
Abstract
ZIKV infection is associated with testicular damage and abnormal spermatogenesis. However, the molecular mechanisms underlying these pathogenic processes remain unclear. Here, we demonstrate that ZIKV disrupts Leydig cells’ ability to produce testosterone, leading to decreased sperm counts and motility. Specifically, the non-structural protein NS2A of ZIKV downregulates testosterone production by directly binding to mRNA of CYP17A1, a key enzyme in testosterone synthesis, thereby inhibiting its translation. Notably, the sole membrane-traversing segment and its flanking loops of NS2A are crucial for this interaction with CYP17A1 mRNA. Scanning mutagenesis studies within this sequence identified amino acid residues critical for NS2A binding and the suppression of CYP17A1 mRNA translation. Testicular inoculation of adeno-associated virus (AAV) delivering ZIKV-NS2A or its mutant showed that ZIKV-NS2A alone is sufficient to affect steroidogenesis and spermatogenesis in vivo. Moreover, a mutant virus generated by reverse genetics, containing a single amino acid mutation that abolishes NS2A’s binding to CYP17A1 mRNA, exhibited significantly lower inhibition of steroidogenesis and spermatogenesis compared to the wild-type virus in mouse models. These findings enhance our understanding of how ZIKV impacts male reproductive health and provide crucial insights for future preventive and therapeutic strategies.
Similar content being viewed by others
Introduction
Zika virus (ZIKV) is a mosquito-borne flavivirus that can cause severe neurological symptoms, including microcephaly in newborns and Guillain-Barre syndrome in adults1,2. In addition, ZIKV infection adversely affects male reproductive health, observed both in ZIKV-infected patients and mouse models3,4,5. The male reproductive system comprises the testes, epididymis, prostate gland, and seminal vesicles. The testes, primarily responsible for spermatogenesis and steroidogenesis, have been identified as a direct target of ZIKV. Indeed, some male patients with ZIKV exhibited symptoms of the reproductive system, such as oligospermia, hematospermia, and prostatitis6,7. Moreover, testosterone levels in ZIKV-infected men also showed lower values8. Nevertheless, the mechanism by which ZIKV infection leads to impaired testosterone and sperm production remains elusive.
In the testicular tissue, various cells, including Germ cells (GCs), Sertoli cells (SCs), Leydig cells (LCs), and peritubular-myoid cells (MCs), are susceptible to ZIKV infection4,9,10,11. Given that LCs serve as the primary site for the synthesis and secretion of male sex hormones, particularly testosterone, ZIKV infection of LCs may potentially be a crucial factor in reducing testosterone levels. Testosterone plays a pivotal role in sustaining spermatogenesis and fertility12,13. Spermatogenesis occurs in the seminiferous tubules of the testes. The seminiferous tubules are composed of two major cell types: SCs and GCs. GCs are reproductive cells that develop into sperm, while SCs provide the nutritional and structural support to GCs, facilitating their maturation. Within the seminiferous tubules, only SCs have receptors for testosterone. GCs do not express androgen receptors (AR)14,15. Consequently, testosterone produced by LCs diffuses into the seminiferous tubules and regulates the expression of proteins involved in spermatogenesis through its action on SCs16,17, thus maintaining normal sperm production. Sperm then mature and are stored in the epididymis, where high testosterone levels are vital for supporting sperm maturation18. Thus, maintaining normal testosterone levels is crucial for the entire spermatogenic process. For instance, withdrawal of testosterone in the testis leads to a substantial decline in sperm production19,20,21. Spermatogenesis is also regulated by the hypothalamic-pituitary-gonadal (HPG) axis. ZIKV infection in male suckling mice has been shown to cause long-term disruptions to the HPG axis22 and, ultimately, fertility. However, ZIKV-induced functional abnormalities in HPG axis have not been observed in adult mice or human patients. Thus, our hypothesis posits a strong correlation between ZIKV infection of LCs and the resultant decline in testosterone levels and sperm counts.
Testosterone production in LCs starts with cholesterol transfer into mitochondria, mediated by the steroidogenic acute regulatory (STAR) protein23. Cholesterol is then converted to pregnenolone by the cholesterol side-chain cleavage enzyme (CYP11A1) in the mitochondria24. Pregnenolone is then transported to the endoplasmic reticulum (ER) and converted sequentially to progesterone by 3-beta-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase (3β-HSD), to 17-hydroxyprogesterone, and then to androstenedione by 17α-hydroxylase/C17–20 lyase (CYP17A1), and finally to testosterone by 17-beta-hydroxysteroid dehydrogenase (17β-HSD)25,26. The synthesis of androgens, particularly testosterone, is a crucial aspect of testicular metabolism and is influenced by various factors27. ZIKV-induced low testosterone levels reveal metabolic abnormalities in ZIKV-infected mouse testes. Notably, virus-induced metabolic changes are often critical for viral survival and host pathogenesis28. For example, ZIKV infection can lead to abnormalities in NAD+ metabolism in the brains of fetal mice, ultimately resulting in microcephaly29. To investigate the molecular mechanisms of ZIKV-induced testosterone reduction, we employed unbiased metabolomics and quantitative proteomics analyses in the testes of both ZIKV-infected and Mock-infected mice, aiming to allow for a multi-dimensional understanding of how ZIKV affects testicular metabolism, facilitating the identification of critical pathways and molecular targets potentially involved in the disruption of testosterone production and steroidogenesis. The proteomics profiling showed a significant downregulation of CYP17A1, a finding supported by the metabolomics analysis. CYP17A1, a membrane-bound dual-function monooxygenase, plays a critical role in the synthesis of many human steroid hormones30. Functional deficits in CYP17A1 can disrupt hormone synthesis, including glucocorticoids and sex hormones, leading to underdeveloped male physical characteristics and reduced fertility31,32,33. Clinically, CYP17A1 inhibitors, like abiraterone acetate, are utilized to lower testosterone levels in patients with advanced prostate cancer34,35,36.
The ZIKV genome encodes a polyprotein that is cleaved into three structural proteins (C, prM/M, E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The structural proteins form viral particles. The nonstructural proteins play critical roles in various stages of the virus life cycle, including replication, assembly, and evasion of host immune responses. NS1 is involved in the formation of the viral replication complex and immune evasion37. NS2A is crucial for the formation of replication vesicles, coordinating membrane rearrangements for RNA synthesis, and assembling viral particles38. It also suppresses interferon signaling, enhancing immune evasion. NS2B serves as a cofactor for NS3, forming a protease complex that facilitates the cleavage of the polyprotein precursor39. NS3 functions as both a protease and helicase, facilitating protein cleavage and RNA replication39. NS4A and NS4B collaborate to regulate the formation of replication vesicles and inhibit the host interferon response37. NS5, as an RNA-dependent RNA polymerase and methyltransferase, is responsible for viral RNA replication and stabilization, while also suppressing host immune responses40.
Here, we show that ZIKV infection impairs CYP17A1 mRNA translation via its non-structural protein NS2A (ZIKV-NS2A) binding, reducing CYP17A1 expression and consequently testosterone production. Further experiments confirmed the direct impact of ZIKV-NS2A expression on reduced testosterone and abnormal sperm production in mice. Our results reveal a previously unidentified pathogenic mechanism and potential interventions for ZIKV-related symptoms.
Results
Metabolomics profiling reveals disruption in steroidogenesis in the testes of ZIKV-infected mice
The testes are integral components of the male reproductive system, responsible primarily for spermatogenesis and hormone synthesis. ZIKV infection leads to disrupted testicular structure5, leakage of blood-testis barrier41, diminished testosterone secretion, and culminates4 in male infertility in mice, strongly suggesting a disturbance on steroid homeostasis. In order to investigate the impact of ZIKV infection on steroidogenesis in testes, we performed untargeted metabolomics analyses on mouse testes from Mock- and ZIKV-infected A129 mice (IFNα/β receptor-deficient), a well-established model for studying ZIKV pathogenesis5. The infected mice displayed evident viremia two days post-ZIKV infection, followed by gradual clearance of the virus (Fig. 1a). The viral load in testes increased with the duration of infection (Fig. 1b), which is consistent with previous reports41. Principal component analysis (PCA) indicated well-defined clustering of individual samples within each group, signifying low variation among replicates, and revealed distinct separation of metabolite profiles between the Mock- and ZIKV-infected groups at 10 dpi (Fig. 1c), suggesting significant metabolic changes. OPLS-DA further confirmed the distinct separation between the two groups (Supplementary Fig. 1a), with Q2 and R2 values showing no overfitting. The metabolomics analysis identified a total of 693 metabolites, of which 335 were differentially represented and mainly enriched in 35 metabolic pathways. Notably, steroid hormone biosynthesis is shown as one of the top 10 enriched pathways among the altered metabolites (Fig. 1d).

a, b Viral RNA copy numbers in both whole blood (a) and testes (b) were quantified in ZIKV-infected (days 0, 2, 4, 6, 8, or 10) mice using real-time quantitative PCR (qRT-PCR), and statistical significance was determined by comparing each infected group to the 0-day group. c PCA of metabolomics data of testes from Mock- (n = 8) and ZIKV-infected (10 dpi) (n = 8) mice. d Altered Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways enriched by significantly altered metabolites (FDR < 0.05) in the testes of ZIKV-infected (10 dpi) compared with the testes of Mock-infected mice. Stars represented pathways include Phenylalanine, tyrosine, and tryptophan biosynthesis (*1), and Alanine, aspartate, and glutamate metabolism (*2). The top 20 enriched pathways are shown. e Volcano plot indicating differential metabolites associated with steroid hormone biosynthesis in the testes of Mock- and ZIKV-infected (10 dpi) mice. Upregulated and downregulated metabolites (FDR < 0.05 and fold change > 2) are colored in red and blue, respectively; others are colored in gray. f A schematic diagram of testosterone production in Leydig cells. g, h Testosterone levels of the serum (g) or testis homogenates (h) were analyzed in ZIKV-infected (days 0, 6, or 10) mice. i, j Progesterone levels of the serum (i) or testis homogenates (j) were analyzed in ZIKV-infected (days 0, 6, or 10) mice. k, l Computer-assisted sperm analysis [total (k) and motile (l)] on samples obtained from the cauda epididymis of ZIKV-infected (days 0, 6, or 10) mice immediately after euthanasia. m Histological analysis of the epididymis collected from Mock- or ZIKV-infected mice (10 dpi) stained with hematoxylin and eosin. Stars indicate spermatozoa in the cauda epididymis. Scale bar, 100 μm. The ocular lens was set to 10×, and the magnification values correspond to the objective lens used. n Representative light micrographs of sperms obtained from the cauda epididymis of Mock- or ZIKV-infected mice (10 dpi). Scale bar, 200 μm. The images in m, n are representative of three independent experiments. The data shown in a, b, g–l are the mean ± s.d (a, b, g–j: n = 5; k, l: n = 7); Statistical analysis of (a, b, g–l) was performed using a one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test. Source data are provided in the Source Data file.
To gain a deep insight into identified metabolites involved in the steroid hormone biosynthesis, we examined the differential metabolites (FDR < 0.05) and identified several prominently upregulated and downregulated metabolites (Fig. 1e). Of note, a substantial decrease in testosterone and androstenedione levels, accompanied by elevated levels of their common precursor, progesterone, was observed. All three metabolites are implicated in testosterone production in Leydig cells (Fig. 1f), indicating a potential blockage in testosterone biosynthesis at intersections downstream of progesterone due to ZIKV infection.
We next performed enzyme-linked immunosorbent assays (ELISA) to quantify testosterone and progesterone levels in both mouse serum and testes. The results revealed a significant decrease in testosterone levels in both serum and testes following ZIKV infection (Fig. 1g, h), with an accumulation of progesterone (Fig. 1i, j), aligning with the observed metabolic profiles. Additionally, we observed a significant reduction in sperm counts and motility in mice after ZIKV infection (Fig. 1k, l). Histological analysis with hematoxylin and eosin (H&E) staining of the epididymis revealed both a decrease in sperm count and structural abnormalities, such as constriction of the epididymal lumen and thickening of inter-luminal tissue (Fig. 1m). Microscopic analysis of sperms further confirmed these observations (Fig. 1n). Together, these data indicate that ZIKV infection contributes to dysfunction in steroidogenesis and spermatogenesis in mouse testes, by hindering testosterone biosynthesis at reactions downstream from progesterone.
ZIKV infection decreases CYP17A1 at the protein level
To elucidate the molecular mechanisms by which ZIKV influences testosterone biosynthesis, we conducted proteomic profiling on mouse testes infected with ZIKV. The results revealed that, after 6 days of infection, 400 proteins displayed significant differences between ZIKV-infected and Mock-infected groups [ZIKV (6 dpi) vs Mock]. Following 10 days of infection, 647 proteins exhibited significant differences compared against the Mock-infected group [ZIKV (10 dpi) vs Mock]. A total of 239 proteins were jointly enriched in both infection groups (Fig. 2a). Through gene-annotation-independent analysis of the differentially expressed proteins, we identified a series of proteins associated with cellular metabolism and reproductive development (Fig. 2b). Hierarchical clustering analysis of these proteins revealed a significant downregulation of the key enzyme CYP17A1, integral to testosterone biosynthesis, in both ZIKV (6 dpi) and ZIKV (10 dpi) groups (Fig. 2c). To examine whether ZIKV infection leads to a downregulation of CYP17A1 expression, we confirmed a decrease in the protein abundance of CYP17A1 in the testes following ZIKV infection by immunofluorescence (Fig. 2d) and Western blotting (Fig. 2e), which was consistent with the results obtained from mass spectrometry analysis, despite the differences in fold change at 6 dpi and 10 dpi. However, there was no change in the mRNA levels of CYP17A1 (Fig. 2f). Other proteins related to testosterone synthesis, such as CYP11A1, 3β-HSD, and STAR, showed no significant differences at both the protein and mRNA levels (Fig. 2e, g and Supplementary Fig. 1b–d). Additionally, we generated a stable A549 cell line expressing CYP17A1 (CYP17A1-A549 cells), where CYP17A1 is strongly expressed in ZIKV-uninfected cells (short white arrows), but its expression is nearly absent in ZIKV-infected cells (long white arrows), demonstrating the loss of stable transfected CYP17A1 expression in ZIKV-infected cells (Fig. 2h).

a Venn diagrams showing the numbers of identified proteins and the overlap of protein identification in the testes from ZIKV-infected mice (6 dpi) vs Mock-infected mice and ZIKV-infected mice (10 dpi) vs Mock-infected mice. b Enriched KEGG pathways based on the overlap of differential proteins described in (a) (FDR < 0.05 and fold change > 2) are shown. c Heat map of altered proteins (FDR < 0.05 and fold change > 2) related to steroid metabolic process and male germ cell proliferation in downregulated pathways in the testes of ZIKV-infected mice. d Representative immunofluorescence images of the testes of ZIKV-infected (days 0, 6, or 10) mice by staining for ZIKV-E and CYP17A1. DAPI staining indicates the nuclei. Scale bar, 50 μm. The ocular lens was set to 15×, with the magnification values corresponding to the objective lens used. e, f Protein levels of CYP17A1/CYP11A1/STAR/3β-HSD and ZIKV-E in the testes of ZIKV-infected (days 0, 6, or 10) mice (e), and CYP17A1 mRNA levels (f) were measured by Western blotting and qRT-PCR, respectively. g Representative immunofluorescence images of the testes of ZIKV-infected (days 0, 6, or 10) mice by staining for ZIKV-E and CYP11A1. DAPI staining indicates the nuclei. Scale bar, 50 μm. The ocular lens was set to 15×, with the magnification values corresponding to the objective lens used. h Representative immunofluorescence images of ZIKV-infected CYP17A1-A549 cells by staining for ZIKV-E and CYP17A1. DAPI staining indicates the nuclei. Scale bar, 25 μm. i, j Primary mLCs were infected with ZIKV at the indicated MOIs for 24 h (i) or were infected with ZIKV at an MOI of 1 for various time courses (j). The expression of endogenous CYP17A1/CYP11A1/STAR/3β-HSD was detected by Western blotting. k Primary mLCs were infected with ZIKV at the indicated MOIs for 24 h. Viral RNA (vRNA) and CYP17A1 mRNA were detected by qRT-PCR. The images in d, e, g–j are representative of three independent experiments. The data shown in f, k are the mean ± s.d (f: n = 5; k: n = 3); Statistical analysis of (f, k) was performed using one-way ANOVA, followed by Tukey’s multiple comparison test; ns, not significant. Source data are provided in the Source Data file.
Leydig cells are the only testicular cell type that can express CYP17A1 and synthesize testosterone42. We isolated and cultured primary mouse Leydig cells (mLCs) in vitro to assess the impact of ZIKV infection. The endogenous marker protein CYP11A1 exhibited well expression in mLCs (Supplementary Fig. 1e). Additionally, we analyzed the levels of testosterone and progesterone in the culture supernatant of mLCs at different time points and observed that the secretion levels of these hormones peaked at 24 h after isolation and subsequently declined over time (Supplementary Fig. 1f, g). We found that ZIKV efficiently infected mLCs without significantly compromising their viability, even at high viral infection doses (Supplementary Fig. 1h–k). ZIKV infection in mLCs led to a time- and dose-dependent reduction in CYP17A1 protein expression (Fig. 2i, j), with no significant changes in mRNA expression levels (Fig. 2k). Other testosterone synthesis-related proteins such as STAR, CYP11A1, and 3β-HSD showed no significant differences in both protein and mRNA levels (Fig. 2i, j and Supplementary Fig. 1l–n). We also performed ELISA assays to quantify testosterone and progesterone levels in the supernatant of ZIKV-infected mLCs. The results revealed a significant decrease in testosterone levels, with an accumulation of progesterone (Supplementary Fig. 1o, p). In summary, these findings demonstrate that ZIKV infection downregulates the protein expression levels of endogenous CYP17A1 in both cells and testes.
ZIKV-NS2A inhibits the production of CYP17A1 protein
To further investigate the mechanism behind the reduction of CYP17A1 production due to ZIKV infection, we individually co-transfected 10 ZIKV-encoded proteins with human CYP17A1 (hCYP17A1) or mouse CYP17A1 (mCYP17A1) into HEK293T cells. NS2A, but not other viral proteins, disrupted hCYP17A1/mCYP17A1 expression (Fig. 3a, b) and this reduction in CYP17A1 levels was not attributable to cell death, as NS2A showed no significant cytotoxicity in HEK293T cells under the conditions tested (Supplementary Fig. 2a). Similarly, NS2A exhibited no substantial cytotoxic effects on A549 or mLCs (Supplementary Fig. 2b, c). The expression of hCYP17A1/mCYP17A1 was inhibited by NS2A in a dose-dependent manner (Fig. 3c, d). We also verified this phenomenon in CYP17A1-A549 cells (Fig. 3e), which was in line with immunofluorescence results indicating the loss of CYP17A1 expression in NS2A-positive cells (Fig. 3f). Furthermore, NS2A decreased the endogenous CYP17A1 protein in mLCs in a dose-dependent manner (Fig. 3g), with no impact on CYP17A1 mRNA levels (Fig. 3h). Notably, NS2A had no effect on the protein and mRNA levels of other testosterone synthesis-related proteins (Fig. 3g and Supplementary Fig. 2d–f). To assess specificity, we evaluated the impact of different flavivirus NS2As on CYP17A1 expression, including West Nile virus (WNV), Japanese Encephalitis virus (JEV), and Dengue virus (DENV). Only ZIKV-NS2A showed the most pronounced effectiveness on CYP17A1 expression (Supplementary Fig. 2g). Additionally, we selected the endoplasmic reticulum protein Calnexin for validation and found that neither ZIKV infection nor ZIKV-NS2A overexpression had any impact on Calnexin expression levels (Supplementary Fig. 2h–k). In summary, our results demonstrate that ZIKV-NS2A induces a downregulation of CYP17A1 expression.

a, b HEK293T cells were co-transfected with plasmids encoding hCYP17A1-HA/mCYP17A1-HA and either ZIKV structural proteins C/E/M or nonstructural proteins NS1/2A/2B/3/4A/4B/5, fused with Flag or empty vector as control. The expression of hCYP17A1-HA/mCYP17A1-HA and individual ZIKV proteins was detected by Western blotting at 36 h post-transfection. The red font highlighted the ZIKV-NS2A protein. c, d HEK293T cells were co-transfected with plasmids encoding hCYP17A1-HA/mCYP17A1-HA and various amounts of NS2A-Flag. The expression of CYP17A1 and NS2A were detected by Western blotting at 36 h post-transfection. e CYP17A1-A549 cells were transfected with various amounts of NS2A-Flag. The expression of CYP17A1 and NS2A were detected by Western blotting at 36 h post-transfection. f CYP17A1-A549 cells were transfected with the NS2A-Flag or empty vector as control. At 36 h post-transfection, representative immunofluorescence images of these cells were shown by staining for NS2A-Flag and CYP17A1. DAPI staining indicates the nuclei. Scale bar, 10 μm. g, h Primary mLCs were transfected with various amounts of the NS2A-Flag. At 36 h post-transfection, the expression of endogenous CYP17A1/CYP11A1/STAR/3β-HSD (g) and CYP17A1 mRNA levels (h) were detected by Western blotting and qRT-PCR, respectively. The images in a–g are representative of three independent experiments. The data shown in h are the mean ± s.d (n = 4 independent experiments); Statistical analysis of (h) was performed using one-way ANOVA, followed by Tukey’s multiple comparison test; ns not significant. Source data are provided in the Source Data file.
ZIKV-NS2A sequestrates CYP17A1 mRNA and reduces its translation
To investigate if the decrease in CYP17A1 protein levels induced by ZIKV-NS2A is attributed to specific protein degradation pathways, we co-transfected CYP17A1 with ZIKV-NS2A in the presence of distinct pathway inhibitors. However, the inhibitors, including the autophagy inhibitor bafilomycin A1 (Baf-A1) and Chloroquine (CQ), the proteasome inhibitor MG-132, and the caspase inhibitor Z-VAD-FMK, failed to prevent the ZIKV-NS2A-induced reduction in CYP17A1 levels (Supplementary Fig. 3a, b). Additionally, we conducted protein stability assays in HEK293T cells expressing CYP17A1 with or without ZIKV-NS2A. These cells were treated with cycloheximide (CHX), a translation inhibitor, for up to 3 or 6 h. The results reinforced that ZIKV-NS2A had no effect on CYP17A1 at the post-translational level (Fig. 4a and Supplementary Fig. 3c), strongly indicating that ZIKV-NS2A may exert its inhibitory function during the translation of CYP17A1 mRNA, given ZIKV-NS2A had no effect on CYP17A1 mRNA levels. To further explore the impact of ZIKV-NS2A on the translation of CYP17A1 mRNA, we transfected ZIKV-NS2A into HEK293T cells, followed by transfection with in vitro synthesized CYP17A1 mRNA. The results showed a downregulation of CYP17A1 expression in the presence of ZIKV-NS2A, further supporting its suppressive effect on CYP17A1 mRNA translation (Fig. 4b).

a HEK293T cells were co-transfected with plasmids encoding CYP17A1-HA and NS2A-Flag or empty vector as control. At 24 h post-transfection, cells were treated with 40 μg/mL CHX for 0, 3 h. The expression of CYP17A1 and NS2A were detected by Western blotting. b HEK293T cells were transfected with NS2A-Flag for 20 h, followed by a subsequent transfection with in vitro synthesized CYP17A1 mRNA for 4 h. The expression of CYP17A1 and NS2A were detected by Western blotting. c CYP17A1-A549 cells were infected with HA-NS2A ZIKV for 24 h. RIP-qPCR were performed to determine the binding between CYP17A1 mRNA and HA-NS2A. The levels of CYP17A1 and GAPDH mRNA was calculated by comparing the amount of RNA precipitated by specific antibody (viral E, claudin-2, or HA) with the amount of the same type of RNA precipitated by isotype control IgG. d MST workflow. See text for detail. e Binding affinities of GFP-NS2A to CYP17A1 mRNA were measured by MST as described in the methods. f The workflow of the in vitro RNA-protein pulldown assay. g The amounts of GST-NS2A in the input and eluates from the pulldown assay were detected by Western blotting. h Competition assay was used to determine the binding affinity between CYP17A1 mRNA-biotin and GST-NS2A in the presence of increasing amounts of competitors. After pulldown, GST-NS2A in the eluates was detected by Western blotting. i EMSA was used to determine the binding affinity of CYP17A1 mRNA-biotin with ZIKV-NS2A. CYP17A1 mRNA-biotin was incubated with increasing amounts of GST-NS2A, with GST used as a control. CYP17A1 mRNA-biotin migration was performed in 0.4% agarose gel without sodium dodecyl sulfate (SDS) and detected using Streptavidin-HRP Conjugate. j Quantification of ribosome loading on to CYP17A1 transcripts. CYP17A1-HA was co-transfected into HEK293T cells with empty vector, NS2A-Flag, and E-Flag, respectively. At 48 h post-transfection, cells were treated with 100 μg/mL CHX for 10 min and used to analyze both the total and ribosome-associated levels of GAPDH, CYP17A1 and Calnexin mRNA. k CYP17A1 mRNA was introduced into rabbit reticulocyte lysates along with increasing amounts of GST-NS2A or GST, and Lysates without CYP17A1 mRNA served as a blank control in the in vitro translation system. The mixtures were incubated at 30 °C for 90 min, and the expression of CYP17A1 in the lysates was detected by Western blotting. The bar plot shows quantification of the band intensity from the western blot image. The images in a, b, g–i, k are representative of three independent experiments. The data shown in c, j, k are the mean ± s.d (n = 3 independent experiments); Statistical analysis of (c, j, k) was performed using two-way ANOVA, followed by Tukey’s multiple comparison test (c, j); one-way ANOVA, followed by Tukey’s multiple comparison test (k); ns not significant. Source data are provided in the Source Data file.
Previously, it has been reported that ZIKV-NS2A can bind a 3’-UTR stem-loop in the viral genome and present viral RNA to the structural proteins for virion assembly38. This led us to hypothesize that ZIKV-NS2A might also bind to CYP17A1 mRNA, thereby affecting its translation. To validate our hypothesis, we investigated the potential binding of ZIKV-NS2A to CYP17A1 mRNA using an RNA immunoprecipitation (RIP) assay. However, one major challenge in studying flavivirus NS2A is the lack of specific and reliable antibodies against this protein. Therefore, we used a hemagglutinin (HA)-tagged NS2A ZIKV (HA-NS2A ZIKV) in the context of an infectious cDNA clone of Cambodian strain FSS1302538. We performed RIP assays by HA, viral E protein, or cellular protein claudin-2 antibodies to precipitate cell lysates of HA-NS2A ZIKV-infected CYP17A1-A549 cells (Supplementary Fig. 3d). Quantitative RT-PCR was used to measure the amount of CYP17A1 mRNA co-precipitated with HA-NS2A, viral E or claudin-2, and GAPDH mRNA was quantified as a control from the precipitated samples. The results showed approximately a 2.5-fold enrichment of CYP17A1 mRNA in the HA-NS2A-precipitated samples, while no enrichment was observed for cellular GAPDH mRNA. Viral E or host claudin-2 protein did not enrich CYP17A1 or GAPDH mRNA (Fig. 4c), suggesting a specific interaction between ZIKV-NS2A and CYP17A1 mRNA in ZIKV-infected cells. The same phenomenon was observed in HEK293T cells co-transfected with plasmids encoding CYP17A1-HA and NS2A-Flag (Supplementary Fig. 3e). Additionally, we examined the affinity between ZIKV-NS2A and CYP17A1 mRNA through microscale thermophoresis technology (MST) assay43, indicating that ZIKV-NS2A binds to CYP17A1 mRNA (Kd = 58.2 nM) (Fig. 4d, e). To test whether ZIKV-NS2A has a direct interaction with CYP17A1 mRNA, we preformed the RNA-protein pulldown experiment, indicating the CYP17A1 mRNA-biotin efficiently pulled down GST-NS2A but not GST (Fig. 4f, g). In contrast, a biotinylated RNA control (an iron response element RNA) failed to pull down GST or GST-NS2A (Fig. 4g). The result demonstrated that ZIKV-NS2A could directly bind to the CYP17A1 mRNA. Furthermore, an RNA competition assay was performed to examine the specificity of GST-NS2A and CYP17A1 mRNA interaction. Increasing amounts of unbiotinylated CYP17A1 mRNA competed away the GST-NS2A/CYP17A1 mRNA interaction, whereas yeast tRNAs did not (Fig. 4h), confirming the specificity of the interaction between GST-NS2A and CYP17A1 mRNA. Additionally, the electrophoretic mobility shift assay (EMSA) results further supported that only the NS2A-GST, but not the GST, formed a stable complex with CYP17A1 mRNA and retarded its migration (Fig. 4i). If cytoplasmic CYP17A1 mRNA levels are constant in the presence of ZIKV-NS2A, yet less CYP17A1 protein is produced, this implies reduced translation. This, in turn, should be reflected in less efficient loading of ribosomes onto CYP17A1 mRNA. To determine whether this is the case, we used a published procedure44 to measure the ribosome association of CYP17A1 mRNA in the presence or absence of ZIKV-NS2A. The results indicated that the relative level of ribosome-associated CYP17A1 mRNA was significantly decreased in the presence of ZIKV-NS2A, but not ZIKV-E (Fig. 4j). As controls, we also measured the ribosome loading of cellular GAPDH and Calnexin mRNAs (Fig. 4j). These results confirm that the reduced ribosome association observed with CYP17A1 mRNA in the presence of ZIKV-NS2A is specific to CYP17A1 mRNA and not due to a global reduction in ribosome loading onto all mRNAs. Furthermore, we observed that the addition of GST-NS2A decreased the synthesis of CYP17A1 protein in a dose-dependent manner by in vitro translation assays using rabbit reticulocyte lysates (Fig. 4k), indicating that ZIKV-NS2A inhibits translation of CYP17A1 mRNA. Collectively, these results suggest that ZIKV-NS2A interacts with CYP17A1 mRNA and reduces its translation
The 1200-1400 nucleotides of CYP17A1 mRNA is responsible for binding to ZIKV-NS2A
To identify the specific binding region between ZIKV-NS2A and CYP17A1 mRNA, we performed a photo-activatable, ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) experiment. The hCYP17A1 mRNA-containing photoreactive 4-thiouridine (4SU) was prepared through in vitro transcription in the presence of 4-S-UTP. Human CYP17A1 mRNA was incubated with GST-NS2A in vitro, forming RNA-protein complex following UV-irradiation. The 4-thiouridine in the hCYP17A1 mRNA was cross-linked to ZIKV-NS2A under UV light, resulting in U to C conversions during reverse transcription of hCYP17A1 mRNA (Fig. 5a). Subsequently, the hCYP17A1 mRNA was then converted into a cDNA library and analyzed by deep sequencing. A summary of the T to C mutations in the hCYP17A1 mRNA when complexed with ZIKV-NS2A is shown in (Fig. 5b). The highest mutation sites mainly occurred within the 200–300 and 1200–1400 nucleotides of hCYP17A1 mRNA (Fig. 5b).

a PAR-CLIP workflow71. See text for detail. b PAR-CLIP result summary. Uridines in the CYP17A1 mRNA that may participate in NS2A-binding were indicated. c, d A competition assay was performed by adding various unbiotinylated CYP17A1 mRNA fragments to the mixture of GST-NS2A and CYP17A1 mRNA-biotin. The remaining amounts of GST-NS2A pulled down by CYP17A1 mRNA-biotin were detected by Western blotting. e Schematic representation of distinct truncated hCYP17A1 mRNA. The nucleotides positions of each truncated hCYP17A1 mRNA are indicated. f HEK293T cells were co-transfected with plasmids encoding distinct truncated hCYP17A1-HA and NS2A-Flag. After 36 h post-transfection, the expression of hCYP17A1-HA and NS2A were detected by Western blotting. The images in c, d, f are representative of three independent experiments.
To investigate the binding of these two regions to ZIKV-NS2A, we conducted a competition assay using in vitro transcribed hCYP17A1 mRNA segments of 200–300 nt and 1200–1400 nt. The full-length hCYP17A1 mRNA served as a positive control. The result indicated 1200–1400 nt segment, but not 200–300 nt segment, competed away the ZIKV-NS2A and hCYP17A1 mRNA interaction (Fig. 5c), indicating that 1200–1400 nt segment binds to ZIKV-NS2A. Next, we aimed to explore whether mCYP17A1 mRNA also interacts with ZIKV-NS2A and whether the structural characteristics of the region on mCYP17A1 mRNA that binds to ZIKV-NS2A are similar to the segment 1200–1400nt of hCYP17A1 mRNA. To address this, we predicted the secondary structures of hCYP17A1 and mCYP17A1 mRNA, revealing that the ZIKV-NS2A binding region, 1200–1400 nt segment on hCYP17A1 mRNA, contains a large number of stem-loop structures, and both hCYP17A1 and mCYP17A1 mRNA form similar secondary structures in this region (Supplementary Fig. 4a, b). Further experiments revealed that the 1200–1400nt segment of mCYP17A1 mRNA also competed away the ZIKV-NS2A and mCYP17A1 mRNA interaction (Fig. 5d), indicating that mCYP17A1 mRNA can bind ZIKV-NS2A, and its segment 1200–1400 nt is responsible for binding to ZIKV-NS2A. Furthermore, we generated several truncated forms of hCYP17A1 mRNA: Δ1 involved the removal of 200–300 nt, Δ2 involved the removal of 1200–1400 nt, and Δ3 involved the removal of both 200–300 nt and 1200–1400 nt (Fig. 5e). These constructs were then individually co-transfected with ZIKV-NS2A into HEK293T cells, indicating the deletion of the 1200–1400 nt segment abolished the inhibitory effect of ZIKV-NS2A on CYP17A1 expression (Fig. 5f). The findings further support the ZIKV-NS2A binds to CYP17A1 mRNA and the 1200–1400 nt of CYP17A1 mRNA is responsible for binding to ZIKV-NS2A.
Mutations at residues 67E/101P abolish the binding between ZIKV-NS2A and CYP17A1 mRNA and the inhibition of CYP17A1 translation
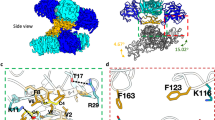
ZIKV-NS2A was reported to be a transmembrane protein with a transmembrane structure anchored on the endoplasmic reticulum, closely associated with the assembly of viral particles on the ER38 (Fig. 6a). Based on the topology of ZIKV-NS2A, we generated various truncated forms of ZIKV-NS2A (Fig. 6b), to pinpoint critical amino acid residues responsible for its binding and subsequent suppression of CYP17A1 mRNA translation. The truncated NS2A protein 1-56aa (NS2A1-56) had no effect on the expression levels of hCYP17A1 or mCYP17A1. Whereas the truncated NS2A protein 1-103aa (NS2A1-103), 1-123aa (NS2A1-123), 1-147aa (NS2A1-147), 1-196aa (NS2A1-196), continued to exhibit downregulation of hCYP17A1 or mCYP17A1 expression (Fig. 6c, d). We tentatively concluded that the residues on ZIKV-NS2A responsible for affecting CYP17A1 expression lie between amino acids 56 and 103, which includes a membrane-traversing segment and two flanking loops. We introduced mutations in the amino acid residues within the 56-103 region that could potentially affect NS2A’s RNA binding ability (Fig. 6e). These mutations involved hydrophilic or positively charged residues, including glutamic acid (67E/103E), aspartic acid (73D), histidine (76H), lysine (84 K), and arginine (86 R/96 R/102 R). Additionally, we included the proline (P101) due to its unique properties: the inability to form main-chain hydrogen bonds and the ability to adopt either the cis or trans peptide bond conformation, which can influence protein conformational changes and folding processes45,46. We found that mutations of residue E67 or P101 on ZIKV-NS2A minimally affected the expression levels of hCYP17A1 or mCYP17A1 (Fig. 6f, g). Subsequent mutations at positions 84, 86, 102, or 103 on ZIKV-NS2A revealed partial compensation for the inhibition of hCYP17A1 or mCYP17A1 (Fig. 6f, g). Additionally, we validated these findings through RIP and RNA pulldown assays, wherein we demonstrated that the mutant NS2AE67A / NS2AP101A failed to interact with hCYP17A1 mRNA, unlike the mutant NS2AD73A (Fig. 6h, i). These mutations (NS2AK84A, NS2AR86A, NS2AR102A, and NS2AE103A) weakened the interaction between NS2A and hCYP17A1 mRNA compared to the NS2AWT (Fig. 6h). Moreover, NS2AE67A/NS2AP101A was unable to disrupt translation of hCYP17A1 mRNA, as observed by an in vitro translation assay (Fig. 6j, k). Collectively, these data indicate that the sole transmembrane segment of NS2A and its flanking loops are crucial for binding CYP17A1 mRNA and inhibiting its translation. Mutations at amino acid residues 67E or 101 P in this region abolish NS2A’s ability to bind CYP17A1 mRNA and inhibit its translation.

a A model of ZIKV NS2A membrane topology47. b Schematic representation of C-terminally truncated ZIKV-NS2A. c, d HEK293T cells were co-transfected with plasmids encoding hCYP17A1-HA/mCYP17A1-HA and each truncated NS2A-Flag. After 36 h post-transfection, the expression of h/mCYP17A1 and NS2A were detected by Western blotting. e Schematic representation of distinct NS2A mutants. f, g HEK293T cells were co-transfected with plasmids encoding hCYP17A1-HA/mCYP17A1-HA and distinct NS2A-Flag mutants. After 36 h post-transfection, the expression of h/mCYP17A1 and NS2A were detected by Western blotting. h Binding of distinct NS2A mutants with hCYP17A1 mRNA were analyzed by RIP-qPCR, and statistical significance was determined by comparing the results of each mutant protein group to the WT group. i hCYP17A1 mRNA-biotin was incubated with distinct mutant forms of GST-NS2A. After pulldown, the amounts of GST-NS2A in the input and eluates was detected by Western blotting. j, k hCYP17A1 mRNA was introduced into rabbit reticulocyte lysates along with distinct mutant forms of GST-NS2A or GST, and Lysates without hCYP17A1 mRNA served as a blank control in the in vitro translation system. The mixtures were incubated at 30 °C for 90 min, and the expression of hCYP17A1 in the lysates was detected by Western blotting. The bar plot shows quantification of the band intensity from the western blot image. The images in c, d, f, g, i, j are representative of three independent experiments. The data shown in h, k are the mean ± s.d (h: n = 2; k: n = 3); Statistical analysis of (h, k) was performed using one-way ANOVA, followed by Tukey’s multiple comparison test. Source data are provided in the Source Data file.
ZIKVE67A mutant virus exhibits significantly lower inhibition on steroidogenesis and spermatogenesis than ZIKVWT
To assess whether ZIKV-NS2A can impede steroidogenesis and spermatogenesis during ZIKV infection, we introduced the E67Aor P101A mutation into a ZIKV infectious clone based on Cambodian strain FSS13025 and attempted to rescue ZIKVE67A or ZIKVP101A mutant viruses from Vero cells. The original infectious clone construct, which produces wild-type viruses (ZIKVWT), was used as a control. We confirmed the full length of the genome sequence of the mutant viruses (Supplementary Fig. 5a, b). ZIKVE67A exhibited similar plaque morphologies to the ZIKVWT, and growth kinetic analysis in BHK21 cells showed no significant difference between ZIKVE67A and ZIKVWT, suggesting that the E67A mutation in NS2A did not affect viral replication in vitro (Supplementary Fig. 5c, d), consistent with previous reports47. However, the P101A mutant genomic RNAs produced neither E-positive cells nor infectious viruses in the transfected cells, as indicated by immunofluorescent assay (Supplementary Fig. 5e), plaque assay (Supplementary Fig. 5f), and qRT-PCR (Supplementary Fig. 5g), whereas the WT RNA did. Incubation of naive Vero cells with culture supernatants from the P101A-transfected cells did not generate any viral E-positive cells (Supplementary Fig. 5h). Subsequently, we intraperitoneally infected A129 mice with ZIKVE67A or ZIKVWT at the same dose for 6 days. ZIKVE67A demonstrated the same infection efficiency as ZIKVWT in vivo (Fig. 7a). We collected mouse serum and testes for testosterone and progesterone measurements. In contrast to ZIKVWT, ZIKVE67A failed to affect testosterone and progesterone levels in both mouse serum and testes (Fig. 7b–e). Similarly, ZIKVE67A resulted in a less significant reduction in the protein levels of CYP17A1 in the testes, without affecting its mRNA production, compared to ZIKVWT (Fig. 7f, g). ZIKVE67A did not influence the protein and mRNA levels of other key enzymes involved in testosterone synthesis (Fig. 7f and Supplementary Fig. 5i–k). This phenomenon was also observed through immunofluorescence (Fig. 7h). Additionally, we noted diminished total and motile sperm counts in fluid collected from the cauda epididymis of ZIKVE67A -infected mice, whereas ZIKVE67A did not inhibit total and motile sperm production (Fig. 7i, j). Co-immunofluorescence staining with anti-DDX4, a marker for cells at various stages of spermatogenesis, revealed that ZIKVWT significantly reduced the number of germ cells, while ZIKVE67A had no inhibitory effect on germ cells (Fig. 7k). This trend was further supported by H&E staining of testes, showing that ZIKVWT decreased the number of germ cells in seminiferous tubules, while testicular architecture remained intact in ZIKVE67A-infected mice (Fig. 7l). Next, we determined whether ZIKV-NS2A alone is sufficient to affect steroidogenesis and spermatogenesis in vivo. Single mutations of ZIKV-NS2A at positions 101, 102, or 103 each attenuated the inhibitory effect of NS2A on mCYP17A1. Previous studies have also reported that the NS2A mutants (R102A) and (E103A) significantly attenuate viral replication and block virion assembly, respectively, indicating critical roles of these residues in NS2A fuction47. Therefore, we speculated that a combined mutation of residues 101-103 (NS2APRE-AAA) would result in a more significant loss of NS2A’s inhibitory effect on mCYP17A1 compared to single mutations. As expected, NS2APRE-AAA exhibited a significantly reduced inhibitory effect on the expression of mCYP17A1 (Supplementary Fig. 6a). We then used adeno-associated virus (AAV) to deliver ZIKV-NS2AWT, ZIKV-NS2APRE-AAA, or the control EGFP into the mouse testes through in situ injection, and observed abnormal steroidogenesis and spermatogenesis after a 5-day AAV-NS2AWT infection compared with AAV-NS2APRE-AAA (Supplementary Fig. 6). In conclusion, our data demonstrate that NS2A can suppress sperm production by inhibiting testosterone synthesis. Additionally, the residue 101P on NS2A that regulates its binding to CYP17A1 mRNA is also critical for viral propagation.

a Viral RNA copy numbers in ZIKV-infected mouse testes (days 6) were quantified using qRT-PCR. b–e Testes were infected with ZIKVWT or ZIKVE67A for 6 days. The levels of testosterone and progesterone in the serum (b, c) or testis homogenates (d, e) were tested by ELISA. f, g Protein levels of CYP17A1/CYP11A1/STAR/3β-HSD and ZIKV-NS2A in ZIKV-infected mouse testes (f), and CYP17A1 mRNA levels (g) were measured by Western blotting and qRT-PCR, respectively. h Representative immunofluorescence images of ZIKV-infected mouse testes by staining for CYP17A1 and NS2A. DAPI staining indicates the nuclei. Scale bar, 100 μm. i, j Computer-assisted sperm analysis [total (i) and motile (j)] on samples obtained from the cauda epididymis of ZIKV-infected mice immediately after euthanasia. k Representative immunofluorescence images of ZIKV-infected mouse testes by staining for DDX4. DAPI staining indicates the nuclei. Scale bar, 100 μm. l Histological analysis of the epididymis collected from ZIKV-infected mice stained with hematoxylin and eosin. Stars indicate loss of spermatogenic cells in testis. Scale bars, 100 μm. The images in f, h, k, l are representative of three independent experiments. The ocular lens was set to 15× in panel h, and to 10× in panels k and l, with the magnification values corresponding to the objective lens used. The data shown in a–e, g, i, j are the mean ± s.d (n = 5 independent experiments); Statistical analysis of (a–e, g, i, j) was performed using one-way ANOVA, followed by Tukey’s multiple comparison test; ns, not significant. Source data are provided in the Source Data file.
Discussion
To date, ZIKV is the only flavivirus that can get across the BTB and establish long-term infection in testis41. The persistence of ZIKV particles in male semen for an extended duration after viremia clearance, along with its sexual transmission from male to female and vertical transmission exacerbate the concern of fetal infection in resulting pregnancies3. Moreover, related reproductive symptoms, including hematospermia and oligospermia, have been reported in male ZIKV patients, implying its harmful effects on spermatozoa production and fertility. The testis functions as the primary organ for spermatogenesis, which is tightly regulated by hormones, particularly testosterone. Considering the testis is a potential reservoir for ZIKV and major steroidogenesis organ, there exists a theoretical basis for investigating the impact of ZIKV infection on testosterone production. Indeed, early clinical observations in male patients infected with ZIKV exhibited notably diminished testosterone levels8, consistent with the observations both in ZIKV-infected IFNα/β receptor-deficient mice and immunocompetent macaques, suggesting the decrease in testosterone was independent of ZIKV-induced interferon activation, and some other mechanisms are involved. However, the absence of IFN signaling in A129 mice presents limitations for studying viral pathogenesis and antiviral immunity. For instance, IFN-α/β-dependent restrictions on viral tropism or effects on B and T cell priming will not be evident in this model48. Although ZIKV infection in immune-deficient mice develop pronounced genital pathological changes, such as substantial inflammation in the testes and testicular atrophy, severe symptoms such as orchitis are rarely reported in humans49, implying virus direct infection rather than the immune-mediated damage play more important roles in the decreased sex hormone production and oligospermia manifested in ZIKV-infected humans. However, the specific mechanisms by which ZIKV infection decreases testosterone secretion and impairs sperm production remain largely unknown.
Current studies on ZIKV proteins have partially answered questions about viral evolution, virulence, tissue tropism, immune evasion, and pathogenicity, with a specific focus on the mechanisms whereby ZIKV induces neurodevelopmental diseases50,51,52,53. Nevertheless, there is too limited knowledge of the molecular-level mechanisms underlying male reproductive consequences of virus infection. Here, from an unbiased and systematic screening of ZIKV-encoded proteins, we identified ZIKV-NS2A as a causal factor for ZIKV-induced low testosterone levels. Mechanistically, ZIKV-NS2A sequestrates CYP17A1 mRNA and inhibits its translation by diminishing the recruitment of ribosomes, leading to a decrease in testosterone synthesis, which in turn suppresses spermatogenesis. Our study, along with others54, revealed that ZIKV infection does not significantly impact the viability of Leydig cells—even at high viral infection doses (Supplementary Fig. 1g–j). As Leydig cells are the exclusive site of CYP17A1 expression in the testes, this finding indicates that the reduced testosterone levels observed during ZIKV infection are not a result of Leydig cell death. This underscores a direct correlation between the decreased testosterone levels resulting from ZIKV infection and the inhibition of CYP17A1 mRNA translation by ZIKV-NS2A. Additionally, we performed a comparative analysis of the impact of different flavivirus NS2As on CYP17A1 expression, revealing that only ZIKV-NS2A exhibited a notably inhibitory effect on CYP17A1 expression. We postulated that this outcome is likely attributed to high sequence diversity and distinct membrane topology observed across various flavivirus NS2As47,55,56. Indeed, a series of studies by Shi et al. have unveiled that, in stark contrast to the topologies of NS2As in other flavivirus, ZIKV-NS2A has a single transmembrane segment traversing the ER membrane, with the remaining six pTMSes peripherally associated with the ER membrane, forming three cytoplasmic loop regions47,56,57. Through truncation experiments, we determined that the amino acid sequence spanning residues 56 to 103 predominantly inhibits the expression of CYP17A1 by NS2A. Notably, scanning mutagenesis studies within this sequence identified amino acid residues 67E and 101P as pivotal for facilitating NS2A binding and suppressing CYP17A1 mRNA translation. Subsequently, we sought to determine if the incapability of NS2AE67A or NS2AP101A to block the translation of CYP17A1 could also be observed in the context of an authentic viral infection. To investigate this, we generated mutant viruses through reverse genetics based on the infectious clone of the ZIKV strain FSS13025. Interestingly, ZIKVE67A infection, compared to ZIKVWT, resulted in the loss of its capacity to suppress testosterone synthesis and spermatogenesis. We also revealed that amino acid residues 84K, 86R, 102R, and 103E exhibited partial inhibition of CYP17A1 expression by NS2A. However, these mutants can reduce viral production and abolish virion assembly47. Functionally, these charged or hydrophilic amino acid residues could potentially influence the conformation and subcellular localization of the NS2A protein within the endoplasmic reticulum. Consequently, we hypothesize that the loss of CYP17A1 expression inhibition by the E67A mutant results from mutations altering the topology and localization of NS2A. However, the precise mechanism by which ZIKV-NS2A influences CYP17A1 remains unclear, and other contributing factors may be involved. It is not yet understood how NS2A 56-103, which compose the pTMS3 domain flanked by two loops oriented towards the ER lumen and the cytosol, interacts with CYP17A1 mRNA while NS2A is recruited with other viral proteins into the ER membrane-associated viral replication complexes. In this study, NS2A was observed to be inherently unstable compared to other flavivirus NS2A proteins (Supplementary Fig. 2g). Our recent study has reported that NS2A undergoes ubiquitination and proteolytic degradation processes58 that may facilitate the release of NS2A fragments and their subsequent exposure to the cytosolic CYP17A1 mRNA. Additionally, the impact of mutations at positions E67, K84, R86, P101, R102, or E103 in NS2A 56-103 on CYP17A1 mRNA binding and translation remains unknown. Further research is needed to clarify these interactions by analyzing the dynamic structure of the NS2A with CYP17A1 mRNA during viral replication.
On the other hand, the RNA secondary structures, such as hairpins and stem loops, play a pivotal role in influencing gene expression, translation, as well as protein recognition and binding59,60,61,62. Integration of PAR-CLIP analysis with predicted secondary structure analysis of hCYP17A1 mRNA disclosed that the region spanning nucleotides 1200–1400, consisting of numerous stem-loops, is responsible for binding to ZIKV-NS2A. Similar secondary structures are also predicted on mCYP17A1 mRNA in the same region. Through in vitro binding assays, we confirmed the binding of 1200–1400 nt segment to ZIKV-NS2A, and the removal of this segment annulled the inhibitory effect of ZIKV-NS2A on CYP17A1 expression. Interestingly, ZIKV-NS2A binds to a stem-loop structure in the 3′ UTR of the ZIKV genome, serving as an “RNA recruitment signal” for ZIKV assembly38. Based on this information, we propose that the presence of these structures may be the primary reason for the specific binding of ZIKV-NS2A to CYP17A1 mRNA.
RNA-binding proteins (RBPs) are commonly defined as proteins that bind RNA through one or multiple RNA-binding domains (RBDs) and change the fate or function of the bound RNAs63,64,65. Flavivirus NS2As, including KUNV-NS2A, ZIKV-NS2A, and DENV-NS2A, have been shown to bind RNA molecules of various species, such as the viral genomic RNA38,57,66 and dsRNA67. Additionally, our study also identified binding to a host mRNA by ZIKV-NS2A. This prompts an investigation into whether these flaviviruses NS2As, especially ZIKV-NS2A, function as RNA-binding proteins. Future work should explore if these interactions are mediated by consensus motifs, represented as linear sequence motifs, or RNA structural motifs, or the structural context in which linear motifs are embedded. This could be achieved through a systematic combination of advanced biochemical methods, like PAR-CLIP, enabling the identification of RNAs bound by RBPs and the corresponding protein binding sites, and computational analysis based on an expanded dataset of RNA secondary structures62. Taken together, our study reveals a unique molecular mechanism by which ZIKV inhibits steroidogenesis and spermatogenesis, offering potential therapeutic targets for mitigating ZIKV-induced male productive system injury and low testosterone secretion. These findings expand our comprehension of virus-host cell interactions in viral pathogenesis and would represent an exciting avenue for future studies.
Methods
Ethics statement
All the animal experiments were performed according to the Regulations of Hubei Province Laboratory Animal Management and approved by Wuhan University Animal Experiment Ethics Committee under project licence WP20230062. All animal experiments were conducted under isoflurane anesthesia to minimize animal suffering. Studies with ZIKV were conducted under biosafety level 2 and Animal Biosafety Level 3 (BSL3) containment.
Cells and viruses
A549 cells (Adeno-carcinomic human alveolar basal epithelial cells), HEK293T cells (Human embryonic kidney cells), Vero cells (African green monkey kidney epithelial cells) and BHK-21 cells (Baby hamster kidney) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (P/S) at 37 °C in an atmosphere containing 5% CO2. Mosquito C6/36 cells (Aedes albopictus mosquito cells) were grown at 28 °C in RPMI 1640 medium containing 10% FBS, 1% P/S. The CYP17A1-A549 cells were maintained in DMEM supplemented with 10% FBS 1% P/S. Primary mouse Leydig cells (mLCs) were isolated from C57BL/6 mice. ZIKV strain ZIKA-SMGC-1 (GenBank accession number: KX266255) was kindly provided by Dr. George F. Gao (Institute of Microbiology, Chinese Academy of Sciences, Beijing, China). HA-NS2A ZIKV (HA-tagged NS2A ZIKV using an infectious cDNA clone of Cambodian strain FSS13025) was kindly provided by Dr. Gong Cheng (Tsinghua University-Peking University Joint Center for Life Sciences, School of Medicine, Tsinghua University, Beijing, China). Viruses were propagated in C6/36 cells, and viral titers were determined by crystal violet-based plaque assay.
Mouse experiments
A129 mice (IFNα/β receptor-deficient) were obtained from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences, and Peking Union Medical College. C57BL/6 mice were purchased from the Hubei Provincial Center for Disease Control and Prevention (Wuhan, China). All mice were housed on a 12 light /12 dark cycle, at ~18–23 °C with 40%–60% humidity. For ZIKV infection, 6–8-week-old male A129 mice were intraperitoneally infected with 1 × 106 plaque-forming units (PFU) of ZIKV. Mice administered 1 × PBS served as a control. Mice were euthanized at the indicated time points post-infection. Whole blood was collected for qRT-PCR (n = 5 per group) and hormone measurements (n = 5 per group), while testes were harvested for metabolomics analysis (n = 8 per group), proteomics analysis (n = 3 per group) and other assays, such as qRT-PCR (n = 5 per group), hormone measurements (n = 5 per group), immunofluorescence analysis (n = 3 per group), and Western blotting analysis (n = 3 per group). For AAV infection, 10 μL (equivalent to 1 × 109 vg) of AAV8-NS2AWT, AAV8-NS2APRE-AAA, or AAV8-GFP was injected into testes. Mice administered 1 × PBS served as a control. At the indicated time points post-infection, the mice were euthanized, and both whole blood and testes were collected for qRT-PCR (n = 5 per group), hormone measurements (n = 5 per group), immunofluorescence analysis (n = 3 per group), and Western blotting analysis (n = 3 per group).
Plaque assay
BHK-21 cells were seeded in 6-well plates and grown to 80%–90% confluence. Viral samples were ten-fold serially diluted six times in DMEM and added to each well, followed by incubation at 37 °C for 2–3 h. The viral solution was then replaced with 2 mL of 1.2% methylcellulose, and the plates were incubated for 5 days and then fixed and stained using formaldehyde-crystal violet, and the visible plaques were counted and viral titers (PFU/mL) were calculated as described previously68.
Antibodies
Rabbit monoclonal anti-CYP17A1 (Cat. no. 94004, 1:1000), rabbit monoclonal anti-CYP11A1(Cat. no. 14217, 1:1000), rabbit monoclonal anti-β-actin (Cat. no. 4970, 1:1000), mouse monoclonal normal IgG (Cat. no. 5415, 1:200), rabbit monoclonal anti-Cleaved Caspase-3 (Cat. no. 9664, 1:1000), and rabbit monoclonal anti-LC3 (Cat. no. 12741, 1:1000) were purchased from Cell Signaling Technology. Mouse monoclonal anti-GAPDH (Cat. no. G9295, 1:5000), mouse monoclonal anti-FLAG M2 (Cat. no. F1804, 1:1000), mouse monoclonal anti-HA (Cat. no. H9658, 1:5000), mouse monoclonal anti-GST (Cat. no. G1160, 1:5000), goat anti-mouse IgG conjugated with HRP (Cat. no. A5278, 1:5000), and goat anti-rabbit IgG conjugated with HRP (Cat. no. AP307P, 1:5000) were purchased from Sigma-Aldrich. Rabbit polyclonal anti-ZIKV-E (Cat. no. GTX133314, 1:5000) and mouse anti-ZIKV antibody 4G2 (Cat. no. GTX57154, 1:1000) were purchased from GeneTex. Rabbit polyclonal anti-GFP (Cat. no. 50430-2-AP, 1:1000), rabbit polyclonal anti-p62 (Cat. no. 18420-1-AP, 1:10000), and rabbit polyclonal anti-Calnexin (Cat. no. 10427-2-AP, 1:20000) were purchased from Proteintech. Rabbit polyclonal anti-STAR (Cat. no. ab96637, 1:1000) and rabbit polyclonal anti-DDX4 (Cat. no. ab13840, 1:1000) were purchased from Abcam. Alexa Fluor 488 goat anti-mouse IgG antibody (Cat. no. 406406, 1:200) and Alexa Fluor 488 goat anti-mouse IgG antibody (Cat. no. 405319, 1:200) were purchased from BioLegend. Mouse monoclonal anti-3β-HSD (Cat. no. sc-515120, 1:1000) was purchased from Santa Cruz Biotech. Fluorescein-Conjuated Affinipure Goat anti-human IgG (Cat. no. ZF-0308, 1:200) was purchased from ZSGB-BIO. Anti-ZIKV antibody (FITC-Z6, 1:1000) was Kindly gifted by Dr. George F. Gao.
Plasmid construction
A mammalian expression vector pcDNA3.1 (Thermo Fisher Scientific) with a cytomegalovirus (CMV) promoter was used to express various viral proteins (C, prM, E, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) in HEK293T cells. To construct the pcDNA3.1-ZIKV-C/prM/E/NS1/NS2B/NS3/NS4A/NS4B/NS5-FLAG plasmid, the genes encoding these ZIKV proteins (strain FSS13025) were synthesized and fused with a C-terminal FLAG tag sequence, followed by cloning into the pcDNA3.1 vector. For constructs pcDNA3.1-SPG-C16-NS2A-FLAG, a DNA fragment containing the Gaussia luciferase signal peptide (SPG), the last 16 amino acids of NS1 protein (C16) to ensure the correct processing and membrane topology of NS2A, and NS2A tagged with a C-terminal FLAG tag sequence was synthesized and cloned into pcDNA3.1 vector. DNA fragments that encode different truncated NS2A segments were amplified from the above pcDNA3.1-SPG-C16-NS2A-FLAG vector and cloned into the pcDNA3.1, resulting in the pcDNA3.1-SPG-C16-ctNS2A-FLAG plasmids (ctNS2A represents C-terminal truncated NS2A). Similarly, pcDNA3.1-SPG-C16-mutNS2A-FLAG plasmids (mutNS2A indicating NS2A mutants) were constructed using the same method. human and mouse CYP17A1(hCYP17A1/mCYP17A1) genes were fused with a C-terminal human influenza hemagglutinin (HA) tag and cloned into the pcAGGS vector under a CMV promoter, resulting in the pcAGGS-hCYP17A1/mCYP17A1-HA plasmid.
pGEX-4T-1 (GE Healthcare Life Sciences) was used as the vector for expressing all N-terminally glutathione S-transferase (GST)-tagged NS2A fusion proteins. The DNA fragments containing various NS2A mutants were cloned into pGEX-4T-1 through restriction enzyme sites BamHI and XhoI. To generate an A549 cell line constitutively expressing CYP17A1, the CYP17A1 gene was constructed by overlap PCR and inserted into the pWPI vector via PmeI and SpeI restriction sites and co-transfected into HEK293T cells with pMD2.G and pSPAX2 plasmids to produce lentivirus.
All plasmids were propagated in E. coli DH5α and verified by DNA sequencing (Tsingke Biotechnology Co., Ltd). All restriction enzymes were purchased from New England Biolabs. Primers were synthesized by Genecreate Biotech.
Adenovirus-associated virus (AAV) production
GFP, HA-NS2AWT, or HA-NS2APRE-AAA were cloned into AAV vector (pAAV-CMV), and then transfected into HEK293T cells with pAAV2/8-RC and pHelper plasmid (gifted from Dr. Yuchen Xia) for 3 days to produce AAVs. AAVs-containing supernatants were harvested and filtered with 0.22 μm filter, then concentrated with Amicon Ultra-15 centrifugal filter unit (Sigma-Aldrich). The concentration of purified AAVs was determined by qRT-PCR as described previously in ref. 69 and finally AAVs at the concentration of 1 × 1011 vg/mL were obtained.
Isolation and culture of primary mouse Leydig cells (mLCs) and cell viability assay
The procedure for isolation of mouse Leydig cells was refined from a procedure described previously in ref. 70. Briefly, 6-week-old male mice were anesthetized with CO2 and then killed by decapitation. Mice were soaked in 75% alcohol for 5 min, and bilateral testicles were removed through abdominal asepsis. The white membrane was removed, and the testicular tissue of the mice was kept intact. Testes obtained from these mice were dispersed by treatment with 0.5 mg/mL combined type II and IV collagenase (Sigma-Aldrich, C2-BIOC and C4-BIOC) at room temperature for 30 min under gentle oscillation. This suspension was then filtered through 100 μm copper meshes and centrifuged with centrifugation at 1000 × g for 5 min. The cells were then cultured in a medium DMEM/F-12 (Gibco) supplemented with 10% FBS, 1% P/S at 37 °C with 5% CO2. After 2 h of incubation, spermatogenic cells and cell debris were removed by washing twice with phosphate-buffered saline (1 × PBS). Primary mLCs that adhere to culture dishes were used in this study. The purity of these Leydig cells was assessed by staining for the cholesterol side-chain cleavage enzyme (CYP11A1) (a marker of Leydig cells). The viability of the primary mLCs were measured using the Alamar Blue (resazurin) fluorescent dye (Sigma-Aldrich).
Enzyme-linked immunosorbent assay (ELISA)
A129 male mice were intraperitoneally challenged with 106 PFU of ZIKV. Mice administered 1 × PBS served as a control. At different time points, the mouse testes were collected by carefully removing the fascia and large visible blood vessels from the surface of each testis to minimize blood contamination. The testes were then rinsed three times with chilled 1 × PBS, followed by homogenization. Blood samples were collected via retro-orbital bleeding and centrifuged to separate the serum. The concentrations of testosterone and pregnenolone in the serum and testis homogenates were measured using mouse testosterone ELISA Kits and mouse pregnenolone ELISA Kits (Beyotime) following the manufacturer’s instructions.
H&E analyses and immunofluorescence
Testes from ZIKV-infected and Mock-infected mice were collected at different time points, fixed in 4% paraformaldehyde (PFA) solution overnight, then dehydrated and paraffin-embedded. For immunofluorescence, 6 μm testis sections were stained with specific antibodies, including CYP17A1, CYP11A1, ZIKV-E, or DDX4, followed by fluorophore-labeled secondary antibodies. For H&E staining, 5 μm testis sections were used. Images were then captured with a Zeiss LSM510 laser scanning confocal microscope.
Confocal microscopy
The cells (Vero cells, CYP17A1-A549 cells, or mLCs) were fixed in 4% PFA at room temperature for 20 min. After three washes with 1 × PBS, the cells were permeabilized using wash buffer (1 × PBS with 0.1% Triton X-100) for 5 min, and subsequently blocked with 1 × PBS containing 5% bovine serum albumin (BSA) for 1 h. The cells were then incubated overnight at 4 °C with the primary antibody, followed by incubation with secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 649. Finally, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) solution for 5 min and subjected to analysis using a confocal laser scanning microscope (Fluo View FV1000; Olympus, Tokyo, Japan).
Real-time quantitative RT-PCR (qRT-PCR)
Total RNA was extracted with TRIzol reagent (Invitrogen), and the concentration was detected by NanoDrop (Thermo Fisher Scientific). Quantitative RT-PCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories) on a Bio-Rad CFX96 Real-Time PCR System. A probe-based assay was performed to quantify viral RNA copy number by qPCR amplification of ZIKV-E gene. A standard curve was generated using serially diluted expression plasmids containing the coding sequence of ZIKV-E. mRNA expression levels of the indicated genes were normalized to the expression levels of GAPDH as an endogenous reference control.
The following primers were used:
Human CYP17A1 forward: 5′-GGACCTAGTCCCCTGGTTGA-3′
Human CYP17A1 reverse: 5′-CACTCCGGAATTTCTCCTTGT-3′
Human GAPDH forward: 5′-ATGACATCAAGAAGGTGGTG-3′
Human GAPDH reverse: 5′-CATACCAGGAAATGAGCTTG-3′
Mouse CYP17A1 forward: 5′-TTCGCCTGGGTACCACAACTGC-3′
Mouse CYP17A1 reverse: 5′-TAGAGTCACCATCTGGGGCCGA-3′
Mouse CYP11A1 forward: 5′-GGGGCAACAAGCTGCCCTTCAA-3′
Mouse CYP11A1 reverse: 5′-TGCAGGGTCATGGAGGTCGTGT-3′
Mouse GAPDH forward: 5′-TGTGTCCGTCGTGGATCTGA-3′
Mouse GAPDH reverse: 5′-TTGCTGTTGAAGTCGCAGGAG-3′
Mouse STAR forward: 5’-AAAGCCAGCAGGAGAACGGGGA-3’
Mouse STAR reverse: 5′-GCCTCCATGCGGTCCACAAGTT-3′
Mouse 3β-HSD forward: 5’-GCGGCTGCTGCACAGGAATA-3′
Mouse 3β-HSD reverse: 5′-GACGCATGCCTGCTTCGTGA-3′
ZIKV-E forward: 5′-TGAYAAGCARTCAGACAC-3′
ZIKV-E reverse: 5′-TCACCARRCTCCCTTTGC-3′
Western blotting analysis
The total amount of sample protein is determined by BCA Protein Assay Kit. The samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 μm pore size polyvinylidene difluoride (PVDF) membranes (Sigma-Aldrich) as described previously41.
Microscale thermophoresis technology (MST)
MST-experiments were performed using Monolith NT.115 (Blue/Red) instrument (NanoTemper Technologies GmbH, Germany). The cell lysates of HEK293T cells expressing GFP-NS2A were used as a source of fluorescently labeled NS2A. HEK293T cells were transfected with GFP-fused NS2A and lysed 24 h after transfection. To evaluate binding of CYP17A1 mRNA to NS2A, cell lysates were diluted with MST buffer (10 mM Na-phosphate buffer, pH 7.4, 1 mM MgCl2, 3 mM KCl, 150 mM NaCl, 0.05% Tween-20, and RNase Inhibitor) to provide optimal level of fluorescence. The lysates of GFP-transfected HEK293T cells were used as negative control. Cell lysates were incubated with titration series of CYP17A1 mRNA (0–100 mM). Samples were loaded into Premium Coated Capillaries, and MST measurements were performed using 20% MST power and 40% LED power at 25 °C. Laser-on and -off times were 30 and 5 s respectively. The MO Affinity Analysis v.1.6.1 software (NanoTemper Technologies) was used to fit the data and to determine the Kd values.
Electrophoretic mobility shift assay (EMSA)
EMSA-experiments were performed to evaluate the binding affinity of GST-NS2A to CYP17A1 mRNA. In brief, 20 ng biotinylated CYP17A1 mRNA was incubated with various concentration of recombinant GST-NS2A proteins in 50 μL binding buffer (50 mM Tris-HCl, pH 7.5,10 mM 2-mercaptoethanol, 2 mM MgCl2, 50 mM KCl) for 10 min at 24 °C, and the protein-RNA complex migrated in 0.4% agarose gel without sodium dodecyl sulfate (SDS) and was transferred to a BrightStar-Plus positively charged nylon membrane (Thermo Fisher Scientific) according to the instructions of the NorthernMax-Gly kit (Ambion). After ultraviolet cross-linking treatment (200 mJ/cm2 of 25 nm UV light), the nylon membrane was incubated with Streptavidin-HRP Conjugate and detected by chemiluminescence analysis.
Purification of GST recombinant protein
GST-NS2AWT, GST-NS2AE67A, GST-NS2AD73A, and GST-NS2AP101A were expressed in E. coli strain BL21 (DE3) at 18 °C overnight in the presence of 0.5 mM IPTG. The cells were pelleted down by centrifugation at 9000 × g for 10 min at 4 °C and resuspended in a lysis buffer [pH 7.5, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 400 mM NaCl, 1 mM EDTA, 10 U/mL Benzonase nuclease (Sigma-Aldrich), and EDTA-free protease inhibitor cocktail (Roche)], followed by ultrasonication at 4 °C. The lysates were clarified by centrifugation at 20,000 × g for 10 min. The supernatants were further centrifuged at 39,000 × g for 1.5 h in an SW40Ti rotor (Beckman Coulter) at 4 °C to pellet down the membrane fractions. Afterward, the pellet was resuspended in a solubilization buffer (pH 7.5, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 1 M NaCl, 1 mM EDTA, 1.5% DDM) with agitation for 2 h at 4 °C. After centrifugation at 39,000 × g for 30 min to remove undissolved debris, the proteins in the supernatant were purified by Glutathione Beads (Smart-Lifesciences) according to the manufacturer’s protocol, and the purified proteins were quantified by BCA protein assay Kit.
RNA-protein pulldown assay
CYP17A1 cDNAs and truncated constructs were transcribed in vitro using the HiScribe T7 ARCA mRNA Kit (New England Biolabs). Biotinylated CYP17A1 mRNA (CYP17A1 mRNA-biotin) was synthesized by Pierce™ RNA 3’End Desthiobiotinylation Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The biotinylated RNAs were purified using the Monarch RNA Cleanup Kits (New England Biolabs). Briefly, the RNA samples were transferred into an RNase-free tube and mixed with two volumes of RNA Cleanup Buffer. Subsequently, one volume of ethanol (≥95%) was added, and the mixture was thoroughly mixed by pipetting up and down or flicking the tube (do not vortex). The sample was loaded onto an RNA cleanup column placed in a collection tube, and the cap was closed. The column was spun for 1 min, and the flow-through was discarded. The column was re-inserted into the collection tube, and 500 μL of RNA Cleanup Wash Buffer was added. After a 1-min spin, the flow-through was discarded, and the wash step was repeated. The column was then transferred to an RNase-free tube. Finally, the purified RNAs were eluted in nuclease-free water and stored at −80 °C. RNA-protein pulldown assay was performed using the Pierce magnetic RNA-protein pulldown Kit (Thermo Fisher Scientific) according to the manufacturer’s guideline.
Quantification of ribosome loading on to CYP17A1 transcripts
Ribosome-loaded RNA was isolated essentially as previously described44. Briefly, CYP17A1 was co-transfected into HEK293T cells with ZIKV-NS2A, ZIKV-E, or empty vector as control. At 48 h post-transfection, cells were then treated with 100 μg/mL of cycloheximide for 10 min at 37 °C, washed in PBS and lysed in ribosome lysis buffer (10 mM Tris-HCl pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% Triton X-100, protease inhibitor, 2 mM DTT, 100 μg/mL cycloheximide and RNase inhibitor). One-tenth of the lysate was then used for RNA isolation for the “input” sample. The rest of the lysates were sheared by passage through a 26-gauge needle before clarification by centrifugation at 1300 × g for 10 min. The supernatants were brought up to 10 mL in lysis buffer and overlaid on a 30% sucrose cushion in lysis buffer, and centrifuged at 164,000 × g for 2 h at 4 °C. RNAs were then extracted from the ribosomal pellet using TRIzol. RNA isolated from the input and ribosome loaded fractions then underwent reverse transcription followed by qRT-PCR.
Photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP)
PAR-CLIP-experiments were performed according to the protocol as described previously57,71. Briefly, 4-thiouridine labeled CYP17A1 mRNA was prepared by adding 4-Thio-UTP (TriLink BioTechnologies) using the HiScribe T7 ARCA mRNA Kit (New England Biolabs). Recombinant GST-NS2A protein was expressed in E. coli strain BL21 (DE3). Ten μg of 4-thiouridine-labeled CYP17A1 mRNA was incubated with 5 μg recombinant GST-NS2A and then irradiated on ice with 150 mJ/cm2 at UV light at 365 nm. An aliquot of the same reaction without UV irradiation was used as the uncross-linked control. The RNA-protein mixtures were immunoprecipitated using BeyoMag Anti-GST Magnetic Beads (Beyotime). The beads were washed and resuspended in proteinase K digestion buffer (100 mM Tris, pH 7.5, 50 mM NaCl, 10 mM EDTA, and 1% SDS) supplemented with proteinase K at a final concentration of 2 mg/mL. Subsequently, the RNA in the reaction was purified using the Monarch RNA Cleanup Kits (New England Biolabs) and resuspended in nuclease-free water. The extracted RNA was reverse transcribed, amplified, and the PCR product was separated on 10% agarose gels, followed by purification using E.Z.N.A. Gel Extraction Kit (Omega BioTek). The purified PCR product was turned into a cDNA library using a TransNGS Tn5 Plasmid DNA Library Prep Kit (TransGen Biotech) and subjected to sequencing using the Illumina MiSeq platform. The indexed sequencing reads of both forward and reverse strands were proceeded with the following bioinformatic pipelines: first, an initial quality filtering was conducted with Fastp to remove low quality reads. Then, the remaining reads underwent adapter trimming with TrimGalore 0.6.10 automaticly (https://github.com/FelixKrueger/TrimGalore/). Subsequently, a paired-end alignment was conducted using the Bowtie2(2.5.2), followed by sorting using SAMtools v1.4 with command line parameters sort, index, and mpileup. Finally, mutation calling for variant positions was performed using varscan (https://github.com/dkoboldt/varscan/)(--min-var-freq0--min-avg-qual10), and the results were visualized using the R4.2.3 with ggplot2 (3.4.4).
Untargeted metabolism analysis
Male A129 mice were infected with ZIKV to form the ZIKV group, while mice administered 1 × PBS served as the Mock group. At 10 days post-infection, all mice were euthanized, and testes were collected for metabolomic analysis (n = 8 per group). Briefly, 20 mg of the testis sample was thawed on ice and homogenized in 400 μL of ice-cold methanol: water (70%, V/V). The homogenized mixtures were incubated on ice for 15 min and centrifuged at 4 °C, 12,000 g for 10 min. The supernatant (300 μL) was collected, incubated at −20 °C for 30 min, and then centrifuged again at 4 °C, 12,000 × g for 3 min. The final supernatant was collected for LC-MS/MS analysis. In brief, the sample extracts were analyzed using a LC-MS/MS system (Waters Acquity SM-FTN-H; Applied Biosystems, TripleTOF 6600 + ) with a Phenomenex Kinetex C18 column (2.6 µm, 2.1 mm × 100 mm). Column temperature, flow rate, and injection volume were set 40 °C, 0.3 mL/min, and 2 μL, respectively. Mobile phase was composed of water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The gradient program initiated from 5% B increased to 95% B in 11.0 min, and held for 5 min, and then decreased 5% B for re-equilibrium. For the quality control (QC) of metabolomic analysis, we pipette 10 μL of each sample to pool a QC sample. When running sample sets on column, one QC sample was injected after 8 samples in the sequence.
Data acquisition was performed using the information-dependent acquisition (IDA) mode, implemented with Analyst TF 1.7.1 software (Sciex). The source operation parameters were as follows: ion source gas 1 and gas 2 were set at 55 psi, curtain gas at 35 psi, and source temperature at 500 °C for positive ion mode and 450 °C for negative ion mode. The declustering potential was configured to 80 V for positive ion mode and −80 V for negative ion mode, while the ion spray voltage was set to 5500 V for positive and −4500 V for negative ion modes.
Data processing and quantification were carried out using the Analytics module of SCIEX OS 3.0 software (AB SCIEX, USA). For metabolite identification, MS/MS spectra were matched to the SCIEX Metabolite_HRMS/MS library (version 1.0), with additional annotations obtained from public databases including HMDB and MetDNA2. The mass tolerance values applied for both MS1 and MS2 ions during metabolite identification were 5ppm. MS-DIAL software also used for metabolite annotation with library ESI (+/−)-MS/MS from authentic standards. The mass tolerance value used in MS-DIAL for MS1 was 0.01 Da and for MS2 was 0.025 Da. Minimum peak height was 1000 amplitude, and mass slice width was 0.1 Da. Peaks with a detection rate below 80% in any sample group were excluded from further analysis.
Finally, statistical analysis was carried out by the R program, with both univariate analysis (Student’s t test) and multivariate analysis (principal component analysis, PCA, and orthogonal partial least-squares discriminant analysis, OPLS-DA). The PCA was employed to visualize the clustering of metabolite profiles and to assess the intrinsic variation between ZIKV-infected and Mock-infected groups. The OPLS-DA model was evaluated with the relevant R2 and Q2, which assess the model’s goodness of fit and predictive ability, respectively. VIP (Variable Importance in Projection) values were extracted from the OPLS-DA results. Only metabolites with VIP > 1 and P value < 0.05 were considered statistically significant. Metabolites with significant differences were subjected to KEGG pathway enrichment analysis using MetaboAnalyst v.4.0, with metabolic pathways with P value < 0.05 deemed significant. Volcano plots were used to filter metabolites of interest which based on log2(FC) and -log10(P-value) of metabolites.
iTRAQ-LC-MS/MS analysis
The iTRAQ experiment was performed in triplicates, with each replicate containing three testicular samples from a Mock-infected mouse, a ZIKV-infected mouse (6 dpi), and a ZIKV-infected mouse (10 dpi). In each replicate, the Mock group was compared with both the 6 dpi and 10 dpi ZIKV-infected groups. Total proteins were extracted from the testicular samples. The testes samples were homogenized in lysis buffer (2.5% SDS, 100 mM Tris-HCl, pH 8.5). The lysates were boiled for 15 min and then sonicated in an ice-water bath to denature the proteins, shear the DNA, and enhance cell disruption. After centrifugation at 4 °C, 12,000 × g for 30 min, the protein in the supernatant was precipitated with four volumes of cold acetone at 4 °C overnight. After centrifugation, the protein pellets were washed twice in cold acetone, air-dried, and dissolved in a buffer consisting of 8 M urea and 100 mM Tris-HCl, pH 8.0. The protein samples were reduced with 10 mM DL-dithiothreitol reducing agent at 37 °C for 1 h and alkylated with 40 mM iodoacetamide at room temperature for 1 h in the dark. Protein concentrations were determined by the Bradford method. Then, the urea concentrations in the samples were reduced to <2 M with 100 mM Tris-HCl (pH 8.0). The proteins were digested overnight at 37 °C with trypsin at an enzyme/protein ratio of 1:50 (w/w). The digestion was stopped by adding TFA until the pH reached 6.0. The supernatant of the digest was subjected to peptide purification using a Sep-Pak C18 desalting column, and the eluate was vacuum dried and stored at −20 °C until use. For iTRAQ labeling, 100 μg of each trypsin-digested peptide sample was reconstituted in iTRAQ reagent buffer, and separately labeled with different channels of 4plex-iTRAQ labeling kit: Mock-infected mouse was labeled with iTRAQ-114, ZIKV-infected mouse for 6 days (6 dpi) with iTRAQ-116, ZIKV-infected mouse for 10 days (10 dpi) with iTRAQ-117. The labeled samples in each replicate were mixed together, desalted using a C18 column, and vacuum dried. Each replicate mixture was fractionated by high pH reverse chromatography using a C18 5 μm 4.6 × 150 mm column (TechMate) on an Agilent 1260 HPLC operated at flow rate of 0.2 mL/min. Buffer A (10 mM ammonium formate, pH 10.0) and Buffer B (10 mM ammonium formate, 80% ACN, pH 10.0) were used to establish the separation gradient. For each replicate, 60 fractions were collected and merged into 15 fractions. Each fraction was vacuum dried and stored at −80 °C.
The LC-MS/MS analysis was performed on a TripleTOF 5600 mass spectrometer (Sciex), as described previously in ref. 72. All mass spectrometry analyses were conducted by SpecAlly Life Technology Co., Ltd, Wuhan, China. In brief, samples were reconstituted in 0.1% formic acid and injected by an autosampler onto a trapping column (C18, 5 µm, 5 × 0.3 mm) for desalting, followed by injection onto a C18 analytical column (75 μm × 150 mm, 3 μm particle size, 100 Å pore size, Eksigent/Sciex). Samples were run using a 100 min gradient from 5% to 80% solvent B (solvent A: 3% DMSO, 97% H2O, 0.1% formic acid; solvent B: 3% DMSO, 97% ACN, 0.1% formic acid), with a flow rate of 300 nL/min. The mass spectrometer was operated in information-dependent acquisition mode with an MS survey range of 350–1500 amu, followed by MS/MS scans of the 40 most intense ions. Dynamic exclusion was set for a period of 18 s and a tolerance of 120 cps. The MS/MS spectra were acquired in high sensitivity mode with the rolling collision energy and iTRAQ reagent collision energy adjustment on.
MS raw data were analyzed with ProteinPilot V4.5 using the Paragon database search algorithm (Sciex). Spectra files were searched against the mouse protein sequence database downloaded from UniProt using the following parameters: Sample Type was iTRAQ 4plex (peptide labeled); Cys alkylation was set to iodoacetamide; digestion was set to trypsin; quantitate was set to bias correction; background correction was enabled for specific processing; search effort was set to Rapid ID. Mass tolerance parameters were automatically set and optimized by ProteinPilot software (for the SCIEX TripleTOF 5600 data, the precursor mass tolerance was set to 0.05 Da and the fragment mass tolerance was set to 0.1 Da). A strict cutoff for an unused protein score ≥ 1.3 was used as the filter criterion for protein identification, which corresponds to less than 1% FDR on protein level (as indicated by ProteinPilot software). Protein quantification matrix was calculated by averaging the protein ratios in 3 replicates of Protein Summary table. P values of T test were calculated using log2-transformed ratios from 3 replicates. Significantly differentially expressed proteins (DEPs) were determined by the P value from Student’s t-test and fold change thresholds. Functional annotations were performed using Gene Ontology (GO), the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. A Fisher’s Exact Test was performed to reveal annotations and pathways. Hierarchical clustering in the heatmap was performed on log2-transformed and normalized intensities. The clusterProfiler package (v3.14.3) was employed for functional enrichment analysis.
RNA immunoprecipitation (RIP) assay
The cells were harvested and subjected to RNA-binding protein immunoprecipitation (RIP) with the RIP Kit (Genecreate) using specific antibodies or IgG isotype control, according to the manufacturer’s instructions. The precipitated RNA was reverse transcribed into cDNA and subjected to qRT-PCR analysis.
Generation of ZIKV mutants
The infectious clone of ZIKV FSS13025 was kindly provided by Dr. Pei-Yong Shi. All mutants (E67A and P101A) were introduced into infectious clone of FSS13205 individually by using a Mut Express II Fast Mutagenesis Kit V2 (Vazyme Biotech). Briefly, the plasmid is amplified by reverse PCR using a pair of primers (P101A-Forward: 5′-AATTGGACAGCCCGTGAGAGCATGCTGCTGGC-3′, P101A-Reverse: 5′-TCACGGGCTGTCCAATTAGCTCTGAAAATGAAA-3′, E67A-Forward: 5′-CTTCGCGGCAATGAACACTGGAGGAGATGTTGC, E67A-Reverse: 5′-TGTTCATTGCCGCGAAGGTGGCACCCATCAAA-3′), followed by digestion of the amplification product using Dpn1, and cyclization recombination reaction of the amplification product using Exnase II. The recombinant product is transformed into DH5α competent cells and the correct clone is selected. The substitution sites were confirmed by DNA sequencing. The infectious clone plasmids were linearized with restriction enzyme ClaI (engineered immediately downstream of the 3′ end of the viral genomic sequence) digestion and purified using the Gel Extraction Kit (Omega). In vitro transcribed genome-length RNA was prepared using HiScribe T7 ARCA mRNA Kit (New England Biolabs) and purified using Monarch RNA Cleanup Kit (New England Biolabs). The RNA was then transfected into Vero cells using Lipofectamine 3000 reagent (Thermo Fisher Scientific) to generate the virus. Six hours later, culture fluids were replaced with fresh medium to remove extracellular input RNAs. After incubation at 37 °C for 24 h, cells were cultured in DMEM with 2% FBS at 30 °C for another 4 days. At various time points, the culture fluids from viral RNA-transfected cells were collected and filtered with 0.22 μm filter, then stored at −80 °C. Extracellular viral titers were determined by plaque assay on BHK-21 cells. Further, viral antigen expression in ZIKV RNA-transfected Vero cells was detected with Confocal microscopy. The culture fluids were used to extract extracellular viral RNA using the QIAamp viral RNA minikit (Qiagen). The total cellular RNAs were extracted by RNeasy minikit (Qiagen). Quantitative RT-PCR was performed according to the protocol described previously in ref. 47.
Statistical analyses
All analyses were performed using GraphPad Prism software (Version 8.0). Measurement data are presented as the mean ± s.d. Statistical differences of normally distributed data were determined using one-way analysis of variance (ANOVA) or with two-way ANOVA statistical tests, followed by Tukey’s post hoc test. P values less than 0.05 were considered significant. ns not significant. Quantitation of western blotting was measured by Image J software (1.52p/java 1.8.0 172).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The full-genome sequencing data for ZIKVWT, ZIKVE67A, and ZIKVP101A generated in this study have been deposited to the GenBank database under accession number: ZIKVWT (PQ849442), ZIKVE67A (PQ849443), and ZIKVP101A (PQ849444). The mass spectrometry proteomics data have been submitted to the ProteomeXchange Consortium with the dataset identifier PXD055433, with differentially expressed proteins detailed in Supplementary Data 1. Metabolomics data have been deposited in the MetaboLights database under the accession number MTBLS12549 [https://www.ebi.ac.uk/metabolights/reviewerb80bfff7-4d2c-4882-a1f0-17859a1ed9d4], and the output results are provided in Supplementary Data 2. Source data are provided with this paper.
References
Petersen, L. R., Jamieson, D. J., Powers, A. M. & Honein, M. A. Zika virus. N. Engl. J. Med. 374, 1552–1563 (2016).
Pierson, T. C. & Diamond, M. S. The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581 (2018).
D’Ortenzio, E., Matheron, S. & Yazdanpanah, Y. Evidence of sexual transmission of Zika virus. N. Engl. J. Med. 374, 2195–2198 (2016).
Govero, J. et al. Zika virus infection damages the testes in mice. Nature 540, 438–422 (2016).
Ma, W. et al. Zika virus causes testis damage and leads to male infertility in mice. Cell 168, 542–542 (2017).
Kurscheidt, F. A. et al. Persistence and clinical relevance of Zika virus in the male genital tract. Nat. Rev. Urol. 16, 211–230 (2019).
Calvet, G. A. et al. Study on the persistence of Zika virus (ZIKV) in body fluids of patients with ZIKV infection in Brazil. BMC Infect. Dis. 18, 49 (2018).
Joguet, G. et al. Effect of acute Zika virus infection on sperm and virus clearance in body fluids: a prospective observational study. Lancet Infect. Dis. 17, 1200–1208 (2017).
Ma, W. et al. Zika virus causes testis damage and leads to male infertility in mice. Cell 167, 1511–1524 e1510 (2016).
Matusali, G. et al. Zika virus infects human testicular tissue and germ cells. J. Clin. Investig. 128, 4697–4710 (2018).
Mlera, L. & Bloom, M. E. Differential Zika virus infection of testicular cell lines. Viruses 11, 42 (2019).
Smith, L. B. & Walker, W. H. The regulation of spermatogenesis by androgens. Semin. Cell Dev. Biol. 30, 2–13 (2014).
Wang, R.-S., Yeh, S., Tzeng, C.-R. & Chang, C. Androgen receptor roles in spermatogenesis and fertility: lessons from testicular cell-specific androgen receptor knockout mice. Endocr. Rev. 30, 119–132 (2009).
Walker, W. H. Non-classical actions of testosterone and spermatogenesis. Philos. Trans. R. Soc. B Biol. Sci. 365, 1557–1569 (2010).
Sar, M., Lubahn, D. B., French, F. S. & Wilson, E. M. Immunohistochemical localization of the androgen receptor in rat and human tissues. Endocrinology 127, 3180–3186 (1990).
Walker, W. H. Testosterone signaling and the regulation of spermatogenesis. Spermatogenesis 1, 116–120 (2011).
Griswold, M. D. The central role of Sertoli cells in spermatogenesis. Semin. Cell Dev. Biol. 9, 411–416 (1998).
Robaire, B. & Hamzeh, M. Androgen action in the epididymis. J. Androl. 32, 592–599 (2011).
Odonnell, L., McLachlan, R. I., Wreford, N. G., deKretser, D. M. & Robertson, D. M. Testosterone withdrawal promotes stage-specific detachment of round spermatids from the rat seminiferous epithelium. Biol. Reprod. 55, 895–901 (1996).
Wong, C. H. et al. Regulation of ectoplasmic specialization dynamics in the seminiferous epithelium by focal adhesion-associated proteins in testosterone-suppressed rat testes. Endocrinology 146, 1192–1204 (2005).
Zirkin, B. R., Santulli, R., Awoniyi, C. A. & Ewing, L. L. Maintenance of advanced spermatogenic cells in the adult rat testis: quantitative relationship to testosterone concentration within the testis. Endocrinology 124, 3043–3049 (1989).
Liu, L.-B. et al. Zika virus infection leads to hormone deficiencies of the hypothalamic-pituitary-gonadal axis and diminished fertility in mice. J. Virol. 97, e0100623 (2023).
Clark, B. J. & Stocco, D. M. Steroidogenic acute regulatory protein: the StAR still shines brightly. Mol. Cell. Endocrinol. 134, 1–8 (1997).
Payne, A. H. & Hales, D. B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25, 947–970 (2004).
BurgosTrinidad, M. et al. Repression of cAMP-induced expression of the mouse P450 17 alpha-hydroxylase/C17-20 lyase gene (Cyp17) by androgens. Mol. Endocrinol. 11, 87–96 (1997).
Payne, A. H. & Youngblood, G. L. Regulation of expression of steroidogenic enzymes in Leydig cells. Biol. Reprod. 52, 217–225 (1995).
Li, L. et al. Hormone regulation in testicular development and function. Int. J. Mol. Sci. 25, 5805 (2024).
Olive, A. J. & Sassetti, C. M. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat. Rev. Microbiol. 14, 221–234 (2016).
Pang, H. et al. Aberrant NAD+ metabolism underlies Zika virus-induced microcephaly. Nat. Metab. 3, 1109–1124 (2021).
Miller, W. L. & Auchus, R. J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 (2011).
Van Den Akker, E. L. T. et al. Differential inhibition of 17α-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J. Clin. Endocrinol. Metab. 87, 5714–5721 (2002).
Kardelen, A. D. et al. A rare cause of congenital adrenal hyperplasia: clinical and genetic findings and follow-up characteristics of six patients with 17-hydroxylase deficiency including two novel mutations. J. Clin. Res. Pediatr. Endocrinol. 10, 206–215 (2018).
Ma, L. Z. et al. Combined 17 alpha-hydroxylase/17,20-lyase deficiency with short stature: case study. Gynecol. Endocrinol. 32, 264–266 (2016).
Danila, D. C. et al. TMPRSS2-ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration-resistant prostate cancer patients treated with abiraterone acetate. Eur. Urol. 60, 897–904 (2011).
Grist, E. & Attard, G. The development of abiraterone acetate for castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 33, 289–294 (2015).
Thibault, C. & Massard, C. New therapies in metastatic castration resistant prostate cancer. Bull. Cancer 102, 501–508 (2015).
Shi, Y. & Gao, G. F. Structural biology of the Zika virus. Trends Biochem. Sci. 42, 443–456 (2017).
Zhang, X. et al. Zika virus NS2A-mediated virion assembly. Mbio 10, e02375–19 (2019).
Zhang, Z. et al. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 354, 1597–1600 (2016).
Ren, W. et al. Zika virus NS5 protein inhibits type I interferon signaling via CRL3 E3 ubiquitin ligase-mediated degradation of STAT2. Proc. Natl Acad. Sci. USA 121, e2403235121 (2024).
Hui, L. et al. Matrix metalloproteinase 9 facilitates Zika virus invasion of the testis by modulating the integrity of the blood-testis barrier. PLos Pathog. 16, e1008509 (2020).
Zirkin, B. R. & Papadopoulos, V. Leydig cells: formation, function, and regulation. Biol. Reprod. 99, 101–111 (2018).
Jerabek-Willemsen, M. et al. MicroScale thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 1077, 101–113 (2014).
Subtelny, A. O., Eichhorn, S. W., Chen, G. R., Sive, H. & Bartel, D. P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508, 66 (2014).
Macarthur, M. W. & Thornton, J. M. Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412 (1991).
Zhou, X. et al. Mutations linked to neurological disease enhance self-association of low-complexity protein sequences. Science 377, eabn5582 (2022).
Zhang, X. et al. Genetic and biochemical characterizations of Zika virus NS2A protein. Emerg. Microbes. Infect. 8, 585–602 (2019).
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Miner, J. J. & Diamond, M. S. Zika virus pathogenesis and tissue tropism. Cell Host Microbe 21, 134–142 (2017).
Zeng, J. et al. The Zika virus capsid disrupts corticogenesis by suppressing dicer activity and miRNA biogenesis. Cell Stem Cell 27, 618–623.e9 (2020).
Zhou, J. et al. Zika virus degrades the omega-3 fatty acid transporter Mfsd2a in brain microvascular endothelial cells and impairs lipid homeostasis. Sci. Adv. 5, eaax7142 (2019).
Yoon, K. J. et al. Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell 21, 349–358 e346 (2017).
Guo, M., Hui, L., Nie, Y., Tefsen, B. & Wu, Y. ZIKV viral proteins and their roles in virus-host interactions. Sci. Chin. Life Sci. 64, 709–719 (2021).
de La Vega, M.-A. et al. Zika-induced male infertility in mice is potentially reversible and preventable by deoxyribonucleic acid immunization. J. Infect. Dis. 219, 365–374 (2019).
Vossmann, S., Wieseler, J., Kerber, R. & Kuemmerer, B. M. A basic cluster in the N terminus of yellow fever virus NS2A contributes to infectious particle production. J. Virol. 89, 4951–4965 (2015).
Xie, X., Gayen, S., Kang, C., Yuan, Z. & Shi, P.-Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 87, 4609–4622 (2013).
Xie, X. et al. Dengue NS2A protein orchestrates virus assembly. Cell Host Microbe 26, 606–602.e8 (2019).
Zhang, L. et al. AMFR-mediated flavivirus NS2A ubiquitination subverts ER-phagy to augment viral pathogenicity. Nat. Commun. 15, 9578 (2024).
Garneau, N. L., Wilusz, J. & Wilusz, C. J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8, 113–126 (2007).
Warf, M. B. & Berglund, J. A. Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem. Sci. 35, 169–178 (2010).
Kozak, M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361, 13–37 (2005).
Wan, Y., Kertesz, M., Spitale, R. C., Segal, E. & Chang, H. Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 12, 641–655 (2011).
Ramanathan, M., Porter, D. F. & Khavari, P. A. Methods to study RNA-protein interactions. Nat. Methods 16, 225–234 (2019).
Castello, A., Fischer, B., Hentze, M. W. & Preiss, T. RNA-binding proteins in Mendelian disease. Trends Genet. 29, 318–327 (2013).
Hentze, M. W., Castello, A., Schwarzl, T. & Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 (2018).
Mackenzie, J. M., Khromykh, A. A., Jones, M. K. & Westaway, E. G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 245, 203–215 (1998).
Qiu, Y. et al. Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci. Adv. 6, eaax7989 (2020).
Nie, Y. et al. Rearrangement of actin cytoskeleton by Zika virus infection facilitates blood-testis barrier hyperpermeability. Virol. Sin. 36, 692–705 (2021).
Teng, Y. et al. Novel function of SART1 in HNF4α transcriptional regulation contributes to its antiviral role during HBV infection. J. Hepatol. 75, 1072–1082 (2021).
Sun, J., Zhong, L., Zhu, Y. & Liu, G. Research on the isolation of mouse Leydig cells using differential digestion with a low concentration of collagenase. J. Reprod. Dev. 57, 433–436 (2011).
Hafner, M. et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141 (2010).
Yang, N. et al. Identification and characterization of proteins that are differentially expressed in adipose tissue of olanzapine-induced insulin resistance rat by iTRAQ quantitative proteomics. J. Proteomics 212, 103570 (2020).
Acknowledgements
This work was supported by the National Key Research and Development Plan of China (2021YFC2300200 to Ying Wu) and the National Natural Science Foundation of China (32122008, 82472277 to Ying Wu).
Author information
Authors and Affiliations
Contributions
W.Y. and H.L. performed the experiments. S.W., R.H., Y.Z., M.G., R.Y., S.L., D.Z., Y. X., Y.L., M.H., and W.X. analyzed the results. Li.H. and Lx.H. contributed significantly to the metabolomics experiments and data analysis. Y.W. initiated and designed the project and directed the research. W.Y., H.L., and Y.W. wrote the manuscript. All authors discussed the results and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gilberto Domont, Philippe Despres, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, W., Li, H., Wang, S. et al. Zika virus disrupts steroidogenesis and impairs spermatogenesis by stalling the translation of CYP17A1 mRNA. Nat Commun 16, 6756 (2025). https://doi.org/10.1038/s41467-025-62044-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62044-x
