrating arrangement around different loops (coloured individually, strand β16 of both is purple to provide a barrel orientation guide).
The pentameric assembly is radially organized around loop 8, a folded-over edge of the barrel, making a chamber at the centre of the oligomer (Fig. 2c–e). The net effect of oligomerisation creates a ~100 Å wide bowl-like architecture (rather than five grouped cylinders), whose outer rim is formed from a continuous “wall” of strands 5–14 (Fig. 1c); this is also distinct from classical porins5. The extracellular face of the pentamer is strongly negatively charged (Fig. 1c) with a notable ring of lysines beneath (Fig. 1e). In comparison to classical 16-stranded trimeric porins, the closest relative to PopA is an anion transporter from Comamonas acidovorans (PDB 1E5414, Foldseek15 score 4.8 e-5, sequence identity 13%); superposition reveals that the PopA fold has an altered register such that β-strand order differs by 2 e.g., PopA strand 12 sits over trimeric porin strand 14. This has the outcome of roughly aligning two chains of the PopA pentamer with classical porins, with the central chamber replacing the third.

A Lefthand, ribbon diagram of FadL with plug (pink). Righthand, overlay of PopA plug (blue). B Lack of pore lumen in PopA, showing constriction of solvent-accessible volume (pink) as calculated using MOLE17. C Side-on and bottom views of PopA pentamer, demonstrative that the central chamber is located at the phospholipid/periplasmic side of the asymmetric outer membrane bilayer. D (side view), E (end-on) Spacefill of hydrophobic residues lining central chamber (from one monomer), using cutaway-view omitting one chain. F (from periplasm), G (in plane of membrane) Electron density and fit of alkyl chains in central chamber, with surrounding aromatic residues, 2fo-fc map at 1σ. H Fit of bound palmitate (white) under sidewall helix, 2fo-fc map at 1σ. The carboxylate of palmitate co-ordinates with Arg88 and the acyl chain sits in a cleft composed of several hydrophobic residues.
The FadL-like plug (Fig. 2a) is held in place by polar and nonpolar interactions (Consurf16 analysis indicates that the plug is highly-conserved, Supplementary Fig. 4 and 5). The three small α-helices of the plug, along with R32 and R43, prevent any potential channel through the barrel of PopA. This closure can be quantified by analysis with MOLE17, which confirms a constriction point of 0 Å (Fig. 2b).
PopA assembles around a central, trapped lipid monolayer
The pentamer has a central chamber formed by the association of five loop 8 peptides that is open to the periplasmic side of the bilayer (Fig. 2c); the chamber is effectively shaped to the dimensions of a lipid monolayer. Consistent with this, the inside of the chamber is lined with hydrophobic residues contributed by the barrel surface and lower face of loop 8 (Fig. 2d, e). During model building, it became apparent that this feature exhibited strong electron density, into which we could model ten octane molecules (Fig. 2f, g), representative of the tail of βOG detergent. There is no apparent channel for lipids to move laterally into or out of this central chamber. A second region of difference density was located behind the α-helix of loop 2 (Supplementary Fig. 6). While using the Alphafold3 server18 to replicate fatty acid binding at the central chamber, we noticed it would additionally place palmitate within this density. Refinement verified that palmitate fits the density well (Fig. 2h), with the carboxylate contacting the side chain of the conserved R88 and the acyl chain situated opposite the bulge created by the disrupted β-strands around S189 that opens to the bilayer. Fatty acid binding adds to the features (general fold, β-bulge, plug domain) shared between PopA and FadL.
Cryo-EM and molecular dynamics simulations confirm PopA monolayer-trapping features
Having observed our original structural information from refolded PopA, we set out to interrogate the native folded state, using our existing structure to inform on introducing an internal His-tag in loop 2, referred to hereafter as PopAhis. The protein was successfully solubilized in 1% (w/v) DDM, and was amenable to cryo-EM analysis. A final particle set of 365,835 was subjected to ab initio reconstitution followed by non-uniform refinement in C1 symmetry. The resultant map at 2.85 Å resolution (according to the gold-standard Fourier shell correlation curve with a threshold of 0.143, Supplementary Figs. 7, 8) allowed building of a model with correlation coefficient (CC mask) of 0.87 (Supplementary Table 2).
The cryo-EM structure of PopAhis retains the five-fold arrangement of the X-ray models, demonstrating that differences in native:refolding methodology and DDM:βOG usage have no significant outcome on the PopA structure (Fig. 3a, chain rmsd to X-ray ranging from 0.7–1.1 Å). The central chamber in PopAhis also has density for alkyl chains, equivalent to 11 DDM molecules, two per monomer with an additional one at the centre (Fig. 3a).

A Cryo-EM density map of native PopAhis, coloured by local resolution. The gross features match those of x-ray determined refolded PopA; the map (yellow, righthand side) is demonstrative of eleven modelled detergent molecules. B PopA simulated in a model outer membrane (upper), showing moderate deformation of the bilayer (lower). C Comparison of experimental alkyl chains (left), exemplar simulation lipids (centre) and density of lipids averaged over 3 500 ns molecular dynamics simulations (right). D Simulation of PopA with palmitate (PAL) and contact time with residues of pocket. E Liposome swelling assays of PopA (light bars) relative to E. coli OmpF (dark bars), illustrating significant permeability to glycine and alanine only. Liposome swelling assays show an average of three technical replicates.
We subsequently performed multi-scale molecular dynamics simulations of PopA in both asymmetrical OM and symmetrical IM models, as both a monomer and pentamer (Fig. 3b, c). Based on the experimental density, we inserted five phosphatidylethanolamine lipids into the central chamber and calculated the simulated densities over three repeats of 500 ns MD simulations. The simulated density agrees favourably with the densities observed from the structural data. As a monomer, PopA shows further fluctuations in the extracellular loops, illustrating that the individual monomers are stabilised in the pentamer complex (Supplementary Fig. 9–11). The cryo-EM structure also retains density behind the helix of loop 2 (Supplementary Fig. 6b), and we were able to validate the x-ray and cryo-EM attribution of this to palmitate (and thus potential FadL-like role) by demonstrating high contact residency time for this ligand during simulations (Fig. 3d).
PopA is not a porin but can induce defects in E. coli prey membranes
We undertook transport assays to monitor if PopA demonstrates any specificity for size or class of substrate. We utilised liposome swelling assays, comparing swelling rates to those of proteoliposomes containing the well-characterized E. coli OmpF porin19. Relative to L-arabinose transport through OmpF (set as 100% swelling rate), PopAhis was able to act as a pore, but only able to permit the passage of small substrates, notably glycine and alanine (Fig. 3e). These data are in agreement with the lack of a sizable channel in our structures. We were unable to test a FadL-like role for PopA because these assays are performed in-vivo20 knowing the fatty acid transport profile of the native strain, which is currently obscure for B. bacteriovorus. To test PopA function via a different route, we attempted to delete the bd0427/popA gene. There was no observable deficit in E. coli predation or lifecycle progression upon bd0427 deletion from B. bacteriovorus HD100 when using host-dependent cultures. We also attempted bd0427 deletion in a host-independent background21, but were unable to obtain this despite a reasonable degree of persistence (see methods) – the cells are thus likely using PopA for an OM function, but further experimentation is needed to outline a potential role for PopA in this conditional context.
To evaluate the potential impact of PopA in the prey cell envelope, we induced the production of PopA, with its native signal peptide, in E. coli MG1655 in which the OM and IM were differentially labelled with fluorescent proteins. Imaging of these cells by phase contrast and fluorescence time-lapse microscopy revealed strong envelope defects, including bulging, unlike those of an E. coli OmpF control (Fig. 4a–c). Notably, among the bulging events observed in 16% of PopA-producing cells (Fig. 4b), IM disruptions (or simultaneous IM and OM defects) were significantly more prevalent compared to the few bulging (4.5%) OmpF–producing cells (Fig. 4c), suggestive of a potential interaction of PopA with the E. coli IM.

Representative time-lapse fluorescence microscopy of the E. coli MG1655 envelope reporter strain (MG1655ER) carrying lopB-mCherry (for OM labelling, red) and msfgfp-glpT (for IM labelling, green), and producing the OmpFE. coli(A) or the PopA protein (B). Examples of envelope defects (including IM and OM bulging events) are shown (white arrowheads). Note that envelope bulging events were observed at various timepoints throughout the movie. Scale bar, 5 μm. C Boxplot representation of the bulging events frequency in E. coli cells producing OmpFE. coli, PopA or PopA-intermChy. The black dot corresponds to an outlier. The bold horizontal bar represents the median value. Means are represented by the empty circle. The bottom and top boundaries of the internal boxplot correspond to the 25th and 75th percentiles, respectively; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles. Pairwise comparisons from at least three biological replicates are indicated above the plots (NS, nonsignificant; **p ≤ 0.01; ***p ≤ 0.001; two-tailed Wilcoxon’s t test). OmpF vs PopA: p = 1.5e−6; OmpF vs PopA-intermChy: p = 0.0011; PopA vs PopA -intermChy: p = 0.52. The number of analysed cells (n) is indicated below for each condition. D Boxplot representation of the frequency of IM, OM, or IM and OM bulging events for E. coli MG1655ER producing OmpFE. coli (blue) or PopA (orange) protein. Based on the subset of bacteria that exhibited bulging events, as quantified previously (related to Fig. 4C). Pairwise comparisons from at least three biological replicates are indicated above the plots (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; two-tailed Wilcoxon’s t test). Inner membrane: p = 0.04374; Outer membrane: p = 0.00047; Inner and outer membrane: p = 0.00164.
To assess the localization of B. bacteriovorus PopA when induced in E. coli, we fused the mCherry protein to the same position as the previously used his-tag, resulting in the PopA-intermChy fusion. We first confirmed that the production of PopA-intermChy results in the same percentage of envelope defects as observed with untagged PopA (Fig. 4c). When induced in E. coli, PopA-intermChy localizes in distinct patches, randomly distributed along the cell envelope (Fig. 5); envelope defects were only observed in cells producing PopA (i.e., with a detectable mCherry fluorescent signal). The non-negligible fraction of cells without PopA-intermChy (33.2% ± 2.8, Supplementary Fig. 12) implies that the frequency of PopA-induced bulging calculated in Fig. 4b, c is likely underestimated.

A Representative time-lapse fluorescence microscopy of E. coli MG1655 producing the PopA-intermChy fusion protein (top panel, phase contrast; middle panel, mCherry channel; bottom panel, overlay of the phase contrast and the mCherry channels). Examples of envelope defects are shown (white arrowheads). Scale bar, 5 μm. Live imaging was performed three times with similar results. B The outer and the inner membrane of PopA-producing E. coli were separated by centrifugation in a two-step sucrose density gradient (see Methods). PopA was found in two different variants: The PopA protein is primarily localized in the outer membrane of E. coli with an expected size of approximately 40 kDa. However, it also exhibits localization in the inner membrane, where it appears as a higher molecular weight form (~65 kDa). Control markers include the major outer membrane lipoprotein Lpp (rabbit polyclonal antibody, Collet lab) and the inner membrane lipoprotein-releasing system LolC (rabbit polyclonal antibody, Collet lab). Goat anti-rabbit IgG-peroxidase antibody (Sigma) was used as a secondary antibody (0.2 μg/mL final). Molecular weight markers (kDa) are indicated on the left. Fractionation experiments were performed in duplicate with identical results. C Western blot of whole-cell protein extracts from B. bacteriovorus in attack phase (AP) or in growth phase (GP, 2 hours post-infection) was probed with α-PopA antibodies. The presence of asterisks (*) indicates the different forms of PopA that are exclusively present in the presence of the prey (i.e., in GP). Molecular weight markers (kDa) are indicated on the left. Western blot analysis was independently performed on two biological replicates, yielding consistent results.
In line with the microscopy data, a sucrose gradient separation of the membrane fraction showed that PopA, when heterologously produced in E. coli, localizes in the OM as expected for a β-barrel (PopA, migrating at a size of ~40 kDa) but also in the IM fraction, although with a slower migration (PopA*, ~70 kDa) (Fig. 5). This result is consistent with a native blot analysis of PopA in B. bacteriovorus attack phase cells (i.e., in the absence of prey) and during the growth phase inside prey (2 hours post-infection) (Fig. 5).
Oligomerisation and chamber formation is conserved in PopA relatives and beyond
Because the validated pentamer was distinct from all previously described OMPs, we were interested to compare whether AlphaFold18 would model this in line with our observations – indeed, this occurs with high confidence (Supplementary Fig. 13). PopA was previously identified as the major OMP of strain HD100, with homologues (23-52 % sequence identity) also representing the major OMPs of related strains11. These also model confidently as pentamers, including an operon of two putative pentameric OMPs, those of B. stolpii (Fig. 6a, differing by 100aa). Beyond predatory strains, FoldSeek15 identifies homologues in several broad, dispersed bacterial groupings (Supplementary Table 3, and discussion). Intriguingly, some of these homologues expand the scope of the superfamily, modelling confidently as tetramers and hexamers (a selection is shown in Fig. 6a, confidence metrics for all models are presented in Supplementary Fig, 14–21, and models have been deposited at modelarchive.org). Hexamers typically adapt to fill the relatively larger central cavity with longer loops, often including α-helical regions, whereas tetramers pack around a smaller loop 8 (Fig. 6a). Regardless of chamber size, all superfamily members retain the hydrophobicity patterns of lipid interaction observed for PopA.

A Alphafold18/Collabfold62 models for select PopA superfamily members. Gene name, source organism and protein size annotated below. Hexamers from Fibrobacter, Ignavibacter and Treponema demonstrate a larger central chamber than pentameric homologues. Equivalents to the PopA oligomer loop 8 (white, 19aa) are coloured as per monomers, demonstrating that the Halothermothrix orenii tetramer (blue) packs around a smaller loop, but hexamers (IPM56_03505, yellow; TDE_2508, green) pack around a larger loop, inclusive of alpha helical regions. The larger size of H. orenii Hore_23180 results in a similar barrel, but “taller” outer wall (95 Å high). B The smaller, 8-stranded OMP BT1926 models as a radial pentamer (one chain in white, with central loop coloured green). C Fold of BT1926 monomer, membrane plane indicated. The large central loop (green) travels “upward” rather than outward. D Homologues of BT1926 model as tetramer (red, D. gadei HMPREF9455_00941) and hexamer (purple, C. hutchinsonii CHU_0007). E Cutaway view of central chamber of BT1926, one chain coloured as in C, hydrophobic residues lining chamber shown in stickform.
From manual inspection of membrane proteomes, we were able to find OMPs from a second, vastly-divergent superfamily that models as radial oligomers with a central hydrophobic cavity. These too model confidently as predominantly tetramers-hexamers (Supplementary Table 4, at least one nonamer), and include Bacteroides thetaiotaomicron BT1926 (Fig. 6b–e). These are 8-stranded, closed β-barrels (no plug) with no sequence or structure link to the PopA superfamily, that use loop 1 as the central symmetry-forming element. We therefore conclude that oligomerization (via an outer composite β-rim, with seam formation) and subsequent central chamber formation is a general feature found in more than one OMP class.
Discussion
The pentameric nature of Bd0427/PopA redefines expectations of OMP assembly and interaction with the lipid bilayer. In comparison to trimeric OMPs, PopA does not retain the same ordering of loops and loop function, i.e., trimers use loop 2 for oligomerisation and loop 3 for stabilization of the outer barrel wall, distinct from loop 8 and loop 2 of PopA, respectively (Fig. 1j, Supplementary Fig. 22). The central chamber of PopA, into which we have modelled detergent alkyl chains, likely houses inner leaflet lipids in vivo. The residues lining the cavity show a high degree of conservation (Supplementary Fig. 4). The density maps from both the x-ray and cryo-EM suggest tight packing of eleven alkyl chains (the central “extra” chain having less defined density in the x-ray map), with chain:chain distances of 4.5 Å commensurate with a crystalline monolayer. Multiple bound lipids are common in several classes of protein, but the ringfence-like trapping of these in PopA is vastly different from the often peripheral groupings of lipids observed in OMPs and closer to the closed-symmetry IM proteins yeast PMA122 and the H. pylori urea channel23. There may be a theme in both IM and OM lipid pools – PMA1 has 115 densities which fit 57 diacylglycerides with 1 random space remaining to ensure free diffusion22; our observation of 11 densities for 5 lipids also matches this pattern.
Because the PopA structure is so distinct, it is interesting to consider oligomer assembly into the OM. The surface bowl-shaped cavity is formed by a continuous β-sheet wall created by the hydrogen bonding between β-barrel strand 5 with strand 14 of the adjacent subunit. Such a structure implies that exclusion of LPS from this space would occur early during PopA subunit assembly by the barrel assembly machine protein A (BamA) and that oligomerisation outweighs the energetic cost of LPS exclusion. Immediately adjacent to L8 is the C-terminal strand of PopA, which contains a β-signal motif that, as shown in canonical OMPs24, is involved in initiation of folding and membrane insertion upon its hybridisation to the first strand of the BamA β-barrel25. The monolayer cavity-forming PopA L8 would therefore be positioned within the OM early in the subunit assembly process and cause significant membrane disruption. Membrane disruption by L8 could lower the energy cost of LPS expulsion and would be assisted by the membrane-thinning properties previously defined for BamA26.
The power of structure-based methods to identify distant homologues enabled us to confidently discover other proteins that we propose to form a radial oligomer OMP superfamily distributed throughout bacteria (Supplementary Table 3). Most interestingly, these include prospective tetramers and hexamers as well as pentamers, including some strains that encode multiple types (e.g., Fibrobacter succinogenes FSU_2404 hexamer and FSU_0230 pentamer). The size of loop 8 in PopA homologues is a good indicator of oligomerisation state, with tetramer loops typically small (often tight turns) and hexamer loops typically larger with helical content. PopA superfamily members have consistent features: 16 β-strands, a high “outer” wall, a tight plug and loops that create the oligomerisation and bowl effect observed in PopA –making a composite β-sheet wall around the outer rim of the oligomer. Homologues retain the hydrophobicity on loop 8 and strands β12-16 needed to form the central chamber. The ability to use our experimentally-derived PopA structure as a predictive tool is high – even monomeric Alphafold models retain the features described above – i.e., setting the correct oligomerisation state is not a prerequisite to discern PopA-like features. Superfamily members from Fibrobacter were observed to be highly expressed during OM vesicle formation (FSU_240427) and cellulose degradation (FSU_023028). Family member TDE_2508 from the human pathogen Treponema denticola has been associated with adhesion and biofilm formation29, and was identified to form high molecular weight oligomers via both chemical crosslinking and blue native PAGE30 - consistent with modelling as a hexamer (Fig. 6a). One PopA homologue, Hore_23180 (Fig. 6a). occurs in the firmicutes (predominantly Gram-positive), in Halothermothrix orenii, which is known to have a Gram-negative diderm phenotype31. Additionally, there may even be a distal relative in the Archaea – from DPANN Candidatus Altiarchaeales (Supplementary Table 3).
The oligomerization and chamber formation of the PopA superfamily is mirrored by a second, unrelated 8-strand superfamily (Fig. 6b–e, Supplementary Table 4). Representatives have attributed roles in the literature including BT1926 implicated in phase-variable modification of B. thetaiotaomicron to alter phage susceptibility32,33, CHU_0007 implicated in C. hutchinsonii adhesion to cellulose34, and Tanf_04855 found in OM vesicles from the pathogen Tannerella forsythia35. Remarkably, the thermophilic bacterium Caldithrix abyssi has both pentameric and hexameric 16-strand representatives (Cabys_3254 and 2908) and a 8-strand representative (Cabys_3215). It is interesting to note that the two superfamilies together propose a general feature for oligomer formation and lipid binding at the periplasmic chamber. Upon insertion to the OM there will be two consequences – displacing more lipids from the outer leaflet than the inner leaflet may effect physical properties of the OM, but more importantly, cells expressing these OMPs will have a lower density of lipopolysaccharide. This latter outcome may assist processes like adhesion by creating some clearance for other factors to function.
When heterologously produced in E. coli, PopA associated with both the OM and the IM, the latter likely mediating the IM blebbing specifically induced by this OMP. IM association may arise during or following secretion to the OM. We favour the latter as it is in keeping with the observed transfer from the Bdellovibrio OM during native predation3,4 and may be an intrinsic property of PopA. It is important to note that under native predation conditions, the proposed transfer of PopA into the prey envelope3,4 does not result in similar IM defects, hence other factors produced during predation may have stabilizing effects36,37 or defects could be magnified by PopA overexpression levels. The behaviour of PopA in liposome swelling assays argues that any role (prior to inter-membrane transfer) in the predator would be unrelated to small-molecule transport. Hence, it may be best to refer to PopA and homologues as porin-like until assays conclusively demonstrate porin behaviour. PopA conformation in the inner membrane of prey has the potential to be influenced by the proton motive force (PMF), but is unlikely to be key for function because the prey PMF has been shown to be dissipated at a very early stage of the predatory lifecycle38. We suggest that the reported PopA transfer into prey may occur via the vesiculation observed in tomograms7. With PopA possessing FadL-like features and several models of homologues instead having more pore-like features (e.g., TDE_2508 has a clear solvent channel inside the barrel), it is likely that individual oligomeric porins have roles specific to themselves, with the above common physical features of lipid-sequestration and lipopolysaccharide exclusion as an additional functional outcome.
Together, our data reveal that a porin-like protein constituting the major OMP of B. bacteriovorus is a representative for a distinct class of membrane proteins. The non-standard features of the pentameric assembly allow for stability in both outer and inner membranes, and the self-enclosed central lipid pool is likely to be a conserved feature of diverse OMP homologues across diverse lifestyles that await further functional characterization.
Methods
Cloning and expression
Constructs were generated via restriction-free cloning39, using primers detailed in Supplementary Table 5. Briefly, PopAΔSP (for refolding) was cloned into pET28a and mature PopAhis (for native expression) with an internal His tag (with a pelB leader sequence at start and coding sequence SSHHHHHHGSS inserted after A103) was synthesized as an insert into pET29b (Twist Biosciences).
For large-scale expression of PopAΔSP in BL21(DE3) cells, 1 litre of LB media was inoculated with 10 mL overnight culture, incubated at 37 °C until an OD600nm of 0.6, then induced with 1 mM IPTG for a subsequent period of 4 hours. Selenomethionyl-Bd0427ΔSP was produced using the method of Doublie et al. 40.
Native expression of PopAhis used BL21(DE3) RIPL cells (New England Biolabs), as per PopAΔSP, except 0.5 mM IPTG and overnight expression were chosen for optimal expression levels.
Purification of PopAΔSP
Cells were harvested by centrifugation, resuspended in Buffer 1 (50 mM Tris pH 8.0, 5 mM EDTA) and lysed via sonication. The insoluble material was pelleted (43,380 g for 1 h), then resuspended in Buffer 1 plus 2% w/v TritonX-100 for 1 h at room temperature (plus stirring). The pelleting and wash procedures were repeated a further four times (two initial washes with TritonX-100, two subsequent washes without). The isolated pellet of inclusion body material was weighed out and 200 mg tumbled in 10 mL of 50 mM Tris pH 8.0, 100 mM NaCl, 8 M Urea (2 hours at room temperature). Insoluble material was pelleted at 43,380 g for 20 min, protein adjusted to 0.1 mM, and added dropwise to 150 mL refolding buffer (50 mM Tris pH 8.0, 100 mM NaCl and a choice of detergent – either 1% w/v DDM or 2% w/v β-OG). Aggregated protein was removed by filtration and correct refolding assessed by semi-native PAGE. Samples were further purified/analysed via size exclusion chromatography (in varying buffers – the optimal buffer for crystallization sample was 50 mM tris-HCl (pH 8), 150 mM NaCl, 50 mM β-OG, 3 mM β-mercaptoethanol).
Purification of PopAhis
Cell pellets were resuspended in 1x PBS, 5 mM EDTA and lysed using an Emulsiflex C3 disruptor (Avastin) at 17,000 psi for 3 cycles. Sequential centrifugation steps were used to isolate cell debris (10,000 g 30 min) and membrane fraction from subsequent supernatant (150,000 g 1 h). Membrane fractions were resuspended in a ratio of 40 mg membrane to 1 ml buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% w/v DDM), homogenized by hand homogenizer, and tumbled at 4 °C overnight. Unsolubilized material was pelleted at 150,000 g for 30 min and the supernatant purified by conventional nickel-NTA chromatography using resuspension buffer containing a lower concentration of 0.3% w/v DDM, eluted with 200 mM imidazole. Size exclusion chromatography was performed using a Superdex 200 column.
PopAΔSP crystallization and structure determination
Purified protein was concentrated to 4.5 mg/ml and screened against a selection of commercial sparse matrix conditions (equal protein:reservoir in drops). Crystals with appreciable diffraction were obtained from MidasPlus C9 (0.1 M NaCl, 25% v/v pentaerythritol propoxylate (5/4 PO/OH), 10% v/v DMSO; form A, C2221 spacegroup), MemGold G12 (1 mM Zinc sulphate, 50 mM HEPES pH 7.8, 28% v/v PEG 600; form B, P22121 spacegroup), and MemGold D5 (50 mM Mg acetate, 50 mM Na acetate pH 5.4, 24% PEG 400; form C, C2 spacegroup). Data were collected at the Diamond Light Source synchrotron (beamline IO4), and processed using ISPyB41 automated software. The CRANK2 pipeline in CCP4 cloud42 was used to phase and build selenomethionyl form A, which was iteratively refined and built using PHENIX43 and COOT44. Monomer models from form A were used as search models in PHENIX_MR to determine forms B and C via molecular replacement. Data collection and refinement statistics are provided in Supplementary Table 1.
Cryo-Electron Microscopy of PopAhis
Quantifoil R1.2/1.3 200-mesh grids were glow discharged for 30 s using a GloQube (Quorum) discharge unit (180 sec at 40 mA) followed by graphene oxide coating. Cryo-EM grids were prepared with a Vitrobot Mark IV (Thermo Fisher) operated at 100% humidity and 4 °C. A 3 µl aliquot of PopA103his (0.02 mg/mL in 50 mM Tris pH 8, 150 mM NaCl, 0.03% w/v DDM) was applied to the grids, blotted 3 s and plunge frozen into liquid ethane.
The samples were imaged using a FEI Titan Krios (300 kV) equipped with a K3 detector and energy filter (20-eV slit size) (Gatan) using automated data collection (ThermoFisher EPU).
Movies were acquired at 105,000x magnification (0.835 Å/pixel, 100 μm objective aperture, 2 s exposure, 50 frames, total dose on specimen ~55.3 e−/Å2 and defocus range of -2.4 to -1.2 in 0.3 μm intervals). A total of 10,308 movies were collected. All data processing was done in cryoSPARC45 v4.2.1 with workflow displayed in supplementary Fig 7. Raw movies were processed using patch motion correction (multi), with dose weighting. The Contrast Transfer Function (CTF) was estimated by patch CTF estimation (multi). Particles were blob picked, 2D classes were inspected and used as a reference for complete particle picking. A total number of 992,500 particles were subjected to ab initio model generation. Three models were generated in C1 symmetry, one model (365,835 particles) was selected for further processing. The consensus map was refined with non-uniform refinement to a 2.85 Å resolution in C1 symmetry and 2.66 Å in C5 symmetry. Resolution was reported from the gold-standard Fourier shell correlation using the 0.143 criterion. Local resolution was estimated using cryoSPARC45. The FSC curve is provided in Supplementary Fig. 7 and local resolution estimation is provided in Supplementary Fig. 8); model-to-data correlation coefficients are provided in Supplementary Table 2.
Liposome swelling assays
Proteoliposomes were prepared from 80 μl of liposome stock solution (100 mg/ml phosphatidylcholine, 2.3 mg/ml dihexadecyl phosphate in chloroform) dried individually under air flow. The lipid film was left to fully dry under vacuum for 2 hours, then resuspended in 100 μl water before protein addition (15 mg OmpF, 15 mg PopA, 1:1 with detergent buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.03% w/v DDM)), followed by a vortex step. Samples were sonicated in a water bath for 90 seconds to clarify, then transferred to a 24-well plate to be dried overnight under vacuum. Proteoliposomes were resuspended in 200 μL buffer (10 mM Hepes pH 7.0, 12 mM stachyose) and left to stand for 1–2 hours. Liposome swelling was monitored by following OD400 over 30 seconds, with measurements taken every second. Assays contained 15 μl of liposomes/proteoliposomes and 200 μL of substrate solution, with each assay repeated in triplicate. Relative permeability was compared to OmpF in the presence of L-arabinose as a percentage.
Bacterial strains, growth conditions and chemicals
All strains used in this study are listed in Supplementary Table 5. Escherichia coli MG1655 strains were routinely grown in Lysogeny Broth (LB, MP Biomedicals) at 37 °C and constant shaking. Plasmids were maintained by adding ampicillin (100 µg.mL−1) and chloramphenicol (15 µg.mL−1).
Plasmid construction
All primers used in this study are listed in Supplementary Table 6. Standard molecular cloning methods were used, and DNA assembly was performed using the NEBuilder HiFi mix (New England Biolabs). To construct pBAD18 derivative plasmids, the Bd0427-coding sequence from B. bacteriovorus HD100 (taxon: 264462) and the OmpF-coding sequence from E. coli MG1655 were PCR amplified with primers oGL1727/oGL1728 or oGL1765/oGL1766, respectively, and cloned in the opened pBAD18 vector (using oGL1487/oGL1488) by DNA assembly. The mCherry-coding sequence was then amplified with oGL2261/oGL2262 from the pTNV215-parBBb-mCherry plasmid46 and cloned in the opened pBAD18-PopA vector (using oGL1488/oGL2256) by DNA assembly.
ΔpopA strain generation
The scarless allelic replacement of bd0427/popA into the HD100 chromosome was performed using a strategy based on the two-step recombination with pK18mobsacB-derived suicide vector, as in Steyert and Pineiro47, screened by PCR and verified by DNA sequencing. For homology, 500 bp upstream and downstream of the loci of interest were used.
We also screened 10, 26 and 29 colonies from three independent pK18mobsacB:bd0427mcherry conjugations into host/prey-independently (HI) grown B. bacteriovorus exconjugants again without success. Attempts were also made to construct a C-terminal mCherry fusion to Bd0427 in predatory B. bacteriovorus HD100. Although it was possible to construct and verify by sequencing, the fusion plasmid in E. coli S17-1, when that was conjugated three times into B. bacteriovorus HD100, this resulted in failure to isolate any fluorescently tagged strains despite screening 367, 676 and 346 individual exconjugant plaques. Taken together, these results indicate to us that Bd0427/PopA, is important for viability of B. bacteriovorus HD100 in prey-independent growth.
Image acquisition
Phase contrast and fluorescence images were acquired on a fully motorized Ti2-E inverted epifluorescence microscope (Nikon) equipped with a CFI Plan Apochromat λ DM 100 × 1.45/0.13 mm Ph3 oil objective (Nikon), a Sola SEII FISH illuminator (Lumencor), a Prime95B camera (Photometrics) or a PrimeBSI camera (Photometrics), a temperature-controlled light-protected enclosure (Okolab), and filter-cubes for mCherry and GFP (Nikon). Multi-dimensional image acquisition was controlled by the NIS-Ar software (Nikon). Pixel size was 0.074 µm for the Prime95B camera (using the 1.5X built-in zoom lens of the Ti2-E microscope) or 0.065 µm for the PrimeBSI camera (with 1X zoom lens). Identical LED illumination power and exposure times were applied when imaging several strains and/or conditions in one experiment and across replicates of the same experiment. They were set to the minimum for time-lapse acquisitions to limit phototoxicity.
Live-cell imaging
To conduct time-lapse imaging, E. coli cells were cultured in LB medium supplemented with appropriate antibiotics at 37 °C. In the case of E. coli strains producing envelope reporters, 10 µM of IPTG was added to the medium after 30 minutes of initial growth to induce the envelope reporters. Once OD600 reached 0.4, cells were incubated with 0.2% L-arabinose for 1 hour to induce the expression of PopA or PopA-intermChy. Subsequently, the cells were harvested at room temperature by centrifugation at 2600 x g for 5 minutes, resuspended in LB medium and then imaged on 1.2% DNB-agarose pads supplemented with 10 µM IPTG and 0.2% L-arabinose. The same fields of view on the pad were imaged at 5-minute intervals with the enclosure temperature set to 28 °C.
Envelope bulging observation and quantification
Envelope bulging was defined as events in which envelope extrusion can be observed. Because envelope bulging can lead to rapid loss of cell integrity and cell lysis, which can happen within the 5-minute interval of our time-lapse experiment, this includes both classical membrane bulging and partial membrane detachment, involving either the inner, outer or both membranes. The frequency of bulging events in each field of view was quantified for each replicate, and then normalized by the total number of cells. In addition, the bulging events were categorized into three categories based on envelope reporters: inner membrane bulging only, outer membrane bulging only, and inner and outer membrane bulging (related to Fig. 4).
Image processing
For figure preparation, images were processed with FIJI48 keeping contrast and brightness settings identical for all regions of interest in each figure. Figures were assembled and annotated using Adobe Illustrator (Adobe Inc.).
Molecular dynamics simulations
All MD simulations were performed using GROMACS v202249. Coarse Grained (CG) MD simulations (martini 2.250) were constructed using Insane51, using the MemProtMD pipeline52 and run for 1 μs to permit the equilibration of the OMP within either a symmetrical inner membrane, at a 7:2:1 ratio of 1-palmitoyl-2-oleoyl-phosphatidyl-ethanolamine (POPE): 1-palmitoyl-2-oleoyl-phosphatidyl-glycerol (POPG):Cardiolipin, or an asymmetric outer membrane, comprised of a 7:2:1 POPE:POPG:Cardiolipin leaflet and a pure lipid A core oligosaccharide leaflet53. The membrane-assembled systems were then converted to atomic detail54. Three independent systems were equilibrated for 10 ns with the protein restrained before repeats of 500 ns of unrestrained atomistic MD simulations using the CHARMM36m force field55. Simulations were performed at 310 K, coupled to the V-rescale thermostat56. In all cases, the systems were coupled to the stochastic rescaling barostat57, with the lateral pressure (Pxy) and normal pressure (Pz) coupled independently using semi-isotropic pressure coupling at 1 bar in each dimension. Simulations were analysed using MDAnalysis58.
Fractionation assay
Cell fractionation was performed as described herein59 with some modifications. Cells were grown at 37 °C in LB medium. Once OD600 reached 0.4, cells were incubated with 0.2% L-arabinose for 1 hour to induce the expression of PopA. Cells were then collected by centrifugation at 6000 g at 4 °C for 15 min, washed and resuspended in PBS buffer. DNase I (1 mg, Roche), RNase A (1 mg, Thermo Scientific) and a half tablet of a protease inhibitor cocktail (Complete EDTA-free Protease Inhibitor Cocktail tablets, Roche) were added to cell suspensions. Cells were broken through a French pressure cell at 1500 psi. After adding 2 mM MgCl2, the lysate was centrifuged at 5000 g at 4 °C for 15 min to remove cell debris. Then, clarified supernatant was placed on top of a two-step sucrose gradient (2.3 mL of 2.02 M sucrose in 10 mM HEPES pH 7.5 and 6.6 mL of 0.77 M sucrose in 10 mM HEPES pH 7.5). The samples were centrifuged at 180,000 g for 3 h at 4 °C in a 55.2 Ti Beckman rotor. After centrifugation, the soluble fraction and the membrane fraction (insoluble fraction) were collected. The membrane fraction was diluted four times with 10 mM HEPES pH 7.5. To separate the inner and outer membranes, the diluted membrane fraction was loaded on top of a second sucrose gradient (10.5 mL of 2.02 M sucrose, 12.5 mL of 1.44 M sucrose, 7 mL of 0.77 M sucrose, in 10 mM HEPES pH 7.5). The samples were then centrifuged at 112,000 g for 16 h at 10 °C in a SW28.1 Beckman rotor. Approximately 30 fractions of 1.5 mL were collected and odd-numbered fractions were subjected to analysis.
Native PopA production analysis
To assess the PopA production during the predation cycle, wild-type B. bacteriovorus HD100 predators were grown overnight in DNB medium (Dilute Nutrient Broth, Becton, Dickinson, and Company; supplemented with 2 mM CaCl2 and 3 mM MgCl2 salts) in the presence of E. coli prey at 30 °C with continuous shaking as described60. Attack phase (AP) predator cells were collected from the cleared overnight culture and prepared following the method described by Kaljević46. For the growth phase (GP) sample, a 1:1 volume ratio of E. coli prey to predator mixture was utilized to minimize the presence of AP cells. Samples from the mixed culture were collected 2 hours post-infection. Western blot analysis of whole-cell protein extracts from B. bacteriovorus in AP and in GP was probed with polyclonal α-PopA/ Bd0427 antibodies (produced by Eurogentec) (1/200). Signal for antibody binding was visualized as described in the Immunoblotting section.
Immunoblotting
Protein samples were separated on 4–12% NuPage Bis-Tris SDS precast polyacrylamide gels (Life Technologies) and ran at 180 V for 1 h in NuPAGE MES SDS running buffer. Western blotting was performed using standard procedures with the following primary antibodies: polyclonal LolC (1/10,000) and Lpp (1/6000) antibodies for LolC and Lpp protein detection, respectively59, polyclonal Bd0427 antibody (produced by Eurogentec) (1/2000) for Bd0427Bb detection. Signal from antibody binding was visualized by detecting chemiluminescence from the reaction of horseradish peroxidase with luminol and chemiluminescence was imaged with an Image Quant LAS 500 camera (GE Healthcare). Goat anti-rabbit IgG-peroxidase antibody (RABHRP1, Sigma) was used as a secondary antibody. Figures were prepared using ImageJ and assembled and annotated in Adobe Illustrator.
Statistical analysis
All analyses of microscopy images were performed using several representative fields of view from at least three independent biological replicates. Statistical parameters (means and standard deviations) were calculated in R using the base R functions61. Pairwise nonparametric comparisons of datasets were performed using the unpaired Wilcoxon rank-sum test. Significance was defined by p > 0.05 (NS), p ≤ 0.05 (*), p ≤ 0.01 (**) and p ≤ 0.001 (***).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates have been deposited in with accession codes 9GA1 (x-ray structure), and 9GF0 and EMD-51308 for the cryoEM structure. Molecular dynamics input, output and parameter files are available at https://doi.org/10.5281/zenodo.15696220. Deposited models for each of the homologues are available at ModelArchive (modelarchive.org) with the accession codes ma-n3ds4 (IPM56_03505 hexamer), ma-e64a6 (C0V70_02135 pentamer), ma-uceig (C0V70_02140 pentamer), ma-rkx5z (Tde_2508 hexamer), ma-lgpuq (Hore_23180 tetramer), ma-c773r (Bt_1926 pentamer), ma-zp266 (HMPREF9455_00941 tetramer) and ma-czwi5 (CHU_0007 hexamer). Source data are provided as Source Data files. Source data are provided with this paper.
References
Otzen, D. E. & Andersen, K. K. Folding of outer membrane proteins. Arch. Biochem. Biophys. 531, 34–43 (2013).
Zeth, K. et al. Structure and function of the PorB porin from disseminating Neisseria gonorrhoeae. Biochem. J. 449, 631–642 (2013).
Tudor, J. J. & Karp, M. A. Translocation of an outer membrane protein into prey cytoplasmic membranes by Bdellovibrios. J. Bacteriol. 176, 948–952 (1994).
Barel, G. et al. Fate of Predator and Prey Proteins during Growth of Bdellovibrio bacteriovorus on Escherichia coli and Pseudomonas syringae Prey. J. Bacteriol. 187, 329–335 (2005).
Vergalli, J. et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 18, 164–176 (2020).
Caulton S. G. & Lovering A. L. Moving toward a better understanding of the model bacterial predator Bdellovibrio bacteriovorus. Microbiology. 169, 001380 (2023).
Kaplan, M. et al. Bdellovibrio predation cycle characterized at nanometre-scale resolution with cryo-electron tomography. Nat. Microbiol. 8, 1267–1279 (2023).
Schwudke, D. et al. The obligate predatory Bdellovibrio bacteriovorus possesses a neutral lipid A containing alpha-D-Mannoses that replace phosphate residues: similarities and differences between the lipid As and the lipopolysaccharides of the wild type strain B. bacteriovorus HD100 and its host-independent derivative HI100. J. Biol. Chem. 278, 27502–27512 (2003).
Rendulic, S. et al. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 303, 689–692 (2004).
Beck, S. et al. Bdellovibrio bacteriovorus strains produce a novel major outer membrane protein during predacious growth in the periplasm of prey bacteria. J. Bacteriol. 186, 2766–2773 (2004).
Beck, S. Characterization of outer membrane protein fractions of Bdellovibrionales. FEMS Microbiol. Lett. 243, 211–217 (2005).
van den Berg, B. et al. Crystal structure of the long-chain fatty acid transporter FadL. Science 304, 1506–1509 (2004).
Vandeputte-Rutten, L. et al. Crystal structure of the outer membrane protease OmpT from Escherichia coli suggests a novel catalytic site. EMBO J. 20, 5033–5039 (2001).
Zeth, K. et al. Crystal structure of Omp32, the anion-selective porin from Comamonas acidovorans, in complex with a periplasmic peptide at 2.1 Å resolution. Structure 8, 981–992 (2000).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Yariv, B. et al. Using evolutionary data to make sense of macromolecules with a ‘face-lifted’ ConSurf. Protein Sci. 32, e4582 (2023).
Sehnal, D. et al. MOLE 2.0: advanced approach for analysis of biomacromolecular channels. J. Cheminform. 5, 39 (2013).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Nikaido, H. & Rosenberg, E. Y. Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J. Bacteriol. 153, 241–252 (1983).
Lepore, B. W. et al. Ligand-gated diffusion across the bacterial outer membrane. Proc. Natl. Acad. Sci. USA 108, 10121–10126 (2011). Jun 21.
Cotter, T. W. & Thomashow, M. F. Identification of a Bdellovibrio bacteriovorus genetic locus, hit, associated with the host-independent phenotype. J. Bacteriol. 174, 6018–6024 (1992).
Zhao, P. et al. Structure and activation mechanism of the hexameric plasma membrane H+-ATPase. Nat. Commun. 12, 6439 (2021).
Cui, Y. pH-dependent gating mechanism of the Helicobacter pylori urea channel revealed by cryo-EM. Sci. Adv. 5, eaav8423 (2019).
Struyve, M. et al. Carboxy-terminal phenylalanine is essential for the correct assembly of a bacterial outer membrane protein. J. Mol. Biol. 218, 141–148 (1991).
Doyle, M. T. et al. Cryo-EM structures reveal multiple stages of bacterial outer membrane protein folding. Cell 185, 1143–1156.e13 (2022).
Liu, J. & Gumbart, J. C. Membrane thinning and lateral gating are consistent features of BamA across multiple species. PLoS Comput Biol. 16, e1008355 (2020).
Yeoman, C. J. et al. In Vivo Competitions between Fibrobacter succinogenes, Ruminococcus flavefaciens, and Ruminoccus albus in a Gnotobiotic Sheep Model Revealed by Multi-Omic Analyses. mBio 12, e03533-20 (2021).
Raut, M. P. et al. Influence of substrates on the surface characteristics and membrane proteome of Fibrobacter succinogenes S85. PLoS One 10, e0141197 (2015).
Abiko, Y. et al. Major membrane protein TDE2508 regulates adhesive potency in Treponema denticola. PLoS One 9, e89051 (2014).
Veith, P. D. et al. Localization of outer membrane proteins in Treponema denticola by quantitative proteome analyses of outer membrane vesicles and cellular fractions. J. Proteome Res 18, 1567–1581 (2019).
Mavromatis, K. et al. Genome analysis of the anaerobic thermohalophilic bacterium Halothermothrix orenii. PLoS One 4, e4192 (2009).
Taketani, M. et al. A phase-variable surface layer from the gut symbiont bacteroides thetaiotaomicron. mBio 6, e01339-15 (2015).
Porter, N. T. et al. Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat. Microbiol. 5, 1170–1181 (2020).
Zhao, D. et al. A Disulfide Oxidoreductase (CHU_1165) is essential for cellulose degradation by affecting outer membrane proteins in Cytophaga hutchinsonii. Appl. Environ. Microbiol. 86, e02789-19 (2020).
Friedrich, V. et al. Outer membrane vesicles of Tannerella forsythia: biogenesis, composition, and virulence. Mol. Oral. Microbiol 30, 451–473 (2015).
Huru, E. et al. Fluorescent D-amino-acids reveal bi-cellular cell wall modifications important for Bdellovibrio bacteriovorus predation. Nat. Microbiol. 2, 1648–1657 (2017).
Ruby, E. G. & Rittenberg, S. C. Attachment of diaminopimelic acid to bdelloplast peptidoglycan during intraperiplasmic growth of Bdellovibrio bacteriovorus 109J. J. Bacteriol. 158, 597–602 (1984).
Rittenberg, S. C. & Shilo, M. Early host damage in the infection cycle of Bdellovibrio bacteriovorus. J. Bacteriol. 102, 149–160 (1970).
van den Ent, F. & Löwe, J. RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem Biophys. Methods 67, 67–74 (2006).
Doublié, S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523–530 (1997).
Fisher, S. J. et al. SynchWeb: a modern interface for ISPyB. J. Appl Crystallogr 48, 927–932 (2015).
Krissinel, E. et al. CCP4 Cloud for structure determination and project management in macromolecular crystallography. Acta Crystallogr D. Struct. Biol. 78, 1079–1089 (2022).
Zwart, P. H. et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435 (2008).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60, 2126–2132 (2004).
Punjani, A. et al. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kaljević, J. et al. Chromosome choreography during the non-binary cell cycle of a predatory bacterium. Curr. Biol. 31, 3707–3720.e5 (2021).
Steyert, S. R. & Pineiro, S. A. Development of a novel genetic system to create markerless deletion mutants of Bdellovibrio bacteriovorus. Appl. Environ. Microbiol. 73, 4717–4724 (2007).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Gronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Monticelli, L. et al. The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput 4, 819–834 (2008).
Wassenaar, T. A. et al. Computational lipidomics with insane: a versatile tool for generating custom membranes for molecular simulations. J. Chem. Theory Comput 11, 2144–2155 (2015).
Stansfeld, P. J. et al. MemProtMD: Automated insertion of membrane protein structures into explicit lipid membranes. Structure 23, 1350–1361 (2015).
Hsu, P.-C. et al. CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput Chem. 38, 2354–2363 (2017).
Vickery, O. N. & Stansfeld, P. J. CG2AT2: an enhanced fragment-based approach for serial multi-scale molecular dynamics simulations. J. Chem. Theory Comput 17, 6472–6482 (2021).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Bussi, G. et al. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Bernetti, M. & Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 153, 114107 (2020).
Michaud-Agrawal, N. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput Chem. 32, 2319–2327 (2011).
El Rayes, J. et al. Disorder is a critical component of lipoprotein sorting in Gram-negative bacteria. Nat. Chem. Biol. 17, 1093–1100 (2021).
Remy, O. et al. An optimized workflow to measure bacterial predation in microplates. STAR Protoc. 3, 101104 (2022).
R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Acknowledgements
SGC, ALL RES PR were funded by the Wellcome Trust Investigator Award in Science (209437/Z/17/Z), RP was funded by a Wellcome Trust AAMR studentship. GR was jointly funded by the BBSRC and the University of Birmingham (through the Midlands Integrative Biosciences Training Partnership - Grant No. BB/M01116X/1). TK is supported by BBSRC Research Grant No. BB/S017283/1. PS acknowledges Wellcome (208361/Z/17/Z), MRC (MR/S009213/1), BBSRC (BB/P01948X/1, BB/R002517/1, BB/S003339/1 and BB/Y003187/1), and the Howard Dalton Centre for funding. MD simulations made use of time on ARCHER and JADE granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (http://hecbiosim.ac.uk), supported by EPSRC (grant no. EP/R029407/1). PJS acknowledges Sulis at HPC Midlands+, which was funded by the EPSRC on grant EP/T022108/1, and the University of Warwick Scientific Computing Research Technology Platform for computational access. MTD is supported by the National Institute of Allergy and Infectious Diseases (R21AI180112), seed funding from the University of Sydney Infectious Diseases Institute and the University of Sydney Drug Discovery Initiative. The research of BvdB was supported by a Wellcome Trust Investigator award (214222/Z/18/Z) and the BBSRC (SPB; BB/R004366/1). YGS was an EMBO post-doctoral fellow and is currently a post-doctoral fellow from the F.R.S.-FNRS. GL is a WELBIO investigator of the WEL Research Institute and a Research Associate of the F.R.S.-FNRS. Work in the Laloux lab is supported by the European Research Council (ERC) under the European Union Horizon 2020 and Horizon Europe research and innovation programmes (ERC StG PREDATOR, Grant agreement No. 802331; ERC CoG VAMPIRE, Grant agreement No. 101171143). We acknowledge the Midlands Regional CryoEM Facility at the Leicester Institute of Structural and Chemical Biology (LISCB), major funding from the MRC (MC_PC_17136). We thank Michaël Deghelt for providing the pTHV037-lpoB-mCherry_msfgfp-glpT, Jessica El Rayes for advice and technical assistance during the fractionation assay, and Jovana Kaljević for providing whole-cell samples of B. bacteriovorus proteins. We are grateful to Satya Bhamidimarri for early assistance with the transport assays. We thank Diamond Light Source for beamtime (proposal mx19880) and for facility support during data collection at I04. For the purposes of open access, the corresponding author has applied a CC BY public copyright licence to the Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
A.L.L. and G.L. independently conceived the project and obtained funding (working on the biochemical and in-vivo aspects, respectively). R.J.P. purified, crystallized and solved the structures of the proteins and investigated function (with transport assays undertaken with AS and BvdB) with supervision from A.L.L. and assistance from S.G.C. and L.M. R.E.S. contributed to the manuscript and supervised predator experimentation. PR investigated gene knockout function in the host-independent strains and investigated the feasibility of mCherry gene tagging in the predator. G.R. helped determine the cryo-EM structure of PopAhis under the supervision of T.K. M.J. trialled molecular dynamics simulations with (and under supervision from) P.S.; P.S. and D.G. performed the MD simulations and analysis. M.T.D. provided insight into the likely folding ramifications of PopA. Y.G.S. investigated PopA expression and properties in-vivo under the supervision of GL.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parr, R.J., Santin, Y.G., Ratkevičiūte, G. et al. A porin-like protein used by bacterial predators defines a wider lipid-trapping superfamily. Nat Commun 16, 6213 (2025). https://doi.org/10.1038/s41467-025-61633-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61633-0
