
Abstract
Primary amines are highly ubiquitous functional groups found in diverse natural products and building blocks. Despite their widespread application as nucleophiles, the potential for facile deaminative functionalization utilizing primary amines, particularly sterically hindered α-tertiary amines, has remained less explored. Herein, we report catalytic direct synthesis of aliphatic diazenes from sterically hindered α-tertiary amines. The catalytic diazene synthetic method exhibits wide functional group tolerance, allowing for expeditious access to a wide array of hitherto-inaccessible, highly congested diazenes in a short time. Noteworthy is that the present catalytic method enables the synthesis of various hetero-diazenes using distinct α-tertiary amines in just one step for the first time. The catalytic process could also be readily incorporated into Fmoc solid-phase peptide synthesis, enabling the synthesis of elastin-derived diazene, which contains 12 amino acid residues. The catalytic diazene synthetic method enables efficient deaminative transformation of C–N bonds into C–halogen, C–H, C–O, C–S, C–Se, and C–C bonds through carbon-centered radical formation.
Similar content being viewed by others

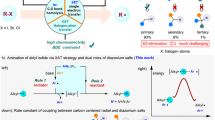
Introduction
Primary aliphatic amines are commonly present in a wide variety of natural molecules, including those with biological activity, and have long served as essential elements in pharmaceutical discovery and molecular design1,2,3. However, despite their natural abundance, these nitrogen-containing functionalities have been significantly underexploited as versatile synthetic platforms for molecular framework modification4,5,6,7. The highly nucleophilic nature of amines allows for coupling reactions with electrophiles, but deaminative functionalization remains a significant challenge due to the low heterolytic nucleofugality and high homolytic C–N bond dissociation energy of amines (Fig. 1a)8,9,10. Furthermore, the characteristic basic nature of amines limits the catalytic use of metal reagents, hindering the effective implementation of deaminative cross-coupling reactions.

a Utility of primary amine for conventional nucleophilic amination and deaminative functionalization. b State-of-the-art deaminative functionalization of primary amine. c Multi-step diazene synthesis for deaminative functionalization via sulfonamide. d Multi-step diazene synthesis for deaminative functionalization using hydroxylamine derivatives. e This work: catalytic synthesis of diazenes from α-tertiary amines for deaminative functionalization.
Several approaches to generate alkyl precursors of primary amines through C–N bond activation have been investigated (Fig. 1b)11,12,13. Among the widely adopted methods is a system utilizing amine-derived Katritzky salts14,15,16,17. The broad popularity of Katritzky salts stems from the ease of their synthesis and accessible reduction potential, enabling various transition metal-catalyzed and photoredox-catalyzed deaminative transformations. However, Katritzky salts cannot be synthesized from sterically hindered α-tertiary amines, which limits their applicability. New strategies that differ from conventional deamination reactions involve the combination of Schiff bases and Ir photoredox catalysts18,19,20,21, or isodiazene from anomeric amide22,23. The method using Schiff bases achieved deaminative functionalization of α-tertiary amines through one-step preactivation. The strategy of isodiazene efficiently functionalized the α-primary and secondary amines using anomeric amide24. Despite intensive efforts to develop efficient deaminative functionalization reactions, challenges remain, such as limitations in functional group transformation and the requirement for stoichiometric amounts of metal activators25,26,27,28.
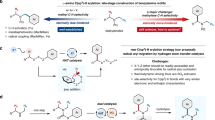
To overcome these limitations, we focused on sterically minimized aliphatic diazenes, which are typically synthesized from ketones and hydrazines through multi-step processes29,30,31,32. Although aliphatic diazenes can generate alkyl radicals by releasing nitrogen gas under heated or light-irradiation conditions, commonly serving as a radical initiator in polymer synthesis, investigations of their application in deaminative functionalization are highly limited. Recently reported synthetic methods for deaminative C–C coupling reactions of diazenes still require multi-step sequences for the preparation of diazenes, as exemplified by sulfonamides (Fig. 1c), and hydroxylamine derivatives (Fig. 1d)33,34,35. Additionally, the range of applicable primary amines is primarily restricted to α-secondary amines. Direct synthesis of aliphatic diazenes from primary aliphatic amines remains scarce compared to the numerous methods available for synthesizing aromatic diazenes. These methods require hazardous reagents36,37,38,39,40, cumbersome multi-step preparations33,35,41,42,43,44,45,46,47, and have a limited substrate scope48,49,50, thus, no catalytic methods have been successfully developed.
Here, we develop a catalytic direct synthesis of aliphatic diazenes from sterically hindered amines under mild and rapid conditions (Fig. 1e). The present catalytic method enables the transformation of C–N bonds into C–halogen, C–H, C–O, C–S, C–Se, and C–C (Csp3, Csp2, Csp) bonds through a deaminative carbon-centered radical formation utilizing diazenes with a broad substrate scope.
Results
We first investigated solvents and reaction times using α-tertiary amine 1a in the presence of a copper catalyst (Table 1). The desired product 2a was obtained in a remarkably short reaction time, only 1 min in dimethylformamide (DMF) using readily available CuOAc, 1,3-dibromo-5,5-dimethylhydantoin (DBDMH), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (entry 1). Acetonitrile (MeCN) also delivered 2a with the same efficiency as DMF. Without the catalyst, only 7% of 2a was observed (entry 2). Various catalysts, including Cu(OAc)2, Fe(OAc)2, and AgOAc, exhibited low catalytic performance (entries 3–5). Various oxidants were evaluated next. Oxygen gas and peroxides did not afford 2a (entries 6–9). In contrast, the use of halogenated oxidants, 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), and 1,3-diiodo-5,5-dimethylhydantoin (DIDMH), afforded 2a, albeit with low chemical yields (entries 10 and 11). N-Bromosuccinimide (NBS) exhibited the same efficiency as DBDMH (entry 12). Base screening revealed that DBU was crucial for efficient reaction progress. Both potassium carbonate and potassium tert-butoxide provided 2a with low efficiency (entries 13 and 14). Although triethylamine delivered 2a in low chemical yield (entry 15), a moderate chemical yield was afforded using 1,4-diazabicyclo[2.2.2]octane (DABCO) (entry 16).
Having developed an efficient diazene synthetic method, we next explored the substrate scope using various α-tertiary amines at a 10-min reaction time (Fig. 2). The catalyst loading and equivalents of reagents could be reduced without detrimental effects (5 mol% CuOAc; DBDMH, 1.2 equiv; DBU, 1.0 equiv), and 2a was isolated in 93% yield. The reaction was successful on a gram scale. A readily available amine hydrochloride salt was also applicable with slight modification of the reaction conditions (2b). Substrates with a 5- or 6-membered alkyl ring at the α-position also afforded the product in high yields (2c, 2d). Cyano and Weinreb amide functionalities were well tolerated, delivering the products with high yields (2e–2g). An amine trifluoroacetic acid (TFA) salt having ketone functionality afforded product 2h in moderate yield. The highly congested adamantyl group was applicable to the present catalysis, although the chemical yield was only moderate (2i). Various functional groups, including Boc-protected tertiary amine, cyclic ether, sulfonyl group, and an indanyl group, were also incorporated without any problem (2j–2m). It is noteworthy that a sterically hindered unnatural amino acid having a 7-membered alkyl ring at the α-position successfully afforded product 2n in synthetically useful yield51,52. β-Amino acids, having no activating group at the α-position, were applicable, delivering product 2o in 66% yield. The reaction proceeded smoothly even when 1-adamantylamine, which has no functional group at all, was used (2p), indicating that an electron-withdrawing group was not crucial for the present catalysis. Amino alcohol derivatives were also transformed into the diazenes 2q and 2r. A variety of dipeptides, such as glycine, alanine, phenylalanine, valine, and asparagine, were used, and the products were obtained in high yields (2s–2w). α-Secondary amines are difficult to use as substrates under oxidative conditions due to undesired imine formation. However, α-secondary amines were also applicable under slightly modified reaction conditions, resulting in diazenes being isolated in synthetically useful yields (2x–2z). Although increasing the amount of copper catalyst effectively suppressed the formation of the imine byproduct, the observation of imine formation even under stoichiometric copper conditions indicates that copper-mediated pathways may still contribute to imine formation.

Isolated yields are shown. Diastereomeric ratios were determined by 1H NMR analysis of crude mixture. a1•HCl salt (1.0 equiv), DBU (2.0 equiv). b1•TFA salt (1.0 equiv), DBU (2.0 equiv). cDMF (0.4 ml), 3 h. dDCDMH was used instead of DBDMH. eCuOAc (20 mol%). fCuOAc (50 mol%). gCuOAc (100 mol%). 1.0 mmol scale. hThree equivalents of the amine on the right side were used. 0.4 ml of DMF. iReaction time was 1 h. j0.50 mmol scale. kCuOAc (100 mol%). lA small amount of impurity was still present after silica gel column chromatography. Boc tert-butoxycarbonyl, Cbz benzoxycarbonyl TBDPS tert-butyldiphenylsilyl, TBS tert-butyldimethylsilyl.
The present catalysis was not confined to homo-diazene synthesis. We further investigated the substrate scope involving hetero-coupling with two distinct α-tertiary amines. Hetero-diazene synthesis from distinct amines has been regarded as highly challenging34,35. In particular, direct hetero-diazene synthesis using sterically hindered α-tertiary amines has never been achieved, even under stoichiometric conditions. When 3.0 equivalents (0.60 mmol) of the α-tertiary amine 1b were used, the hetero-diazene 3ab was isolated in 71% yield (0.15 mmol), which is nearly consistent with the statistical theoretical maximum, along with the concomitant formation of homo-diazenes (2a: 0.028 mmol, 2b: 0.23 mmol). The overall material balance was satisfactory, confirming that the reaction cleanly proceeded. The use of 2.0 equivalents (0.40 mmol) of 1b also delivered hetero-diazene 3ab in 66% yield, consistent with the statistical ratio. These results indicate that, although an excess amount of amine is required, the present catalysis is highly effective for the direct synthesis of hetero-diazenes from different α-tertiary amines. α-Tertiary amine without electron-withdrawing groups could be incorporated into the hetero-diazene without any detrimental effects (3ap, 3dq). Dipeptides could also be incorporated into hetero-diazene, providing the peptide-amino acid-derived diazenes 3sb and 3tb in synthetically useful yields. A diverse range of hetero-diazenes could be synthesized using various combinations of α-tertiary amines (3cp, 3dp, 3kp, 3dk, 3jd). Cross-coupling of α-tertiary amine with α-secondary amine successfully provided the hetero-diazene 3py, although the chemical yield was modest, and a small amount of impurity remained. The wide substrate scope evident in the present catalysis clearly indicates the potential to offer a range of hitherto inaccessible highly congested hetero-diazenes in just one step.
We investigated thermal stability using thermogravimetric analysis (TGA) for several representative compounds, namely 2a, 2r, 2t, 2y, and 3ap, which possess different hindered structures (Fig. S6). The thermal stability of these compounds was comparable to, or significantly greater than, that of a typical diazene compound (i.e., azobisisobutyronitrile, AIBN), which shows an onset of thermal decomposition at approximately 100–125 °C53. Therefore, the synthesis reported here may enhance the availability of thermally stable diazene compounds, which could also be valuable in the field of polymer science, as these compounds are commonly used for radical polymerization.
The rapid and broad substrate scope observed in the present catalysis prompted us to further investigate diazene synthesis using larger functionalized molecules. To assess the applicability of the catalysis to peptide-derived diazene synthesis, we applied our method to Fmoc solid-phase peptide synthesis using elastin, a fibrous protein primarily responsible for linking collagen molecules (Fig. 3). The repetitive sequence peptide of elastin was synthesized through Fmoc solid-phase peptide synthesis. Subsequently, peptide 1α attached to resin was subjected to the optimal reaction conditions, followed by resin cleavage using a TFA cocktail. Peptide-derived diazene 2α was isolated in 11% yield following 15 steps in Fmoc solid-phase peptide synthesis, as a pure form after HPLC purification, showcasing the high applicability of the present method in the solid-phase system.

Catalytic diazene synthesis applied to solid-phase peptide synthesis.
Finally, the present catalysis was applied to deaminative functionalization (Fig. 4). Direct transformation of α-tertiary amine into tertiary alkyl halides was successfully achieved for the first time. Under catalytic conditions for synthesizing diazenes, a simple temperature increase from room temperature to 70 °C enabled a remarkable one-step deaminative bromination 4. Deaminative chlorination from 5 was also achieved using DCDMH instead of DBDMH, clearly demonstrating that the present protocol allows for the use of α-tertiary amines as an alkyl source4,5,51,54. The direct deaminative bromination reaction also proceeded using 1d and 1p, although the yields were moderate (6, 7).

Various deaminative functionalizations were performed, including halogenation, hydrogenation, hydroxylation, thiolation, selenation, and C–C bond formation.
Further utility of the present deaminative functionalization strategy was demonstrated by one-pot transformations. Deaminative iodination was accomplished by synthesizing the diazene followed by adding NaI to the crude mixture (8). The addition of triisopropylsilanethiol under a similar protocol enabled one-pot removal of the α-tertiary amine (9). The addition of TBHP achieved deaminative hydroxylation, delivering tertiary alcohol 10 in synthetically useful yield. Diazene 2a also served as 2 equivalents of a tertiary carbon donor for versatile radical coupling reactions. Treatment of iodine under heated conditions provided iodinated product 8 in high yield55. Deaminative peroxidation also proceeded in the presence of TBHP (11). The introduction of TEMPO was achieved under blue LED light irradiation (12). The use of sulfonyl reagents enabled deaminative thioetherification and selenylation (13, 17, respectively). Diazene 2a turned out to be a suitable substrate for C–C coupling reactions, including Csp3, Csp2, and Csp. Coupling reactions with silyl enolate provided alkylated product 14 in the presence of a copper catalyst56. The NHC-catalyzed method delivered carbonylated compound 15 in high yield32. Alkynylated product 16 was obtained through treatment with a hypervalent iodine reagent.
Hetero-diazenes, which could be directly synthesized in one step from different amines using our catalytic method, were applicable to radical coupling reactions involving denitrogenating. Under blue light irradiation, hetero-diazenes constructed highly congested contiguous quaternary carbon centers, which are generally challenging to assemble, through solvent-cage directed radical cross-coupling (18-21)57, demonstrating the utility of our catalytic diazene synthesis method.
Next, we performed a series of control experiments (Fig. 5). Treatment with DBDMH in the absence of CuOAc and DBU exclusively afforded the N-monobromo and dibromo derivatives (22, 23) without forming 2a (Fig. 5a). The addition of CuOAc and DBU to the mixture of 22 and 23 delivered 2a in quantitative yield, indicating that 22 and 23 serve as intermediates for the generation of diazene 2a. Omission of CuOAc delivered 2a in 4% yield (Fig. 5b). Without DBU, a low chemical yield of 2a was also observed (Fig. 5c). In the course of investigating bases, DABCO, known to form complexes with NBS or NIS through halogen interactions confirmed by X-ray crystallography58,59,60,61, exhibited high performance for generating 2a (Table 1, entry 16), suggesting that DBU would weaken the N–Br bond or stabilize the bromine radical62,63,64,65,66,67,68.

a Intermediate formations of 17 and 18 with DBDMH. b Control experiments without copper suggested that a copper catalyst significantly facilitated the formation of 2a. c Control experiments without DBU indicated that DBU was crucial for efficient reaction progress. d EPR analysis of DMPO adduct 24. e Proposed catalytic cycle. The halogen atom abstraction would first proceed with intermediate A to generate copper (II) species and aminyl radical species C. Copper(III) species B formation would be feasible, but aminyl radical generation would be unfavorable. The N–N bond formation proceeds through aminyl radical species C with D to afford intermediate E.
To further investigate the formation of the aminyl radical, we performed electron spin resonance (EPR) measurements using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trapping agent (Fig. 5d). When the reaction was conducted under the standard conditions using NBS as the oxidant in the presence of DMPO, HRMS peak corresponding to the DMPO adduct 24 was observed. Furthermore, EPR measurements successfully detected the signal of the DMPO adduct 24, suggesting that an aminyl radical species would be generated under the present reaction conditions.
On the basis of a series of control experiments and DFT calculation, the proposed catalytic cycle is shown in Fig. 5e. Without a copper catalyst, the N–N bond formation would proceed through a two-electron mechanism, which was sluggish due to the significant steric hindrance by the two α-tertiary amines. In the presence of a copper catalyst, the reaction rate was significantly enhanced, indicating a potentially distinct reaction pathway compared to the reaction without a copper catalyst. Although the formation of a copper(III) species via oxidative addition of the copper catalyst to intermediate A is feasible, DFT calculations suggest that the subsequent generation of aminyl radical species C is energetically unfavorable. In contrast, the halogen atom abstraction pathway is more likely and is considered to be the predominant route leading to aminyl radical formation, as supported by EPR analysis. Furthermore, the reaction pathway involving aminyl radical species C followed by the N–N bond formation to afford intermediate E, which is less impacted by steric hindrance, would be operative (calculated ΔG‡ = 8.8 kcal/mol, ΔG = −38.8 kcal/mol. See Supplementary Information section 9)69,70,71,72,73,74,75,76. The formed halogenated hydrazine E would subsequently be converted into the diazene product 2 by DBU without the involvement of copper species77.
In summary, we developed a catalytic deaminative protocol through direct synthesis of aliphatic diazenes from sterically hindered α-tertiary amines under mild and rapid conditions. A variety of α-tertiary amines could be incorporated into the corresponding diazenes78, not only homo-diazene but also hetero-diazenes, delivering highly congested diazenes that had not been previously synthesized. The exceptional catalytic efficiency allowed for application to Fmoc solid-phase peptide synthesis, affording elastin-derived diazene. The catalytic diazene synthetic method enabled efficient transformation of C–N bonds into C–halogen, C–H, C–O, C–S, C–Se, and C–C bonds through carbon-centered radical formation. We believe that these results demonstrate the high potential of the present catalytic diazene synthetic method using sterically hindered amine as an alkyl source.
Methods
General procedure for catalytic homo-diazene synthesis
To a 4-ml vial equipped with a magnetic stir bar, CuOAc (1.2 mg, 10 µmol, 5.0 mol%) was added in a glovebox followed by the addition of cold DMF (0.20 ml, 1.0 M), 1,3-dibromo-5,5-dimethylhydantoin (68.6 mg, 0.24 mmol, 1.2 equiv), amine (0.20 mmol, 1.0 equiv), and 1,8-diazabicyclo[5.4.0]undec-7-ene (30 μl, 0.20 mmol, 1.0 equiv) under Ar atmosphere. The reaction mixture was stirred at 25 °C for 10 min and diluted with CH2Cl2. The diluted solution was filtered through a silica short column and washed with EtOAc. After evaporation of the organic solvent under reduced pressure, the resultant mixture was purified by silica gel flash chromatography to obtain the desired compound.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
McGrath, N. A., Brichacek, M. & Njardarson, J. T. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ. 87, 1348–1349 (2010).
Wang, Y., Haight, I., Gupta, R. & Vasudevan, A. What is in our kit? An analysis of building blocks used in medicinal chemistry parallel libraries. J. Med. Chem. 64, 17115–17122 (2021).
Njardarson Web site (accessed Oct 2023). https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster.
Juliá, F., Constantin, T. & Leonori, D. Applications of halogen-atom transfer (XAT) for the generation of carbon radicals in synthetic photochemistry and photocatalysis. Chem. Rev. 122, 2292–2352 (2022).
Sachidanandan, K., Niu, B. & Laulhé, S. An overview of α-aminoalkyl radical mediated halogen-atom transfer. ChemCatChem 15, e202300860 (2023).
Tibbetts, J. D. et al. Decarboxylative, radical C–C bond formation with alkyl or aryl carboxylic acids: recent advances. Synthesis 55, 3239–3250 (2023).
Sandford, C. & Aggarwal, V. K. Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem. Commun. 53, 5481–5494 (2017).
Jaramillo, P., Domingo, L. R. & Pérez, P. Towards an intrinsic nucleofugality scale: The leaving group (LG) ability in CH3LG model system. Chem. Phys. Lett. 420, 95–99 (2006).
Blanksby, S. J. & Ellison, G. B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 36, 255–263 (2003).
Tshepelevitsh, S. et al. On the basicity of organic bases in different media. Eur. J. Org. Chem. 2019, 6735–6748 (2019).
Ouyang, K., Hao, W., Zhang, W. X. & Xi, Z. Transition-metal-catalyzed cleavage of C-N single bonds. Chem. Rev. 115, 12045–12090 (2015).
Wang, Q., Su, Y., Li, L. & Huang, H. Transition-metal catalysed C-N bond activation. Chem. Soc. Rev. 45, 1257–1272 (2016).
Berger, K. J. & Levin, M. D. Reframing primary alkyl amines as aliphatic building blocks. Org. Biomol. Chem. 19, 11–36 (2021).
Bapat, J. B. et al. Pyridines as leaving groups in synthetic transformations: Nucleophilic displacements of amino groups, and novel preparations of nitriles and isocyanates. Tetrahedron Lett. 17, 2691–2694 (1976).
M. Correia, J. T. et al. Photoinduced deaminative strategies: Katritzky salts as alkyl radical precursors. Chem. Commun. 56, 503–514 (2020).
Li, Y., Xiao, F., Guo, Y. & Zeng, Y. Recent developments in deaminative functionalization of alkyl amines. Eur. J. Org. Chem. 2021, 1215–1228 (2021).
Yousif, A. M., Colarusso, S. & Bianchi, E. Katritzky salts for the synthesis of unnatural amino acids and late-stage functionalization of peptides. Eur. J. Org. Chem. 26, e202201274 (2023).
Ashley, M. A. & Rovis, T. Photoredox-catalyzed deaminative alkylation via C–N bond activation of primary amines. J. Am. Chem. Soc. 142, 18310–18316 (2020).
Dorsheimer, J. R., Ashley, M. A. & Rovis, T. Dual nickel/photoredox-catalyzed deaminative cross-coupling of sterically hindered primary amines. J. Am. Chem. Soc. 143, 19294–19299 (2021).
Dorsheimer, J. R. & Rovis, T. Late-stage isotopic exchange of primary amines. J. Am. Chem. Soc. 145, 24367–24374 (2023).
Marchese, A. D., Dorsheimer, J. R. & Rovis, T. Photoredox-catalyzed generation of tertiary anions from primary amines via a radical polar crossover. Angew. Chem. Int. Ed. 63, e202317563 (2024).
Berger, K. J. et al. Direct deamination of primary amines via isodiazene intermediates. J. Am. Chem. Soc. 143, 17366–17373 (2021).
Dherange, B. D. et al. Direct deaminative functionalization. J. Am. Chem. Soc. 145, 17–24 (2023).
Berger, K. J. et al. N-(Benzyloxy)-N-(pivaloyloxy)−4-(trifluoromethyl)- benzamide. Org. Synth. 100, 113–135 (2023).
Huang, H., Ji, X., Wu, W., Huang, L. & Jiang, H. Copper-catalyzed formal C–N bond cleavage of aromatic methylamines: assembly of pyridine derivatives. J. Org. Chem. 78, 3774–3782 (2013).
Fang, H. & Oestreich, M. Reductive deamination with hydrosilanes catalyzed by B(C6F5)3. Angew. Chem. Int. Ed. 59, 11394–11398 (2020).
Li, Y. et al. Direct activation of the C(sp3)–NH2 bond of primary aliphatic alkylamines by a high-valent CoIII,IV2 (μ-O)2 diamond core complex. J. Am. Chem. Soc. 145, 2690–2697 (2023).
Thennakoon, D. S., Talipov, M. R. & Yi, C. S. Scope and mechanism of the ruthenium-catalyzed deaminative coupling reaction of enones with amines via regioselective Cα –Cβ bond cleavage. Organometallics 42, 2867–2880 (2023).
Overberger, C. G., O’Shaughnessy, M. T. & Shalit, H. The preparation of some aliphatic azo nitriles and their decomposition in solution. J. Am. Chem. Soc. 71, 2661–2666 (1949).
Simpkins, N. S. Azobisisobutyronitrile. in Encyclopedia of Reagents for Organic Synthesis vol. 4 11–12 (John Wiley & Sons, Ltd., 2001).
Elliott, Q. & Alabugin, I. V. AIBN as an electrophilic reagent for cyano group transfer. J. Org. Chem. 88, 2648–2654 (2023).
Wang, J.-M., Chen, T., Yao, C.-S. & Zhang, K. Synthesis of β-ketonitriles via N -heterocyclic-carbene-catalyzed radical coupling of aldehydes and azobis(isobutyronitrile). Org. Lett. 25, 3325–3329 (2023).
Chattapadhyay, D. et al. Harnessing sulfur(VI) fluoride exchange click chemistry and photocatalysis for deaminative benzylic arylation. ACS Catal. 13, 7263–7268 (2023).
Steiniger, K. A., Lamb, M. C. & Lambert, T. H. Cross-coupling of amines via photocatalytic denitrogenation of in situ generated diazenes. J. Am. Chem. Soc. 145, 11524–11529 (2023).
Doktor, K., Vantourout, J. C. & Michaudel, Q. A unified synthesis of diazenes from primary amines using a SuFEx/electrochemistry strategy. Org. Lett. 26, 7501–7506 (2024).
Nelsen, S. F., Bartlett, P. D. & Azocumene, I. Preparation and decomposition of azocumene. Unsymmetrical coupling products of the cumyl radical. J. Am. Chem. Soc. 88, 137–143 (1966).
Timberlake, J. W. & Martin, J. C. Synthetic routes to cyclopropyl-substituted azoalkanes. Some reactions of cyclopropylcarbinyl cyanates, isocyanates, benzoates, and p-nitrobenzoates. J. Org. Chem. 33, 4054–4060 (1968).
Martin, J. C. & Timberlake, J. W. Kinetic studies of reactions leading to cyclopropylcarbinyl radicals. Cyclopropyl-substituted azomethanes and hexacyclopropylethane. J. Am. Chem. Soc. 92, 978–983 (1970).
Klein, C. L. et al. Synthesis and crystallography of 2,3,3,5,6,6-hexamethyl-3,6-dihydropyrazine hexahydrate and 3-amino-3-methyl-2-butanone hydrochloride. J. Org. Chem. 49, 1208–1211 (1984).
Luedtke, A., Meng, K. & Timberlake, J. W. Carbonyl group stabilization of radicals: Solvent effects on azoalkane decomposition. Tetrahedron Lett. 28, 4255–4258 (1987).
Stowell, J. C. 1,2-Diazetidinediones. J. Org. Chem. 32, 2360–2362 (1967).
Engel, P. S. & Bishop, D. J. Recombination of unsymmetrical allylic and propargylic radicals produced from azo compounds. J. Am. Chem. Soc. 94, 2148–2149 (1972).
Golzke, V., Groeger, F., Oberlinner, A. & Ruechardt, C. Aliphatic azo compounds. Part XI. The thermolysis of polycyclic bridgehead azo compounds. Nouv. J. Chim. 2, 169–178 (1978).
Schmittel, M. & Ruechardt, C. Aliphatic azo compounds. XVI. Stereoisomerization and homolytic decomposition of cis and trans bridgehead diazenes. J. Am. Chem. Soc. 109, 2750–2759 (1987).
Neuman, R. C., Grow, R. H., Binegar, G. & Gunderson, H. J. cis-Diazenes. Viscosity effects, one-bond scission, and cis-trans isomerization. J. Org. Chem. 55, 2682–2688 (1990).
Lindovska, P. & Movassaghi, M. Concise synthesis of (−)-hodgkinsine, (−)-calycosidine, (−)-hodgkinsine B, (−)-quadrigemine C, and (−)-psycholeine via convergent and directed modular assembly of cyclotryptamines. J. Am. Chem. Soc. 139, 17590–17596 (2017).
Scott, T. Z., Armelin, V. F. & Movassaghi, M. Total synthesis and stereochemical assignment of (−)-psychotridine. Org. Lett. 24, 2160–2164 (2022).
Stetter, H. & Smulders, E. Über Verbindungen mit Urotropin-Struktur, XL VIII Über monofunktionelle stickstoffhaltige Derivate des Adamantans. Chem. Ber. 104, 917–923 (1971).
Bespal’ko, G. K., Khmaruk, A. M., Khimchenko, T. V. & Pinchuk, A. M. New types of nitrogen-containing organolead compounds. Zh. Obs. Khimii 50, 2046–2050 (1980).
Bauer, R. & Wendt, H. Anodic NN linkage by oxidation of anions of primary and secondary amines. Angew. Chem. Int. Ed. Engl. 17, 202–203 (1978).
Matsumoto, Y. et al. Amino acid schiff base bearing benzophenone imine as a platform for highly congested unnatural α-amino acid synthesis. J. Am. Chem. Soc. 142, 8498–8505 (2020).
Tsuji, T. et al. α-Amino acid and peptide synthesis using catalytic cross-dehydrogenative coupling. Nat. Synth. 1, 304–312 (2022).
Zhang, Q., Zuo, M., Wang, Y., Liu, W. & Zuo, X. Preparation and characterization of polyaniline microcapsule loaded with 2,2’-azobis(2-methylpropionitrile) initiator and its controlled release. J. Appl. Polym. Sci. 139, 1–10 (2022).
Matsuda, K., Tanaka, C., Sato, D. & Nishikata, T. Synthesizing complex quaternary carbons by the sequence-regulated additions of tert-alkyl radicals to two different olefins. Org. Lett. 25, 2840–2845 (2023).
Li, B., Shi, Y. & Fu, Z. Schiff base as a novel kind of catalyst for reversible complexation-mediated radical polymerization of methyl methacrylate. J. Polym. Sci. Part A Polym. Chem. 57, 1653–1663 (2019).
Zhang, X. & Huang, H. Copper-catalyzed oxidative coupling of AIBN and ketone-derived enoxysilanes to γ-ketonitriles. Org. Lett. 20, 4998–5001 (2018).
Movassaghi, M., Ahmad, O. K. & Lathrop, S. P. Directed heterodimerization: stereocontrolled assembly via solvent-caged unsymmetrical diazene fragmentation. J. Am. Chem. Soc. 133, 13002–13005 (2011).
Li, J., Kwon, E., Lear, M. J. & Hayashi, Y. Halogen bonding of N -halosuccinimides with amines and effects of brønsted acids in quinuclidine-catalyzed halocyclizations. Helv. Chim. Acta 104, e2100080 (2021).
Crowston, E. H., Lobo, A. M., Parbhakar, S., Rzepa, H. S. & Williams, D. J. X-Ray and SCF–MO model study of the complex formed between N-bromosuccinimide and 1,4-diazabicyclo[2.2.2]octane. J. Chem. Soc., Chem. Commun. 276, 278 (1984).
Castellote, I. et al. Reaction of imines with N-iodosuccinimide (NIS): unexpected formation of stable 1: 1 complexes. Chem. Commun. 1281–1283 (2007).
Raatikainen, K. & Rissanen, K. Breathing molecular crystals: halogen- and hydrogen-bonded porous molecular crystals with solvent induced adaptation of the nanosized channels. Chem. Sci. 3, 1235 (2012).
Wei, Y., Lin, S. & Liang, F. One-pot cascade leading to direct α-imidation of ketones by a combination of N-bromosuccinimide and 1,8-diazabicyclo[5.4.1]undec-7-ene. Org. Lett. 14, 4202–4205 (2012).
Wei, Y., Lin, S., Liang, F. & Zhang, J. N -Bromosuccinimide/1,8-diazabicyclo[5.4.1]undec-7-ene combination: β-amination of chalcones via a tandem bromoamination/debromination sequence. Org. Lett. 15, 852–855 (2013).
Wei, Y., Liang, F. & Zhang, X. N -bromoimide/DBU combination as a new strategy for intermolecular allylic amination. Org. Lett. 15, 5186–5189 (2013).
Li, M., Yuan, H., Zhao, B., Liang, F. & Zhang, J. Alkyne aminohalogenation enabled by DBU-activated N-haloimides: direct synthesis of halogenated enamines. Chem. Commun. 50, 2360 (2014).
Tan, H., Li, M. & Liang, F. Direct α-C-H amination of β-dicarbonyl compounds using DBU-activated N-haloimides as nitrogen sources. RSC Adv. 4, 33765 (2014).
Li, Y., Zhang, L., Yuan, H., Liang, F. & Zhang, J. Rapid α-amination of N-substituted indoles by using DBU-activated N-haloimides as nitrogen sources. Synlett 26, 116–122 (2015).
Li, Y. & Liang, F. With DBU-activated N-bromophthalimide as potential N-sources to achieve P–N cross-coupling of P(O)–H compounds. Tetrahedron Lett. 57, 2931–2934 (2016).
Heuger, G., Kalsow, S. & Göttlich, R. Copper(I) catalysts for the stereoselective addition of N-chloroamines to double bonds: a diastereoselective radical cyclisation. European J. Org. Chem. 1848–1854 (2002).
He, C. et al. Aryl halide tolerated electrophilic amination of arylboronic acids with N -chloroamides catalyzed by CuCl at room. Temp. Angew. Chem. Int. Ed. 47, 6414–6417 (2008).
Kawano, T., Hirano, K., Satoh, T. & Miura, M. A new entry of amination reagents for heteroaromatic C−H bonds: copper-catalyzed direct amination of azoles with chloroamines at room temperature. J. Am. Chem. Soc. 132, 6900–6901 (2010).
Miura, T., Morimoto, M. & Murakami, M. Copper-catalyzed amination of silyl ketene acetals with N -chloroamines. Org. Lett. 14, 5214–5217 (2012).
Li, Y. et al. Directed copper-catalyzed intermolecular aminative difunctionalization of unactivated alkenes. J. Am. Chem. Soc. 141, 18475–18485 (2019).
Zhou, X. et al. Haloamines as bifunctional reagents for oxidative aminohalogenation of maleimides. Org. Lett. 23, 3669–3673 (2021).
Li, Y. et al. Regio-, site-, and stereoselective three-component aminofluorination of 1,3-dienes via cooperative silver salt and copper catalysis. ACS Catal. 13, 2410–2421 (2023).
Zawalski, R. C. et al. Synthesis and decomposition of a β-ketodiazene. Tetrahedron Lett. 21, 425–428 (1980).
Overberger, C. G., Huang, P.-T. & Berenbaum, M. B. 1,1’-Azo-bis-1-cyclohexanenitrile. Org. Synth. 32, 16 (1952).
Brook, K. B., Sahoo, S. R. & Uyeda, C. Dicobalt-catalyzed N=N coupling reactions of tertiary alkyl azides to form azoalkanes. Chem 102437 (2025).
Acknowledgements
This work was financially supported by the JST-FOREST program (JPMJFR2229), KAKENHI grant numbers JP24H01777 for the Grant-in-Aid for Transformative Research Areas (A) Latent Chemical Space from MEXT, JP21A204, JP21H05207, and JP21H05208 for the Grant-in-Aid for Transformative Research Areas (A) Digitalization-driven Transformative Organic Synthesis (Digi-TOS) from MEXT, a Grant-in-Aid for Scientific Research (B) (JP21H02607), a Grant-in-Aid for Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) (JP20am0101091, JP22ama121031) and AMED (JP21ak0101167, JP22ak0101167, JP23ak0101167). T.T. thanks the JSPS for a predoctoral fellowship. T.O. thanks NAGASE Science Technology Foundation for financial support. R.Y. thanks the Takeda Science Foundation, the Noguchi Institute. and the Nakatani Foundation, and the Japan Association for Chemical Innovation, for financial support. We are grateful to Prof. Ken Yamazaki (Okayama University) for fruitful discussion about DFT calculation, Prof Kenichi Yamada, Dr. Masami Abe, and Mr. Eisho Kozakura for EPR measurement.
Author information
Authors and Affiliations
Contributions
T.T. and R.Y. conceived the work, analyzed the data, discussed the results, and all authors co-wrote the manuscript. T.O. checked the manuscript and Supplementary Information. T.T. designed and carried out the experiments with I.F. and T.H. T.T. and A.O. performed DFT calculation. M.H. performed thermogravimetric analysis (TGA).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qing-Hai Deng and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsuji, T., Fukumoto, I., Hario, T. et al. Catalytic diazene synthesis from sterically hindered amines for deaminative functionalization. Nat Commun 16, 6266 (2025). https://doi.org/10.1038/s41467-025-61662-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61662-9
