
Abstract
Recent progress in multiplexed tissue imaging is deepening our understanding of tumor microenvironments related to treatment response and disease progression. However, analyzing whole-slide images with millions of cells remains computationally challenging, and few methods provide a principled approach for integrative analysis across images. Here, we introduce SpatialTopic, a spatial topic model designed to decode high-level spatial tissue architecture from multiplexed images. By integrating both cell type and spatial information, SpatialTopic identifies recurrent spatial patterns, or “topics,” that reflect biologically meaningful tissue structures. We benchmarked SpatialTopic across diverse single-cell spatial transcriptomic and proteomic imaging platforms spanning multiple tissue types. We show that SpatialTopic is highly scalable to large-scale images, along with high precision and interpretability. It consistently identifies biologically and clinically significant spatial topics, such as tertiary lymphoid structures, and tracks spatial changes over disease progression. Its computational efficiency and broad applicability will enhance the analysis of large-scale imaging datasets.
Similar content being viewed by others


Introduction
Recent advancements in multiplexed tissue imaging allow the profiling of RNA and protein expression in situ across thousands to millions of single cells within a whole-slide tissue context1,2,3,4,5. These technologies generate high-dimensional molecular imaging data, offering significant opportunities for a spatially resolved understanding of cellular heterogeneity and organization within tissues. Compared to other single-cell technologies (such as single-cell RNA-seq, flow cytometry), multiplexed imaging provides unique opportunities to examine spatial patterns of diverse cell types and characterize the tissue microenvironment of interest, which may play an essential role in understanding disease progression, tissue development, and mechanisms of treatment response1,2,4,5,6,7. One recent discovery in cancer, partly enabled by multiplexed spatially resolved omics data, is the presence of tertiary lymphoid structures (TLSs) in tumor tissues and its role in the adaptive antitumor immune response8,9,10,11. TLSs have been identified in a wide range of human cancers9 and have demonstrated a promising positive association with improved outcomes in cancer patients who underwent immunotherapy8.
While promising, the complex cellular architecture revealed by whole-slide multiplexed tissue imaging presents significant analytical challenges. Pathology images of tissue samples affected by certain diseases, such as cancer, are particularly complex, displaying abnormal cellular structures and significant variation between tumor samples. Currently, most analyses focus on individual images, examining elements such as cell densities and inter-cellular distances1,2,6, or conducting basic spatial domain analyses that primarily focus on binarized tissue compartments, such as tumor versus stroma12,13,14. Associating these features with outcomes requires manual and heuristic aggregation across images. While promising, a significant hurdle in spatial pattern analysis is deciphering biologically and clinically relevant patterns from the complex architecture within tissue across various slides.
In recent literature, cell neighborhood (niche) analysis has emerged as a popular approach. This analysis pipeline typically consists of two primary steps by first identifying neighborhood features for each single cell using either a K-nearest-neighbor (KNN) graph or a defined radius, and then applying a clustering algorithm, such as k-means, Louvain, or Latent Dirichlet Allocation (LDA)2,6,7,15,16,17. Seurat v518, for instance, clusters cells using k-means based on similar cell type compositions, offering a straightforward niche analysis method. There are different variants of the approach depends on how to incorporate spatial information into the clustering process. UTAG15 averages marker expression within the neighborhood for clustering, while BankSY19 further refines this by combining local mean expression with individual cell expression. Spatial-LDA16 incorporates spatial priors into clustering to allow proximity-closed cells to share similar cell neighborhoods. More recently, graph neural networks have been employed to discern cell neighborhood patterns, such as CytoCommunity20. However, deep learning methods like CytoCommunity require significant computational resources, posing challenges for individual labs, particularly for large-scale image analysis. Other studies adapt computational methods designed for spatial transcriptomics to analyze tissue imaging data21,22,23, such as those intended for 10× Visium, face limitations due to high computational costs19 and are generally restricted to single tissue sections with fewer spots15,21. These methods struggle with large-scale images, like whole-slide multiplexed images containing millions of cells, and are challenging to adapt for modern imaging platforms like Nanostring CosMx and 10× Xenium.
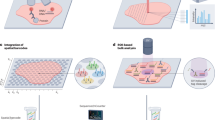
Highly interpretable and scalable machine learning methods are in great need for analyzing molecular tissue imaging data. In this work, we propose SpatialTopic, a Bayesian topic model designed to identify and interpret spatial tissue architecture across various multiplexed images by considering both the cell types and their spatial arrangement (Fig. 1A). We adapt an approach originally developed for image segmentation in computer vision24, incorporating spatial information into the flexible design of regions (image partitions, analogous to documents in language modeling). Unlike standard image pixels, the basic units of analysis in multiplexed tissue images are cells, which are not uniformly distributed due to the complexity of human tissue samples, posing a unique challenge. To address these challenges, we refined the original model used for image segmentation by using a nearest-neighbor kernel function to boost computational efficiency, as well as a unique initialization strategy for increasing robustness. In addition, we also provide an efficient C++ implementation of the spatial topic model in our R package SpaTopic.

A Overview of SpatialTopic. SpatialTopic identifies biologically relevant topics across multiple images, while each topic is a distribution of cell types, reflecting the spatial tissue architecture across images. B Image analysis pipeline designed for multiplexed immunofluorescence images. SpatialTopic is designed as a critical step for spatial analysis after cell phenotyping. C Graphic representation for SpatialTopic. D SpatialTopic groups cells in an unsupervised manner based on spatially co-occurrent cell types, similar to image segmentation based on spatially co-occurrent colors in the photo.
SpatialTopic offers a scalable solution for cell neighborhood and domain analysis for large-scale, multi-image datasets, efficiently handling millions of cells without the need to extract cell neighborhood information for each individual cell—a process that becomes computationally demanding and inefficient at scale. In contrast, SpatialTopic completes analysis of an image with 100,000 cells within 1 min on a laptop. Moreover, unlike the rigid clustering strategies of other methods, SpatialTopic identifies “topics”—tissue microenvironment features—through a probabilistic distribution over cell types and across diverse tissue images using a generative model. We demonstrate our method can accurately identify and quantify interpretable and biologically meaningful topics from imaging data without human intervention. We also present multiple case studies encompassing tissue images from mouse spleen, non-small cell lung cancer (NSCLC), healthy lung, and melanoma tissue samples. Finally, we highlight an example of a TLS-like topic and its correlation with outcomes from SpatialTopic analysis across different platforms, as well as a multi-stage example showing dynamic changes in spatial tissue architecture across varying disease stages. With computational efficiency and broad applicability, SpatialTopic provides a scalable framework that will enhance the analysis of large-scale tissue imaging studies.
Results
Overview of SpatialTopic, a Bayesian probabilistic model for highly scalable and interpretable spatial topic analysis across multiplexed tissue images
SpatialTopic is designed as a flexible spatial analysis module within the current imaging analysis workflow (Fig. 1B). Its main objective is to identify biologically meaningful topics across multiplexed images using unsupervised learning. Here, “topics” refer to latent spatial features defined by distinct cell type compositions within tissue microenvironment neighborhoods. SpatialTopic incorporates spatial data into an LDA model, assuming that each cell in an image arises from a mixture of spatially resolved topics, with each topic being a distribution over distinct cell types. Combining cell-type information with their spatial layout, this method enables the automated and simultaneous detection of immunological patterns across multiple images. Subsequent analyses can further link these topics with patient data, such as treatment response and survival.
We adopt a Bayesian approach for model inference, considering the uncertainties inherent in tissue spatial patterns. SpatialTopic requires cell types and their locations as input, with the cell types determined by the users’ preferred phenotyping algorithm tailored to the specific marker panel of the dataset. The algorithm generates two key statistics for further analysis: (1) topic content, a spatially resolved topic distribution over cell types, and (2) topic assignment for each cell within the images. After Gibbs sampling, the topic assignment of each cell is determined by the topic with the highest posterior probability. Cell types enriched in the same topic tend to be spatially correlated across images, leading to the identification of recurrent patterns of cell–cell interactions.
We developed an R package SpaTopic to efficiently implement the SpatialTopic algorithm as outlined in Fig. 1A, which details the primary steps of the algorithm (See the “Methods” section). Figure 1C displays a graphical representation of the spatial topic model. The key inputs for SpatialTopic are the cell type annotations \({\boldsymbol{\mathcal{C}}}\) and their locations \({\boldsymbol{\mathcal{X}}}\) across all images. Here, Zgi denotes the topic assignment, and Dgi indicates the region assignment of cell i in image g. Analogous to how computer vision algorithms segment images by spatially co-occurring pixel patterns with similar color, intensity or texture for object detection, SpatialTopic identifies topics as clusters of spatially co-occurring cell types (shown in Fig. 1D), potentially corresponding to biologically meaningful cellular structures (e.g., TLSs). The process involves the following steps:
-
Initialization: anchor cells are chosen as regional centers via spatially stratified sampling. For each image, a KNN graph is constructed between anchor cells and all other cells: for each cell, we retrieve its top m closest anchor cells. The initial region assignments of cells are made based on proximity to region centers.
-
Collapsed Gibbs Sampling: each cell undergoes two main steps per iteration:
-
Sample topic assignment Zgi conditional on its region assignment Dgi and cell type cgi, as well as the topic composition of the region Dgi and the cell type composition of the topic Zgi.
-
Sample region assignment Dgi conditional on current topic assignment Zgi, distance of the cell \({{\boldsymbol{x}}}_{gi}^{c}\) to the region center \({{\boldsymbol{x}}}_{{D}_{gi}}^{d}\), and the topic composition of the region Dgi. The spatial information is weakly incorporated with a kernel function.
-
-
After Gibbs sampling, the output includes the posterior probabilities of Zgi of each cell and the per-topic cell type distribution \(\{{\hat{{\boldsymbol{\beta }}}}_{k}\}\). Each cell in the image is assigned to a topic with the highest posterior probability \(P({Z}_{gi}| {\boldsymbol{\mathcal{C}}},{\boldsymbol{\mathcal{X}}})\).
We applied SpatialTopic to multiple datasets from diverse imaging platforms, including spatial proteomics data from Co-detection by Indexing (CODEX), Multiplexed ImmunoFluorescence (mIF), and Imaging Mass Cytometry (IMC) platforms, as well as spatial transcriptomics data from Nanostring CosMx platform (Supplementary Table 1). In the next few sections, we apply SpatialTopic to analyze tissue imaging data from a variety of spatial molecular profiling platforms and benchmark SpatialTopic with other popular algorithms for spatial domain/niche analysis, including Seurat v518, Spatial-LDA16, CytoCommunity20, UTAG15, and BankSY19 (Supplementary Table 2). The benchmark datasets contain between 0.1 and 1 million cells; making it challenging to apply methods with high computational costs. In contrast, SpatialTopic processes these large-scale images within just a few minutes.
SpatialTopic identifies global and local spatial features of human lung cancer tissue with higher precision and interpretability
We applied our method to a single NSCLC tissue image generated using a 960-plex CosMx RNA panel on the Nanostring CosMx Spatial Molecular Imager platform, which is publicly available on the Nanostring website25. We selected a Lung5-1 sample containing ~100,000 cells, with 38 cell types annotated using Azimuth26 based on the human lung reference v1.0 (Fig. 2A).

A We compare SpatialTopic, Seurat v5, Spatial-LDA, CytoCommunity, BankSY, and UTAG results on the human lung tumor tissue sample. We also visualize the distribution of the top 10 most abundant cell types and five unique mRNA molecules (KRT17, C1QA, IL7R, TAGLN, MS4A1), showing the tissue architecture. We only show up to a total of 20k molecules due to limitations in visualization. The two black rectangles (1 and 2) mark the zoom-in areas shown in (D, E), respectively. The red rectangle marks the zoom-in area shown in (F). B Dot plots showing gene marker expression across all 38 annotated cell types. KRT17, C1QA, IL7R, TAGLN, and MS4A1 are marker genes for tumor, macrophage, CD4 T, stroma, and B cells, respectively. C Heatmap shows per-topic cell type composition. Topic 2 represents tumor regions. The other topics represent distinct immune-enriched stroma regions, including topic 3, which captures the lymphoid structure in the lung tissue consisting of B cells and CD4 T cells, and topic 4, which is a macrophage-enriched stroma region. D, E SpatialTopic can better capture the local structures of the lung tumor tissue. F Topic 3 captures the lymphoid structures, consistent with the distribution of IL7R (CD4 T cell marker, red) and MS4A1 (B cell marker, blue). G We compare the consistency of different results, presenting the percentage of cells in the identified tumor domains expressing the KRT17 gene. SpatialTopic and UTAG generally show higher consistency compared to other methods. Source data are provided as a Source Data file.
To illustrate the general tissue architecture, Fig. 2A displays the distribution of the top 10 main cell types and the expression patterns of key genes, including KRT17, C1QA, IL7R, TAGLN, and MS4A1. These genes serve as markers for tumor cells, macrophages, CD4 T cells, stroma cells, and B cells, respectively (Fig. 2B). Our results demonstrate that SpatialTopic identified seven distinct topics from the complex image (Fig. 2A), with each topic representing a unique spatial niche characterized by a specific cell-type composition, as detailed in Fig. 2C. For example, Topic 2 is predominantly composed of tumor cells, indicating the tumor region in the image. Topic 4 represents a stromal region with a high proportion of macrophages, as well as dendritic cells. Topic 6 consists of a mixture of myofibroblast and smooth muscle cells spatially located tightly around the tumor, with dense cellular structure limiting the interactions between tumor and immune cells. Topics 1, 5, and 7 are fibroblast-concentrated stroma regions, each enriched with a different immune cell type: macrophages, dendritic cells, and plasma cells, respectively.
Notably, Topic 3 captures lymphoid aggregate structures in this lung cancer tissue sample, consisting of B cells, CD4 T cells, and smaller proportions of dendritic cells and CD8 T cells. This composition aligns with the current understanding of cell types in TLSs, recognized as a promising biomarker for cancer immunotherapy across various cancer types, including NSCLC8,9,10,11,27. TLSs are generally characterized as aggregates of B cells and other types of immune cells found in nonlymphoid tissue, and the presence of TLSs in tumor biopsy has shown to be highly correlated with a better prognosis and clinical outcome upon immunotherapy28. In a recent publication, we reported B-cell aggregates strongly associated with progression-free survival in patients with unresectable melanoma treated with immune checkpoint inhibitors14. Despite the importance of TLSs, challenges remain to reliably detect them to be used in clinical applications due to their complex cell type composition and variation in size and spatial location. SpatialTopic provides a flexible and efficient computational tool to address this need.
Cancer-associated fibroblasts (CAFs) are also an important component of tumor microenvironment. A recent publication by Liu et al.29 found four spatially distinct CAF subtypes, each exhibiting different transcriptomic profiles as a result of cellular interactions with their unique neighbors. On the Nanostring CosMx lung cancer dataset, the topics identified by SpatialTopic are highly correlated with the four distinct CAF spatial subtypes reported in the paper29 (Supplementary Fig. 1). Specifically, Topic 6 is associated with s1-CAFs adjacent to the tumor; Topics 5 and 7 correspond to s2-CAFs within the stromal niche; Topic 1 is linked to s3-CAFs in the myeloid niche; and Topic 3, primarily comprising B cells and CD4 T cells along with some myofibroblasts, corresponds to s4-CAFs in the TLS niche. In addition, SpatialTopic offers a more comprehensive analysis by encompassing all cell types, not just CAFs, and is scalable to larger datasets. This allows for further exploration, such as testing for differential gene expression among cell types, like CAFs, across various tumor microenvironments (topics).
Moreover, we benchmarked the results from SpatialTopic with Seurat v5, Spatial-LDA, CytoCommunity, BankSY, and UTAG. BankSY and UTAG directly use cell-level gene expression as input, whereas the other four methods, including SpatialTopic, rely on cell-type annotations (Fig. 2A, D, E). All methods can detect the global structure of the lung cancer tissue and classify tumor and stromal regions. However, BankSY and UTAG appear to miss the TLSs, likely because they did not take advantages of the cell-type annotations. Moreover, both methods assume homogeneous gene expression patterns in the neighborhoods, while TLSs are mixture of B cell and T cell subsets with highly mixed and variable transcriptomic profiles, thus making it difficult for methods that rely on homogeneous gene expression assumptions. Reference-based cell type annotation typically offers more detailed information and can be more robust for noisy data when matched with a single-cell reference30,31. SpatialTopic distinctly identified the lymphoid structure as Topic 3, comprising a mix of CD4 T cells and B cells (Fig. 2F). Additionally, when we focused on two local tumor tissue regions (Fig. 2D, E), SpatialTopic identified the tumor region with higher precision (Topic 2), more consistently matching the expression pattern of KRT17, a lung cancer marker gene. SpatialTopic and UTAG are the only two methods showing consistency (between tumor domain and KRT17 expression) higher than 0.8 across the entire image (Fig. 2G), which aligns with the visual measure in Fig. 2D, E. Additionally, we can evaluate the posterior probabilities of each cell’s assignment to various topics (Supplementary Fig. 2), as SpatialTopic employs a soft-clustering approach. Unlike hard clustering methods such as k-means, which categorically assign cells to clusters/domains, SpatialTopic quantifies uncertainty, providing a probabilistic measure of the statistical confidence for each cell’s topic assignment, and enriching the interpretation of the cell-topic relationships by revealing nuanced details that might otherwise be overlooked.
SpatialTopic identifies tertiary lymphoid structures from whole-slide melanoma tissue imaging
We applied SpatialTopic to a whole-slide melanoma tissue image obtained from our previous published mIF imaging datasets, with a 12-plex marker panel14. This analysis covered a whole-slide soft tissue image containing 0.4 million cells, annotated into seven major cell types (CD4 T cells, Tumor/Epithelial, B cells, CD8 T cells, Macrophages, Regulatory T (Treg) cells, and Others). The categorization was based on the expression of six lineage markers: PanCK/SOX10, CD3, CD8, CD20, CD68, and FoxP3. Cells were annotated as “Other” if they showed negative expression for all six markers.
Despite fewer identified cell types (due to fewer markers) compared to the Nanostring CosMx platform, SpatialTopic identified five distinct topics (Fig. 3A): Topic 1 (tumor), Topic 2 (CD4 immune zone), Topic 3 (stroma), Topic 4 (immune-enriched tumor-stroma boundary), and Topic 5 (TLSs). The tissue structures revealed by these topics visually correspond to the histological pattern seen in the co-registered Hematoxylin and Eosin (H&E) image (Fig. 3B) and the merged raw mIF images (Fig. 3C) with three key markers: CD3 (T cells), CD20 (B cells), and PanCK/SOX10 (tumor cells). Figure 3D demonstrates that topic 5 (TLSs) mainly consists of B cells, CD4 T cells, a few CD8 T cells, and Treg cells, consistent with the TLS-like pattern identified in the Nanostring dataset discussed earlier. Due to the lack of a dendritic cell marker in the mIF dataset, dendritic cells could not be identified and included in topic 5. This analysis demonstrates that SpatialTopic can consistently detect the same biologically relevant patterns across various tumor tissues and imaging platforms, which may be clinically significant, as TLS has been recognized as a promising biomarker for cancer immunotherapy28.

A Pseudocolor plot showing spatial topic assignments from SpatialTopic, identifying five topics across the whole-slide melanoma tissue sample: topic 1 (tumor region), topic 2 (CD4 T cell region), topic 3 (stroma), topic 4 (immune-enriched stroma-tumor boundary), and topic 5 (putative tertiary lymphoid structures). Three regions of interest (ROIs) are highlighted. This panel is a pseudocolor representation of the same tissue shown in (C). B Hematoxylin and Eosin (H&E)-stained images of the whole-slide tissue and three ROIs49. B corresponds to an adjacent section of the tissue shown in (A, C). C Merged multiplexed immunofluorescence images of the same tissue section as in (A), showing PanCK/SOX10 (red), CD3 (blue), and CD20 (green) across the full slide and selected ROIs. D Heatmap shows per-topic cell type composition for the five topics identified by SpatialTopic. Topic 5 (tertiary lymphoid structures) mainly consists of B cells and CD4 T cells, with a small proportion of CD8 T cells and regulatory T (Treg) cells. Source data are provided as a Source Data file.
SpatialTopic recovers spatial domain from cell type spatial organization in healthy lung tissue
We further demonstrate that SpatialTopic can effectively distill signals from noisy cell type annotations and identify clear tissue architecture based solely on the spatial arrangement of cells. To illustrate this, we applied SpatialTopic to the IMC dataset from the UTAG paper15, which includes 26 small regions of interest (ROIs) images from healthy lung tissue. For comparison, we used the UTAG result provided in the paper15 without rerunning UTAG.
Our analysis shows that SpatialTopic can recover tissue architectures directly from the spatial distribution of cell type annotations, yielding results consistent with manual annotations (Fig. 4A). SpatialTopic performs comparably to UTAG using only cell type annotations (Fig. 4B), as indicated by the adjusted Rand index, which shows similar performance levels. Additionally, Fig. 4C illustrates the topic content and the cell type composition for each topic identified by SpatialTopic. This demonstrates SpatialTopic’s capability to perform domain analysis without discarding existing cell-type annotations, offering valuable flexibility for datasets with cell-type annotations or for incorporating any existing cell-type annotation method. Unlike UTAG, which learns spatial tissue architecture directly from cell features due to noisy cell type annotations, we show that SpatialTopic can effectively identify tissue architecture from these annotations. Thus SpatialTopic is a robust alternative that leverages existing data without the need for additional cell-level features. We note that this dataset is challenging and we have to increase number of initialization to find a good start for SpatialTopic.

A Spatial distribution of cell type annotations, manual domain annotations, and spatial domains recovered by SpatialTopic and UTAG from the healthy lung tissue samples. B Comparing manual spatial domain to the cell type annotation, SpatialTopic, and UTAG with and without ad hoc relabel, using Adjusted Rand Index across 26 regions of interest (n = 26). The box plots show the median (center line), 25th and 75th percentiles (box bounds), and whiskers extending to the minimum and maximum values excluding outliers. UTAG results are obtained from the original publication. C Heatmap shows per-topic cell type composition for the four topics identified by SpatialTopic. Source data are provided as a Source Data file.
SpatialTopic identifies disease-specific topics and tracks topic evolution in mouse spleen over disease progression
We also applied SpatialTopic to a CODEX mouse spleen dataset2 to demonstrate its proficiency in identifying spatial topics across multiple images. This dataset includes nine images: three control normal BALBc spleens (BALBc 1–3) and six MRL/lpr spleens (MRL/lpr 4–9) at varying disease stages–early (MRL/lpr 4–6), intermediate (MRL/lpr 7–8), and late (MRL/lpr 9) of systemic autoimmune disease (Fig. 5A). Using a 30-plex protein marker panel, the study identified 27 major splenic-resident cell types across the nine tissue images. We use the cell type annotation provided in the original paper2.

A Six topics were identified by SpatialTopic across nine mouse spleen samples representing normal (BALBc 1-3) and different disease stages of lupus: early (MRL/lpr 4–6), intermediate (MRL/lpr 7–8), and late (MRL/lpr 9). B Heatmap shows per-topic cell type composition for the six main topics identified by SpatialTopic. Based on cell type compositions, the first three topics are labeled as red pulp, PALS (periarteriolar lymphoid sheath), and B-follicle in normal mouse spleen tissue, while the other three topics are unique to the disease stages. C Barplots shows the top 10 per-topic cell types for the six main topics identified by SpatialTopic. D Dynamic change in the topic proportion of the six topics during disease progression. Normal spleen samples are primarily characterized by topics 1 (dark blue), 2 (red), and 3 (light green), which reflect red pulp (mixed of B cells, erythroblasts, and F4/80(+) macrophages), PALS (mixed of CD8 T cells and CD4 T cells), and B-follicle (B cell dominated) respectively. There is an increase in Topic 6 (dark green) and depletion of Topic 1 in lupus-affected spleen (MRL/lpr samples), representing much fewer B cells and F4/80(+) macrophages but more granulocytes and erythroblasts in the red pulp regions. Topic 5 (mainly B220(+) DN T cells and CD4(+) T cells, pink) is enriched in lupus-affected spleen tissue at late disease stage. Topic 4 (light blue) is mainly in lupus-affected spleen (MRL/lpr samples) with high expression of CD106 in the stroma. E Dynamic change of key cell types within each topic, identified jointly by FREX(omega = 0.9) and lift metrics (See Supplementary Fig. 6). Source data are provided as a Source Data file.
SpatialTopic identified six topics from ~0.7 million cells across the nine images, highlighting the dramatic changes in spatial tissue structures associated with disease progression from normal spleen to spleen tissue at different disease stages (Fig. 5A). Figure 5B, C highlights per-topic cell type compositions aiding in labeling each topic. The normal spleen tissue samples predominantly comprised three topics: Topic 1 (red pulp), Topic 2 (periarteriolar lymphoid sheath, PALS), and Topic 3 (B-follicle). Supplementary Figs. 3 and 4 show the cell type distribution and domain annotations from the original paper, demonstrating SpatialTopic’s ability to capture the main structures consistent with these annotations, as compared to other methods (Supplementary Figs. 3 and 5). With an increasing number of topics, SpatialTopic also successfully delineated the marginal zone from the B-follicle (Supplementary Fig. 3).
One key advantage of SpatialTopic is its ability to identify topics jointly across all images, ensuring that topics are comparable across normal and diseased spleens. This allows us to identify condition-specific topics and quantify changes in topic proportions as the disease progresses. Diseased tissues often become disorganized, posing additional challenges to delineate spatial features compared to normal tissues. For instance, in contrast to normal spleens, the red pulp region in lupus-affected spleens (MRL/lpr) shows early signs of reorganization. These spleens exhibit an increase in granulocytes and erythroblasts, indicative of lupus-related splenic hematopoiesis and potentially leading to splenomegaly, in contrast to the B cells and F4/80(+) macrophages typically observed in normal spleens2,32,33. With SpatialTopic, this alteration is marked by the dominance of Topic 6 (Erythromyeloid niche) in lupus spleens, which supersedes Topic 1, the prevalent topic in normal spleen tissue. Other methods, including Spatial-LDA, UTAG, and BankSY, did not detect this change in the original red pulp region (Supplementary Fig. 5), partly due to overcorrection of batch effects. Topic 4 (CD106+ stroma niche), emerging in the lupus spleen, is characterized by a high abundance of CD106+ stroma cells, which attract immune cells to the inflamed areas2,34, and is enriched with CD4 and CD8 T cells. Notably, plasma cell, a disease-related B-cell subtype, is also uniquely enriched in Topic 4 and appears consistently across different disease stages. A plasma cell is a representative cell type for this topic, as identified by both lift and FREX metrics (Supplementary Fig. 6). A high abundance of plasma cells is often observed in lupus-affected tissue, such as the spleen. Therapeutic strategies aimed at eliminating plasma cells have demonstrated efficacy in patients with refractory systemic lupus erythematosus35. Topic 5 (Double Negative T cell niche), also unique to lupus spleens, features an enrichment of B220+ double negative (DN) T cells, as well as conventional CD4 T cells, and is more likely to be seen in the advanced stages of the disease, as compared to Topic 4 and 6. The expansion of DN T cells, is associated with disease progression, not only in MRL/lpr mice but also in patients with systemic lupus erythematosus36,37. These dynamic changes in the spleen tissue architecture indicate a significant reorganization of the immune landscape, reflecting immune dysregulation as systemic lupus erythematosus progresses2.
Furthermore, SpatialTopic identifies topics based on the spatial proximity of cell types, implying that cell types grouped within the same topic are likely co-localized and prone to interaction. Figure 5D illustrates the changes in topic proportions throughout the course of the disease progression. The distinct contributions of cell types to each topic are highlighted in Fig. 5E, mainly selected based on the lift and FREX metrics38,39 (Supplementary Fig. 6), as well as cell type composition. Cell types are clustered into topics that exhibit similar dynamics across different slides, which provides further insights into cell–cell interactions in both normal and diseased tissues.
SpatialTopic is highly scalable on large-scale modern images
To benchmark the scalability of SpatialTopic as the number of cells in images increases, we conducted tests using simulated datasets of varying scales. Figure 6A shows that SpatialTopic significantly outperforms Seurat v5 in terms of scalability with an increasing number of cells within a single image. Moreover, as demonstrated in Fig. 6B, SpatialTopic shows high efficiency on real large-scale imaging datasets, requiring less user time compared to other methods. For example, on the Nanostring CosMx NSCLC image with ~0.1 million cells, SpatialTopic runs within 1 min on a standard MacBook Air, a performance currently unbeatable by other methods.

A Runtime of SpatialTopic (region radius r = 60) and Seurat-v5 (K = 30) on simulated datasets with increasing cell numbers on a single image. n = 5 replicates. The Error bar represents the standard error of the mean. B Runtime of SpatialTopic, Seurat-v5, BankSY, UTAG, Spatial-LDA, and CytoCommunity on large-scale Nanostring NSCLC and CODEX mouse spleen datasets. All methods were benchmarked on a standard MacBook Air (M2, 2022) unless exceeding the memory limitation. Source data are provided as a Source Data file.
Across all datasets, SpatialTopic ranks in the highest tier with Seurat v5, while BankSY and UTAG fall into a second tier due to their reliance on similar but less optimized strategies. CytoCommunity, limited by its dependency on GPU support and memory demands, was run with reduced epochs and CPU-only for the NSCLC dataset, which compromised its performance and underscored its impracticality for labs without extensive computing resources on large-scale imaging analysis. Additionally, on images with cells more than 0.1 million, both UTAG and CytoCommunity required running on high-performance computing servers, due to their high memory demands. In contrast, SpatialTopic is highly scalable on large-scale imaging analysis, with all analysis done within minutes on a standard laptop.
Discussion
In summary, we introduced SpatialTopic, a spatial topic model designed to identify and quantify biologically relevant topics across multiple multiplexed tissue images. This unique computational approach leverages language modeling techniques to decipher the tissue microenvironment from tissue imaging data. SpatialTopic stands out as one of the few unsupervised learning methods capable of discerning clinically relevant spatial patterns15,19,21. Unlike other methods that rely on hard clustering strategies for analyzing samples, SpatialTopic is a probabilistic model-based approach using Bayesian inference to identify complex tissue architectures. The model generates two key outputs: The first of these, the topic content maps the cell type composition in spatial niches, allowing direct interpretation of the corresponding topic (e.g., TLSs); The second output, topic assignment for each single cell allows the quantification of each topic in individual tissue samples for subsequent association analysis with patient outcome. Application to multiple datasets along with benchmark analysis shows that SpatialTopic achieves higher precision in defining global and local spatial niches and higher sensitivity at capturing complex structures such as TLSs. Notably, our method is highly scalable to large-scale imaging data with efficient runtime, handling millions of cells on a standard laptop.
SpatialTopic is designed as a flexible spatial analysis module within the current imaging analysis workflow. A standard image analysis pipeline includes cell segmentation, data normalization/batch correction, cell phenotyping/clustering, and the analysis of cell type content and spatial relationships. Downstream statistical analysis typically starts with cell-level metadata derived from image analysis. Due to varied marker panels and molecular imaging platforms, a one-size-fits-all solution for cell phenotyping across diverse platforms seems unlikely. In practice, we find that reference-based cell annotation works best on single-cell imaging data rather than unsupervised clustering due to high-noisy data. SpatialTopic does not specify any upstream method and thus can be seamlessly integrated with other cell phenotyping modules tailored for datasets from different platforms. This design offers users adaptability, accommodating datasets from different panel designs.
In our proposed analysis pipeline for imaging data, we separate cell phenotyping from cell neighborhood/domain analysis for image-based spatial data, with SpatialTopic directly taking cell types as input. This key difference sets SpatialTopic apart from UTAG and BankSY, which use protein/gene expression as input for niche/domain analysis. UTAG performs dimension reduction before message passing, while BankSY engineers new spatial features for each cell before dimension reduction. We propose that treating cell phenotyping and neighborhood/domain analysis as distinct steps is a better analysis strategy for datasets generated by image-based technology with selected marker panels. Using cell type annotations as input for cell neighborhood analysis enhances the interpretability of different tissue microenvironments and undoubtedly increases the computational efficiency when analyzing large-scale images. The performance of SpatialTopic may rely on the accuracy of cell phenotyping. A better strategy for cell phenotyping is to annotate cells directly from cell images instead of using summary statistics, such as mean marker expression or gene count data. As part of the analysis pipeline, we are developing an image-based deep learning method for cell phenotyping, incorporating subcellular information, as well as domain knowledge40.
For multi-sample analysis, addressing the batch effect is a key challenge. Our proposed analysis pipeline seeks to mitigate the batch effect during cell phenotyping using a reference-based cell phenotyping method. For spatial transcriptomics data, a supervised classification method with a reliable single-cell reference can mitigate batch effects and inherent noises in the imaging data. The Batch effect is more critical for algorithms that directly consider gene expression data as input. When analyzing the mouse spleen dataset, we used Combat41 for batch correction across multiple images before applying UTAG and BankSY. However, Combat appears to over-correct for batch effects (Supplementary Fig. 5), thus failing to distinguish between normal and diseased red pulp tissue and ignoring key players in diseased tissues. This might stem from the substantial differences between normal and diseased tissues.
Modern datasets from platforms, such as 10× Xenium and Nanostring CosMx, require scalable computational methods to handle their size and complexity. Existing spatial domain analysis methods, originally designed for 10× Visium spatial transcriptomics data and optimized for datasets with thousands of cells or spots per slide, find it challenging to handle these more advanced datasets with millions of cells per image. SpatialTopic meets this need by efficiently managing neighborhood calculations and constructing the KNN graph only among m anchor cells instead of all n cells in the image. This reduces the time complexity of constructing KNN graphs from \(O(n\log n)\) to \(O(m\log m)\), and the time complexity of finding the closest anchor cell for each cell from \(O(\log n)\) to \(O(\log m)\), where m ≪ n. Furthermore, SpatialTopic maintains linear time complexity relative to the number of cells and iterations with collapsed Gibbs sampling and adapts an efficient approach for K nearest neighbor searching. These optimizations ensure SpatialTopic’s computational efficiency, making it accessible on standard laptops and practical for analyzing large-scale imaging data from platforms such as the 10× Xenium and Nanostring CosMx.
Moreover, advances in technology now enable the quantification of immune cell spatial diversity and the characterizing of tumor microenvironments in three-dimensional (3D) tissues42. While SpatialTopic can be adapted to infer immunological topics from 3D tissue, a refined strategy is needed to select anchor cells in the 3D spaces, as the spatial information obtained by SpatialTopic primarily relies on the relationships between anchor cells and other cells. In Supplementary Fig. 7, we demonstrate the applicability of SpatialTopic to a 3D spleen tissue image reconstructed from multiple tissue sections. To further improve the performance and applicability of SpatialTopic, several strategies could be pursued in the future. For instance, incorporating a hierarchical Dirichlet prior to topic distributions across regions would allow regions within the same image to share priors while allowing variability across different images. Furthermore, optimizing the initialization strategy is essential for applying SpatialTopic to population-scale datasets comprising hundreds or even thousands of images. These improvements would broaden the applicability and robustness of SpatialTopic.
Methods
SpatialTopic
Notations
We assume there are total V cell types that contribute to K different tissue microenvironments (topics) across G multiplexed images. Let cgi be the ith cell at the location \({{\boldsymbol{x}}}_{gi}^{c}=({x}_{gi1}^{c},{x}_{gi2}^{c}),g=1,2,\ldots,G,i=1,2,\ldots,{n}_{g}\), on the gth image with total ng cells. Let cgi = v if the cell has been classified to the vth cell type. Let \({\boldsymbol{\mathcal{C}}}={\{{c}_{gi}\}}_{i=1,2,\ldots,{n}_{g}}^{g=1,2,\ldots,G}\) and \({\boldsymbol{\mathcal{X}}}^{c}={\{{{\boldsymbol{x}}}^c_{gi}\}}_{i=1,2,\ldots,{n}_{g}}^{g=1,2,\ldots,G}\) denote all observed cell types and cell locations across all G images.
Model
In a conventional LDA model, each image is treated as an individual document, employing a bag-of-words approach without accounting for spatial information. This approach is similar to our prior work on longitudinal flow cytometry data analysis38. Here, in order to incorporate spatial information within images, we introduce a spatial topic model, SpatialTopic, integrating spatial data into the foundational LDA framework. This spatial topic framework was originally proposed for image segmentation24, instead of viewing each image as a singular document, we treat each image consisting of densely placed overlapping regions (documents). Unlike the conventional LDA model where relationships between documents and words are known and fixed, the word-document relationship here is unknown: each cell (word) is flexible to be assigned to all possible regions (documents). This flexible region (document) design allows us to identify spatial structure with irregular shapes.
For SpatialTopic, we introduce an additional hidden variable, Dgi, to denote cell region (document) assignment. Thus, each cell is associated with two hidden variables: the latent topic assignment Zgi ∈ {1, 2, …, K } and the latent region assignment Dgi ∈ {1, 2, …, M}, M = ∑gMg, where Mg denotes the number of regions on the image g. During the initialization, we pre-selected anchor cells as region centers. Let \({{\boldsymbol{\mathcal{X}}}}^{d}={\{{{\boldsymbol{x}}}_{d}^{d}\}}_{d=1,2,\cdots,M}\) be the set of all M region centers across all images. Let θd be the proportion of region d over K topics and βk be the proportion of topic k over V cell types. Hyperparameters ψ and α specify the nature of the Dirichlet priors of {βk} and {θd}, respectively.
Then we are ready to describe our generative model:
-
For each topic k, sample βk (topic weights over V cell types) from a Dirichlet prior βk ~ Dir(ψ).
-
For each image region d (centered at \({{\boldsymbol{x}}}_{d}^{d}\)), sample topic proportion θd ~ Dir(α)
-
For each cell, the ith cell in the image g:
-
Sample its region assignment Dgi from a uniform prior over possible documents (regions) in the image g.
-
Sample the location \({{\boldsymbol{x}}}_{gi}^{c}\) conditional on its region assignment Dgi with a kernel function based on the distance between the cell location \({{\boldsymbol{x}}}_{gi}^{c}\) and the region center \({{\boldsymbol{x}}}_{d}^{d}\).
$${{\boldsymbol{x}}}_{gi}^{c}| {D}_{gi}=d\propto K({{\boldsymbol{x}}}_{gi}^{c},{{\boldsymbol{x}}}_{d}^{d}).$$ -
Sample topic assignment Zgi∣Dgi = d ~ Multi(θd, 1).
-
Sample cell type cgi∣Zgi = k ~ Multi(βk, 1).
-
Hyperparameters α and ψ should be chosen based on the belief on {θd} and {βk} in a Bayesian perspective. In our application, both α and ψ are set very small by default (default: αk = 0.01, ∀k; ψv = 0.05, ∀v) to encourage the sparsity in region-topic distributions {θd} and topic-celltype distributions {βk}.
Nearest-neighbor exponential kernel
The flexible relationships between regions and cells in SpatialTopic allow each cell to be assigned to any one of its proximate regions. We employ a nearest-neighbor Gaussian kernel to capture the spatial correlation between cells and their respective regions, as previously used in the nearest-neighbor Gaussian process43. For computational efficiency, especially with large-scale images, we restrict our consideration to the top nearest-neighbor regions for each cell. Let \({\mathcal{N}}({{\boldsymbol{x}}}^c_{gi})\subset {{\boldsymbol{\mathcal{X}}}}^{d}\) be the collection of m closed region centers to the cell \({{\boldsymbol{x}}}_{gi}^{c}\) (default: m = 5). In practice, the commonly used squared exponential Gaussian kernel function decays too rapidly. This rapid decay often results in cells predominantly being linked to their closest region, irrespective of their cell types. Let σ be the length scale that controls how fast correlation decays with distance in the kernel function. Thus, drawing inspiration from ref. 44, instead of the squared exponential kernel, we used the following exponential kernel,
where \(| | {{\boldsymbol{x}}}_{gi}^{c}-{{\boldsymbol{x}}}_{d}^{d}| {| }_{2}\) represents the Euclidean distance between the cell location \({{\boldsymbol{x}}}_{gi}^{c}\) and the region center \({{\boldsymbol{x}}}_{d}^{d}\). We fix σ for computational efficiency, but it may be sampled during the Gibbs sampling. Increasing σ would reduce the strength of the spatial correlation, resulting in a diminished spatial effect when assigning cells to regions.
Collapsed Gibbs Sampling
We use collapsed Gibbs sampling for model inference. The collapsed Gibbs sampling algorithm was originally introduced as the Bayesian approach of LDA45. This method’s comprehensive derivation and implementation can be found in the paper46. Similar to ref. 24, we further adapted and extended the algorithm for our proposed spatial topic model. It’s noteworthy that during the collapsed Gibbs sampling process, the parameters βk and θd are integrated out and are not explicitly sampled. Instead, our focus is on the two hidden variables associated with each cell: the topic assignment Zgi and the region (or document) assignment Dgi. These variables undergo iterative sampling using the collapsed Gibbs sampler:
-
1.
Sample topic assignment Zgi conditional on region assignment Dgi with45
$$P({Z}_{gi}= k\;|\; {D}_{gi}=d,{c}_{gi}=v,{{\boldsymbol{\mathcal{D}}}}_{-gi},{{\boldsymbol{\mathcal{Z}}}}_{-gi},{{\boldsymbol{\mathcal{C}}}}_{-gi},{\boldsymbol{\psi }},{\boldsymbol{\alpha }})\\ \propto \frac{{n}_{k,-gi}^{(v)}+{\psi }_{v}}{\mathop{\sum }\nolimits_{t=1}^{V}{n}_{k,-gi}^{(t)}+{\psi }_{t}}\frac{{n}_{d,-gi}^{(k)}+{\alpha }_{k}}{\mathop{\sum }\nolimits_{{k}^{{\prime} }=1}^{K}{n}_{d,-gi}^{({k}^{{\prime} })}+{\alpha }_{{k}^{{\prime} }}}$$(2)where \({n}_{k,-gi}^{(v)}\) refers the number of times that cell type v has been observed with topic k and \({n}_{d,-gi}^{(k)}\) refers the number of times that topic k has been observed in region d, both excluding the current cell gi, the ith cell on the gth image. The first ratio expresses the probability of cell type v under topic k, and the second ratio expresses the probability of topic k in region d. \({{\boldsymbol{\mathcal{D}}}}_{-gi}\), \({{\boldsymbol{\mathcal{Z}}}}_{-gi}\), and \({{\boldsymbol{\mathcal{C}}}}_{-gi}\) denote collections of \({\boldsymbol{\mathcal{D}}}\), \({\boldsymbol{\mathcal{Z}}}\), and \({\boldsymbol{\mathcal{C}}}\) excluding cell gi.
-
2.
Sample Dgi conditional on Zgi with
$$P({D}_{gi}=\, d\;|\; {Z}_{gi}=k,{{\boldsymbol{\mathcal{D}}}}_{-gi},{{\boldsymbol{\mathcal{Z}}}}_{-gi},{{\boldsymbol{x}}}_{gi}^{c},{{\boldsymbol{x}}}_{d}^{d},{\boldsymbol{\alpha }},\sigma )\\ \propto \, P({Z}_{gi}=k\;|\; {{\boldsymbol{\mathcal{Z}}}}_{-gi},{D}_{gi}=d,{{\boldsymbol{\mathcal{D}}}}_{-gi},{\boldsymbol{\alpha }})P({{\boldsymbol{x}}}_{gi}^{c}\;|\; {D}_{gi}=d,{{\boldsymbol{x}}}_{d}^{d},\sigma )P({D}_{gi}=d)$$According to ref. 46, \(P({Z}_{gi}=k\;|\; {{\boldsymbol{\mathcal{Z}}}}_{-gi},{D}_{gi}=d,{{\boldsymbol{\mathcal{D}}}}_{-gi},{\boldsymbol{\alpha }})\) can be obtained by integrating out θd, that
$$P({Z}_{gi}=k\;|\; {{\boldsymbol{\mathcal{Z}}}}_{-gi},{D}_{gi}=d,{{\boldsymbol{\mathcal{D}}}}_{-gi},{\boldsymbol{\alpha }})=\frac{{n}_{d,-gi}^{(k)}+{\alpha }_{k}}{\mathop{\sum }\nolimits_{{k}^{{\prime} }=1}^{K}{n}_{d,-gi}^{({k}^{{\prime} })}+{\alpha }_{{k}^{{\prime} }}}.$$We can further omit P(Dgi = d) due to uniform prior. Thus Dgi can be sampled based on the following conditional distribution:
$$P({D}_{gi}=\, d\;|\; {Z}_{gi}=k,{{\boldsymbol{\mathcal{D}}}}_{-gi},{{\boldsymbol{\mathcal{Z}}}}_{-gi},{{\boldsymbol{x}}}_{gi}^{c},{{\boldsymbol{x}}}_{d}^{d},{\boldsymbol{\alpha }},\sigma )\\ \propto \, K({{\boldsymbol{x}}}_{gi}^{c},{{\boldsymbol{x}}}_{d}^{d})\,\frac{{n}_{d,-gi}^{(k)}+{\alpha }_{k}}{\mathop{\sum }\nolimits_{{k}^{{\prime} }=1}^{K}{n}_{d,-gi}^{({k}^{{\prime} })}+{\alpha }_{{k}^{{\prime} }}}$$(3)
Initialization
During initialization, we employ a spatially stratified sampling approach to randomly select anchor cells from each image, which serve as region centers. The number of anchor cells per image is determined by a predefined region radius r (default: r = 400) and the image size. The choice of r should take into account both image resolution and tissue complexity. To ensure an accurate estimation of the topic distribution θd, each region should contain a sufficient number of cells. In practice, for whole-slide imaging, we expect at least 100 cells per region on average, which guides our selection of region radius r for each dataset.
Since different imaging platforms may report spatial coordinates in either pixels or microns, users are advised to adjust parameters accordingly. In addition, the length scale σ, another critical parameter controlling the strength of the spatial effect, should be tuned in conjunction with the region radius r. Empirically, we have found that setting \(\sigma \approx \sqrt{r}\) often works well. However, the optimal value may vary depending on the structure complexity of the imaging data. A smaller σ benefits the identification of local structure, while a larger σ supports global structure. We recommend users to turn the two parameters on a small subset of images before applying to the whole datasets. For each image, we construct an m-nearest-neighbor graph linking all cells to the selected anchor cells. Specifically, for each cell, its m-closest anchor cells are identified. For computational efficiency, distances between each cell and its top m-nearest anchor cells are pre-computed before Gibbs sampling.
The performance of SpatialTopic may be sensitive to the initialization of anchor cells, especially on images with highly complex spatial organization. To address this, we take a warm start strategy instead of starting Gibbs sampling from a single random initialization. This involves running multiple short Gibbs sampling chains during initialization (default: ninit = 10), each with a unique set of randomly-selected anchor cells. After a few iterations (default: niter_init = 100), only the one with the highest log-likelihood is retained and continued.
Implementation
We implemented SpatialTopic in Rcpp and made it an R package SpaTopic (officially available on CRAN after Jan 17, 2024). The complete algorithm is shown in Box 1. For the Gibbs sampling, we have set the default parameters as follows: iter = 200, burnin = 1000, thin = 20 (200 Gibbs sampling draws are made with the first 1000 iterations discarded and then every 20th iteration kept). We can infer topic distributions across all images using the posterior samples drawn from the Gibbs sampling. For each of these posterior samples, both parameters {βk} and {θd} can be estimated as follows:
Moreover, we also keep the posterior distribution of Zgi from all posterior samples for each individual cell. Notably, Dgi has been marginalized during this process and each cell in the end is assigned to the topic with the highest posterior probability. Thus we are also able to visualize the spatial distribution of cell topics in the images.
Model selection
The likelihood of the topic model is intractable to compute in general, but we can approximate the model log-likelihood in terms of model parameters {βk} and {θd}47. With the law of total probabilities, we take into account uncertainties both in cells’ region and topic assignment, then the log-likelihood of the spatial topic model can be presented as
where \({\eta }_{gi}^{d}=P({{\boldsymbol{x}}}_{gi}^{c}\;|\; {D}_{gi}=d,{{\boldsymbol{x}}}_{d}^{d})P({D}_{gi}=d)\propto K({{\boldsymbol{x}}}_{gi}^{c},{{\boldsymbol{x}}}_{d}^{d})\).
We use the Deviance Information Criterion (DIC)48 to select the number of topics, a generalization of the Akaike Information Criterion (AIC) in Bayesian model selection:
where the Deviance is defined as \(D({\boldsymbol{\mathcal{C}}},{\boldsymbol{\mathcal{X}}})=-2ll({\boldsymbol{\mathcal{C}}},{\boldsymbol{\mathcal{X}}})\) and \({p}_{D}=\frac{1}{2}\overline{Var(D({\boldsymbol{\mathcal{C}}},{\boldsymbol{\mathcal{X}}}))}\).
DIC requires calculating the log-likelihood for every posterior sample, which is time-consuming. To determine the optimum number of topics, we run SpatialTopic with a varied number of topics (2–9 in practice) and collect a few posterior samples (such as the first 20 posterior samples) after convergence (with trace=TRUE). The number of topics was selected based on DIC with (7). Otherwise, we only output the deviance and the log-likelihood of the final posterior sample (default: trace=FALSE). In Supplementary Fig. 8, we show the convergence of the Gibbs sampling algorithm on the Nanostring NSCLC dataset. The number of topics was selected as seven based on DIC.
Comparing to other methods
We compared the performance of SpatialTopic with other five niche analysis methods: spatial-LDA, Seurat-v5, UTAG, CytoCommunity, and BankSY. BankSY and UTAG used protein or gene expression data and cell spatial coordinates as inputs, while the other methods used existing cell-type annotations and cell spatial coordinates. We followed the pre-processing procedures and parameters described in the original papers and tutorials for each method, with some hyperparameters slightly adjusted for computational efficiency on large datasets or when clear guidelines for tuning parameters were available. Details of these adjustments and the rationale for not using the default settings are described in this section.
All methods were initially run using R Studio (for R-based methods) or Jupyter Lab (for Python-based methods) on a standard MacBook Air (M2, 2022). If a method could not be run on a standard Mac due to memory constraints, we used our high-performance computing server with a single-core CPU and 200 GB of assigned memory. For the Nanostring CosMx NSCLC dataset, both CytoCommunity and UTAG were run on the server due to high memory usage. Additionally, for the CODEX mouse spleen dataset, UTAG can be run on the Mac only without the default parallel mode due to memory constraints.
SpatialTopic (SpaTopic R package v1.1.0)
We ran SpatialTopic with region_radius = 400, 150, 300 for the Nanostring CosMx NSCLC, the CODEX mouse spleen, and the mIF melanoma datasets, respectively, allowing around 100 cells per region on average during initialization, which is necessary for accurately estimating the topic-region distribution. We chose length-scale sigma = 20 for the mouse spleen dataset while using the default parameters for the NSCLC and the melanoma dataset. Posterior samples were collected after the convergence of the Gibbs sampling chain, with a burn-in period of 2000 iterations for the NSCLC and the melanoma dataset, and 1500 iterations for the mouse spleen dataset. For the healthy lung dataset with 26 small ROIs, SpatialTopic was run with sigma = 5 and region_radius = 60 to identify the complex local structures. Only on this dataset, we increased the number of initializations to 200 times to increase the robustness of identifying consensus patterns across ROIs while increasing the running time.
Seurat-v5 (v5.0.2)
We used the default niche analysis in Seurat v5, specifically the BuildNicheAssay() function in the Seurat R package. Seurat v5 employs k-means clustering to group cell neighborhood features, which are derived from the shared-nearest-neighbor graph (default neighbors.k = 30), a variant of the KNN graph, as part of its image-based spatial data analysis pipeline. We ran BuildNicheAssay() with all default parameters except for the Nanostring CosMx NSCLC datasets, for which we set neighbors.k = 100. Because we found that increasing neighbors.k from 10 to 100 (testing neighbors.k = 10, 30, 50, 100) significantly improved the algorithm’s performance on this dataset, with results presented in Supplementary Fig. 9.
Spatial-LDA (v0.1.3)
For the CODEX mouse spleen datasets, we used the same parameters as the authors used in the original methodology paper, though we now use neighborhoods of all cells as the input, not only B cells. For the Nanostring CosMx NSCLC datasets, we also use neighborhoods of all cells as the input but set radius = 400 to extract neighborhood cell type compositions, consistent with region_radius r = 400 in SpatialTopic on this dataset. To reduce the computational complexity for both datasets, we set the threshold = 0.01 for ADMM Primal-Dual optimizer. Finally, we output the topic weights for every cell and assign every cell to a topic with the maximal weight.
CytoCommunity (v1.1.0)
CytoCommunity (unsupervised mode) was run on a CPU with 200 GB of assigned memory and evaluated only on the Nanostring CosMx NSCLC dataset due to its demand for large memory and its unsupervised mode’s inability to learn Tissue Cell Neighborhoods across multiple images. We set KNN-K = 300 for 0.1M cells, as suggested in the original paper. For large-scale image data, the second step of CytoCommunity is time-consuming when trained on a CPU. Therefore, we greatly reduced num_RUN to 10 and Num_Epoch to 100 per run while ensuring the final loss was less than −0.2 for each run. Other parameters were set to the default.
UTAG (v0.1.1)
UTAG was primarily developed for protein expression data with limited marker channels. For the Nanostring CosMx NSCLC datasets with 960 genes, we used typical pre-processing steps suggested by Scanpy (v1.9.8) for analyzing scRNA-seq datasets. These steps included filtering low-prevalence genes, log transformation, and retaining only highly variable genes. We then performed z-score normalization, truncated at 10 standard deviations, followed by PCA. Only the top 50 principal components were used as input for UTAG. UTAG was run under multiple clustering resolutions [0.05, 0.1, 0.3, 0.5] and mix_dist = 60, with an image resolution of 0.18 microns per pixel, since the authors suggested setting mix_dist between 10 and 20 µm in the user manual. For the CODEX mouse spleen dataset (with intensity values already transformed), we performed z-score normalization truncated at 10 standard deviations, followed by Combat batch correction41 and a second z-score normalization truncated at 10 standard deviations, a similar procedure as introduced in the UTAG paper for preprocessing IMC data15. We also set mix_dist = 60, with an imaging resolution of 0.188 microns per pixel.
BankSY (v0.99.9)
In contrast to UTAG, BankSY is specifically designed to analyze spatial transcriptomics datasets. We ran BankSY with lambda = 0.8 to identify spatial domains, as recommended, with other parameters set to default, as described in the Github tutorial. For the Nanostring CosMx NSCLC dataset, we followed the same pre-processing procedures outlined in the domain analysis tutorial, using k_geom = 30, npcs = 50, and clustering resolutions of 0.1, 0.2, 0.3, and 0.5. For the CODEX mouse spleen datasets, we used the same input as UTAG, after batch correction and normalization. We followed the tutorial for multi-sample analysis, running the results under npcs = 30 since the dataset has only 30 markers.
Data preprocessing
Nanostring CosMx human NSCLC
The Nanostring CosMx NSCLC dataset is available on the Nanostring Website (https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/nsclc-ffpe-dataset/). For our analysis, we selected Lung5-1 sample and annotated about 0.1M cells into 38 cell types using Azimuth26 with a human lung reference v1.0 (https://azimuth.hubmapconsortium.org/references/). We used the same cell type annotations from the Seurat image analysis pipeline tutorial (https://satijalab.org/seurat/articles/seurat5_spatial_vignette_2.html). Since healthy lung tissue was used as the reference, the “basal” cells were re-labeled as tumor cells since they are the most closed cell type. We checked that the tumor locations indicated by the reference-based cell annotations are consistent with the tumor region labeled by the Nanostring company.
CODEX mouse spleen
We used the cell type annotation, marker expression level, and imaging coordinates from the original paper2. The image dataset can be downloaded from https://data.mendeley.com/datasets/zjnpwh8m5b/1. For cell coordinates, we only use the X and Y axes of the samples, ignoring Z axis. However, the result is similar when considering all three dimensions.
IMC healthy lung
We used the cell type annotation, marker expression level, cell imaging coordinates, and cell UTAG domain labels in the original paper15. This image dataset can be downloaded from https://zenodo.org/records/6376767.
mIF melanoma
The 12-plex whole-slide mIF image on Melanoma tissue sample is one of the imaging datasets that have been published from our group14. The dataset contains following 12 markers: CD8, PD-1, PD-L1, CD68, CD3, CD20, FoxP3, pancytokeratin+SOX10 (panCK/SOX10), TCF1/7, TOX, Ki67, LAG-3. The processed dataset can be downloaded from Mendeley data with link https://data.mendeley.com/datasets/syfmgsv3d9/1. Here, we only used cell phenotypes (classified based on marker expression of CD8, panCK/SOX10, CD68, CD3, CD20, FoxP3) and cell locations as the input of SpatialTopic.
Simulation
We tested methods on simulated datasets of different scales to benchmark the scalability of SpatialTopic with an increasing number of cells in images. We randomly sampled 10 k, 40 k, 90 k, 160 k, and 250 k pixels from an image, similar to the simulation method described in ref. 23, to represent cell locations. We did not simulate gene expression levels for every individual cell. Instead, for each domain, we randomly sampled cells with domain-specific cell type distributions, with parameters simulated from Dirichlet(1, 1, 1, 1, 1), anticipating five distinct cell types per domain (a simulated example is shown in Supplementary Fig. 10A). Five unique datasets were generated for each simulation scenario. We also scaled the X and Y axes to maintain consistent cell densities across all simulation scenarios.
In addition, we also assessed other aspects of SpatialTopic, including the recovery of the number of topics and topic distributions in our simulation studies. In Supplementary Fig. 10, we evaluated SpatialTopic across 20 simulated datasets (each with 10 k cells) and showed that SpatialTopic not only achieves high clustering accuracy (evaluated using Adjusted Rand Index) but also effectively recovers the topic distributions (evaluated by sum of residual squared error between the true topic content matrix {βk} and the estimated \(\{{\hat{{\boldsymbol{\beta }}}}_{k}\}\)), which highlights our model’s capability to recover the underlying topic structure.
Statistics and reproducibility
All statistical calculations were implemented in R (v4.3.3). No statistical method was used to predetermine sample size. The experiments were not randomized. This study does not involve group allocation that requires blinding.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All datasets we used in the study are publicly available and can be downloaded online, with analysis details described in the Method section. The Nanostring CosMx NSCLC dataset is available on the Nanostring Website (https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/nsclc-ffpe-dataset/). The CODEX Mouse spleen dataset can be downloaded from Mendeley Data with link https://data.mendeley.com/datasets/zjnpwh8m5b/1. The IMC Healthy Lung dataset can be downloaded from Zenodo with link https://zenodo.org/records/6376767. The mIF Melanoma image dataset14 present in the manuscript can be downloaded from Mendeley Data with link https://data.mendeley.com/datasets/syfmgsv3d9/1. Source data for each individual figure are provided with this paper. Source data are provided with this paper.
Code availability
The R package is available on Github (https://github.com/xiyupeng/SpaTopic/) with a tutorial (https://xiyupeng.github.io/SpaTopic/). The R package is also available on CRAN (https://cloud.r-project.org/package=SpaTopic) with https://doi.org/10.32614/CRAN.package.SpaTopic. The first version of the R package was officially released on CRAN on Jan 17, 2024. Analysis codes are available on Github (https://github.com/xiyupeng/SpatialTopic_Analysis_codes) with https://doi.org/10.5281/zenodo.15588176.
References
Keren, L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387.e19 (2018).
Goltsev, Y. et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15 (2018).
Ko, J. et al. Spatiotemporal multiplexed immunofluorescence imaging of living cells and tissues with bioorthogonal cycling of fluorescent probes. Nat. Biotechnol. 40, 1654–1662 (2022).
Hoch, T. et al. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci. Immunol. 7, eabk1692 (2022).
Moldoveanu, D. et al. Spatially mapping the immune landscape of melanoma using imaging mass cytometry. Sci. Immunol. 7, eabi5072 (2022).
Nirmal, A. J. et al. The spatial landscape of progression and immunoediting in primary melanoma at single-cell resolution. Cancer Discov. 12, 1518–1541 (2022).
McCaffrey, E. F. et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat. Immunol. 23, 318–329 (2022).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Vanhersecke, L. et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2, 794–802 (2021).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Fridman, W. H. et al. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 19, 441–457 (2022).
Feng, Y. et al. Spatial analysis with SPIAT and spaSim to characterize and simulate tissue microenvironments. Nat. Commun. 14, 2697 (2023).
Vanguri, R. S. et al. Integration of peripheral blood- and tissue-based biomarkers of response to immune checkpoint blockade in urothelial carcinoma. J. Pathol. 261, 349–360 (2023).
Smithy, J. W. et al. Quantitatively defined stromal B cell aggregates are associated with response to checkpoint inhibitors in unresectable melanoma. Cell Rep. 44, 115554 (2025).
Kim, J. et al. Unsupervised discovery of tissue architecture in multiplexed imaging. Nat. Methods 19, 1653–1661 (2022).
Chen, Z., Soifer, I., Hilton, H., Keren, L. & Jojic, V. Modeling multiplexed images with spatial-LDA reveals novel tissue microenvironments. J. Comput. Biol. 27, 1204–1218 (2020).
Patrick, E. et al. Spatial analysis for highly multiplexed imaging data to identify tissue microenvironments. Cytometry A 103, 593–599 (2023).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Singhal, V. et al. BANKSY unifies cell typing and tissue domain segmentation for scalable spatial omics data analysis. Nat. Genet. 56, 431–441 (2024).
Hu, Y. et al. Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes. Nat. Methods 21, 267–278 (2024).
Li, Z. & Zhou, X. BASS: multi-scale and multi-sample analysis enables accurate cell type clustering and spatial domain detection in spatial transcriptomic studies. Genome Biol. 23, 1–35 (2022).
Chidester, B., Zhou, T., Alam, S. & Ma, J. SpiceMix enables integrative single-cell spatial modeling of cell identity. Nat. Genet. 55, 78–88 (2023).
Shang, L. & Zhou, X. Spatially aware dimension reduction for spatial transcriptomics. Nat. Commun. 13, 1–22 (2022).
Wang, X. & Grimson, E. Spatial latent Dirichlet allocation. In Proc. Advances in Neural Information Processing Systems 20 (NIPS, 2007).
He, S. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 40, 1794–1806 (2022).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Rakaee, M. et al. Tertiary lymphoid structure score: a promising approach to refine the TNM staging in resected non-small cell lung cancer. Br. J. Cancer 124, 1680–1689 (2021).
Schumacher, T. N. & Thommen, D. S. Tertiary lymphoid structures in cancer. Science 375, eabf9419 (2022).
Liu, Y. et al. Conserved spatial subtypes and cellular neighborhoods of cancer-associated fibroblasts revealed by single-cell spatial multi-omics. Cancer Cell. 43, 905–924 (2025).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Meng, G. et al. imply: improving cell-type deconvolution accuracy using personalized reference profiles. Genome Med. 16, 65 (2024).
Golub, R., Tan, J., Watanabe, T. & Brendolan, A. Origin and immunological functions of spleen stromal cells. Trends Immunol. 39, 503–514 (2018).
Zervopoulou, E. et al. Enhanced medullary and extramedullary granulopoiesis sustain the inflammatory response in lupus nephritis. Lupus Sci. Med. 11, e001110 (2024).
El-Jawhari, J. J., El-Sherbiny, Y., McGonagle, D. & Jones, E. Multipotent mesenchymal stromal cells in rheumatoid arthritis and systemic lupus erythematosus; from a leading role in pathogenesis to potential therapeutic saviors? Front. Immunol. 12, 643170 (2021).
Schett, G., Mackensen, A. & Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 402, 2034–2044 (2023).
Li, H. et al. Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat. Commun. 11, 2859 (2020).
Crispín, J. C. et al. Expanded double negative t cells in patients with systemic lupus erythematosus produce il-17 and infiltrate the kidneys. J. Immunol. 181, 8761–8766 (2008).
Peng, X. et al. A topic modeling approach reveals the dynamic T cell composition of peripheral blood during cancer immunotherapy. Cell Rep. Methods 3, 100546 (2023).
Roberts, M. E., Stewart, B. M. & Tingley, D. stm: An R package for structural topic models. J. Stat. Softw. 91, 1–40 (2019).
Yosofvand, M. et al. Spatial immunophenotyping from whole-slide multiplexed tissue imaging using convolutional neural networks. Preprint at bioRxiv https://doi.org/10.1101/2024.08.16.608247 (2024).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2006).
Kuett, L. et al. Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat. Cancer 3, 122–133 (2022).
Datta, A., Banerjee, S., Finley, A. O. & Gelfand, A. E. Hierarchical nearest-neighbor Gaussian process models for large geostatistical datasets. J. Am. Stat. Assoc. 111, 800–812 (2016).
Weber, L. M., Saha, A., Datta, A., Hansen, K. D. & Hicks, S. C. nnSVG for the scalable identification of spatially variable genes using nearest-neighbor Gaussian processes. Nat. Commun. 14, 4059 (2023).
Griffiths, T. L. & Steyvers, M. Finding scientific topics. Proc. Natl. Acad. Sci. USA 101, 5228–5235 (2004).
Heinrich, Gregor. Parameter estimation for text analysis. Darmstadt, Germany: Technical report, 2005.
Newman, D., Asuncion, A., Smyth, P. & Welling, M. Distributed algorithms for topic models. J. Mach. Learn. Res. 10, 1801–1828 (2009).
Gelman, A., Carlin, J. B., Stern, H. S. & Rubin, D. B. Bayesian Data Analysis 2nd edn. in Texts in Statistical Science (CRC Press, 2004).
Peng, X. et al. 1303 Spatial topic modeling of tumor microenvironment with multiplexed imaging. J. Immunother. Cancer 11, A1448–A1448 (2023).
Acknowledgements
We would like to thank the computational support from MSK-MIND. This work is supported in part by the MSKCC Society and NIH/NCI Cancer Center Support Grant NIH P30 CA008748 for MSKCC (to X.P., J.W.S., C.E.K., F.E., J.L., M.B., M.Y., M.A.P., M.K.C., R.S., and K.S.P.), the V Foundation (to M.K.C.), the Parker Institute for Cancer Immunotherapy (to X.P.), NIH/NCI grant R01 CA276286 (to R.S., K.S.P., M.K.C., and J.L.), and the MSK-MIND consortium (to R.S. and M.K.C.).
Author information
Authors and Affiliations
Contributions
X.P. contributed to the original draft, developed the statistical model, and wrote the software. X.P., J.W.S., R.S., and K.S.P. developed the initial study concept. X.P., R.S., K.S.P., and J.L. developed the algorithm. X.P., C.E.K. contributed to the R package. X.P., J.W.S., C.E.K., F.E., M.Y., and M.B. analyzed the data. R.S., K.S.P., M.A.P., and M.K.C. oversaw all data generation and analysis. X.P., J.W.S., F.E., J.L., M.B., R.S., and K.S.P. edited the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.W.S.: Research funding—IO Biotech (Inst), Regeneron (Inst), Daiichi Sankyo (Inst); Consulting or advisory role—IO Biotech, Iovance, Bristol Myers Squibb, Daiichi Sankyo; Travel—Immatics; M.A.P.: Consulting or Advisory Role—Bristol-Myers Squibb; Cancer Expert Now; Chugai Pharma; Eisai; Erasca, Inc; Intellisphere; Merck; MJH Associates; Nektar; Novartis; Pfizer; WebMD; Research Funding—Array BioPharma (Inst); Bristol-Myers Squibb (Inst); Infinity Pharmaceuticals (Inst); Merck (Inst); Novartis (Inst); Rgenix (Inst); K.S.P. Stock ownership in 23 and Me, Vincerx, Eyepoint, and Kyverna; C.E.K.: Stock ownership in Johnson & Johnson; M.K.C.: BMS—Research support (Inst), advisory role/consulting; Medimmune—advisory role/consulting; Immunocore—advisory role/consulting; Merus—family member employee; M.B.: Regeneron—collaborator; Sanofi—direct contractor; X.P., J.L., M.Y., R.S., and F.D.E.: No disclosures;
Peer review
Peer review information
Nature Communications thanks Ellis Patrick, who co-reviewed with Elijah Willie, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peng, X., Smithy, J.W., Yosofvand, M. et al. Scalable topic modelling decodes spatial tissue architecture for large-scale multiplexed imaging analysis. Nat Commun 16, 6619 (2025). https://doi.org/10.1038/s41467-025-61821-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61821-y
