
Abstract
Polytetrafluoroethylene (PTFE) and other fluoropolymers are widely used because of their exceptional chemical resistance and thermal stability. However, their disposal poses a significant environmental challenge. Conventional methods for degrading PTFE either require high temperatures or rely on complex reagents and often neglect efficient fluorine recovery. Herein, we present an approach for the room-temperature defluorination of PTFE using sodium dispersion, enabling the conversion of PTFE into sodium fluoride (NaF) under mild conditions. This method not only eliminates the need for elevated temperatures, but also demonstrates high yields of fluoride ion recovery, reaching up to 97% under optimized conditions. We further extend the application of this method to non-polymer, per- and polyfluoroalkyl substances (PFAS), including perfluorononanoic acid (PFNA), perfluorooctanoic acid (PFOA), perfluorobutanesulfonic acid (PFBS) and trifluoroacetic acid (TFA), achieving similarly high yields of NaF with appropriate adjustments of the reaction time and reagent amounts.
Similar content being viewed by others

Introduction
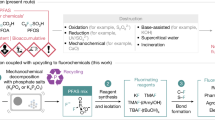
PTFE and other fluoropolymers have been used in various industries owing to their outstanding chemical resistance, low friction, and remarkable thermal stability1,2. These properties make PTFE indispensable in a wide range of applications, from household products such as nonstick cookware to advanced technologies in the automotive, semiconductor, and telecommunications sectors, where it is used in components such as electrical wire coatings and optical fiber cables. Despite the undeniable utility of PTFE, its extreme durability presents significant challenges at the end of its lifecycle, particularly in terms of disposal and environmental impact. The disposal of PTFE is particularly problematic because its high thermal stability requires incineration at high temperatures. This process not only consumes large amounts of energy but also generates corrosive hydrogen fluoride (HF) gas as a byproduct, which poses serious risks to incineration infrastructure1. Other methods, such as landfilling, where it remains inert and undegraded, contribute to the growing environmental burden of persistent polymer waste3. The defluorination of PTFE represents a significant chemical process that converts PTFE into its constituent fluorine compounds, providing a promising method for the recycling and degradation of this highly resistant polymer (Fig. 1A)4,5. This process not only addresses environmental concerns but also provides crucial insights for advancing chemical recycling technologies3.

A Industrial utility of PTFE and end of its lifecycle, such as incineration, landfilling, defluorination and potential recycling to PTFE. B Defluorination approaches: (a–d) High-temperature methods. (e–h) Low-temperature methods (surface modification of PTFE). C State-of-the-art by (a) Yanagihara and Katoh (2020), (b) Crimmin (2023), (c) Shibata (this work).
Traditional laboratory methods for defluorinating PTFE are generally classified into two categories based on the reaction temperature: high-temperature reactions above 500 °C, and low-temperature reactions below 100 °C (Fig. 1B). In most cases, fluorine is recovered as inorganic fluorides. High-temperature methods include defluorination using a) metallic magnesium in supercritical CO26, b) zinc powder7, c), magnesium silicide8, and d) calcium hydroxide in supercritical water9 (Fig. 1B, a–d). Low-temperature methods, on the other hand, employ reagents such as e) reducing agents like benzoin dianions or alkyl lithium compounds10,11, f) alkali metal/liquid ammonia or amines systems12,13,14,15, g) alkali metal vapors16, and h) alkali metal naphthalenides17,18,19 (Fig. 1B, e–h). Despite their success in defluorinating PTFE, these approaches primarily focus on the surface modification of the polymer and residue analysis without addressing the crucial aspect of fluorine recovery. Consequently, although fluorine compounds are formed, the efficiency and practicality of recovering fluorine resources remains unexplored (note; During the review process of this manuscript the following work was reported20,21,22).
Recent advancements in defluorination techniques have opened new possibilities for PTFE recycling (Fig. 1C). Yanagihara and Katoh demonstrated that PTFE could be mineralized into calcium fluoride (CaF₂) using molten sodium hydroxide (NaOH) at elevated temperatures (500 °C), achieving a yield of 73.8% and offering a more environmentally friendly recycling method (Fig. 1C, a)4. More recently, Crimmin et al. reported the room-temperature defluorination of PTFE using a magnesium reagent, achieving an 85% yield of molecular magnesium fluoride (Fig. 1C, b)5. However, this process requires a large excess of PTFE, resulting in a low fluorine recovery efficiency of only approximately 5%. Additionally, the use of benzene as a solvent—a known carcinogen—poses significant environmental and safety concerns, severely limiting its feasibility for industrial applications. Despite the promise of these methods, they still depend on either high temperatures or complex reagent systems, which hinders their practical application in large-scale industrial settings.
In this study, we introduce an approach for the room-temperature defluorination of PTFE using sodium dispersion (Fig. 1C, c)23,24. This method offers a simple pathway for breaking down PTFE and converting it into NaF under mild conditions. By avoiding the need for high temperatures and harsh reagents, our approach presents an attractive alternative to managing PTFE waste. Moreover, the process is not limited to PTFE but can be applied to other small molecular PFAS, such as PFNA, PFOA, PFBS and TFA, enabling the conversion of various PFAS into sodium fluoride.
Results and discussion
Optimization of reaction conditions
We optimized the defluorination of PTFE (particle size: 2.63 µm) using metallic sodium dispersion under low-temperature conditions (Table 1). After quenching the reaction with water, followed by washing with ether and water, the aqueous phase and solid black material were recovered, and the aqueous phase was freeze-dried to give the white solid, which was analyzed by ion chromatography and 19F NMR. We could not detect any byproducts in ether and water phases using techniques such as 1H and 19F NMR (for further details, see Supplementary analysis of reaction products of the defluorination of PTFE). Initially, using five equivalents of dispersion to PTFE in 1,4-dioxane at 80 °C for 24 h, we achieved 69% yield of NaF (entry 1). When switching solvent to triglyme and diglyme, the yields increased to 88% and 87%, respectively (entries 2 and 3). The reason for the improved yields should be mixing efficiency. While PTFE does not mix well in 1,4-dioxane, triglyme and diglyme form a heterogeneous mixing solution. Lowering the temperature to 50 °C with 1,4-dioxane resulted in a moderate yield of 74%, whereas triglyme and diglyme maintained high yields of 86% and 85% (entries 4–6). Tetrahydrofuran (THF) as the solvent at 50 °C afforded the highest yield (94%, entry 7).
We further refined the process using THF at 25 °C to obtain NaF in 90% yield (entry 8). By testing various reaction times at 25 °C with THF, we found that 12 h was optimal, achieving 98% yield of NaF. Shorter times of 1, 3, and 6 h resulted in lower fluoride ion recovery rates of 49%, 63%, and 74%, respectively (entries 9–12). Using sodium dispersion (2.0 equivalents) in THF at 25 °C for 12 h afforded a high yield of 97% (entries 13–15). However, reducing it further to 1.5 and 1.0 equivalent significantly decreased the yields to 65% and 22%, respectively (entries 16 and 17). Increasing the amount of PTFE to 6.0 mmol under the same conditions initially lowered the yield to 53% (entry 18); however, extending the reaction time to 24 h improved the yield to 80% (entry 19). These findings demonstrate that our optimized conditions effectively enhanced the yield of NaF from PTFE using sodium dispersion at low temperatures. Larger PTFE particle size (200 µm) was also effective for the defluorination reaction but required a longer reaction time to achieve comparable yields (67% yield after 12 h and 95% after 24 h; entries 20 and 21). The stirring speed was critical, with an optimal magnetic stirrer speed identified at 350 rpm. Slower stirring (200 rpm) significantly reduced the reaction efficiency, yielding only 53% after 12 h (entry 22). Furthermore, the reaction yield was sensitive to the type of sodium dispersion used. The results described above were obtained using commercially available sodium dispersion (TCI, SD Super Fine™, 25 wt% sodium dispersion in mineral oil). When another sodium dispersion (Aldrich, sodium, 25–35 wt% dispersion in paraffin) was employed without further optimization, a notably lower yield of 33% was observed (entry 23). The reaction was inhibited in the presence of TEMPO (2.0 equiv), indicating the radical species involved (entry 24). To further investigate the beneficial role of THF as a solvent, we conducted comparative reactions using non-polar solvents such as hexane and decane. Under these conditions, significantly reduced yields (64%, 68%, entries 25 and 26) were observed. These results indicate that ether-type solvents, which can coordinate and stabilize intermediate species, are particularly advantageous for this transformation. This observation aligns well with the mechanism of the Birch reduction, in which liquid ammonia effectively solvates free electrons25,26.
Spectral analyses
We next conducted an elemental analysis of the solid black residue obtained under optimal conditions (entry 15, Table 1) to discuss the fluoride ion recovery from PTFE (Table 2)12. The comparison of the elemental composition of PTFE and the residue is presented in Table 2. Entries 1 to 4 show the data for PTFE (analyzed four times), revealing carbon and fluorine contents of approximately 24% and 76%, respectively. These values correspond to a C/F ratio of 1/2, which aligns with the molecular formula of PTFE, (C1F2)n, indicating no loss of fluorine from the PTFE samples. In contrast, the residue analysis (tested four times, entries 5–8) shows carbon and fluorine contents of 56.25% (56.47%) and 11.60% (11.77%), respectively, resulting in a C/F ratio of approximately 1/0.13. This suggests a fluorine loss of about 93.5% from the original PTFE, which correlates well with the fluoride ion recovery determined via ion chromatography (97%, entry 15, Table 1).
Additionally, the X-ray diffraction (XRD) pattern of the dried white precipitate obtained (entry 15, Table 1) displayed distinct peaks at 2θ values of 33.45°, 38.78°, 56.05°, 66.85°, and 70.26°, corresponding to the (111), (200), (220), (311), and (222) crystallographic planes of NaF, respectively. These diffraction peaks provide clear evidence of NaF formation as a result of the treatment (Fig. 2A). As 2 equivalents of sodium were used for PTFE decomposition, NaF was co-isolated with NaOH in a molar ratio of approximately 1:1 after aqueous work-up. A detailed analysis is given in supplementary pH measurements.

A XRD pattern of the NaF obtained. The product was identified using the ICDD PDF-2 database (International Centre for Diffraction Data, Newtown Square, PA, USA). B Raman spectrum of PTFE (red-line) and the black residue (purple-line). C IR spectrum of PTFE (red-line) and the black residue (purple-line). D 19F SS-MAS NMR of PTFE (red-line) and the black residue (purple-line). E 1H SS-MAS NMR of black residue. F XRD of PTFE (red-line) and the black residue (purple-line). G SEM-EDS of PTFE and black residue.
The residue obtained after the degradation reaction exhibited a black appearance, visually consistent with amorphous carbon. Elemental analysis of this residue (Table 2) revealed an oxygen content of approximately 28%. Trace levels of nitrogen (below 1%) were also detected; however, these are likely due to minor contamination and considered insignificant for interpreting the primary results. To further characterize the residue, Raman and IR spectroscopic analyses were performed. The Raman spectrum of the black residue (entry 15, Table 1) was directly compared to that of the initial PTFE. As illustrated in Fig. 2B, the Raman spectrum of the residue (purple line) lacked characteristic PTFE signals, notably the CF2 group vibrations. Instead, two prominent new peaks were observed at 1419 cm−1 and 1583 cm−1, corresponding to the D band (disordered sp2 carbon) and G band (graphitic sp2 carbon), respectively27,28,29. These bands clearly indicate effective cleavage of the original C–F bonds in PTFE and confirm the formation of an amorphous carbon structure. In contrast, the Raman spectrum of pristine PTFE (Fig. 2B, red line) displayed characteristic peaks at 294 cm−1 and 388 cm−1 (torsional and deformation vibrations of CF2 groups), 734 cm−1 (symmetric CF2 stretching), 1216 cm−1 (antisymmetric CF2 stretching), as well as peaks at 1301 cm−1 and 1380 cm−1 corresponding to C–C stretching vibrations30,31. These observed spectral features align well with literature values, confirming the absence of CF2 groups in the black residue.
Infrared (IR) spectroscopic analysis of the residue (Fig. 2C, purple line) revealed four significant absorption bands distinctly different from PTFE: a broad peak at approximately 3441 cm−1 (O–H stretching), a peak at 2958 cm−1 (alkyl C–H stretching), a sharp band at 1715 cm−1 (C=O stretching), and another at 1621 cm−1 (aromatic C=C stretching). These spectral features corroborate the elemental analysis, confirming the incorporation of oxygen- and hydrogen-containing functional groups into the residue structure (Table 2). It should be pointed out that we observe that the residue consistently exhibit IR absorption bands corresponding to hydroxyl (O–H) and carbonyl (C=O) functional groups, regardless of the solvent used during PTFE decomposition (e.g., THF, triglyme, or hexane, see Supplementary Fig. 19 and 20). This indicates that the oxygen content is not derived from the solvent or the PTFE starting material itself. Our interpretation, consistent with prior studies (e.g., Polymer, 1978, 19, 856), is that the as-formed carbonaceous material is highly reactive due to the presence of carbon-centered radicals and conjugated polyene structures (see the later part of Mechanistic Investigations)32. These features are prone to rapid oxidation upon exposure to atmospheric oxygen or water during workup and isolation.
Solid-state magic-angle spinning (SS-MAS) NMR analysis was subsequently conducted to further characterize the residue. The 19F SS-MAS NMR spectrum of the black residue showed no detectable fluorine-related peaks, apart from minor signals attributed to trace amounts of unreacted PTFE, aligning well with the elemental analysis results (Fig. 2D, black residue: purple line; PTFE: red line). Furthermore, the 1H SS-MAS NMR spectrum of the residue exhibited three broad peaks at approximately 1.06 ppm, 4.23 ppm, and 7.0 ppm, which are consistent with alkyl hydrogens, hydroxyl hydrogens, and aromatic hydrogens, respectively (Fig. 2E). The 13C SS-MAS NMR spectrum of the residue shows prominent broad signals in the range of approximately 80–180 ppm, which are characteristic of sp2-hybridized carbon species. Additionally, smaller broad resonances are observed around 10–30 ppm and 190–210 ppm, corresponding to sp3-hybridized carbon and carbonyl carbon functionalities, respectively. These spectral features strongly support the formation of amorphous carbon structures. A minor sharp singlet at 111.6 ppm is also present, indicating the presence of a small amount of residual unreacted PTFE (Supplementary Figs. 5 and 7). Powder X-ray diffraction (XRD) analysis of pristine PTFE (Fig. 2F, red line) revealed characteristic reflections at 2θ = 18° (crystalline regions) and approximately 40° (amorphous regions). In contrast, the black residue displayed only a weak reflection at 18°, indicating significant disruption of both crystalline and amorphous structures of PTFE (Fig. 2F, purple line). Scanning electron microscopy coupled with energy-dispersive X-ray spectroscopy (SEM-EDS) analysis (Fig. 2G) showed that PTFE consists of dense, irregularly shaped grains with predominantly smooth surfaces. After degradation, the black residue exhibited a pronounced morphological transformation, presenting a highly irregular surface with cracks and rough textures. Elemental mapping demonstrated a substantial increase in carbon signal intensity and a corresponding significant reduction in fluorine content, strongly confirming the extensive decomposition of PTFE. Additionally, an increased oxygen signal observed in the residue suggests the formation of oxygen-containing functional groups (C–O bonds), likely due to interaction with moisture or oxygen during the washing process. Collectively, these analyses confirm the transformation of PTFE into an amorphous, carbon-rich material (for further details, see Supplementary analysis of reaction products of the defluorination of PTFE).
Defluorination of PFAS
Building on our successful transformation of PTFE into NaF, we extended the sodium dispersion defluorination approach to nonpolymer, small-molecule PFAS. We selected commercially available, representative nonpolymer PFAS: PFNA, PFOA and PFBS (Table 3). Initial reactions were conducted at 25 °C for 12 h in THF, using two equivalents of sodium dispersion per fluorine atom, following the optimized PTFE defluorination conditions (Table 1). Under these conditions, we observed NaF formation with PFNA, achieving yields of 67% after 12 h and 78% after 24 h (entries 1 and 2). Increasing the sodium dispersion quantity to 5 equivalents further improved NaF yields to 89% and 90% for 24 h and 48 h reactions, respectively (entries 3 and 4). The method was also applicable for 10 times diluted PFNA conditions (entry 5, 88%). PFOA was also defluorinated into NaF under the same and diluted conditions (88%, 97%, entries 6 and 7). With PFBS, the initial conditions yielded NaF at 46% (Entry 8), which increased significantly to 97% when the reaction time was extended to 24 h and the sodium dispersion was raised to 5 equivalents (entry 9). These findings highlight the versatility and efficacy of the sodium dispersion-defluorination method for degrading both perfluoropolymers (such as PTFE) and nonpolymer PFAS (PFNA, PFOA and PFBS) with high yields.
Additionally, we explored sodium dispersion-mediated defluorination of TFA, a frequently used organic acid in laboratory and industrial settings. The environmental impact of TFA, due to its high acidity and persistence, has raised concerns33. Under the conditions of 2 equivalents of sodium dispersion at 25 °C for 24 h, NaF was obtained in a low yield of 24%, which increased to 45% after 24 h (entries 10 and 11). When the sodium dispersion quantity was increased to 5 equivalents, NaF yields improved to 70% and 73% after 24 h and 48 h, respectively (entries 12 and 13).
These results demonstrate that the sodium dispersion defluorination method provides an effective pathway for defluorinating a broad spectrum of PFAS, including both polymeric and nonpolymeric types, as well as environmentally persistent organofluorine compounds like TFA, highlighting its potential for PFAS degradation. Although the defluorination reaction typically requires 12 h to 24 h, depending on the specific PFAS substrate—likely due to the heterogeneous nature of the reaction system—the method achieves nearly complete defluorination under mild reaction conditions within a practical timeframe.
Mechanistic investigations
Our reaction conditions resemble those of the Birch reduction, a classical method involving alkali metals in liquid ammonia to reduce aromatic compounds. Previous studies have shown that reductive approaches, including electrochemical methods, effectively cleave C–F bonds, generating fluoride ions (F⁻). Recently, Qu and Kang et al. reported photocatalytic defluorination of PTFE at 60 °C over 48 h, yielding amorphous carbon and fluoride salts as major products20. Their method involves single-electron transfer (SET) from a photoexcited catalyst, forming PTFE radical anions; however, they did not clearly describe the initial C–F bond cleavage step. Additionally, Xue et al. (2024) proposed a mechanism involving Lewis base-boryl radicals that selectively activate C(sp3)–F bonds via a concerted electron-fluoride transfer (cEF-T) process34. Supported by experimental data and density functional theory (DFT) calculations, their mechanism involves direct fluorine atom abstraction, rather than simply weakening the C–F bond through coordination, forming alkyl radicals without detectable intermediates.
The proposed reaction mechanism is shown in Fig. 3A, and DFT calculations were employed to provide further insights into the energetics of these pathways. Perfluorohexane (F-hexane, CF3-CF2-CF2-CF2-CF2-CF3) was used instead of PTFE for computational simplicity to reduce computational cost (Fig. 3B). All calculations were performed with Gaussian 16 at the ωB97XD/Def2-TZVPP-SMD(THF) level of theory35. Considering these previous studies and established reductive defluorination mechanisms, as well as our own experimental observations—where no fluorinated by-products were detected by careful analysis using 1H and 19F NMR spectroscopy of ether and aqueous extracts, or through direct NMR monitoring of the reaction mixture—we propose the following mechanism for the sodium dispersion-mediated defluorination of PTFE to yield amorphous carbon and NaF (Fig. 3A). Initially, electron transfer from Na to PTFE generates radical intermediate I and releases fluoride ions (F⁻), immediately captured as NaF. The computational results support that the formation of the radical intermediate I under the studied reaction conditions proceeds via an enthalpically barrierless process and this step was found to be highly exergonic (−60.7 kcal mol−1) (for F-hexane, Fig. 3B). The presence of radical intermediate I is supported by TEMPO radical-trapping experiments (entry 24, Table 1). We analyzed the spin-center distribution in radical intermediate I by DFT calculations, and as indicated in Fig. 3B, the spin density is mainly located on the carbon atom (0.72) and in the α-bonded fluorine atom (0.12). Intermediate I then accepts an additional electron from Na, forming anion intermediate II, which subsequently undergoes further fluoride elimination. The formation of anion intermediate II-F-hexane from radical intermediate I-F-hexane was calculated to be highly exergonic (−44.2 kcal mol−1). The highly endergonic nature of this fluoride elimination step played a leading role in driving the degradation of PTFE. The initial C–F bond cleavage to form radical intermediate I from PTFE likely proceeds through a concerted electron-fluoride transfer (cEF-T) mechanism (Fig. 3A). Specifically, electron transfer from Na into the antibonding σ* orbital of the C–F bond weakens it, facilitating simultaneous fluoride transfer to Na⁺ and forming radical intermediate I via C-I and C-I’. Following the formation of intermediate II, three possible pathways are proposed for subsequent fluoride elimination: pathway (a) involves fluoride ion elimination from carbene intermediate III, which dimerizes to produce unsaturated perfluoropolymer intermediate IV; pathway (b) involves β-elimination directly generates unsaturated perfluoropolymer intermediate V; and lastly, pathway (c) based on δ-elimination with β-C-C bond cleavage generates two unsaturated perfluoropolymer intermediate VI and VI’. When comparing the different possible fluoride elimination pathways, calculations (Fig. 3B) indicated that carbene intermediate III-F-hexane formation and subsequent dimerization (pathway a) was energetically unfavorable (34.8 kcal mol−1). The β-elimination pathway providing V-F-hexane (pathway b), consistent with findings by Qu and Kang, showed a significantly lower activation barrier (−38.2 kcal mol−1), making pathway (b) clearly the more feasible mechanism. The computed activation barrier for β-elimination was ΔG‡ = 5.4 kcal mol−1. On the other hand, the δ-elimination with β-C-C bond cleavage via pathway c was also thermodynamically favorable (−24.6 kcal mol−1), however, it proceeds via a transition state with a high Gibbs activation barrier (ΔG‡ = 36.2 kcal mol−1), making it not feasible at room temperature.

A Proposed reaction mechanism. B DFT calculations of the reaction mechanism using perfluorohexane (F-hexane) for computational simplicity to reduce computational cost. Gibbs energies and ooptimized structures at ωB97XD/Def2-TZVPP -SMD(THF) level of theory are displayed. C Gibbs activation barriers and ooptimized structures of transition states for fluoride elimination ωB97XD/Def2-TZVPP-SMD(THF) level of theory. Model compounds C5F11CO2H for PFNA and C4F9SO3H for PFBS were used for simplicity.
All unsaturated intermediates IV, V and VI (VI’) in Fig. 3A are electron-deficient and rapidly accept electrons from Na. Repeated defluorination cycles produce polyene structures that ultimately cyclize and aromatize into amorphous carbon. DFT calculations showed that Gibbs barriers for defluorination increased as the number of fluorine atoms decreased. Detailed 1H, and 19F NMR analyses revealed no detectable intermediates other than the initial PTFE and final NaF products, indicating that intermediates IV, V and VI have extremely short lifetimes and low activation barriers, being rapidly reduced further by sodium.
In addition, perfluorohex-2-ene (V-F-hexane) is more electron-deficient compared to the initial F-hexane, i.e, perfluorohexane, allowing it to readily accept electrons from sodium (Fig. 3B). The subsequent polyene intermediate, perfluorohexa-2,4-diene (VII-F-hexane), exhibits even greater electron deficiency than perfluorohex-2-ene, further accelerating the electron-transfer reaction cycles. This increase in reactivity is supported by calculated Global Electrophilicity Index (GEI) values, which clearly indicate enhanced electrophilicity along the reaction pathway (GEI: 0.776 eV for perfluorohexane, 1.936 eV for perfluorohex-2-ene, and 2.504 eV for perfluorohexa-2,4-diene). The GEI of perfluoroprop-1-ene (VI-F-hexane) is 1.419 eV, which also much higher reactivity than that of perfluorohexane. These results also support the experimental facts that no clear observation of reaction intermediates.
DFT calculations were conducted to further evaluate and compare the reactivity of different organofluorine compounds, specifically PFNA, PFBS, and TFA, under our degradation conditions. Model compounds C5F11CO2H for PFNA and C4F9SO3H for PFBS were used for simplicity. These calculations, performed at the ωB97XD/Def2-TZVPP-SMD (THF), identified transition states corresponding to fluoride elimination, each with distinct activation barriers that align closely with experimental observations (Fig. 3C). For PFNA, calculations showed that after initial defluorination, fluoride elimination from the resulting anion intermediate proceeded via a transition state with an activation Gibbs energy of 9.2 kcal mol−1. In contrast, the analogous step for PFBS exhibited a slightly higher activation Gibbs energy of 9.6 kcal mol−1. For TFA, the activation barrier was notably higher (13.1 kcal mol−1), consistent with its experimentally observed slower defluorination rate. This occurs because the negative charge (anion) resides on the same carbon atom from which fluoride elimination takes place. Although PFNA, PFBS, and TFA contain acidic functional groups such as carboxylic acid or sulfonic acid moieties, which could potentially undergo alternative decomposition pathways, the differences in computed activation Gibbs energies closely correspond to the experimentally observed relative decomposition efficiencies (PFNA > PFBS ≫ TFA). This agreement confirms that computational predictions effectively capture the primary defluorination pathway in our experiments.
This research introduces a significant advance in defluorination, effectively decomposing PTFE and non-polymeric PFAS utilizing metallic sodium dispersion under mild conditions (25 °C). Our method not only achieved impressive fluoride ion recovery—up to 98% for PTFE—but also demonstrated versatility across non-polymer PFAS, including PFNA, PFOA and PFBS. By optimizing reaction parameters such as time and sodium dispersion quantity, we established that this adaptable method can effectively process a broad range of fluoropolymer and PFAS substrates, including environmentally concerning compounds like TFA, which persist across Europe36. Fluorine-containing compounds are also extensively used in both the pharmaceutical and agrochemical industries37,38. From the perspective of a sustainable society, this raises important considerations, particularly regarding the environmental impact of agrochemicals that are widely distributed in agricultural fields. This innovative approach circumvents the need for high temperatures and aggressive reagents, offering a practical, environmentally responsible solution for fluorine resource recovery. By enabling fluoride recycling from these persistent materials, our method addresses two pressing issues: reducing fluoropolymer waste and decreasing reliance on imported fluorine raw materials, such as fluorite3. Our findings lay essential groundwork for future advancements in fluoropolymer recycling, contributing to global efforts in environmental protection and resource security within fluorine-based industries.
Limitations of the study
Our approach effectively addresses the critical challenge of decomposing stockpiles of fluorinated substances within industrial settings, demonstrating the broad applicability of small-molecule PFAS (e.g., TFA and PFOS) to fluoropolymers such as PTFE. The developed sodium dispersion method achieved high defluorination efficiencies at room temperature, providing alternative to traditional high-temperature incineration processes39. Notably, sodium dispersion is considered safer and easier to handle than metallic sodium, and it has already been employed in some industrial applications40,41. However, several important limitations of this study remain to be acknowledged. First, the production and regeneration of sodium dispersions remain resource-intensive, potentially diminishing the overall environmental benefits when scaled to industrial levels. Energy consumption, waste generation, and reagent regeneration are critical factors that require careful evaluation of other existing and emerging PFAS and PTFE degradation methods. Furthermore, our sodium dispersion approach cannot be applied directly to aqueous solutions contaminated with PFAS because sodium reacts violently with water. Consequently, this method is not suitable for treating environmentally diluted PFAS contaminants, such as groundwater or wastewater, limiting its application primarily to concentrated industrial stockpiles or solid residues.
Methods
General procedure for the degradation of PTFE
Sodium dispersion was added to a solution of PTFE powder in dry solvent with a glass stirrer bar, and the resulting mixture was stirred. The reaction mixture was cooled to 0 °C, and distilled water was added dropwise until no hydrogen gas evolved. After stirring at room temperature for 5 min, the mixture was filtered off, and the solid was washed with Et2O and distilled water. The aqueous layer of the filtrate was separated, and the organic layer was extracted with distilled water three times. The organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under reduced pressure and measured by the 19F NMR (CDCl3) (no fluorine peak was observed). The combined aqueous layer was freeze-dried to give the white precipitate. A portion of the solid was measured by the ion chromatography twice. The resulting solid was dissolved in D2O, which was used for measurement of the 19F NMR.
Entry 15, Table 1: The reaction of sodium dispersion (442 mg, 4.8 mmol, F: Na = 1:2) with PTFE powder (66.3 mg, 0.66 mmol) in dry solvent (2.5 mL) resulted in the formation of a white precipitate (341 mg). Ion chromatography analysis confirmed the precipitate contained 2.58 mmol of NaF (97%).
General procedure for the degradation of PFNA, PFOA, PFBS and TFA
Sodium dispersion was added to a solution of PFNA (0.2 mmol), PFBS (0.2 mmol) or TFA (1.0 mmol) in dry THF (2.5 mL) with a glass stirrer bar, and the resulting mixture was stirred at 25 °C. The reaction mixture was cooled to 0 °C, and distilled water was added dropwise until no hydrogen gas evolved. After stirring at room temperature for 5 min, the resulting mixture was extracted with water three times. The organic layer was washed with brine, then dried over Na2SO4. The solvent was removed under reduced pressure and measured by the 19F NMR (CDCl3) (no fluorine peak was observed). The combined aqueous layer was measured by the ion chromatography twice.
Entry 4, Table 3: Sodium dispersion (1.52 g, 17 mmol, F: Na = 1:5) was added to a solution of PFNA (97.3 mg, 0.21 mmol) in dry THF (2.5 mL). Ion chromatography analysis confirmed 3.21 mmol of NaF (90%).
Entry 6, Table 3: Sodium dispersion (1.37 g, 15 mmol, F: Na = 1:5) was added to a solution of PFOA (84.0 mg, 0.20 mmol) in dry THF (2.5 mL). Ion chromatography analysis confirmed 2.68 mmol of NaF (88%).
Entry 9, Table 3: Sodium dispersion (815 mg, 8.9 mmol, F: Na = 1:5) was added to a solution of PFBS (60.5 mg, 0.20 mmol) in dry THF (2.5 mL). Ion chromatography analysis confirmed 1.76 mmol of NaF (97%).
Entry 13, Table 3: Sodium dispersion (1.40 g, 15 mmol, F: Na = 1:5) was added to a solution of TFA (110.4 mg, 0.97 mmol) in dry THF (2.5 mL) Ion chromatography analysis confirmed 2.12 mmol of NaF (73%).
Computational details
All DFT calculations were performed using the Gaussian16 software35. All the structures were optimized using the long-range corrected hybrid ωB97xD density functional42 in combination with the Def2TZVP basis set43.The effect of the solvent was mimicked by applying the SMD model using ether as solvent44.The nature of the stationary points was confirmed by frequency calculations analysis at the same level of theory (minima were characterized by no imaginary frequencies, whereas transition states had one imaginary frequency). Transition states were further verified by relaxing the imaginary frequency towards the reactant and the product and by means of IRC calculations. 3D structures of optimized stationary points were represented using the CYLview 1.0 program45. Hirshfeld spin distributions were computed with Multiwfn 3.846. The Global Electrophilicity Index (GEI) values were calculated using Rowan Scientific47.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon request. Source data are provided with this paper.
References
Puts, G. J., Crouse, P. & Ameduri, B. M. Polytetrafluoroethylene: Synthesis and characterization of the original extreme polymer. Chem. Rev. 119, 1763–1805 (2019).
Améduri, B. Fluoropolymers as unique and irreplaceable materials: challenges and future trends in these specific per or poly-fluoroalkyl substances. Molecules 28, 7564 (2023).
Améduri, B. & Hori, H. Recycling and the end of life assessment of fluoropolymers: recent developments, challenges and future trends. Chem. Soc. Rev. 52, 4208–4247 (2023).
Yanagihara, N. & Katoh, T. Mineralization of poly(tetrafluoroethylene) and other fluoropolymers using molten sodium hydroxide. Green. Chem. 24, 6255–6263 (2022).
Sheldon, D. J., Parr, J. M. & Crimmin, M. R. Room temperature defluorination of Poly(tetrafluoroethylene) by a magnesium reagent. J. Am. Chem. Soc. 145, 10486–10490 (2023).
Wang, Q., Cao, F. & Chen, Q. Formation of carbon micro-sphere chains by defluorination of PTFE in a magnesium and supercritical carbon dioxide system. Green. Chem. 7, 733–736 (2005).
Chen, X. Y., Cheng, L. X., Deng, X., Zhang, L. & Zhang, Z. J. Generalized conversion of halogen-containing plastic waste into nanoporous carbon by a template carbonization method. Ind. Eng. Chem. Res. 53, 6990–6997 (2014).
Wang, L. et al. Synthesis of silicon carbide nanocrystals from waste polytetrafluoroethylene. Dalton Trans. 46, 2756–2759 (2017).
Yang, X. et al. A chemical route from PTFE to amorphous carbon nanospheres in supercritical water. Chem. Commun. 342, 343 (2004).
Costello, C. A. & McCarthy, T. J. Surface modification of poly(tetrafluoroethylene) with benzoin dianion. Macromolecules 17, 2940–2942 (1984).
Hlavaty, J., Kavan, L. & Bastl, Z. Simultaneous fluorine elimination and alkylation of poly(tetrafluoroethylene). Die Angew. Makromol. Chem. 238, 165–175 (1996).
Miller, M. L., Postal, R. H., Sawyer, P. N., Martin, J. G. & Kaplit, M. J. Conditioning polytetrafluoroethylene surfaces for use in vascular prostheses. J. Appl. Polym. Sci. 14, 257–266 (1970).
Chakrabarti, N. & Jacobus, J. The chemical reduction of poly(tetrafluoroethylene). Macromolecules 21, 3011–3014 (1988).
Chaban, V. V. & Prezhdo, O. V. Electron solvation in liquid ammonia: lithium, sodium, magnesium, and calcium as electron sources. J. Phys. Chem. B 120, 2500–2506 (2016).
Herlem, G. et al. Direct defluorination and amination of polytetrafluoroethylene and other fluoropolymers by lithium alkylamides. Molecules 29, 3045 (2024).
Tasker, S., Chambers, R. D. & Badyal, J. P. S. Surface defluorination of PTFE by sodium atoms. J. Phys. Chem. 98, 12442–12446 (1994).
Nelson, E., Kilduff, T. J. & Benderly, A. A. Bonding of Teflon. Ind. Eng. Chem. 50, 329–330 (1958).
Shiraishi, S. et al. Preparation of porous carbon with defluorination of PTFE by radical anion. TANSO 195, 395–399 (2000).
Yoshino, K. et al. Conducting polymer prepared from teflon. Jpn. J. Appl. Phys. 21, L301–L302 (1982).
Zhang, H., Chen, J.-X., Qu, J.-P. & Kang, Y.-B. Photocatalytic low-temperature defluorination of PFASs. Nature 635, 610–617 (2024).
Liu, X. et al. Photocatalytic C–F bond activation in small molecules and polyfluoroalkyl substances. Nature 637, 601–607 (2025).
Yang, L. et al. Phosphate-enabled mechanochemical PFAS destruction for fluoride reuse. Nature 640, 100–106 (2025).
De, P. B., Asako, S. & Ilies, L. Recent advances in use of sodium dispersion for organic synthesis. Synthesis 53, 3180–3192 (2021).
Ito, S., Takahashi, F. & Yorimitsu, H. Defluorinative diborasodiation of benzotrifluorides with Bis(pinacolato)Diboron and Sodium. Asian J. Org. Chem. 10, 1440–1443 (2021).
Dye, J. L. Electrides: early examples of quantum confinement. Acc. Chem. Res. 42, 1564–1572 (2009).
Lei, P. et al. A practical and chemoselective ammonia-free birch reduction. Org. Lett. 20, 3439–3442 (2018).
Ferrari, A. C. & Robertson, J. Resonant Raman spectroscopy of disordered, amorphous, and diamondlike carbon. Phys. Rev. B 64, 075414 (2001).
Sheka, E. F., Golubev, Y. A. & Popova, N. A. Graphene domain signature of raman spectra of sp2 Amorphous Carbons. Nanomaterials 10, 2021 (2020).
Hamaura, J., Hori, H., Fujishima, A. & Mukae, H. Efficient mineralization of fluoroelastomers using superheated water in the presence of potassium hydroxide. Molecules 28, 7057 (2023).
Firsov, S. P., Zhbankov, G. R., Bakhramov, M., Abdukadyrov, A. & Gafurov, A. Raman spectra and structure of polytetrafluoroethylene subjected to elastic deformation grinding. J. Appl. Spectros. 59, 644–647 (1993).
Schmälzlin, E. et al. Raman imaging with a fiber-coupled multichannel spectrograph. Sensors 14, 21968–21980 (2014).
Barker, D. J., Brewis, D. M., Dahm, R. H. & Hoy, L. R. J. Study of the intercalated carbon formed by electrochemical reduction of polytetrafluoroethylene. Polymer 19, 856–857 (1978).
Freeling, F. & Björnsdotter, M. K. Assessing the environmental occurrence of the anthropogenic contaminant trifluoroacetic acid (TFA). Curr. Opin. Green. Sustain. Chem. 41, 100807 (2023).
Guo, X., Zhang, Y., Lai, X., Pang, Y. & Xue, X.-S. C(sp3)-F bond activation by lewis base-boryl radicals via concerted electron-fluoride transfer. Angew. Chem. Int. Ed. 64, e202415715 (2025).
Frisch, M. J. et al. Gaussian 16, Revision B.01 (Gaussian Inc., 2019).
Itumoh, E. J. et al. Addressing the persistence of per- and polyfluoroalkyl substances (PFAS): current challenges and potential solutions. RSC Sustainability 2, 3183 (2024).
Inoue, M., Sumii, Y. & Shibata, N. Contribution of organofluorine compounds to pharmaceuticals. ACS Omega 5, 10633–10640 (2020).
Ogawa, Y., Tokunaga, E., Kobayashi, O., Hirai, K. & Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 23, 101467 (2020).
Barisci, S. & Suri, R. Occurrence and removal of poly/perfluoroalkyl substances (PFAS) in municipal and industrial wastewater treatment plants. Water Sci. Technol. 84, 3442–3468 (2021).
KI Chemical Co. https://www.ki-chemical.co.jp/english/technology/chemical/index.html (accessed 2025-03-30).
KMR Dispersions, https://www.kmrdispersions.com/ (accessed 2025-03-30).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
6. CYLview20; Legault, C. Y., Université de Sherbrooke, 2020 (http://www.cylview.org).
Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists. Multiwfn. J. Chem. Phys. 161, 082503 (2024).
Rowan Scientific. https://www.rowansci.com (accessed 2025-06-06).
Acknowledgements
The computational resources from the Servei d’Informàtica de la Universidad de Valencia (SIUV) are gratefully acknowledged for providing access to supercomputing resources. CREST program of the Japan Science and Technology Agency, entitled Precise Material Science for Degradation and Stability (grant number: JPMJCR21L1). CMC Research Institute, Japan (Dr. Seiji Motojima). FIU program of the Universitat de València.
Author information
Authors and Affiliations
Contributions
Conceptualization: NS. Methodology: T.A., H.O., Y.M. Investigation: T.A., H.O., Y.M. Analyses: T.A., H.O., Y.M., J.H., H.H. Computational study: J.E. Discussion: T.A., H.O., Y.M., Y.S., H.A., T.K., J.H., H.H. J.E., N.S. Funding acquisition: N.S., H.H. Project administration: N.S. Supervision: N.S. Writing – original draft: N.S. Writing – review & editing: N.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mohamed Ateia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Araki, T., Ota, H., Murata, Y. et al. Room-temperature defluorination of PTFE and PFAS via sodium dispersion. Nat Commun 16, 6526 (2025). https://doi.org/10.1038/s41467-025-61819-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61819-6
This article is cited by
-
Rethinking sustainable pathways for PFAS
Nature Chemistry (2026)
-
Upcycling of PTFE and PVDF to fluorochemicals through mechanochemical process
Nature Communications (2025)
