
Abstract
Although ring-opening reactions of bicyclo[1.1.0]butanes (BCBs) provide a reliable platform for synthesizing functionalized cyclobutanes, current methods frequently encounter challenges such as poor diastereoselectivity, regioselectivity issues, and a lack of α- and β‘-selective transformations. Herein, we report a catalyst-controlled, regiodivergent α- and β‘-selective hydrophosphination of acyl BCBs, which expands the chemical space of tertiary phosphines with multi-substituted cyclobutane backbones derived from identical starting materials. Utilizing a Cu(I) catalytic system, we achieve an α-selective nucleophilic addition to 1,3-disubstituted BCBs. This reaction exhibits a broad substrate scope under mild conditions, yielding valuable 1,1,3-functionalized cyclobutanes predominantly as single diastereoisomers. In contrast, the unusual β‘-selective pathway facilitated by a Cu(II) catalytic system produces 1,2,3-trisubstituted variants with up to >20:1 d.r. The developed method holds promise for accessing structurally diverse cyclobutanes with potential applications in medicinal chemistry and the design of organophosphorus catalysts.
Similar content being viewed by others
Introduction
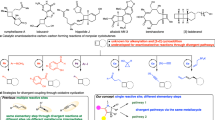
Controlling the selectivity of reactions is a primary goal in the field of synthetic organic chemistry. Among them, controllable regiodivergent synthesis is particularly appealing yet challenging, as it provides a unique platform for obtaining diverse products through regioselective functionalization of the same functional group in identical starting materials1,2,3,4. Multi-substituted cyclobutanes, especially the 1,1,3- and 1,2,3-functionalized variants, are essential components of various biologically active molecules. These molecules demonstrate significant pharmaceutical activities, including antidopaminergic effects, as well as antiviral and anticonvulsive properties (Fig. 1a)5,6,7,8. Furthermore, they could also be employed as conformationally restricted bioisosteres for flexible ethyl or propyl linkers9. Although many state-of-the-art strategies, such as [2 + 2] cycloadditions10,11, ring contractions12,13, ring expansions of cyclopropanes14, and functionalization of existing four-membered ring substrates15,16,17,18, have been developed, constructing multi-substituted cyclobutane frameworks with high stereoselective control remains a significant challenge. This difficulty arises from the inherent ring strain and congested environment compared to their five- and six-membered congeners19,20,21,22,23. Therefore, developing a highly selective and divergent synthesis strategy to expand the chemical space of multi-substituted cyclobutanes from the same starting materials is of significant interest and importance.

a Representative biologically active multi-substituted cyclobutanes. b Substrate-dependent divergent ring-opening of BCB-Bpin. c Palladium catalyzed α-selective alkenylation of BCBs. d Synthesis of cyclobutanes by ring-opening of BCBs. e Catalyst-dependent regiodivergent hydrophosphination of acyl BCBs.
Bicyclo[1.1.0]butanes (BCBs) represent one of the most intriguing classes of “spring-loaded” reagents24,25,26,27,28,29,30, capable of being ring-opened by various nucleophiles31,32,33,34,35,36, electrophiles37,38, as well as by radicals39,40,41,42. Following Baran’s fundamental work on strain-release amination in 201643, the ring-opening reaction of BCBs has been revived as a reliable synthetic toolkit for functionalized cyclobutanes. The highly strained, “bent” central σ-bond in BCBs can participate in conjugation, enabling BCBs substituted with electron-withdrawing groups (EWGs) to function as electrophiles in Michael-type additions or as radical acceptors in Giese reactions. Consequently, β-selective heterolytic and homolytic ring-opening processes involving BCBs have been extensively developed and are now well-established30. For instance, Wipf conducted pioneering research on the base-promoted β-selective phosphination of BCB nitriles using phosphine boranes or H-phosphonates. However, similar to most β-selective ring-opening reactions of BCBs, this reaction suffers from low diastereoselectivity (1.1:1 to 3.3:1)44. In contrast, α-selective ring-opening process of BCBs is rare. In 2021, Aggarwal group developed α-selective ring-opening reactions of BCBs with a variety of O-, S-, and N-nucleophiles45. The application of monosubstituted bicyclo[1.1.0]butyl boronic ester (BCB-Bpin) to form a boronate complex with nucleophiles is essential for enabling a consecutive 1,2-migration process, thereby achieving exclusive α-selectivity. When 1,3-disubstituted BCB was employed as the substrate, only the β-functionalized cyclobutane product was produced (Fig. 1b). Subsequently, Gevorgyan reported an elegant palladium hydride-enabled regioselective hydroalkenylation of monosubstituted BCBs, leading to α-alkenylated 1,1-disubstituted cyclobutanes (Fig. 1c)46. Complementary to the aforementioned two impressive studies, we have recently accomplished the α-selective radical addition to acyl bicyclobutanes, which facilitates the synthesis of cyclobutene products47. Although progress has been made, several challenging issues related to ring opening of BCBs remain (Fig. 1d): (1) How to achieve α-selective ring-opening reactions of 1,3-disubstituted bicyclo[1.1.0]butanes (BCBs) for the synthesis of valuable 1,1,3-trisubstituted cyclobutanes with high diastereoselectivity? (2) How to achieve regiodivergent ring-opening functionalization of BCBs to rapidly expand the chemical space of multi-substituted cyclobutanes derived from the same starting materials. (3) Due to the presence of a fragile central C–C bond, nearly all BCB ring-opening reactions result in the formation of new bonds with the bridgehead carbon of BCB. The ring-opening β‘-selective functionalization of BCBs48, which generates 1,2,3-functionalized cyclobutanes, is rare and is limited to the cyclization of BCBs as reported by Glorius49,50.
Recently, strain-release-driven hydrophosphination reactions have garnered considerable attention in organic synthesis51,52,53,54. This approach is highly valued for its atom-economical and streamlined nature, enabling the construction of organophosphorus architectures that feature cyclopropane55,56,57,58, cyclobutane44,59, and bicyclo[1.1.1]pentane backbones60,61,62,63. Motivated by these advancements, we herein report a catalyst-controlled, switchable unusual α- and β‘- phosphination of acyl bicyclobutanes. This method effectively addresses the three aforementioned challenges associated with BCB ring-opening reactions (Fig. 1e).
Results
Reaction optimization
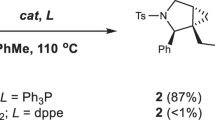
We initiated our investigation using BCB ester 1a as the model substrate and diphenyl phosphine (2) as the nucleophile for the ring-opening reaction (Table 1). We tested various transition metal catalysts, including MgI2, ZnCl2, and copper salts (Table 1, entries 1–7; See the Supplementary Tables 1, 2 for the complete set of data). In contrast to Wipf’s β-selective phosphination of BCB44, the model reaction unexpectedly yielded α-phosphanyl-substituted cyclobutyl ester 3a without the use of a base. While monovalent copper catalysts, including CuI, CuBr and Cu(CH₃CN)₄PF₆ (entries 3–4 and 6), as well as the divalent copper salt Cu(OAc)₂ (entry 7), afforded the cyclobutyl phosphine product 3a with low to moderate diastereoselectivity, the use of CuCl as the catalyst exclusively produced the desired product with a 95% yield and a diastereomeric ratio (d.r.) of 16:1 (entry 5). A range of solvents were evaluated, among which tetrahydrofuran, an ether-type solvent, and toluene, a nonpolar solvent, exhibited superior performance in terms of both yield and selectivity (entry 5 and entries 11–13). While the addition of Cs₂CO₃ resulted in a slight decrease in yield and diastereoselectivity (entry 14), elevating the reaction temperature to 60 °C was found to effectively enhance both the yield and diastereomeric ratio of the product (entry 15). Control experiments demonstrated that the desired product was undetectable by ¹H NMR spectroscopy when the reaction was conducted at room temperature (entry 16), whereas a 22% yield of the desired diastereoisomer of 3a was observed when the reaction was performed at 60 °C for 48 h (entry 17). Eventually, a 97% NMR yield of product 3a, a d.r. value exceeding 20:1, and exclusive α-selectivity were achieved under optimal conditions A: the reaction of BCB (1.0 equiv), diphenyl phosphine (1.2 equiv), in the presence of CuCl (10 mol%), in THF at 60 °C (entry 15). To our surprise, during the screening of copper catalysts, we observed that the presence of copper(II) halides in the current ring-opening reaction resulted in the formation of both α-phosphinated product 3a and β‘-phosphinated product 4a (entries 8–10). In comparison to nonpolar solvents (e.g. toluene), halogenated solvents (e.g. CH2Cl2), and polar protic solvents (e.g. MeOH), the polar aprotic solvents such as DMSO, DMA, and DMF yielded a higher amount of product 4a (entries 18–22). Altering the stoichiometry of substrates 1a and 2 has substantially influenced the distribution of products (entry 23 versus entry 24). After further optimization of additives and reaction temperature (entries 25–26), the desired β‘-phosphinated product 4a was obtained in 78% NMR yield with >20:1 d.r. (entry 26). The reaction employed substrates 1 (1.0 equiv) and diphenyl phosphine (2.0 equiv), CuBr2 (10 mol%) as the catalyst, and LiBr as the additive in DMF at 60 °C (conditions B).
Substrate scope
With the optimized conditions in hand, we first investigated the generality of this protocol for constructing trans-1,1,3-trisubstituted cyclobutanes via the α-selective ring-opening reaction of BCBs, and the results are summarized in Fig. 2. The variation in ester groups, including methyl (3a), ethyl (3b), benzyl (3 d), phenyl (3e), and the sterically hindered tert-butyl (3c), led to the formation of the corresponding cyclobutylated products with excellent yields (81–99%) and diastereoselectivity (>20:1). Notably, the scale-up synthesis of 3a (1.0 mmol) was performed almost without loss of efficiency and selectivity (92% yield, d.r. > 20:1). Both para- and meta-substituted aryl moieties were smoothly engaged in the ring-opening reaction, yielding the expected products (3f–3n) with good yield. Notably, BCBs bearing electron-donating (e.g. methyl group) and electron-withdrawing (e.g. CF3 group) bridgehead aryl substituents afforded the corresponding cyclobutanes 3g and 3j in yields of 79% and 91%, respectively. The experimental results indicated that the current α-selective ring-opening reaction may occur without the formation of a benzyl cation intermediate. The structure and relative stereochemistry of 3l were unambiguously determined by X-ray crystallography analysis. The BCB substrate containing a substituent at the ortho position of the phenyl group was tolerated, although it resulted in a lower yield (3o). In addition, not only the phenyl moiety but also the 2-thienyl derived BCB (as in 3p) produced the expected product with a high yield and excellent diastereomeric ratio (>20:1). BCB 1q, derived from the natural product L-menthol, furnished the desired phosphine 3q in 97% yield. Besides BCB esters, the substituted phenyl BCB ketones (3r–3u) reacted efficiently with diphenyl phosphine (2) to yield the corresponding products in satisfactory yields ranging from 80% to 97%, although with moderate d.r. values. The alkyl-substituted BCB ketone is a suitable substrate for the selective formation of the desired phosphine 3v with >20:1 d.r. The methylsubstituted phenyl BCB ketone also exhibited smooth reactivity, yielding 51% of product 3w under the conditions A. While the BCB amide substrate (1x) is suitable for this reaction, BCB substrates modified with sulfonyl (1y) and nitrile (1z) groups are incompatible with the α-selective ring-opening reaction.

aConditions A: 0.2 mmol BCB 1, 1.2 equiv. diphenyl phosphine, and 10 mol% CuCl in 2.0 mL THF at 60 °C for 24 h. bIsolated yield. c1.0 mmol scale.
In parallel, the β‘-selective functionalization of BCBs was employed to synthesize phosphines containing 1,2,3-trisubstituted cyclobutane scaffolds (Fig. 3). The regio- and diastereoselective ring-opening reaction was not restricted to methyl ester-derived BCBs; ethyl (as in 4b), tert-butyl (as in 4c), benzyl (as in 4d) and phenyl (as in 4e) substituted BCB esters also produced the target products in good yields and excellent d.r. value. The molecular connectivity of 4e has been confirmed by X-ray crystallographic analysis. Notably, the reaction between 1a and 2a could be scaled up to yield 4aa at 72% efficiency using 1.0 mmol of 1a. Aryl-substituted BCB ester 1d successfully yielded phosphine 4d, whereas BCB 1bb, which contains a methyl group in the β-position of the BCB, did not produce the desired product. This emphasizes the critical role of the β-phenyl ring in the ring-opening reaction. BCBs, which contain various substituents including OCF3 (4f), alkyl groups (methyl in 4g), and halogen groups (4-F in 4h, 4-Br in 4i, 3-F in 4k, 3-Cl in 4l), at the meta- or para-position of the phenyl ring, are compatible with standard conditions B. These reactions yielded the corresponding phosphines with moderate to good yields and excellent stereoselectivity. For BCB substrates 1j and 1m, lower reaction temperature (25 °C) are necessary to achieve improved diastereoselectivity. As a trend, BCBs bearing electron-rich aryl substituents generally afforded the corresponding products in higher yields compared to those with electron-poor aryl substituents (4g versus 4j; 4m versus 4n). Moreover, the BCB ester containing a 2-thienyl group yielded the desired product 4p at a 50% yield as a single diastereoisomer. The use of L-menthyl as a chiral auxiliary resulted in the formation of the desired chiral phosphine 4q, containing six chiral carbon centers, in 75% yield. Furthermore, the substituents at the meta- and para-positions of the phenyl ring in BCB ketones were systematically examined, and each successfully delivered the desired β’-ring-opening products (4r–4u) while maintaining high diastereoselectivity. Notably, BCB amide (1aa) was found to be compatible with this reaction system, affording the cyclobutane product (4aa) in 74% yield with excellent diastereoselectivity. In contrast, BCB derivatives containing an acyl pyrazole unit (1x) or a sulfonyl group (1y) failed to undergo the desired ring-opening process.

aConditions B: 0.2 mmol BCB 1, 2.0 equiv. diphenyl phosphine, 1.0 equiv. LiBr and 10 mol% CuBr2 in 2.0 mL DMF at 60 °C for 24 h. bIsolated yield. c1.0 mmol scale, 4a: d.r. = 15:1.dRun at 25 °C.
Synthetic applications & proposed mechanism
To demonstrate the practical applicability of this methodology in expanding the chemical diversity of organophosphines, we performed a series of transformations on the ring-opening products (Fig. 4). Due to the oxygen sensitivity of the phosphine group in the ring-opening product, Me₂S•BH₃ and S₈ were introduced during the work-up phase, respectively, to form the corresponding phosphine-borane complexes (5 and 6) and phosphine sulfides (7 and 8) in a one-pot sequence. The cyclobutane-derived phosphine-borane 6 can also be efficiently prepared through the reduction of phosphine oxide 4a. The treatment of compounds 3a, 4a, 5, and 6 with LiBH4 effectively reduced the ester groups, resulting in the formation of the corresponding primary alcohols 24, 21, 9, and 18. The Swern oxidation of compound 9 afforded the corresponding aldehyde 10 in 80% yield. The reaction of 3a with Grignard reagent afforded tertiary alcohol 25 in quantitative yield and unharmed d.r. value. Hydrolysis of 3a and 4a produced the corresponding carboxylic acids 26 and 22, with yields of 86% and 95%, respectively. This result facilitates subsequent esterification. For instance, the distinctive cyclobutyl phosphine scaffold can be integrated into bioactive molecules, such as estrone (22 → 23), and chiral urea catalysts (26 → 27). Phosphine-nitrogen-based ligands, particularly phosphine oxazolines, represent a versatile class of ligands that have been widely employed in various transition-metal-catalyzed asymmetric reactions64. While phosphines incorporating cyclopentane or cyclohexane backbones are commonly utilized in ligand design, phosphine ligands featuring a cyclobutane backbone are relatively rare. This may be due to the challenges associated with their synthesis. Therefore, an efficient mothod was developed for the synthesis of phosphine oxazoline ligands with multisubstituted cyclobutane backbones (26 → 28 and 29). The deprotection of compounds 6 and 9 with diethylamine effectively removed the borane groups, affording the free tertiary phosphines 15 and 11 in quantitative yields. The transformation of tertiary phosphines to iminophosphoranes (13 and 16), as well as phosphonium methyl iodides (14 and 17), is also feasible. Notably, based on the current selective ring-opening reaction, the synthesis of diphosphine ligands containing cyclobutyl phosphine scaffolds has been achieved, as exemplified by the efficient synthesis of compound 19 and chiral diphosphine ligand 12. Compound 19 was treated with CpRuCl(PPh₃)₂ to yield ruthenium complex 20 with a 60% yield. Moreover, ligand 19 demonstrated efficacy in the palladium-catalyzed hydroamidation of isoprene65. The preliminary application of chiral diphosphine ligand 12 in asymmetric catalysis was explored through a palladium-catalyzed enantioselective allylic alkylation reaction, yielding the corresponding product 37 with 67% ee (Fig. 5). The aforementioned results highlight the potential of regiodivergent hydrophosphination of BCBs as a strategy for designing new phosphine ligands and advancing asymmetric catalysis.

a LiBH4, THF. b Oxalyl dichloride, DMSO, Et3N, CH2Cl2, −78 °C. c Et2NH, 65 °C. d (1) Et2NH, 65 °C; (2) (S)-BINOL, PCl3, DMAP, Et3N. e Et2NH then TsN3. f Et2NH then MeI. g (1) PPh3, CBr4, CH2Cl2, 0 °C to RT; (2) Ph2PH, nBuLi, THF, −78 °C to RT; (3) Et2NH, 65 °C. h CpRuCl(PPh3)2, toluene, 90 °C. i Cu(OTf)2, TMDS, toluene then Me2S·BH3. j LiOH, THF/H2O. k Estrone, EDC·HCl, DMAP, DMF. l MeMgBr, THF, RT. m (R)-1-(3,5-bis(trifluoromethyl)phenyl)-3-(2-hydroxy-1-phenylethyl)urea, DMAP, EDC·HCl, Et3N, CH2Cl2. n (1) oxalyl dichloride, DMF, CH2Cl2; (2) Amino alcohol, Na2CO3, CH2Cl2; (3) TsCl, Et3N, CH2Cl2.

Compounds 12 and 19, serving as novel phosphine ligands, are applied to the palladium-catalyzed hydroamidation of isoprene and the enantioselective allylic alkylation reaction.
To gain insight into this transformation, we conducted a series of control experiments designed to probe the reaction pathway. Initially, radical quenching experiments were conducted by introducing various radical inhibitors, specifically 2,6-di-tert-butyl-4-methylphenol (BHT) and 1,1-diphenylethylene, into the α-phosphination reaction system under controlled conditions. The addition of these scavengers had a minimal impact on the progression of the reaction, suggesting that a radical pathway may not be involved in this process. In contrast, when BCB 1a was treated with diphenyl phosphine (2), LiBr, and CuBr2 in the presence of BHT or 1,1-diphenylethylene at room temperature, the yield and diastereomeric ratio of 4a decreased; however, the yield of 3a increased (Fig. 6a). Furthermore, no corresponding radical trapping products were detected by 1H NMR and HRMS.

a Radical quenching experiments. b Isomerization of BCB. c Hydrophosphination of cyclobutene. d Hydrophosphination of monosubstituted BCB ketone.
Heating BCB 1a in THF for 24 h in the presence of CuCl did not yield cyclobutene 38a. In contrast, mixing BCB 1a with CuBr2 in DMF at room temperature resulted in the isomerization of 1a to cyclobutene 38a, achieving a 27% NMR yield. The yield of cyclobutene can be enhanced to 70% through the addition of LiBr (Fig. 6b). These results prompt an investigation into whether cyclobutene 38a serves as the key intermediate for the synthesis of the current cyclobutylphosphine products. The CuCl-catalyzed hydrophosphination of cyclobutene did not produce the product 3a; however, it yielded 4a with 40% NMR yield and >20:1 diastereomeric ratio. A comparable phenomenon was observed when CuBr2 was employed as a catalyst (Fig. 6c). Under standard conditions A and B, the hydrophosphination of monosubstituted BCB ketone regioselectively yielded the 1,3-disubstituted cyclobutane 40, while the corresponding α- or β‘-phosphinated cyclobutane products were not detected (Fig. 6d).
Based on the experimental results presented above and previous studies on copper-catalyzed hydrophosphination reactions57,58, we propose a possible mechanism for the selective ring-opening reaction of 1 with 2, as illustrated in Fig. 7. Initially, the diarylphosphorus group is transferred to the copper(I) catalyst, resulting in the formation of a nucleophilic copper(I)-diphenylphosphide intermediate (INT-I). Subsequently, this intermediate INT-I coordinates with acyl bicyclobutane 1 to generate the intermediate INT-II. When R1 is hydrogen (R1 = H), a β-selective nucleophilic addition occurs, followed by protonolysis with HX present in the system. This sequence of events leads to the formation of 1,3-disubstituted cyclobutane 40’ and concomitantly regenerates the original copper catalyst. However, when a sterically hindered group is present at the β-position of BCB 1, the transition state for the α-selective ring-opening pathway (TS-II) is favored. This preference arises due to steric repulsion from the R1 substituent (TS-I) as the diarylphosphorus group on the copper atom approaches the β-position of the BCB 1. Consequently, the ring-opening reaction proceeds with exclusive α-selectivity, yielding 1,1,3-trisubstituted cyclobutane 3′.

BCBs undergo α-, β, and β‘-selective ring-opening reactions with HPPh2 under copper catalysis.
Regarding the β‘-selective ring-opening functionalization of BCBs, the divalent copper salt CuBr2 was employed as a catalyst, which facilitated the Lewis-acid-catalyzed isomerization of BCBs and led to the formation of cyclobutenes 38 through the carbocation species INT-IV37,66,67. Stabilization of the carbocation intermediate via an aryl group (R1 = aryl) through resonance facilitates the subsequent intramolecular E1 elimination. In the presence of HPPh₂, the copper(II) salts can be reduced to copper(I), which serves as the active catalyst68,69. LiBr can act as a mediator to facilitate the reduction of divalent copper salt70. Subsequently, 1,2,3-trisubstituted cyclobutane 4′ is generated through a copper(I)-catalyzed regioselective hydrophosphination of cyclobutene.
Discussion
In conclusion, we report a catalyst-controlled regiodivergent synthesis of multi-substituted cyclobutanes from BCBs. By modulating the catalytic system with varying copper oxidation states, we have achieved precise control over the regioselective ring-opening pathways of acyl BCBs. The α-selective hydrophosphination pathway delivers 1,1,3-trisubstituted cyclobutane-derived phosphines, while the unusual β′-selective process affords 1,2,3-trisubstituted variants, both with excellent regio- and diastereoselectivity control (up to >20:1 d.r.). This strategy addresses long-standing challenges in BCB chemistry, including the limited availability of α-selective transformations for 1,3-disubstituted BCBs and the rarity of β′-selective functionalization. Moreover, the reaction system demonstrates good functional group tolerance, underscoring its robustness and versatility. The scalability of the process, along with the successful derivatization of products, particularly facilitates the preparation of new phosphine ligands that incorporate cyclobutane frameworks and allow for late-stage modification of bioactive compounds. The (chiral) diphosphine ligands, synthesized through the divergent hydrophosphination of BCBs, have shown promising potential in regioselective reactions and asymmetric catalysis. This further emphasizes the practical utility and synthetic value of the protocol. This divergent ring-opening phosphination strategy for BCBs paves the way for the development of innovative methodologies aimed at the selective synthesis of valuable cyclobutane derivatives and tertiary phosphine compounds.
Methods
General procedure for the α-selective ring-opening functionalization of BCBs
Under an atmosphere of N2, to a 25 mL oven-dried Schlenk tube were added BCBs 1 (0.20 mmol, 1.0 equiv), CuCl (2.0 mg, 0.020 mmol) and diphenylphosphane (44.7 mg, 42 μL, 0.24 mmol, 1.2 equiv), followed by 2.0 mL of anhydrous THF. The mixture was stirred at 60 °C for 24 h. Upon cooling to room temperature, the reaction was quenched with H2O2 (0.10 mL, 30% w/w) and stirred for an additional 1 h. The reaction mixture was subsequently diluted with ethyl acetate (5 mL) and water (5 mL). The aqueous layer was extracted with EtOAc (10 mL × 2). The combined organic layers were washed with brine, dried over anhydrous MgSO₄, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using a CH2Cl2/MeOH mixture (100:1, v/v) as the eluent, yielding the desired product 3.
General procedure for the β‘-selective ring-opening functionalization of BCBs
Under an atmosphere of N2, to a 25 mL oven-dried Schlenk tube were added BCBs 1 (0.20 mmol, 1.0 equiv), CuBr2 (4.5 mg, 0.020 mmol), LiBr (17.4 mg, 0.20 mmol, 1.0 equiv) and diphenylphosphane (74.5 mg, 70 μL, 0.40 mmol, 2.0 equiv), followed by 2.0 mL of anhydrous DMF. The mixture was stirred at 60 °C for 24 h. Upon cooling to room temperature, the reaction was quenched with H2O2 (0.10 mL, 30% w/w) and stirred for an additional 1 h. The reaction mixture was subsequently diluted with ethyl acetate (5 mL) and water (5 mL). The aqueous layer was extracted with EtOAc (10 mL × 2). The combined organic layers were washed with brine, dried over anhydrous MgSO₄, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using a CH2Cl2/MeOH mixture (75:1, v/v) as the eluent, yielding the desired product 4.
Data availability
The data supporting the findings of this study are available within this article and its Supplementary Information, which contains experimental details, characterization data, copies of NMR spectra and HPLC spectra for all new compounds, and X-ray structural analysis. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2418952 (3l) and CCDC 2418954 (4e). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data are available from the corresponding author upon request.
References
Schreiber, S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964–1969 (2000).
Funken, N., Zhang, Y.-Q. & Gansäuer, A. Regiodivergent catalysis: a powerful tool for selective catalysis. Chem. Eur. J. 23, 19–32 (2017).
Lee, Y.-C., Kumar, K. & Waldmann, H. Ligand-directed divergent synthesis of carbo- and heterocyclic ring systems. Angew. Chem. Int. Ed. 57, 5212–5226 (2018).
Viji, M. et al. Regiodivergent organocatalytic reactions. Catalysts 11, 1013 (2021).
Gaoni, Y. et al. Synthesis, NMDA receptor antagonist activity, and anticonvulsant action of 1-aminocyclobutanecarboxylic acid derivatives. J. Med. Chem. 37, 4288–4296 (1994).
Hanrahan, J. R., Taylor, P. C. & Errington, W. The synthesis of 3-phosphonocyclobutyl amino acid analogues of glutamic acid via diethyl 3-oxycyclobutylphosphonate, a versatile synthetic intermediate. J. Chem. Soc., Perkin Trans. 1, 493–502 (1997).
Wahl, J. M., Conner, M. L. & Brown, M. K. Synthesis of (-)-Hebelophyllene E: an entry to geminal dimethyl-cyclobutanes by [2+2] cycloaddition of alkenes and allenoates. Angew. Chem. Int. Ed. 57, 4647–4651 (2018).
Goetzke, F. W. et al. A catalytic asymmetric cross-coupling approach to the synthesis of cyclobutanes. Nat. Chem. 13, 880–886 (2021).
van der Kolk, M. R. et al. Cyclobutanes in small-molecule drug candidates. ChemMedChem 17, e202200020 (2022).
Xu, Y., Conner, M. L. & Brown, M. K. Cyclobutane and cyclobutene synthesis: catalytic enantioselective [2+2] cycloadditions. Angew. Chem. Int. Ed. 54, 11918–11928 (2015).
Poplata, S. et al. Recent advances in the synthesis of cyclobutanes by olefin [2+2] photocycloaddition reactions. Chem. Rev. 116, 9748–9815 (2016).
Hui, C. et al. Stereoselective synthesis of cyclobutanes by contraction of pyrrolidines. J. Am. Chem. Soc. 143, 18864–18870 (2021).
Qin, H. et al. N-atom deletion in nitrogen heterocycles. Angew. Chem. Int. Ed. 60, 20678–20683 (2021).
Li, J. et al. Recent advances in the total synthesis of cyclobutane-containing natural products. Org. Chem. Front. 7, 136–154 (2020).
Gutekunst, W. R. & Baran, P. S. Applications of C−H functionalization logic to cyclobutane synthesis. J. Org. Chem. 79, 2430–2452 (2014).
Kang, G. et al. Transannular C–H functionalization of cycloalkane carboxylic acids. Nature 618, 519–525 (2023).
Gauvry, N. et al. Substituted cyclobutenes, their preparation, and their versatility in synthesis. Eur. J. Org. Chem. 2006, 5207–5218 (2006).
Wang, M. & Lu, P. Catalytic approaches to assemble cyclobutane motifs in natural product synthesis. Org. Chem. Front. 5, 254–259 (2018).
Fawcett, A., Biberger, T. & Aggarwal, V. K. Carbopalladation of C–C σ-bonds enabled by strained boronate complexes. Nat. Chem. 11, 117–122 (2019).
Wang, H. et al. syn-Selective difunctionalization of bicyclobutanes enabled by photoredox-mediated C−S σ‑bond scission. J. Am. Chem. Soc. 145, 23771–23780 (2023).
Shen, H.-C. et al. Iridium-catalyzed asymmetric difunctionalization of C−C σ‑bonds enabled by ring-strained boronate complexes. J. Am. Chem. Soc. 145, 16508–16516 (2023).
Xiao, C. et al. Stereoselective radical acylfluoroalkylation of bicyclobutanes via N-heterocyclic carbene catalysis. Angew. Chem. Int. Ed. 64, e202416781 (2025).
Kerner, M. J. & Wipf, P. Semipinacol-type rearrangements of [3-(arylsulfonyl)bicyclo[1.1.0]butan-1-yl]alkanols. Org. Lett. 23, 3615–3619 (2021).
Golfmann, M. & Walker, J. C. L. Bicyclobutanes as unusual building blocks for complexity generation in organic synthesis. Commun. Chem. 6, 9 (2023).
Kelly, C. B., Milligan, J. A., Tilley, L. J. & Sodano, T. M. Bicyclobutanes: from curiosities to versatile reagents and covalent warheads. Chem. Sci. 13, 11721–11737 (2022).
Turkowska, J., Durka, J. & Gryko, D. Strain release-an old tool for new transformations. Chem. Commun. 56, 5718–5734 (2020).
Walczak, M. A. A., Krainz, T. & Wipf, P. Ring-strain-enabled reaction discovery: new heterocycles from bicyclo[1.1.0]butanes. Acc. Chem. Res. 48, 1149–1158 (2015).
Bellotti, P. & Glorius, F. Strain-release photocatalysis. J. Am. Chem. Soc. 145, 20716–20732 (2023).
Hu, Q.-Q. et al. Strain-release transformations of bicyclo[1.1.0]butanes and [1.1.1]propellanes. Tetrahedron Chem. 9, 100070 (2024).
Zhan, X., He, H.-X., Peng, Q. & Feng, J.-J. Synthesis of cyclobutanes and cyclobutenes by strain-release-driven ring-opening of bicyclo[1.1.0]butanes. Synthesis 56, 3829–3848 (2024).
Gaoni, Y. Conjugate addition of organocopper reagents to 1-arylsulfonylbicyclobutanes. synthesis of the racemic form of the sex pheromone of the citrus mealybug, Planococcus citri (Risso). Tetrahedron Lett. 23, 5215–5218 (1982).
Panish, R. et al. Enantioselective synthesis of cyclobutanes via sequential Rh-catalyzed bicyclobutanation/Cu-catalyzed homoconjugate addition. J. Am. Chem. Soc. 135, 9283–9286 (2013).
Lopchuk, J. M. et al. Strain-release heteroatom functionalization: development, scope, and stereospecificity. J. Am. Chem. Soc. 139, 3209–3226 (2017).
Guin, A. et al. Lewis acid-catalyzed diastereoselective carbofunctionalization of bicyclobutanes employing naphthols. Chem. Sci. 14, 6585–6591 (2023).
Tokunaga, K. et al. Bicyclobutane carboxylic amide as a cysteine-directed strained electrophile for selective targeting of proteins. J. Am. Chem. Soc. 142, 18522–18531 (2020).
Wölfl, B. et al. Strain-release driven epoxidation and aziridination of bicyclo[1.1.0]butanes via palladium catalyzed σ-bond nucleopalladation. Angew. Chem. Int. Ed. 62, e202217064 (2023).
Dhake, K. et al. Beyond bioisosteres: divergent synthesis of azabicyclohexanes and cyclobutenyl amines from bicyclobutanes. Angew. Chem. Int. Ed. 61, e202204719 (2022).
Bennett, S. H. et al. Difunctionalization of C−C σ‑bonds enabled by the reaction of bicyclo[1.1.0]butyl boronate complexes with electrophiles: reaction development, scope, and stereochemical origins. J. Am. Chem. Soc. 142, 16766–16775 (2020).
Ociepa, M. et al. Polarity-reversal strategy for the functionalization of electrophilic strained molecules via light-driven cobalt catalysis. J. Am. Chem. Soc. 142, 5355–5361 (2020).
Ernouf, G. et al. Photochemical strain-release-driven cyclobutylation of C(sp3)-centered radicals. Angew. Chem. Int. Ed. 59, 2618–2622 (2020).
Majhi, J. et al. Metal-free photochemical imino-alkylation of alkenes with bifunctional oxime esters. J. Am. Chem. Soc. 144, 15871–15878 (2022).
Silvi, M. & Aggarwal, V. K. Radical addition to strained σ‑bonds enables the stereocontrolled synthesis of cyclobutyl boronic esters. J. Am. Chem. Soc. 141, 9511–9515 (2019).
Gianatassio, R. et al. Strain-release amination. Science 351, 241–246 (2016).
Milligan, J. A. et al. Hydrophosphination of bicyclo[1.1.0]butane-1-carbonitriles. Org. Lett. 18, 4300–4303 (2016).
Guo, L., Noble, A. & Aggarwal, V. K. α-Selective ring-opening reactions of bicyclo[1.1.0]butyl boronic ester with nucleophiles. Angew. Chem. Int. Ed. 60, 212–216 (2021).
Zhang, Z. & Gevorgyan, V. Palladium hydride-enabled hydroalkenylation of strained molecules. J. Am. Chem. Soc. 144, 20875–20883 (2022).
Xiao, Y. et al. Photochemical α-selective radical ring-opening reactions of 1,3-disubstituted acyl bicyclobutanes with alkyl halides: modular access to functionalized cyclobutenes. Chem. Sci. 14, 13060–13066 (2023).
McNamee, R. E., Thompson, A. L. & Anderson, E. A. Synthesis and applications of polysubstituted bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 143, 21246–21251 (2021).
Liang, Y. et al. Synthesis of polysubstituted 2‑oxabicyclo[2.1.1]hexanes via visible-light-induced energy transfer. J. Am. Chem. Soc. 144, 20207–20213 (2022).
Zhang, F. et al. Solvent-dependent divergent cyclization of bicyclo[1.1.0]butanes. Angew. Chem. Int. Ed. 64, e202418239 (2024).
Koshti, V., Gaikwad, S. & Chikkali, S. H. Contemporary avenues in catalytic P-H bond addition reaction: a case study of hydrophosphination. Coord. Chem. Rev. 265, 52–73 (2014).
Chew, R. J. & Leung, P.-H. Our odyssey with functionalized chiral phosphines: from optical resolution to asymmetric synthesis to catalysis. Chem. Rec. 16, 141–158 (2016).
Li, Z. & Duan, W.-L. Recent advances in the asymmetric conjugate addition reactions of phosphorus nucleophiles to electron-deficient alkenes. Youji Huaxue 36, 1805–1813 (2016).
Wang, C. et al. Asymmetric synthesis of P‑stereogenic secondary phosphine-boranes by an unsymmetric bisphosphine pincer-nickel complex. J. Am. Chem. Soc. 143, 5685–5690 (2021).
Zhang, Y. et al. Palladium-catalyzed diastereo- and enantioselective desymmetric hydrophosphination of cyclopropenes. Chem. Catal. 2, 3163–3173 (2022).
Lin, X. et al. Diastereo- and enantioselective hydrophosphination of cyclopropenes under lanthanocene catalysis. Angew. Chem. Int. Ed. 62, e202308488 (2023).
Daniels, B. S. et al. Copper-phosphido catalysis: enantioselective addition of phosphines to cyclopropenes. Angew. Chem. Int. Ed. 62, e202306511 (2023).
Zhang, S. et al. Copper(I)-catalyzed asymmetric hydrophosphination of 3,3-disubstituted cyclopropenes. Angew. Chem. Int. Ed. 62, e202218798 (2023).
Krishnan, C. G. et al. Strain-releasing ring-opening diphosphinations for the synthesis of diphosphine ligands with cyclic backbones. JACS Au 4, 3777–3787 (2024).
Wiberg, K. B. & Waddell, S. T. Reactions of [1.1.1]Propellane. J. Am. Chem. Soc. 112, 2194–2216 (1990).
Shin, S. et al. Visible-light-induced 1,3-aminopyridylation of [1.1.1]propellane with N-aminopyridinium salts. Angew. Chem. Int. Ed. 60, 7873–7879 (2021).
Perry, G. & Schley, N. D. Tris(bicyclo[1.1.1]pentyl)phosphine: an exceptionally small tri-tertalkylphosphine and its bis-ligated Pd(0) complex. J. Am. Chem. Soc. 145, 7005–7010 (2023).
Takano, H. et al. Synthesis of bicyclo[1.1.1]pentane (BCP)-based straight-shaped diphosphine ligands. Angew. Chem. Int. Ed. 62, e202303435 (2023).
Helmchen, G. & Pfaltz, A. Phosphinooxazolines-a new class of versatile, modular P,N-ligands for asymmetric catalysis. Acc. Chem. Res. 33, 336–345 (2000).
Banerjee, D., Junge, K. & Beller, M. A general catalytic hydroamidation of 1,3-dienes: atom-efficient synthesis of N-allyl heterocycles, amides, and sulfonamides. Angew. Chem. Int. Ed. 53, 1630–1635 (2014).
Woodward, R. B. & Hoffmann, R. The conservation of orbital symmetry. Angew. Chem. Int. Ed. 8, 781–932 (1969).
Lin, S.-L. et al. Enantioselective synthesis of chiral cyclobutenes enabled by Brønsted acid-catalyzed isomerization of BCBs. J. Am. Chem. Soc. 145, 21152–21158 (2023).
Yi, H. et al. Direct observation of reduction of Cu(II) to Cu(I) by P−H compounds using XAS and EPR Spectroscopy. Organometallics 35, 1426–1429 (2016).
Leyva-Pérez, A. et al. Copper(I)-catalyzed hydrophosphination of styrenes. J. Organomet. Chem. 696, 362–367 (2011).
Lu, Q. et al. Operando X-ray absorption and EPR evidence for a single electron redox process in copper catalysis. Chem. Sci. 6, 4851–4854 (2015).
Acknowledgements
We are grateful to the National Natural Science Foundation of China (22471068 for J.-J.F.) and Fundamental Research Funds for the Central Universities for financial support. 1H, 13C NMR spectra, single crystal X-ray diffraction and HRMS were performed at Analytical Instrumentation Center of Hunan University.
Author information
Authors and Affiliations
Contributions
J.-J.F. conceived the study. H.-X.H. and F.W. carried out the experiments and data analysis work. K.-J.W. and L.W. synthesized the bicyclobutanes. The paper was written by J.-J.F. All authors contributed to discussions. H.-X.H. and F.W. contributed equally.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jet Tsien and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, HX., Wu, F., Wang, KJ. et al. Diastereoselective synthesis of multi-substituted cyclobutanes via catalyst-controlled regiodivergent hydrophosphination of acyl bicyclobutanes. Nat Commun 16, 6639 (2025). https://doi.org/10.1038/s41467-025-61726-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61726-w
