
Abstract
Human guanylate kinase (GMPK) as the sole enzyme for GDP biosynthesis plays pivotal roles in antiviral prodrug activation and tumorigenesis. Despite its biological significance, the catalytic mechanism remains poorly understood. Here, we resolve crystal structures of GMPK in free and GMP-bound form, revealing the interdomain motions of GMPBD and LID relative to the CORE domain. Biochemical assays demonstrate potassium’s dual functionality in substrate recognition and phosphoryl transfer catalysis. Structural analyses uncover intradomain conformational motion within the LID domain and essential interactions for ADP/ATP binding. Notably, the cooperative ATPγS binding potentiated by prior GMP binding are structurally elucidated. Three key complexes, pre-reaction state (GMP/ATPγS), transition state (AlF4- mimic), and post-reaction state (GDP/ADP), collectively delineate the reversible catalytic pathway. This comprehensive structural characterization of GMPK’s dynamic landscape establishes a foundation for developing conformation-specific inhibitors through structure-guided drug design.
Similar content being viewed by others

Introduction
Nucleotide metabolism is a cornerstone of cellular functions serving as the vital source of building blocks for DNA and RNA synthesis1, and thus fueling intracellular energy production to regulate key cellular signaling pathways crucial for cell proliferation and survival2,3. Cellular nucleotide pools are sustained through two main pathways: de novo synthesis via purinosomes4,5 or pyrimidinesomes6, and the salvage pathway that reutilizes existing nucleosides7. These two pathways ultimately produce nucleoside monophosphates (NMPs), which are in turn phosphorylated to nucleoside diphosphates (NDPs) catalyzed by nucleoside monophosphate kinases (NMPKs)8. Notably, NMPKs regulate intracellular nucleotide homeostasis by strategically dictating either nucleotide anabolism or reversible phosphoryl transfer9,10. Dysregulation of nucleotide metabolism has been attributed to various diseases, e.g., tumorigenesis11,12 and immune system disorders13,14.
GMPK, which catalyzes the phosphoryl transfer from ATP to GMP, is the sole known enzyme responsible for GDP synthesis15,16. GMPK harbors a domain architecture conserved among NMPKs, consisting of a CORE domain, a LID domain, and an NMP-binding domain17. Notably, the GMP-binding domain (GMPBD) of GMPK is composed of a four-stranded β-sheet, distinct from the α-helices observed in other NMPKs. This structural arrangement confers GMPK exceptional selectivity over the substrate GMP18. GMPK was initially identified for its role in activating GMP analogues as antiviral prodrugs19,20,21. Most recently, GMPK has emerged as a metabolic liability in ALK+ non-small cell lung cancer, which rewires cellular metabolic networks to support unrestrained proliferation22. Furthermore, increased expression and phosphorylation of GMPK was observed in a subset of patient-derived cell lines with resistance to ALK inhibitors, indicating metabolic reprograming via GMPK may mediate acquired resistance to targeted therapy. Despite the vital role of GMPK in cellular metabolism, structural studies of GMPK have predominantly focused on non-human organisms, e.g., bacteria23,24, yeast25,26 and mouse27, except a solution structure of human GMPK in its free form28. Consequently, it so far remains unknown for a comprehensive profiling of the catalytic conformations at every stage of human GMPK catalysis.
In this work, we comprehensively profile the catalytic conformations of human GMPK. The free and GMP-bound form structures of GMPK reveal remarkable interdomain motions of GMPBD and LID relative to the CORE domain induced by GMP binding. Subsequent mutagenesis studies demonstrate GMP binding is not the sole determinant of GMPK catalytic activity, which is further underpinned by the potassium and sulfate ion containing crystal structures. The sulfate ion partially mimics the ATP/ADP binding states of GMPK as also implicated in the ADP-bound structure of GMPK. The mutual feedback between ATP and GMP binding is depicted by the GMPK pre-reaction complex with GMP and ATPγS, and the abortive complex with GMP and ADP. Finally, the catalytic transition state mimicked by tetrafluoroaluminate ion (AlF4-)29,30 and the post-reaction complex with ADP and GDP complement the full landscape of the GMPK catalytic pathway (Supplementary Fig. 1). The GMPK conformations accompanying every catalytic stage delineate the molecular mechanism underlying the phosphoryl transfer function, and will facilitate subsequent structure-guided discovery of chemical probes targeting GDP synthesis.
Results
GMP induces interdomain motions of GMPK
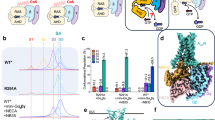
Titration of GMP to the 15N-labeled GMPK induced significant chemical shift perturbations (CSPs) of residues within the CORE and GMPBD domains (Fig. 1a, and Supplementary Fig. 2a), at a dissociation constant (Kd) of 30 μM determined from the dose-dependent CSPs (Supplementary Fig. 2b). The GMP binding surface mapped by residues with significant CSPs to the GMPK NMR structure was far beyond the size of the GMP itself (Supplementary Fig. 2c), indicating an interdomain motion upon substrate binding that is the characteristic feature of the nucleoside triphosphate kinase family of proteins17. To further investigate this, we solved the crystal structure of GMPK in its free form (Fig. 1b, Supplementary Fig. 2d, Supplementary Table 1) and in complex with GMP (Fig. 1c, and Supplementary Fig. 2e, Supplementary Table 1). The crystal structure of the free-form GMPK exhibits a high degree of similarity to the previously reported NMR structure, with a Cα RMSD value of 1.83 Å (Supplementary Fig. 2f). The GMP-bound structure reveals a remarkable conformational rearrangement of GMPK relative to its free form (Supplementary Fig. 2g), while each individual domain remains untouched with a root mean square deviation (RMSD) of the Cα atoms of ~1 Å (Supplementary Fig. 2h, i). These findings indicate that GMP binding induces interdomain motions of the LID and GMPBD domains relative to the CORE domain.

a Histogram of GMP-induced chemical shift perturbations on amino acid residues with a molar ratio of GMP to GMPK of 5:1. b Cartoon representation of the apo form of GMPK: aquamarine for the CORE domain; pink for the GMPBD domain; slate for the LID domain and wheat for the Hinge region. c Cartoon representation of the GMPK-GMP complex with the mFo-DFc omit map density (black mesh) for GMP shown at a contour level of 3σ. d Electrostatic surface representation of the GMPK-GMP complex. e, f Detailed views of the interaction between the GMP and GMPK. Dashed lines indicate interatomic distances. g, h Binding affinities between GMPK mutants and the GMP, measured by ITC. The displayed values represent the fitted Kd and their corresponding errors. i Kinetic parameters (KM, kcat and kcat/KM) for GMP were determined for both the wild-type and mutant GMPK. N.D. indicates no detectable activity.
To investigate the conformational plasticity of GMPK across species, we selected three highly conserved residues to characterize the interdomain motions, i.e., K17, R44, and R148, within the surface of the CORE, GMPBD, and LID domains, respectively (Supplementary Fig. 2j). In general, remarkable interdomain motions between GMPBD and CORE were observed in the free states of GMPK (Supplementary Fig. 2k), while the binding of GMP brings the GMPBD domains into closer proximity to the CORE domain (Supplementary Fig. 2l). A quantitative analysis of the Cα-Cα distances between K17 and R44, or between R44 and R148, revealed that the human GMPK exhibits the most significant interdomain motions upon GMP binding when compared to all other GMPKs with known structures (Supplementary Fig. 2m).
Detailed structural analysis shows that GMP binds to the cleft between the CORE and GMPBD domains. The GMP guanine moiety binds to the acidic pocket while the phosphate moiety binds to the basic pocket (Fig. 1d). More specifically, the guanine moiety forms hydrogen-bonds with residues S37, E72, T83, and D103 (Fig. 1e). The phosphate moiety is recognized by R41, R44, R148 via salt bridges, and by Y53 and Y81 via hydrogen bonds (Fig. 1f). Mutagenesis of these residues demonstrated that S37, R41, Y53, E72, and Y81 were key to the GMP binding (Fig. 1g, h, and Supplementary Fig. 3a, Supplementary Table 3), with over 10-fold affinity reduction relative to the wild type GMPK as assessed by isothermal calorimetric titration (ITC), without compromising the global folding of GMPK (Supplementary Fig. 4a). The binding affinity between the wild-type GMPK and GMP determined by ITC was in good agreement with this determined by NMR CSPs (Supplementary Fig. 2b). Furthermore, we introduced the S37P mutation in the hinge region that significantly restricted the motion of yeast GMPK31,32. The overall architecture of the human GMPKS37P mutant remains almost untouched (Supplementary Fig. 5a, b, and Supplementary Table 1) relative to the wild-type GMPK. However, CSPs of GMPKS37P demonstrated that GMP perturbed residues within GMPBD only (Supplementary Figs. 5c–e), albeit with a reduced affinity, and a similar binding pattern and reduced affinity to GMP was observed for the S37A mutant (Supplementary Figs. 5f–h). Interestingly, S37A retained partial enzymatic activity relative to S37P (Fig. 1i), suggesting that the diminished S37P activity may be attributed to rigidity of proline that restricts GMPK motion and reduced GMP binding. Our data implicated that GMP was initially captured by GMPBD, while subsequent interdomain motions enabled a transition from the open to a closed state.
GMP recognition is not the sole determinant of GMPK Catalytic activity
To gain mechanistic insight into the functional modulation of GMPK, we evaluated the phosphorylation activities of various GMPK mutants (Fig. 1i, and Supplementary Fig. 6a–g). The S37A, Y53A, and Y81A mutants demonstrated over tenfold increase of KM for GMP and over threefold reduction in kcat, when compared to the wild-type GMPK. Furthermore, the S37P, R41A, R44A, and E72A mutants exhibited no detectable activities, in consistent with the significantly reduced GMP binding affinity of these mutants. Meanwhile, the GMPK T83A and D103A mutants exhibited minimal inhibition of the phosphorylation activities, as expected from its modestly reduced GMP binding affinity. Although the R148A mutagenesis only weakened the GMP binding by 1.7 fold, it remarkably suppressed the phosphorylation activities of GMPK. Our findings demonstrate that GMP binding plays an important role in the catalytic activity of GMPK, however, the reaction rate is also dependent on rate-limiting steps that remain to be determined.
Given that the equilibrium dissociation constant between GMP and GMPK is greater than the GMP concentration, which ranges from 1 to 30 μM in lymphocytes33, we speculated that additional factors might facilitate the recognition of GMP. The mouse GMPK structure reveals a potassium ion neighboring the GMP binding site27. Inspired by a previous report of partial activity of GMPK in the presence of sodium ions compared to potassium ions16, we hence determined the GMP binding affinity of human GMPK in a potassium chloride solution using ITC (Fig. 2a, b, and Supplementary Fig. 7, Supplementary Table 4). Notably, the potassium ion enhances GMP binding affinity (Kd = 7.1 μM) by fivefold relative to the sodium ion. The CSPs assay, involving titration of KCl or NaCl into the GMPK-GMP solution, revealed a Kd of 19 mM and 57 mM for KCl and NaCl, respectively (Supplementary Fig. 8). Enzyme activity assays10 under saturating substrate conditions showed that the global reaction rate was lower at a 100 mM potassium ion than that at a 100 mM Na+ concentration, as quantified by the GDP/GMP intensity ratio observed in the 1H NMR (Supplementary Fig. 9a). Furthermore, the NADH based enzyme activity assay indicated that the initial reaction rate under saturating substrate conditions reached its maximum at a K+ concentration of about 7.5 mM. At high concentrations (75–150 mM), the initial reaction rate with potassium ions was lower than that with sodium ions (Supplementary Fig. 9b). Kinetic studies demonstrated that at 150 mM potassium ions, the KM decreased 2.3-fold relative to 10 mM, but the kcat also decreased 1.5-fold, while sodium ions had only a minor effect (Fig. 2c, Supplementary Fig. 9c, d). K+ augments GMPK catalysis relative to Na+, at a low GMP concentration (Supplementary Fig. 9c, d), consistent with the results observed by Agarwal et al.16.

a, b ITC analysis of GMPK binding to GMP in buffers containing 150 mM NaCl (blue curve) and 150 mM KCl (magenta curve), respectively. The displayed values represent the Kd and errors from triplicate experiments. c Kinetic parameters (KM, Ki, kcat and kcat/KM) for GMP obtained under various potassium and sodium ion conditions. d Cartoon representation of the GMPK-GMP-K+ complex, with a detailed view of the potassium ion binding site. Potassium ion is depicted as purple spheres, water molecules as red spheres, and the residues S37, D101, and GMP are shown in stick representation. The mFo-DFc omit map density (black mesh) for the Potassium ion and water molecules is shown at a contour level of 10σ. Dashed lines indicate interatomic distances. e Structural superposition of GMPK-GMP (white) and GMPK-GMP-K+ (aquamarine). Water molecules near the potassium binding site are shown as spheres. D101 and GMP are shown as sticks.
To resolve the discrepancies observed between GMP binding and catalytic activity mediated by potassium ions, we soaked potassium ions into the GMPK-GMP complex crystal (Supplementary Table 1, Supplementary Fig. 9e). A strong peak in the electron density map proximal to residues S37 and D101 was modelled as a potassium ion (Fig. 2d). The potassium ion forms coordination bonds with the hydroxyl group of S37, the backbone carbonyl and the side-chain carboxyl groups of D101, and three adjacent water molecules. This coordination site is proximal to V36 and V99, as also identified from the CSPs of GMPK-GMP induced by potassium ions (Supplementary Fig. 8g). This potassium ion thus bridges the interaction between GMPBD and CORE to facilitate GMP binding. Superimposition of the GMPK-GMP complex structures in the absence and presence of the potassium ion reveals a displacement of the D101 side chain away from the P-loop (Fig. 2e). Although the potassium ion enhances the GMP binding capability of GMPK, it on the other side displaced the carboxyl group of D101 from optimal water-mediated coordination with the magnesium ion34 (Supplementary Fig. 9f). Therefore, these two factors in combination rendered an enhanced catalytic activity of GMPK at a low K+ concentration, while a suppressed GMPK activity at a high K+ concentration (Fig. 2c).
Structural basis for ATP/ADP recognition of GMPK
During structural studies of GMPK in complex with GMP, we unintentionally solved the crystal structure of GMPK in complex with GMP and SO42− at a 1.9 Å resolution (Supplementary Fig. 10a–c, and Supplementary Table 1). Interestingly, superimposition of the two GMPK molecules within an asymmetrical unit reveals an interdomain motion of LID relative to CORE or GMPBD (Supplementary Fig. 10d). These two conformations are hence designated as a closed state and a partially closed state. The sulfate ion forms an intensive hydrogen-bond network with residues G14, G16, S18, and T19 in the P-loop region within these two states (Supplementary Fig. 10e, f). In the closed state, the sulfate ion forms a salt-bridge interaction with R137, whereas in the partially closed state, this residue is displaced 7.1 Å away from SO42−. NMR titration experiments revealed that sulfate ions bind to the p-loop region of GMPK with a Kd of 5.6 mM (Supplementary Fig. 11a–d). Furthermore, enzymatic activity assays suggested the sulfate ion as a competitive inhibitor of ATP (Supplementary Fig. 11e). Our data suggests that the sulfate ion partially mimics the ATP/ADP binding states of GMPK.
To further elucidate the interaction between GMPK and ATP/ADP, we titrated the ATP analog ATPγS and ADP to GMPK (Supplementary Fig. 12a–c). The CSP pattern indicated that ATPγS and ADP predominantly bound to the interface between the LID and the CORE domain (Fig. 3a, b, and Supplementary Fig. 12d, e), and ATPγS binds to GMPK with a Kd of 30 μM determined from the dose-dependent CSPs (Supplementary Fig. 12b). Consistently, the crystal structure of GMPK in complex with ADP (Supplementary Fig. 12f, g, and Supplementary Table 2), despite our initial effort is to resolve the GMPK-ATP complex structure, reveals that ADP binds to the cleft between the LID domain and the CORE domain (Supplementary Fig. 12h). The phosphate moiety of ADP forms extensive hydrogen bonds with residues in the P-loop region, partially mimicked by SO42−. Furthermore, the adenine moiety of ADP forms a cation-π interaction with R133. The ADP-bound form of GMPK adopts an overall architecture similar to that of the free form, while the helix α5 was moderately reoriented upon ADP binding (Supplementary Fig. 12i).

a Histogram of ATPγS-induced chemical shift perturbations in GMPK at a molar ratio of 2:1. b Mapping of ATPγS-induced CSPs larger than mean plus 2σ (red) onto the apo state of the GMPK. c Cartoon representation of the GMPK-GMP-ATPγS complex structure with the mFo-DFc omit map density (black mesh) for GMP and ATPγS contoured at 3σ. d Electrostatic surface potential of the GMPK-GMP-ATPγS complex. e Structural superposition of GMPK-GMP-ATPγS and GMPK-ADP. pink: GMPK-ADP, aquamarine: GMPK-GMP-ATPγS. f Detailed view of the adenine moiety and α/β -phosphate moiety interactions with GMPK with dashed lines indicating interatomic distances. g Detailed view of the γ-phosphate moiety interactions with GMPK. h Binding affinity between GMPK mutants with ATPγS as determined by ITC with fitted values and errors presented. i Kinetic parameters (KM, kcat and kcat/KM) for ATP were determined for both the wild-type and mutant GMPK. N.D. indicates no detectable activity.
The GMPK pre-reaction complex
We further determined the crystal structure of the pre-reaction complex, i.e., GMPK in complex with GMP and ATPγS, diffracted at a resolution of 2.8 Å (Supplementary Table 2, and Supplementary Fig. 12j, Fig. 3c). ATPγS binds to the cleft between the LID and the CORE domain in a pose similar to ADP, while GMP adopts a similar pose in both the pre-reaction complex and the GMP-bound complex (Fig. 3d, e). The adenine moiety of ATPγS forms a cation-π interaction with residue R133 and two hydrogen bonds with residue N171 of GMPK, while the phosphate moiety of ATPγS curated a hydrogen bond network with residues in the P-loop (Fig. 3f). The γ-phosphate of ATPγS forms hydrogen bonds with S13, K17 and S18 of GMPK. The sulfur atom of ATPγS is positioned 3.0, 3.7 and 3.2 Å away from residues R137, R148, R44 of GMPK (Fig. 3g), respectively. We thus speculated that the natural substrate ATP may demonstrate high binding affinity for GMPK through additional charge-charge interactions with residues R137, R148, and R44. The residues of GMPK critical for ATP binding were further validated by mutagenesis studies. ITC assays demonstrated that residues R133, R137 and N171 were essential for ATPγS recognition (Fig. 3h, and Supplementary Fig. 3b, Supplementary Table 3). Furthermore, these mutants exhibited significantly reduced catalytic efficiency (kcat/KM), without compromising GMPK folding (Fig. 3i, and Supplementary Fig. 6h, i, Supplementary Fig. 4b). The S13A, K17A, S18A, and R148A mutagenesis all resulted in approximately threefold reduction of ATP binding capability. Notably, with the exception of S13A, no significant enzymatic activity was detected for the rest mutants.
GMP facilitates ATP binding to GMPK
The pre-reaction complex reveals that the distance between the sulfur atom of ATPγS and the oxygen atom of the GMP phosphate moiety is only 3.1 Å, which may facilitate the following phosphoryl transfer reaction (Fig. 3g). However, previous molecular dynamics simulations suggested that the binding of ATP and GMP to GMPK may influence each other34. ITC experiments demonstrated that the binding of ATPγS to GMPK was enhanced by incubation of GMP a priori (Fig. 4a, b, and Supplementary Table 4, Supplementary Fig. 13). Moreover, ATPγS titration induced a fast exchange of GMPK chemical shift in the absence of GMP, but induced an intermediate exchange phenomenon in presence of GMP (Fig. 4c), which cross validated that the prior binding of GMP promoted the following recruitment of ATP by GMPK. However, the binding affinity of ADP to GMPK was not affected by incubation with GMP a priori, but the heat released after binding increased. (Fig. 4b, d, and Supplementary Table 4, Supplementary Fig. 13).

a Raw ITC binding isotherms of ATPγS binding to GMPK (blue curve) and GMPK-GMP mixtures (magenta curve). b Fitted ITC binding isotherms, with Kd values and associated errors presented from triplicate experiments. c1 H-15N HSQC spectra of ATPγS titration against GMPK and GMPK-GMP mixtures, with colors representing molar ratios of ATPγS to GMPK: red (0), cyan (0.5), pink (1), and purple (2). d Raw ITC binding isotherms of ADP binding to GMPK (gray curve) and GMPK-GMP mixtures (pink curve). e Structural superposition of apo, ADP-bound and GMP-ATPγS-bound GMPK. f Structural superposition of apo, GMP-bound and GMP-ATPγS-bound GMPK.
Superimposition of GMPK structures in its free form, ADP-bound form and pre-reaction state indicated that ATPγS or ADP binding induced a moderate re-orientation of the LID domain (Fig. 4e). Superimposition of the GMPK structures in its free form, GMP-bound form and pre-reaction state (Fig. 4f) indicated that GMP binding induced not only a moderate re-orientation of the LID domain, but also a large interdomain motion that brought R44 proximal to the ATP-binding site. More specifically, R137 was displaced upon GMP binding to a position closer to the γ phosphate of ATPγS. That is to say, despite of the electrostatic repulsion between the phosphate moieties of GMP and ATP (Fig. 3g), the GMP binding induced a rearranged conformation favoring the subsequent ATP binding to compensate for this electrostatic repulsion.
Feedback of ATP/ADP on GMP binding to GMPK
It is hence pertinent to investigate the reverse feedback of ATP/ADP on GMP binding to GMPK. Interestingly, prior incubation of ATPγS attenuated the GMP binding to GMPK from a Kd value of 0.032 to 0.17 mM, while incubation of ADP only slightly reduced the GMPK-GMP binding to 0.06 mM (Supplementary Table 4, Supplementary Fig. 14). To dissect the mechanism underlying this differentiated feedback of ATP and ADP, we further determined the crystal structure of GMPK in complex with GMP and ADP, i.e., the abortive complex (Supplementary Fig. 15a, b, and Supplementary Table 2). The overall structures and substrate-binding site residues of GMPK remain essentially identical in the pre-reaction and abortive complexes (Supplementary Fig. 15c, d). The shortest interatomic distance between the GMP phosphate and the γ-phosphate of ATPγS or the phosphate of ADP was measured to be 3.1 Å and 4.8 Å, respectively (Supplementary Fig. 15e). Notably, the orientation of the GMP phosphate group is subtly altered in the GMPK-GMP-ATPγS complex compared to the GMPK-GMP-ADP complex. Specifically, the distances between the GMP phosphate and residues R41, Y53, and R148 increase from 2.9, 2.7, and 2.8 Å in the GMPK-GMP complex to 3.9, 3.1, and 3.9 Å in the GMPK-GMP-ATPγS complex (Supplementary Fig. 15f). These distances remain less perturbed in the GMPK-GMP and GMPK-GMP-ADP complexes (Supplementary Fig. 15g). These observations suggest that the close proximity of ATPγS induces a subtle conformational shift in the GMP phosphate moiety, thereby weakening its interactions with the residues R41, Y53, and R148 and consequently impacting GMP binding.
The post-reaction GMPK complex
After the formation of pre-reaction complex, the γ-phosphate of ATP is transferred to GMP to produce GDP. To fully understand the catalytic mechanism of GMPK, we further determined crystal structure of the post-reaction complex, i.e., GMPK in complex with GDP and ADP (Supplementary Fig. 16a, b, and Supplementary Table 2). The electron densities of GDP and ADP phosphates were clearly visible in this post-reaction complex, despite of the lack of clear electron density of the adenosine moiety of ADP. This post-reaction GMPK complex adopts a closed conformation, wherein the residues proximal to the GDP and ADP binding site aligned well with those in with those in the pre-reaction complex (Fig. 5a, and Supplementary Fig. 16c). The ADP recognition pattern is in general similar to that of ATPγS, except those residues binding to the γ-phosphate (Supplementary Fig. 16d). Residues key to ADP binding, e.g., K17, T19, R133, R137 and N171, were further validated by ITC assays of the GMPK mutants (Supplementary Fig. 16e). Distinct from the pre-reaction complex, the β phosphate of GDP in the post-reaction GMPK complex forms hydrogen bonds and salt bridges with S13 and R137, respectively (Fig. 5b, and Supplementary Fig. 16f, g). The binding between GDP β phosphate and GMPK was further validated by the reduced affinities of the GMPK S13A and R137A mutants, as determined by ITC assays, which further emphasized the role of S13 and R137 in GDP recognition (Fig. 5c, and Supplementary Fig. 3c, Supplementary Table 3).

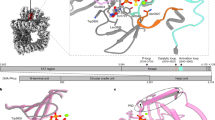
a Structural superposition of the GMPK-GDP-ADP and GMPK-GMP-ATPγS complexes. b Interaction of the GDP β-phosphate with GMPK. c ITC analysis of GMPK mutants binding to GDP. d Reverse reaction recorded at different time points by 1H NMR. e Cartoon representation of the GMPK-GMP-ADP-AlF4--Mg2+-K+ complex with the mFo-DFc omit map density (black mesh) for GMP, ADP, AlF4-, Mg2+ and K+ contoured at 3σ. f AlF4- and magnesium ion binding site. Potassium ion is depicted as purple sphere, water molecule as red sphere, magnesium ion as green sphere and AlF4- is shown in stick representation. g Structural superposition of the transition state analogue and GMPK-GMP-ATPγS-K+ complexes. h Proposed catalysis model of GMPK.
The catalytic transition state of GMPK
The high degree of structural similarity between the pre- and post-reaction states suggests a reversible phosphoryl transfer reaction. To confirm this, we monitored changes in the intensity of characteristic proton NMR signals for the substrates (GMP, GDP, ATP, ADP) over time (Supplementary Fig. 17a). The signal intensity of GDP and ADP increased when ATP and GMP were present, and vice versa (Supplementary Fig. 17b, and Fig. 5d). The equilibrium constant of this reversible phosphoryl transfer reaction was determined to be 2.04 (Supplementary Fig. 17c–h), comparable to that of yeast GMPK10 and adenylate kinase35.
The reversible catalysis, coupled with the close proximity (1.9 Å) between the phosphorus atoms of the γ-phosphate of ATP and the β-phosphate of GDP, suggests a potential sp³d hybridization of either moiety in the transition state like many phosphate-transfer enzymes, e.g., adenylate kinase30. To elucidate the transition state geometry, we employed aluminum fluoride (AlF4-) as a transition-state analog29 and determined the structure of GMPK in complex with GMP, ADP, K+, Mg2+ and AlF4- (Fig. 5e, and Supplementary Fig. 18a, Supplementary Table 2). This transition complex adopts a closed conformation. The planar AlF4- occupies a central position between the β-phosphate of ADP and phosphate group of GMP (Fig. 5f, and Supplementary Fig. 18b, c), positioned nearly perpendicular to the imaginary line connecting the closest oxygen atoms between these two substrates. Additionally, AlF4- forms hydrogen bonds to residues S13, K17, R44, R137 and R148 of GMPK, respectively (Fig. 5f, and Supplementary Fig. 18d–f, Supplementary Table 5). Notably, the magnesium ion coordinates with S18, the phosphate group of ADP, AlF4- and forms three water-bridged interactions with D101, ADP and GMP (Supplementary Fig. 18g–i, and Supplementary Table 5). Superimposition of the transition state and the pre-reaction state reveals a high degree of alignment for residues near the substrates binding site, except S13, whose side chain rotates towards AlF4- (Fig. 5g). The hydrogen network and magnesium ion coordination likely contribute to stabilizing and promoting the formation of the transition state by balancing the charge distribution of phosphate group.
Discussion
The catalytic mechanism of GMPK has long been puzzling regarding the temporal order of the two substrates entering the complex. A random sequential model was proposed based on the similar affinity of each single substrate to yeast GMPK10, as was corroborated by our ITC measurement of affinities of GMP, GDP, ADP and ATPγS to human GMPK. However, a seemingly contradictory model for a sequential binding of GMP and ATP was deduced from the abortive complex of mouse GMPK27. Our competitive ITC experiments demonstrated that pre-incubation of GMP enhances ATPγS binding to GMPK, but not vice versa, as was further addressed by the pre-reaction and abortive complex of human GMPK. The difficulty in obtaining high-quality diffraction data27 has long hampered the structural insights of human GMPK. Based on our comprehensive structural studies of human GMPK in complex with various combinations of substrates, we proposed the following model of the catalytic pathway of human GMPK involving four single-substrate states (ADP, ATP, GDP, and GMP) and three two-substrate states (GDP-ADP, GMP-ATP, and GMP-ADP) (Fig. 5h).
In the single-substrate state, GMP/GDP binding triggers GMPBD domain closure, whereas ATP/ADP binding elicits relative helical movement within the LID domain. The GMPBD displays greater flexibility compared to the LID domain. Recently, the crystal structure of GMPK in complex with an antiviral drug AT800121 also resembles our free-form structure of GMPK, with a Cα RMSD of 1.93 Å (Supplementary Fig. 19a). Integrating these findings with our NMR and structural data, we propose a binding model wherein GMP initially binds to the GMPBD of open-state, progressing through intermediate conformations resembling those observed in GMPK-AT8001 and GMPK-GMP-sulfate complexes, culminating in domain closure (Supplementary Fig. 19a). Furthermore, principal component analysis of all available GMPK crystal structures revealed two dominant modes of conformational dynamics: a global open-closed transition and localized motions of GMPBD and LID domains (Supplementary Fig. 19b–d).
The forward reaction pathway from the apo to pre-reaction state likely involves two distinct substrate binding pathways: GMP-first or ATP-first. The population of the pre-reaction complex relies on the binding affinity of GMP to GMPK and sequentially the affinity of ATP to GMPK-GMP in the former pathway, or the binding affinity of ATP to GMPK and that of GMP to GMPK-ATP in the latter pathway. Therefore, GMPK may prefer to first bind GMP rather than ATP, thus achieves a higher population of the pre-reaction complex, as derived from the distinct feedback of GMP to GMPK-ATP binding and ATP to GMPK-GMP binding (Fig. 4, and supplementary Fig. 13, 14). This hypothesis aligns with molecular dynamics simulations conducted on Mycobacterium tuberculosis GMPK34. The significantly lifted concentration of ATP in tumor cells relative to somatic cells may further promote the forward reaction catalyzed by GMPK, thus synthesized more GDP.
Upon formation of the pre-reaction complex, a phosphate group is transferred from ATP to GMP. Amino acid residues proximal to the phosphate engage in extensive hydrogen bonding, potentially facilitating optimal phosphate transfer by restricting the relative orientation of the phosphate moieties. To date, the mechanism of GMPK catalyzed phosphate transfer remains elusive. Our data supports an associative mechanism of GMPK, similar to adenylate kinase (ADK)36,37. During this process, the γ-phosphate of ATP undergoes a hybridization change to form a planar intermediate state, which is supported by the AlF4--mimicked transition state. The associative mechanism adopted by the enzyme minimizes the window for water attack, mitigating this risk of intermediate hydrolysis. Additionally, the magnesium ion coordinates with surrounding water molecules, effectively restricting their mobility and hindering water attack on the intermediate and subsequent ATP hydrolysis.
Previous studies have indicated that GMPK activity is influenced by both magnesium and potassium ions16. While structural analyses of murine GMPK revealed the presence of potassium ions27, their functional role remained unclear. Our findings demonstrate that potassium ions enhance GMPK binding to GMP. However, at a high potassium ion concentration, a reduced GMPK catalytic rate and substrate inhibition is observed (Fig. 2c, and Supplementary Fig. 9c). We propose a dual regulatory mechanism of potassium ions, on one side, K+ ions enhance enzyme activity by promoting GMP binding, while on the other side, K+ binds to D101 to displace the side-chain carboxy group and consequently disrupts Mg2+ coordination. At low potassium concentrations, the former effect dominates, resulting in enhanced activity, whereas the latter prevails at high K+ concentrations, leading to inhibition of GMPK activity. Given the physiological K+ concentration of 140-150 mM, hGMPK is likely not in an environment that promotes maximum efficiency in vivo. Furthermore, high potassium concentrations distort the D101 side chain, potentially impairing ATP-Mg2+ binding and facilitating ADP competition with ATP, thereby forming abortive complexes and causing substrate inhibition.
Following transition state formation, ATP transfers a phosphate group to GMP, resulting in a post-reaction complex. Notably, the pre-reaction, transition, and post-reaction states exhibit high structural similarity. The equilibrium constant for this reversible reaction is 2.04, suggesting that GMPK favors the forward reaction to produce GDP. Although we have not investigated the order of dissociation in the post-reaction complex, based on the fact that GDP binds to GMPK with an affinity close to that of ADP and is encapsulated within GMPK, and the low occupancy of ADP in the post-reaction complex, we speculate that ADP dissociates first, followed by GDP.
In summary, our work provides structural insights into the GMPK catalytic cycle. Given the critical role of nucleotide metabolism homeostasis in cell growth and proliferation2,11,12, GMPK’s reversible catalytic activity may also impact intracellular nucleotide homeostasis, thereby contributing to tumor cell viability. Analysis of potential ligand binding pockets in both the apo and GMP-bound states revealed that, in addition to the substrate binding site, a potential binding pocket for lead-like compounds is present at the GMPBD-CORE interface in the GMP-bound state (Supplementary Fig. 19e, f) and further facilitate structure-based drug discovery by targeting the dynamic conformation of GMPK.
Methods
Cloning, expression and purification of human GMPK
GMPK gene was amplified using polymerase chain reaction with human cDNA as the template, and then was inserted into the pET28a plasmid, incorporating an N-terminal hexa-histidine tag followed by a sumo tag (Supplementary Table 6, Supplementary Data 1)28. All GMPK mutants were generated (Supplementary Data 1) using a homologous recombination kit (C112, Vazyme). The recombinant plasmid was transformed into E.coli Rosetta (DE3) and subsequently analyzed via Sanger sequencing. For the unlabeled protein, cells were cultured in LB medium at 37 °C until the optical density at 600 nm (OD600) reached 0.8. For 15N-label protein, cells were cultured in 300 mL of LB medium at 37 °C overnight, harvested and resuspended in 1 L of M9 medium. The culture was then incubated at 37 °C until reaching an OD600 of 1.2 to allow for growth recovery. The expression of GMPK was induced by the addition of 0.5 mM IPTG. Cells were harvested after 20 h of growth at 16 °C. The cell pellets were resuspended in lysis buffer (20 mM Tris-HCl, 1 M NaCl, pH 8.0) and subsequently lysed under high pressure, followed by centrifugation at 15000 g. The supernatant obtained was subjected to a nickel column, eluted with elution buffer (20 mM Tris-HCl, 1 M NaCl, pH 8.0, 0.5 M imidazole), and mixed with ULP1 protease for the cleavage of the sumo tag. The reaction product was incubated with nickel column to remove sumo tag, and subsequently purified using size-exclusion chromatography in buffer (20 mM HEPES, 150 mM NaCl, pH 7.5).
Isothermal titration calorimetry assay
Isothermal Titration Calorimetry (ITC) experiments were conducted using a Malvern PEAQ ITC instrument at 25 °C. The initial injection volume was set to 1 μL, followed by 19 subsequent injections of 2 μL each. The reference power was maintained at 5 μcal/s. For the binding assays involving various nucleotides and GMPK wild type or mutants, the protein concentrations were prepared at 50 μM. Different nucleotide solutions, including 1.25 or 2.5 mM GMP (CG5931, Coolaber), 1 mM ADP (CA1271, Coolaber), 1 or 2 mM ATPγS (GC12574, GLPbio), and 1 mM GDP (GDP, HARVEYBIO), were prepared separately and loaded into the syringe. For the GMP binding assays conducted in the presence of either potassium or sodium ions, GMPK and GMP were dissolved in a buffer composed of 20 mM HEPES, pH 7.5, and either 150 mM KCl or 150 mM NaCl. The GMPK concentration was prepared at 50 μM and loaded into the cell, and GMP was prepared at 1 mM and loaded into the syringe. For the GMPK-GMP mixtures titrated with ATPγS or ADP, a mixture of 50 μM GMPK and 0.5 mM GMP was loaded into the cell, while 1 mM ATPγS or ADP was loaded into the syringe. Conversely, for GMPK-ATPγS or GMPK-ADP mixtures titrated with GMP, a mixture of 50 μM GMPK and 0.5 mM ATPγS or ADP was loaded into the cell, and 2 or 2.5 mM GMP was loaded into the syringe. All experimental data were analyzed using the Malvern ITC analysis software.
Enzyme activity assay
An NADH-dependent assay was conducted to measure the initial reaction velocity16. The reaction mixture contained 6 U/ml lactate dehydrogenase (HY-P2807, MCE), 6 U/ml pyruvate kinase (from Escherichia coli PKM2, which is monovalent cation-independent38), 2 nM GMPK, 0.2 mM NADH (N814671, Macklin), 100 mM Tris-HCl (pH 7.5), 150 mM KCl, 10 mM MgCl2, 2.5 mM ATP (A100880, Aladdin), 2.5 mM GMP, 0.1 mM ribose 5-phosphate (HY-W009371A, MCE) and 0.2 mM phosphoenolpyruvate (P816869, Macklin). The effect of monovalent cations on initial reaction velocity was investigated by varying KCl concentrations while maintaining constant ionic strength with tetramethylammonium chloride (T19526, Sigma-Aldrich). To assess the impact of sodium ions, KCl was substituted with NaCl at varying concentrations. GMP KM values at varying KCl or NaCl concentrations were determined by varying GMP concentration while holding ATP concentration constant at 2.5 mM and maintaining constant ionic strength with tetramethylammonium chloride. For determination of KM values for GMP or ATP, the concentration of the respective substrate (GMP or ATP) was varied while keeping the other substrates at 2.5 mM. ATP was pre-incubated with the coupled enzymes and all substrates except GMP to deplete residual ADP. The ATP hydrolysis rate in the absence of GMP was subtracted from the reaction rates measured at different substrate concentrations to correct for background ATP hydrolysis. For relative initial reaction velocities of wild-type and mutants, the reaction mixture contained 2.5 U/ml lactate dehydrogenase, 3.5 U/ml pyruvate kinase, 20 nM GMPK wild-type or mutants, 0.2 mM NADH, 100 mM Tris-HCl, 100 mM KCl, 10 mM MgCl2, 4 mM ATP, 2 mM GMP,1.5 mM phosphoenolpyruvate. The reaction was monitored by measuring the change in absorbance at OD340 using a CLARIOstar instrument. The initial reaction velocities were measured within the first 5 min, during which the OD340 exhibited a linear decrease. The slope of this linear phase was used to calculate the initial reaction rate (v) according to the equation: v = Slope/2ε(NADH), where ε(NADH) represents the molar extinction coefficient of NADH. Initial reaction velocities and KM curves were fitted using linear regression and the Michaelis-Menten equation or substrate inhibition equation, respectively, using GraphPad Prism 8. Each reaction was conducted in triplicate.
1H-NMR spectroscopy was employed to monitor the reactant and product transformations based on their distinctive chemical shifts10. For forward reaction, the mixture contained 10 mM KH2PO4, 100 mM KCl, 5 mM MgCl2, 2 mM ATP, 2 mM GMP and 12 nM GMPK. For backward reaction, the mixture contained 10 mM KH2PO4, 100 mM KCl, 5 mM MgCl2, 2 mM ADP, 2 mM GDP and 20 nM GMPK. To determine the reaction equilibrium constant (Keq), the concentration of GMPK was set at 1 μM, and all other conditions remained unchanged. For the comparison of reaction velocities in the presence of Na+ or K+, KH2PO4 and KCl were replaced with NaH2PO4 and NaCl respectively, and GMPK was set as 10 nM. The reaction was allowed to proceed for one hour, ensuring sufficient signal-to-noise ratio but not reached equilibrium yet. The reaction mixture was transferred into an NMR tube, and a 1H spectrum was recorded on an Agilent 500 MHz spectrometer with 32 scans at 25 °C. The 1H NMR data were processed using MestReNova software.
Crystallization and data collection
Crystals of GMPK were obtained by sitting drop vapor diffusion. For the apo form, GMPK was concentrated to 1 mM and mixed with a cell solution (25% w/v PEG 3350, 0.2 M NaCl, 0.1 M Bis-Tris, pH 6.5) in a 1:1.5 (v/v) ratio. For the GMPKS37P, GMPKS37P was concentrated to 1 mM and mixed with a cell solution (20% w/v PEG 3350, 0.2 M di-ammonium tartrate) in a 1:1 (v/v) ratio. For the GMPK-GMP complex, GMPK was diluted to 50 μM with SEC buffer (20 mM HEPES, 150 mM KCl, pH 7.5), supplemented with 10 mM GMP, and incubated overnight at 4 °C. The mixture was concentrated to 1 mM GMPK and then mixed with a cell solution (35% PEG4000, 0.1 M ammonium acetate, 0.1 M trisodium citrate, pH 6.0) in a 1.5:1 (v/v) ratio. For the GMPK-GMP-K+ complex, GMPK-GMP crystals were soaked in a cell solution containing 20 mM KNO3. For the preparation of the GMPK-GMP-ATPγS-K+ complex, GMPK was diluted to 50 μM in SEC buffer, supplemented with 10 mM GMP, 5 mM ATPγS, 20 mM KCl and 10 mM MgCl2, and incubated at 4 °C overnight. Subsequently, the mixture was concentrated to 1 mM GMPK and mixed with a cell solution (35% PEG4000, 0.1 M ammonium acetate, 0.1 M trisodium citrate, pH 5.0) in a 1.5:1 (v/v) ratio. For the GMPK-GDP-ADP complex, GMPK was diluted to 50 μM with SEC buffer, supplemented with 10 mM GDP and 2.5 mM ADP, and incubated at 4 oC overnight. Subsequently, the mixture was concentrated to 1 mM GMPK and mixed with a cell solution (35% PEG4000, 0.1 M ammonium acetate, 0.1 M trisodium citrate, pH 5.0) in a 1:1.5 (v/v) ratio. For the GMPK-GMP-SO42− complex or GMPK-GMP-ADP complex, GMPK was diluted to 50 µM with SEC buffer, supplemented with 10 mM GMP and 2.5 mM ADP, and incubated at 4 °C overnight. Subsequently, the mixture was concentrated to 1 mM GMPK. GMPK-GMP-SO42− crystals were grown in a reservoir containing 25% PEG4000, 0.2 M ammonium sulfate, 0.1 M sodium acetate, pH 4.6 with a sample-to-reservoir volume ratio of 1:1. GMPK-GMP-ADP crystals were grown in a reservoir containing 40% PEG4000, 0.1 M ammonium acetate, 0.1 M trisodium citrate, pH 5.2 with a sample-to-reservoir volume ratio of 1.5:1. For the GMPK-ADP complex, GMPK was diluted to 50 µM with SEC buffer, supplemented with 2.5 mM ADP, and incubated at 4 °C overnight. Subsequently, the mixture was concentrated to 1 mM GMPK and mixed with cell solution (5% isopropanol, 2.0 M ammonium sulfate) in a 1:1 (v/v) ratio. For the GMPK-GMP-ADP-AlF4--K+-Mg2+ complex, GMPK was diluted to 50 μM with SEC buffer, supplemented with 10 mM GMP, 5 mM ADP, 20 mM LiAlF4 (920355, Sigma-Aldrich), 20 mM KCl and 30 mM MgCl2, and incubated at 4 °C overnight. Subsequently, the mixture was concentrated to 1 mM GMPK and mixed with a cell solution (35% PEG4000, 0.1 M ammonium acetate, 0.1 M trisodium citrate, pH 5.2) in a 1.5:1 (v/v) ratio. To prepare the transition state analogue complex for crystallization at 1.75 Å and 1.80 Å resolution, GMPK was diluted to 50 μM in SEC buffer, then supplemented with 10 mM GMP, 5 mM ADP, 30 mM LiAlF4, and 30 mM MgCl2. This mixture was incubated overnight at 4 °C. Following incubation, the mixture was concentrated to 0.5 mM GMPK and subsequently mixed at a 1.5:1 (v/v) ratio with a precipitant solution consisting of either 35% PEG 4000, 0.1 M ammonium acetate, and 0.1 M trisodium citrate (pH 5.6), or a second precipitant solution of 40% PEG 4000, 0.1 M ammonium acetate, and 0.1 M trisodium citrate (pH 5.6).
All crystal growth conditions were maintained at 4 °C, except for the GMPK-ADP complex and transition state analogue complex (PDB ID 9LO6), which was grown at 20 °C. Prior to data collection, crystals were soaked in cryoprotection buffer containing 20% glycerol and 80% cell solution and flash-frozen in liquid nitrogen. Diffraction data were collected at beamline BL19U1, BL02U1 at the Shanghai Synchrotron Radiation Facility (SSRF).
Structure determination
Diffraction data were indexed and integrated using XDS software39. Subsequently, the data were scaled using Aimless software40. Molecular replacement was performed to solve the structures of all GMPK proteins using the MOLREP software41. The NMR model (PDB ID: 6NUI) served as the search model for determining the structure of the open form. For the closed form structure, the mouse GMPK-GMP-ADP model (PDB ID: 1LVG) was employed as the search model. Refinement was conducted both manually, using Coot42 and automatically, using Phenix.refine43. The density near the p-loop in both the GMPK-GMP-K+ and GMPK-GMP complexes could not be unambiguously assigned to any specific solvent, and thus was not modeled. The composite omit map of ligands was generated using the Phenix.composite_omit_map program. Structural figures were generated using PyMOL (https://pymol.org) and ChimeraX44.
Chemical shift perturbation
The 15N-labeled GMPK was diluted to 50-100 μM with SEC buffer containing 20 mM HEPES, 150 mM NaCl, pH 7.5, and supplemented with 10% D2O (DLM-4, Cambridge isotope laboratories). For the titration of GMPK and GMP with KCl and NaCl, GMP was present in a tenfold excess relative to GMPK. The samples were dissolved in 20 mM HEPES, 150 mM tetramethylammonium chloride, pH 7.5, supplemented with 10% D2O. The 1H-15N HSQC spectrum was acquired at 298 K on an Agilent 700 MHz instrument equipped with a cryoprobe and VnmrJ_3.2 data acquisition software. The HSQC experiments were performed with 1.3 s relaxation delay, 16 or 32 scans, a total number of complex points of 64, and an acquisition time of 24 or 48 min. Details of pulse sequence, sample concentrations and buffer conditions are included in Supplementary Data 2. All 2D spectra were processed and visualized using NMRPipe45 and Sparky46. Chemical shift perturbations relative to the initial state were calculated according to the following Eq. (1)47:
where \({\Delta {\rm{\delta }}}_{{\rm{H}}}\) and \(\Delta {{\rm{\delta }}}_{{\rm{N}}}\) are the chemical shift differences of the 1H and 15N dimensions, respectively. The chemical shifts were transferred from Khan’s assignment data (BMRB entry 27151)48. The assignments were transferred using the read_peak_list function in Sparky software, applying a tolerance of 0.35 ppm for the nitrogen dimension and 0.03 ppm for the proton dimension. In spectra recorded with a buffer containing 20 mM HEPES, deviations were observed in the chemical shifts for residues R5, I170, I169, L100, L9, and L8 compared to Khan’s assignment data. However, when the buffer was supplemented with 16 mM sulfate, the chemical shifts for these residues aligned with those from Khan’s assignment data recorded in a buffer containing 20 mM phosphate. Thus, assignments were confirmed by integrating data from both buffer conditions. Additionally, to minimize transfer errors, overlapped peaks that could not be unambiguously assigned (particularly in crowded regions) were excluded from quantitative analyses. The NMR titration data were globally fitted to a one-site binding model (Eq. 2) using a shared Kd across all analyzed residues:
Where \({\rm{x}}=\frac{{\rm{L}}}{{\rm{P}}}\), L represents the ligand concentration, P is the protein concentration, CSPmax is the maximum chemical shift perturbation, and Kd is the equilibrium dissociation constant.
Circular Dichroism Spectroscopic Analysis
The hGMPK samples were dialyzed against a buffer containing 20 mM NaH2PO4, 50 mM NaCl, pH 7.5. The concentrations of both wild-type (WT) and mutant hGMPK samples were adjusted to 5–10 μM. Circular dichroism spectra were recorded at 25 °C over a wavelength range of 200–260 nm using a Chirascan qCD spectrometer. Three scans were acquired and averaged for each measurement. The averaged CD spectrum of each sample was corrected by subtracting its corresponding buffer spectrum and then smoothed (window size = 3).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates and structure factors for GMPK in its free form and substrate-bound forms generated in this study were deposited to the Protein Data Bank (PDB) under accession codes: 9J8L (apo form of GMPK), 9JAB (Transition state analogue, GMPK in complex with GMP, ADP, Mg2+, K+ and AlF4- at 293 K), 9JAC (GMPK in complex with GMP), 9JAD (GMPK in complex with GMP and K+), 9JAE (GMPKS37P), 9JAF (GMPK in complex with GMP and sulfate), 9JAG (GMP in complex with ADP), 9JAH (GMPK in complex with GMP, ATPγS and K+), 9JAI (GMPK in complex with GMP and ADP), 9JAJ (GMPK in complex with GDP and ADP), 9LO3 (Transition state analogue, GMPK in complex with GMP, ADP, Mg2+, and AlF4- at 277 K), and 9LO6 (Transition state analogue, GMPK in complex with GMP, ADP, Mg2+, and AlF4- at 293 K). The ITC, NMR titration, and enzyme activity data generated in this study are provided in the Supplementary Information file. The source data underlying Figs. 1i, 2c, 3i and Supplementary Fig. 6c–i, 9a–d, 11e are provided as a Source Data file. Previously published data for crystal structures of GMPK are available with PDB accession codes: 1EX6, 1EX7, 1LVG, 1S4Q, 1S96, 1ZNX, 1ZNW, 1GKY, 2ANB, 2ANC, 2QOR, 3TR0, 6WCT, 7LUY, 8PTS. Previously published data for NMR structure of GMPK are available with PDB accession code 6NUI and BMRB code 27151. Source data are provided with this paper.
References
Chandel, N.S. et al. Nucleotide Metabolism. Cold Spring Harb Perspect Biol. 13, a040592 (2021).
Diehl, F. F. et al. Nucleotide imbalance decouples cell growth from cell proliferation. Nat. Cell Biol. 24, 1252–1264 (2022).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–33 (2009).
Pedley, A. M., Pareek, V. & Benkovic, S. J. The purinosome: a case study for a mammalian metabolon. Annu Rev. Biochem 91, 89–106 (2022).
An, S., Kumar, R., Sheets, E. D. & Benkovic, S. J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320, 103–6 (2008).
Yang, C. et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat. Cell Biol. 25, 836–847 (2023).
Tran, D. H. et al. De novo and salvage purine synthesis pathways across tissues and tumors. Cell 187, 3602–3618.e20 (2024).
Van Rompay, A. R., Johansson, M. & Karlsson, A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharm. Ther. 87, 189–98 (2000).
Fujisawa, K. et al. Regulation of adenine nucleotide metabolism by adenylate kinase isozymes: physiological roles and diseases. Int. J. Mol. Sci. 24, 5561 (2023).
Li, Y., Zhang, Y. & Yan, H. Kinetic and thermodynamic characterizations of yeast guanylate kinase. J. Biol. Chem. 271, 28038–44 (1996).
Mullen, N. J. & Singh, P. K. Nucleotide metabolism: a pan-cancer metabolic dependency. Nat. Rev. Cancer 23, 275–294 (2023).
Shi, D. D., Savani, M. R., Abdullah, K. G. & McBrayer, S. K. Emerging roles of nucleotide metabolism in cancer. Trends Cancer 9, 624–635 (2023).
Sprenger, H. G. et al. Cellular pyrimidine imbalance triggers mitochondrial DNA-dependent innate immunity. Nat. Metab. 3, 636–650 (2021).
Vaughn, B. P., Robson, S. C. & Burnstock, G. Pathological roles of purinergic signaling in the liver. J. Hepatol. 57, 916–20 (2012).
Oeschger, M. P. & Bessman, M. J. Purification and properties of guanylate kinase from Escherichia coli. J. Biol. Chem. 241, 5452–60 (1966).
Agarwal, K. C., Miech, R. P. & Parks, R. E. Jr. Guanylate kinases from human erythrocytes, hog brain, and rat liver. Methods Enzymol. 51, 483–90 (1978).
Yan, H. & Tsai, M. D. Nucleoside monophosphate kinases: structure, mechanism, and substrate specificity. Adv. Enzymol. Relat. Areas Mol. Biol. 73, 103–34, x (1999).
Hall, S. W. & Kühn, H. Purification and properties of guanylate kinase from bovine retinas and rod outer segments. Eur. J. Biochem 161, 551–6 (1986).
Miller, W. H. & Miller, R. L. Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase. J. Biol. Chem. 255, 7204–7 (1980).
Boehme, R. E. Phosphorylation of the antiviral precursor 9-(1,3-dihydroxy-2-propoxymethyl)guanine monophosphate by guanylate kinase isozymes. J. Biol. Chem. 259, 12346–9 (1984).
Chazot, A. et al. The activation chain of the broad-spectrum antiviral bemnifosbuvir at atomic resolution. https://doi.org/10.1371/journal.pbio.3002743 (2024).
Schneider, J. L. et al. Abstract 1156: GUK1 is a novel metabolic liability in oncogene-driven lung cancer. Cancer Res. 83, 1156–1156 (2023).
Hible, G. et al. Calorimetric and crystallographic analysis of the oligomeric structure of Escherichia coli GMP kinase. J. Mol. Biol. 352, 1044–59 (2005).
Hible, G. et al. Unique GMP-binding site in Mycobacterium tuberculosis guanosine monophosphate kinase. Proteins 62, 489–500 (2006).
Stehle, T. & Schulz, G. E. Three-dimensional structure of the complex of guanylate kinase from yeast with its substrate GMP. J. Mol. Biol. 211, 249–54 (1990).
Stehle, T. & Schulz, G. E. Refined structure of the complex between guanylate kinase and its substrate GMP at 2.0 A resolution. J. Mol. Biol. 224, 1127–41 (1992).
Sekulic, N., Shuvalova, L., Spangenberg, O., Konrad, M. & Lavie, A. Structural characterization of the closed conformation of mouse guanylate kinase. J. Biol. Chem. 277, 30236–43 (2002).
Khan, N. et al. Solution structure and functional investigation of human guanylate kinase reveals allosteric networking and a crucial role for the enzyme in cancer. J. Biol. Chem. 294, 11920–11933 (2019).
Jin, Y., Molt, R. W. Jr. & Blackburn, G. M. Metal Fluorides: tools for structural and computational analysis of phosphoryl transfer enzymes. Top. Curr. Chem. (Cham) 375, 36 (2017).
Kerns, S. J. et al. The energy landscape of adenylate kinase during catalysis. Nat. Struct. Mol. Biol. 22, 124–31 (2015).
Johnston, C. A., Whitney, D. S., Volkman, B. F., Doe, C. Q. & Prehoda, K. E. Conversion of the enzyme guanylate kinase into a mitotic-spindle orienting protein by a single mutation that inhibits GMP-induced closing. Proc. Natl. Acad. Sci. USA 108, E973–8 (2011).
Whitney, D. S., Volkman, B. F. & Prehoda, K. E. Evolution of a protein interaction domain family by tuning conformational flexibility. J. Am. Chem. Soc. 138, 15150–15156 (2016).
Traut, T. W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem 140, 1–22 (1994).
Delalande, O., Sacquin-Mora, S. & Baaden, M. Enzyme closure and nucleotide binding structurally lock guanylate kinase. Biophys. J. 101, 1440–9 (2011).
Su, S. & Russell, P. J. Adenylate Kinase from Bakers’ Yeast: III. EQUILIBRIA: EQUILIBRIUM EXCHANGE AND MECHANISM. J. Biol. Chem. 243, 3826–3833 (1968).
Matte, A., Tari, L. W. & Delbaere, L. T. How do kinases transfer phosphoryl groups?. Structure 6, 413–9 (1998).
Krishnamurthy, H., Lou, H., Kimple, A., Vieille, C. & Cukier, R. I. Associative mechanism for phosphoryl transfer: a molecular dynamics simulation of Escherichia coli adenylate kinase complexed with its substrates. Proteins 58, 88–100 (2005).
Waygood, E. B., Rayman, M. K. & Sanwal, B. D. The control of pyruvate kinases of Escherichia coli. II. Effectors and regulatory properties of the enzyme activated by ribose 5-phosphate. Can. J. Biochem 53, 444–54 (1975).
Kabsch, W. XDS. et al. Acta crystallogr d biol crystallogr. 66, 125–32 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution?. Acta Crystallogr D. Biol. Crystallogr 69, 1204–14 (2013).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr D. Biol. Crystallogr 66, 22–5 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–93 (1995).
Lee, W., Tonelli, M. & Markley, J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–7 (2015).
Williamson, M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson Spectrosc. 73, 1–16 (2013).
Khan, N. et al. (1)H, (13)C and (15)N resonance assignment of human guanylate kinase. Biomol. NMR Assign. 12, 11–14 (2018).
Acknowledgements
We thank the staff of the BL19U1 and BL02U1 beamline at the National Center for Protein Science Shanghai and Shanghai Synchrotron Radiation Facility for assistance during data collection. We thank the National Key R&D Program of China [2024YFA1306201] (to K.R.), National Natural Science Foundation of China [22377119] (to K.R.), Anhui Provincial Natural Science Foundation [2208085MC50] (to J.G.), Research Funds of Center for Advanced Interdisciplinary Science and Biomedicine of IHM [QYPY20220008] (to K.R.), and USTC Research Funds of the Double First-Class Initiative [YD9100002028] (to K.R.) and [YD9100002036] (to J.G.) for their financial support.
Author information
Authors and Affiliations
Contributions
L.W., Y.D., J.G. and K.R conceptualized the project. L.W. performed the biochemical and structural experiments and analyzed the data. L.W. and K.R. wrote the paper. L.W., Z.L., Y.X., J.Q., S.L., F.Z., Y.Song, K.Y., M.L., F.L., J.Z., Y.P., S.G., Y.Z., Y.Shi, X.L., Y.D., J.G. and K.R. provided technical support, read and analyzed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Clarissa Czekster and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Li, Z., Xuan, Y. et al. Comprehensive profiling of the catalytic conformations of human Guanylate kinase. Nat Commun 16, 6859 (2025). https://doi.org/10.1038/s41467-025-61732-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61732-y
