
Abstract
The mechanisms underlying clear cell renal cell carcinoma (ccRCC) metastasis remain largely unexplored. We demonstrate that Deleted in Split hand/Split foot protein 1 (DSS1), a critical cofactor of BRCA2 in DNA repair, is upregulated in metastatic ccRCC and promotes both tumor growth and distant metastasis. Mechanistically, DSS1 interacts with LC3 and promotes its degradation via TRIM25-mediated Lys63 (K63)-linked polyubiquitination at LC3B-K51. This impairs (macro) autophagic flux and leads to p62 accumulation, thereby stabilizing TWIST1 and facilitating its nuclear translocation, ultimately activating epithelial-mesenchymal transition (EMT). DSS1 highly expressed (DSS1hi) tumor cells are enriched in late-stage tumors and are associated with microvascular invasion within a vascularized invasive niche at the tumor-stromal interface, mediated by SPP1-ITGB1 interactions. Clinically, DSS1hi tumor cells correlate with therapeutic resistance and poorer patient outcomes. Collectively, these findings provide new insights into the mechanisms of ccRCC metastasis and suggest potential avenues for therapeutic intervention.
Similar content being viewed by others


Introduction
Clear cell renal cell carcinoma (ccRCC) accounts for approximately 70% of the kidney tumors1. Patients with distant metastasis have a 5-year survival rate of less than 10%2. Many patients experience substantial drug toxicity or develop resistance following cytokine-based or targeted therapies, including mechanistic target of rapamycin (mTOR) inhibitors3,4 and vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitors (TKIs)2. Although combination therapies involving VEGFR-TKIs and immune checkpoint inhibitors show promise in improving survival, the benefits remain modest5 and only a limited number of patients respond6,7, making the treatment of metastatic ccRCC particularly challenging.
Although many tumor cells can be eliminated during treatment, surviving cancer cell subclones may undergo reprogramming to acquire epithelial-mesenchymal transition (EMT) characteristics, thereby facilitating metastasis8,9. This highlights EMT as a fundamental process through which cancer cells gain metastatic potential10,11. Moreover, tumor metastasis relies on the support of stromal cells (e.g., fibroblasts and endothelial cells) and immune cells (e.g., T cells and macrophages), as reducing this support significantly diminishes the invasive capacity of tumor cells12,13. Recent advances in the study of the tumor microenvironment (TME) have revealed that EMT-enriched tumor niches are spatially localized at the tumor margins8,14. However, the mechanisms by which TME components are organized to promote distant metastasis remain poorly understood. Therefore, dissecting highly invasive niches at the levels of molecular mechanisms and intercellular communication is essential to uncovering the cellular and molecular basis of metastatic ccRCC.
Deleted in Split hand/Split foot protein 1 (DSS1), initially identified as an autosomal dominant candidate gene for split-hand/foot malformation15, was subsequently found to bind directly to breast cancer susceptibility gene 2 (BRCA2), where it plays a critical role in regulating R-loop-associated DNA damage and transcription-related genomic instability16. More recently, DSS1 has been implicated in mRNA transport, RNA splicing, and protein degradation15. DSS1 has also been found to be upregulated in glioblastoma and associated with poor prognosis, although its functional role in this context remains unclear17. Here, we identify DSS1 as a driver gene in ccRCC metastasis. DSS1 interacts with LC3 to promote its ubiquitin-mediated degradation, resulting in impaired (macro)autophagic flux, p62 accumulation, and TWIST1 stabilization, thereby triggering EMT. Distinct from SPP1⁺ macrophages in hypoxic regions18,19, DSS1 highly expressed (DSS1hi) tumor cells engage microvascular cells via SPP1-ITGB1 signaling within a vascularized invasive niche at the tumor-stroma interface, thereby promoting metastasis. The identification of DSS1hi tumor cells may advance our understanding of the mechanisms underlying metastasis in advanced ccRCC.
Results
DSS1 upregulation is associated with tumor metastasis in ccRCC
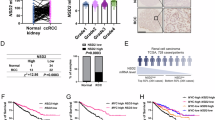
Integrative analysis identified DSS1 as a metastasis-associated gene in ccRCC (Fig. 1a, b). Comparative analysis demonstrated aberrant upregulation of both DSS1 mRNA (Fig. 1c, Supplementary Fig. 1a) and DSS1 protein levels (Fig. 1e) in advanced ccRCC relative to early-stage tumors and normal kidney tissues. This upregulation may not be influenced by patient sex, as no significant sex-specific differences were observed between tumor and normal tissues (Supplementary Fig. 1b). Elevated expression of DSS1 was an unfavorable factor for patient survival of ccRCC (Fig. 1d, f), with no sex-specific differences observed (Supplementary Fig. 1c). DSS1 was broadly upregulated across multiple cancer types and associated with poorer patient survival (Supplementary Fig. 1d, e, Supplementary Fig. 2a, b).

a Schematic workflow for identifying DSS1 as a metastasis-associated driver gene in ccRCC. We leveraged Catalogue of Somatic Mutations in Cancer (COSMIC) cancer gene census due to its expert-curated cancer hallmark annotations (e.g., invasion, metastasis) from experimental evidence. b Scatter plot showing DSS1/SEC31B mRNA levels (log2 transformed count per million [log2CPM]) vs. wound healing scores in TCGA-KIRC cohort (n = 578 distinct samples, two-sided Spearman’s rank correlation test). Dashed lines: linear regression fit; Shaded area: the 95% confidence interval. c DSS1 mRNA expression in ccRCC versus adjacent normal tissues across datasets (n = 560 distinct samples, two-tailed Welch’s t-test). Boxplot: Center line = median; box = 25th to 75th percentiles; whiskers = minima to maxima. d Kaplan-Meier survival curves for DSS1 expression (DESeq2 log2CPM) in TCGA-KIRC cohort (n = 522 distinct patients, Log-rank test). e Immunohistochemistry (IHC) of DSS1 protein in ccRCC tumor and paired normal tissues (n = 74 distinct patients; boxes: zoom-in regions; scale bar: 200×, 100 μm, 400×, 50 μm; error bar: mean (centre) ± Standard Deviation [SD]; two-tailed Welch’s t-test). ISUP, the International Society of Urologic Pathologists. f Survival analysis stratified by DSS1 IHC levels in the in-house cohort (n = 74 distinct patients; Log-rank test). g, i Representative images of metastatic lung lesions from xenograft mouse models (BALB/c-nu, male, 7 weeks before harvest, tail vein injection) using Caki-1 cells (g) or of 786-O cells (i). Scale bar: lung, 5 mm; 5×, 2 mm; 200×, 50 μm. Boxes: zoom-in regions. h, j Quantification of metastatic foci number and total lesion area for Caki-1 based models (h; Lentivirus [Lv]-shNC as control) or 786-O based models (j; Lv-Vector as control). k Subcutaneous tumors (axillary fossa injection) from Caki-1 cells-based mouse models (BALB/c-nu, male, Lv-shNC as control, 4 weeks before harvest). l, m Tumor growth curves (l) and final weights (m) of subcutaneous tumors. Lv-shNC as control. g–m n = 6 mice per group, Error bar: mean ± SD, two-tailed Welch’s t-test. Statistics are provided in the source data. Source data are provided as a Source Data file.
DSS1 expression was associated positively with wound healing scores (Fig. 1b), based on analysis of previously reported gene signatures20,21. To assess the role of DSS1 in regulating the in vivo migratory capacity of ccRCC cells, we injected Caki-1 cells, derived from a cutaneous metastasis of ccRCC, stably expressing either a DSS1 knockdown construct Lentivirus (Lv)-shDSS1 or a negative control (Lv-shNC) into nude mice via the tail vein. Among the shRNAs tested, DSS1 shRNA#3, which exhibited the highest knockdown efficiency, was selected for use in this and subsequent experiments (Supplementary Fig. 2c). Mice in the Lv-shDSS1 group exhibited a marked reduction in both the number and volume of lung metastatic nodules compared to the Lv-shNC group seven weeks post-injection (Fig. 1g, h). To further evaluate whether DSS1 enhances the metastatic potential of non-metastatic ccRCC cells, we selected 786-O cells, which were originally derived from a primary ccRCC tumor. 786-O cells have previously been shown to successfully establish lung metastasis models in vivo4,22. Importantly, DSS1 expression in 786-O cells is lower than that in Caki-1 cells (Supplementary Fig. 2d). To establish metastatic mouse models, we injected 786-O cells stably expressing either the DSS1 overexpression construct (Lv-DSS1) or the empty vector control (Lv-Vector) via the tail vein. The efficacy of DSS1 overexpression is shown in Supplementary Fig. 2e. Mice injected with Lv-DSS1 cells developed a greater number and larger volume of lung metastatic nodules compared to those injected with Lv-Vector cells, as assessed seven weeks after injection (Fig. 1i, j).
The sustained proliferative capacity of cancer cells is essential for tumor progression. DSS1 expression was positively associated with a proliferation-related gene signature (Supplementary Fig. 3a). Cell Counting Kit-8 (CCK-8) and EdU assays demonstrated that DSS1 knockdown suppressed the proliferation of ccRCC cells, whereas DSS1 overexpression enhanced their proliferative capacity (Supplementary Fig. 3b–d). To further evaluate the role of DSS1 in tumorigenicity in vivo, subcutaneous xenograft models were established using Caki-1 cells in nude mice. Compared with the Lv-shNC control group, DSS1 knockdown resulted in the formation of smaller, slower-growing tumors, supporting a pro-tumorigenic role for DSS1 (4 weeks post-injection, Fig. 1k−m).
DSS1 promotes migration and invasion of ccRCC cells by blocking autophagic flux
Pathway enrichment analysis suggested that autophagy may be negatively regulated by DSS1 in ccRCC metastasis, as indicated by a normalized enrichment score (NES) of −1.69 in the kidney renal clear cell carcinoma cohort from The Cancer Genome Atlas (TCGA-KIRC, Fig. 2a). This hypothesis was supported by Gene Set Enrichment Analysis (GSEA, DSS1_hi vs. DSS1_lo, median as a cut-off) of three additional datasets, including GSE25190523 (NES = −1.81), GSE353824 (NES = −1.49), GSE25456625 (NES = −1.20, Supplementary Fig. 4a). In contrast, GSEA results for the mTOR pathway showed inconsistent NES directions across datasets: TCGA-KIRC (NES = −1.30) and GSE251905 (NES = −1.64) showed negative enrichment, whereas GSE3538 (NES = 0.99) and GSE254566 (NES = 1.00) showed positive enrichment (Supplementary Fig. 4a). These discrepancies suggest that DSS1 expression is unlikely to be consistently associated with the mTOR pathway. Since GSEA was based on transcriptomic profiles, we further investigated the correlation between DSS1 expression and the RNA levels of autophagy-related genes. We performed RT-qPCR on 37 genes commonly contributing to autophagy enrichment. Among these, 12 genes were significantly upregulated in DSS1-knockdown cells and are known to regulate key stages of autophagy, including initiation (TRAF6, ULK2), elongation/maturation (TBK1, SMCR8), PI3K-III complex and nucleation (ATG14, STX17, UVRAG), LC3 lipidation (ATG4C, ATG7), and autophagosome-lysosome fusion (STX17, SNAP29, NBR1, CALCOCO2) (Supplementary Fig. 4b). Together, these transcriptomic and experimental findings support a potential inhibitory role of DSS1 in the regulation of autophagy.

a Schematic diagram illustrating KEGG autophagy pathway negatively associated with DSS1 expression (DESeq2 log2CPM). b Transmission electron microscopy (TEM, left) images and quantification (right) of autophagic vacuoles (arrows) in ACHN and Caki-1 cells transfected with siDSS1 or siNC (n = 3 independent experiments; scale bars: 8000×, 2 μm, 20000×, 0.5 μm; boxes: zoom-in regions; error bar: mean ± SD; two-tailed Welch’s t-test, Benjamini-Hochberg [BH] adjustment). c Confocal microscopy images of LC3 puncta (magenta) and LAMP1 (turquoise) co-localization in cells treated with siNC, siDSS1, with or without chloroquine (CQ: 10 μM, control: 0.02% DMSO, 24 h before harvest; n = 3 independent experiments; scale bar: 10 μm; error bar: mean ± SD; two-tailed Welch’s t-test, BH adjustment). COSMIC, Catalogue of Somatic Mutations in Cancer. Statistics are provided in the source data. Source data are provided as a Source Data file.
Transmission electron microscopy revealed an increased accumulation of autophagic vacuoles in DSS1-silenced ccRCC cells (Fig. 2b). DSS1 knockdown also led to a significant increase in LC3 puncta formation and LC3-LAMP1 colocalization, both in the presence and absence of chloroquine (CQ), a lysosomal acidification inhibitor that blocks autophagosome degradation (Fig. 2c). These findings support the role of DSS1 as a negative regulator of autophagic flux. Furthermore, Transwell assays demonstrated that DSS1 silencing markedly suppressed the invasive and migratory abilities of ccRCC cells, whereas DSS1 overexpression enhanced both processes (Fig. 3a, b). However, in the presence of CQ, the differences in invasion and migration between DSS1-silenced and control cells were no longer significant (Fig. 3a, b), suggesting that DSS1 modulates invasion and migration in an autophagy-dependent manner. Morphometric analysis further showed that control Caki-1 cells exhibited a typical spindle-shaped morphology, while DSS1-silenced cells displayed a rounded, pebble-like appearance, indicative of reduced migratory and invasive potential (Fig. 3c).

a, b Transwell migration and Matrigel invasion assays in ACHN and Caki-1 cells treated with shNC, shDSS1, pcDNA3.1, DSS1 plasmids, with or without CQ (Chloroquine, representative images; scale bar: 100 μm). Quantification of migrated/invaded cells (error bar: mean ± SD; two-tailed Welch’s t-test; n = 3 independent experiments). c Morphology images of ccRCC cells between DSS1 knockdown (pebble-shaped) and negative control (spindle-shaped; scale bar: 100 μm; error bar: mean ± SD; two-tailed Welch’s t-test). The average cell aspect ratio was determined from 100 randomly selected cells per group per experiment (n = 3 independent experiments). d, e Immunoblotting (IB) of autophagy markers (LC3-II, p62) and EMT proteins (E-cadherin, N-cadherin, Vimentin) in siNC- and siDSS1-treated (with or without CQ, 0.02% DMSO as control) cells. Endogenous control: β-actin. Densitometry quantification (e, n = 3 independent experiments, error bar: mean ± SD, two-tailed Welch’s t-test). The samples derived from the same experiment were run on parallel gels, with each gel probed for a different antibody. f RT-qPCR analysis of MAP1LC3B and SQSTM1 mRNA levels in siDSS1 vs. siNC cells (normalized to GAPDH; n = 3 independent experiments; error bar: mean ± SD; two-tailed Welch’s t-test). Statistics are provided in the source data. Source data are provided as a Source Data file.
We next investigated the mechanism by which DSS1 blocks autophagic flux and promotes cell migration and invasion. DSS1 silencing increased LC3-II protein levels regardless of CQ treatment, suggesting that DSS1 depletion enhances LC3 induction rather than impairs autophagosome clearance (Fig. 3d, e). In addition, DSS1 knockdown led to upregulation of E-cadherin and downregulation of p62, Vimentin, and N-cadherin (Fig. 3d, e). However, in the presence of CQ, the levels of these proteins did not differ significantly between DSS1-silenced and control cells (Fig. 3d, e), indicating that the observed changes of EMT markers are dependent on functional autophagic flux. Moreover, a low correlation was observed between DSS1 mRNA levels and those of MAP1LC3B or SQSTM1 (Supplementary Fig. 3e). Consistently, no significant differences in MAP1LC3B or SQSTM1 transcript levels were detected between DSS1-silenced cells and controls (Fig. 3f). These findings suggest that DSS1 inhibits autophagic flux in ccRCC cells by suppressing LC3-II induction, thereby promoting EMT activation.
DSS1 promotes EMT activation by regulating TWIST1 in ccRCC
Next, we investigated how DSS1-mediated autophagy suppression leads to EMT activation. Transforming growth factor-β (TGF-β), a canonical upstream inducer of EMT, has also been reported to inhibit autophagy26. We therefore examined whether DSS1 modulates TGF-β expression. However, TGF-β levels were not altered in DSS1-silenced ccRCC cells compared to negative controls (Supplementary Fig. 5a, b). Given that the protein levels of E-cadherin, N-cadherin, and Vimentin were affected by autophagy inhibition (Fig. 3d, e), we hypothesized that DSS1 may promote EMT by regulating EMT-inducing transcription factors, such as ZEB1/2, SNAI1/2, and TWIST1. To identify transcription factors potentially involved in the DSS1-autophagy-EMT regulatory axis, we developed a Consensus Scoring of Genes in Cascade (CSGC) algorithm (Fig. 4a). Candidate transcription factors were expected to meet three criteria: (1) upregulated in ccRCC or metastatic ccRCC; (2) positively correlated with both EMT activity and DSS1 expression; and (3) negatively correlated with autophagy activity. The activities of EMT and autophagy in ccRCC samples were quantified using single-sample gene set enrichment analysis (ssGSEA) based on established gene signatures (Fig. 4a). Among the screened factors, TWIST1 was identified as the top candidate.

a Identification of TWIST1 as a potential downstream effector of DSS1-autophagy axis. Gene signatures (Molecular Signature DataBase v2022.1) were analyzed by single-sample gene set enrichment analysis (ssGSEA) in TCGA-KIRC (n = 522 distinct tumors). b Fold changes (DESeq2) of transcription factor (TF) mRNA expression in metastatic (Met.) vs. primary ccRCC and primary ccRCC vs. normal kidney (TCGA-KIRC). c Immunoblotting of EMT-TFs (ZEB1, ZEB2, SNAI1, SLUG/SNAI2, FOXC2, TCF3) in shDSS1 vs. shNC cells (two-tailed Welch’s t-test, Benjamini-Hochberg [BH] adjustment). β-actin: endogenous control. d Immunoblotting showing TWIST1 protein levels in cells treated with siNC/siDSS1, autophagy inhibitor (CQ, 10 μM; Control: 0.02% DMSO; 24 h before harvest), or both (two-tailed Welch’s t-test). e Immunoblotting showing TWIST1 levels in cells treated with siNC, TWIST1 knockdown, pcDNA3.1 or TWIST1 plasmids (two-tailed Welch’s t-test). f Rescue experiments showing protein levels of EMT markers in ccRCC cells transfected with different combinations of control, DSS1, and TWIST1 siRNAs, and DSS1 and TWIST1 plasmids (two-tailed Welch’s t-test). g Transwell assays following the same treatments as in panel (f) (scale bar: 100 μm, two-tailed Welch’s t-test). h Immunohistochemistry using lung metastatic lesions of xenograft mouse models generated by tail vein injection of Caki-1 cells treated with Lv-shDSS1 or control Lv-shNC (n = 5 independent samples per group; scale bar: 100 μm; two-tailed Welch’s t-test). IOD, Integrated Optical Density. i Caki-1 cells were analyzed by immunoprecipitation with antibody to the LC3 or TWIST1 epitope, followed by SDS-PAGE and immunoblotting (IgG: negative control, input: 5% lysate). j Immunofluorescence microscopy showing the co-localization of Bcl-2 (magenta), p62 (orange), and TWIST1 (turquoise) in ACHN cells treated with DSS1 knockdown or control (scale bar: 5 μm). DAPI (blue): nucleus. c–f The samples derived from the same experiment were run on parallel gels, with each gel probed for a different antibody. c–g, i, j n = 3 independent experiments. c–h Error bars: mean ± SD. Statistics are provided in the source data. Source data are provided as a Source Data file.
Previous studies have reported that both TWIST1 mRNA and TWIST1 protein levels are upregulated in ccRCC27. Immunoblotting confirmed that DSS1 silencing reduced TWIST1 protein levels (Fig. 4d), while the expression of other EMT-related transcription factors that are commonly upregulated in ccRCC or metastatic ccRCC remained unaffected (Fig. 4b, c). Notably, in the presence of CQ, TWIST1 levels were comparable between DSS1-silenced and control cells (Fig. 4d), indicating that DSS1 modulates TWIST1 protein levels through autophagy regulation. To further assess the role of TWIST1 in DSS1-mediated EMT, we performed rescue experiments. TWIST1 silencing in DSS1-overexpressing cells led to increased E-cadherin levels and decreased N-cadherin and Vimentin levels (Fig. 4e, f). Conversely, TWIST1 overexpression in DSS1-silenced cells reversed these effects, resulting in decreased E-cadherin and increased N-cadherin and Vimentin expression (Supplementary Fig. 5c, d). In contrast, TWIST1 overexpression abolished the effects of DSS1 knockdown on these EMT markers, with no significant changes observed in E-cadherin, N-cadherin, or Vimentin levels between DSS1-silenced and control groups (Fig. 4f). Functionally, TWIST1 silencing suppressed the invasive and migratory capabilities of DSS1-overexpressing ccRCC cells, whereas DSS1 knockdown had no significant impact on these behaviors in TWIST1-overexpressing cells (Fig. 4g). These findings indicate that DSS1 promotes EMT, migration, and invasion through upregulation of TWIST1. Consistently, analysis of lung metastatic lesions from xenograft mouse models (tail vein injection of Caki-1 cells) revealed that DSS1 knockdown resulted in lower p62 and TWIST1 protein levels, alongside increased LC3 and E-cadherin levels, compared to lesions from control mice (Fig. 4h). Together, these in vitro and in vivo findings demonstrate that DSS1 suppresses autophagy to promote EMT via TWIST1 regulation.
We next investigated how DSS1-mediated autophagy suppression regulates TWIST1 protein levels. Co-immunoprecipitation (Co-IP) assays did not detect an interaction between LC3 and TWIST1 (Fig. 4i), suggesting that TWIST1 regulation is unlikely to involve selective autophagy. Instead, it may depend on p62-mediated stabilization, as previous studies have shown that p62 accumulation inhibits TWIST1 degradation28. In addition, Bcl-2, a known upstream regulator of both autophagy and apoptosis, has been reported to interact with TWIST1, facilitating its nuclear translocation and promoting angiogenesis and metastasis29. Interestingly, Bcl-2 protein levels were reduced in DSS1-knockdown ccRCC cells, whereas Beclin-1 levels remained unchanged (Supplementary Fig. 5a, b). To explore whether Bcl-2 and p62 are involved in DSS1-mediated EMT activation, we performed immunofluorescence analyses. Compared to control cells, DSS1 knockdown reduced both the nuclear co-localization of Bcl-2 and TWIST1, and the cytoplasmic co-localization of p62 and TWIST1 (Fig. 4j, Supplementary Fig. 5e). These findings suggest that DSS1 suppresses autophagy to promote p62 accumulation, thereby stabilizing TWIST1 and enhancing its association with Bcl-2. This facilitates TWIST1 nuclear translocation and contributes to EMT activation.
DSS1 interacts with LC3 and promotes TRIM25-mediated polyubiquitination degradation of LC3
We next investigated the mechanism by which DSS1 inhibits LC3-II induction. As mTOR is a central regulator of autophagy initiation and the autophagic lysosome reformation cycle30, we examined whether DSS1 modulates autophagy by altering mTOR activity. However, the protein levels of total mTOR and phosphorylated mTOR (p-mTOR) remained unchanged between DSS1-knockdown and control ccRCC cells (Supplementary Fig. 5a, b), indicating that DSS1 suppresses autophagy via an mTOR-independent pathway. LC3 is a key protein involved in the elongation of phagophores and the maturation of autophagosomes31. Given that DSS1 knockdown increased LC3-II levels (Fig. 3d, e), and that DSS1 is a known component of the 19S proteasome regulatory particle complex, we hypothesized that DSS1 facilitates the degradation of ubiquitinated LC3. Co-IP assays demonstrated a physical interaction between DSS1 and LC3 (Fig. 5a, b). This interaction was further validated by glutathione-S-transferase (GST) pulldown assays using recombinantly expressed human proteins (Fig. 5c). GST-tagged DSS1 migrated at ~38 kDa on SDS-PAGE, consistent with previous reports32. Recombinant full-length human LC3B (pro-LC3B) migrated at ~14 kDa, as pro-LC3B runs similarly to LC3B-II in SDS-PAGE33.

a Co-Immunoprecipitation (Co-IP) with antibody to the FLAG epitope, followed by SDS-PAGE and immunoblotting with antibody to LC3 (endogenous, IgG: isotype control; Input: 5% lysate). Short/long: exposure time of the same membrane. b Co-IP with antibody to the LC3 epitope, followed by SDS-PAGE and immunoblotting showing endogenous DSS1. a, b IgG light chain-specific secondary antibody was used. c Glutathione-S-Transferase (GST)-pulldown analysis for DSS1-LC3B interaction using 50 ng recombinantly expressed human full-length LC3B and 1 mg GST/GST-DSS1 (GST: negative control; Purified LC3B: positive control; Glutathione agarose beads: 50 µL). d Cells were lysed with EDTA (10 mM) or not, followed by metal bath at 95 °C for 5 min and immunoblotting with antibody to DSS1. e GST-pulldown analysis showing the interaction of LC3 and DSS1 mutant using 50 ng recombinantly expressed human LC3 and 1 mg GST-DSS1W27GW39GW43GF52A (Glutathione agarose beads: 50 µL). f Caki-1 cells were transfected with plasmids encoding HA-DSS1W27GW39GW43GF52A. Cell lysates and cell immunoprecipitants were immunoprecipitated with antibody to the LC3 epitope and HA epitope, respectively. g Knockdown of DSS1 or treatment with MG-132 (20 μM, 6 h before harvest) elevates LC3 protein levels in HEK293T cells (control: siNC and/or 0.02% DMSO, error bar: mean ± SD, two-tailed Welch’s t-test). h Immunoblotting showing the indicated protein levels in HEK293T cells transfected with control or DSS1 siRNA, and/or treated with 0.02% DMSO or MG-132 (20 μM, 6 h before harvest). i Immunoblotting of cycloheximide (CHX, 50 μg/mL) chase assay in shDSS1 cells compared to shNC cells (error bar: mean ± SD, two-tailed Welch’s t-test). c–e, g–i The samples derived from the same experiment were run on parallel gels, with each gel probed for a different antibody. a–i n = 3 independent experiments. Statistics are provided in the source data. Source data are provided as a Source Data file.
Interestingly, human DSS1 (70 amino acids; predicted molecular weight ~8 kDa) exhibited aberrant migration on SDS-PAGE, appearing at ~18 kDa, ~37 kDa, and higher molecular weights34,35. This aberrant migration is partially attributed to its highly acidic nature, which impairs SDS binding36 (theoretical isoelectric point [pI] ~3.81, calculated using the ExPASy Compute pI/Mw tool: https://web.expasy.org/compute_pi/). The additional bands may represent oligomeric forms or adducts of DSS1. A sharp ~37 kDa DSS1 band was consistently detected in anti-LC3 immunoprecipitates (Fig. 5b). DSS1 has previously been shown to form SDS-resistant oligomers and adducts with cellular proteins, mediated by four critical residues (W27, W39, W43, and F52), which are also implicated in ubiquitination-related degradation34. The 37 kDa oligomeric form has been observed in both endogenous and purified preparations of DSS1, independent of redox status or metal ion availability34,35. Reactive oxygen species (ROS), induced by UV or H₂O₂ treatment, promote DSS1 adduct formation, whereas scavengers such as EDTA or vitamin C suppress adduct formation. Notably, these treatments do not affect the levels or migration pattern of the DSS1 oligomer34. To assess whether the 37 kDa band observed in anti-LC3 IP products represents a DSS1 adduct, we treated cells with EDTA. However, EDTA treatment did not eliminate the 37 kDa band (Fig. 5d), suggesting it is not a ROS-mediated adduct. Furthermore, GST pulldown assays using purified LC3B and mutant GST-DSS1W27GW39GW43GF52A confirmed that LC3B can still bind to DSS1 in the absence of the adduct-forming residues (Fig. 5e), consistent with co-immunoprecipitation results from ccRCC cells (Fig. 5f). Taken together, the ~37 kDa DSS1 band likely represents a DSS1 oligomer, as it lacks features of glycosylation or ubiquitination (e.g., smearing), shows no evidence of methylation, acetylation, or lipidation (e.g., subtle mass shifts), and remains resistant to reductants (e.g., DTT)34 and ROS scavengers.
DSS1 knockdown or treatment with MG-132, a proteasome inhibitor, in HEK293T cells led to increased levels of both total LC3 and ubiquitinated LC3B (Fig. 5g, h), indicating that LC3 degradation was inhibited. In contrast, MG-132 treatment equalized the levels of ubiquitinated LC3 and restored LC3 protein abundance, as proteasomal degradation was blocked regardless of DSS1 expression (Fig. 5g, h). These findings suggest that DSS1 knockdown impairs LC3 degradation, resulting in the accumulation of ubiquitinated LC3. To further strengthen the mechanistic link between DSS1-mediated autophagy suppression and LC3 degradation, we assessed LC3 protein stability following treatment with cycloheximide (CHX), a protein synthesis inhibitor. Time-course analyses revealed that LC3 degradation was significantly slower in DSS1-knockdown cells compared to controls (Fig. 5i). We next examined the subcellular localization of DSS1-LC3 interactions using immunofluorescence. PSMD3 (also known as Rpn3), a subunit of the 19S regulatory particle of the proteasome and a known DSS1 interactor37, was used as a proteasomal marker. Co-localization of DSS1 and LC3 with PSMD3 was observed predominantly in the cytoplasm (Fig. 6a, Supplementary Fig. 6a). Collectively, these results demonstrate that DSS1 binds to LC3 and promotes its degradation via the ubiquitin-proteasome system.

a Representative images of immunofluorescence showing the subcellular localization of DSS1 (magenta), LC3 (yellow), and PSMD3 (azure) in ACHN cells (n = 3 independent experiments). Nuclei: DAPI (blue). Scale bar: 5 μm. b LC-MS/MS analysis of LC3 interactomes identifies E3 ligases potentially associated with LC3 (n = 3 independent experiments; Proteome Discoverer 2.4, Thermo Scientific; False discovery rate <0.01). c The mRNA expression of TRIM25 in ccRCC patients from the TCGA-KIRC dataset (n = 595 distinct samples, tumor vs. normal, two-tailed Welch’s test). Boxplot: Center line = median; box = 25th to 75th percentiles; whiskers = minima to maxima. d Representative images of immunohistochemistry analysis showing TRIM25 protein levels upregulated in ccRCC tissues vs. normal (n = 66 distinct pairs; scale bar: 50 μm; error bar: mean (centre) ± SD; two-tailed paired t-test). e, f Immunoblotting showing TRIM25 knockdown increases LC3 levels, while TRIM25 overexpression reduces LC3 levels (n = 3 independent experiments, error bar: mean ± SD, two-tailed Welch’s t-test). g Co-IP analysis in Caki-1 cells showing the interaction between endogenous TRIM25 and LC3 (n = 3 independent experiments). h GST-pulldown analysis for TRIM25-LC3B interaction using 50 ng recombinantly expressed human full-length LC3B and 1 mg GST-TRIM25 (n = 3 independent experiments; Glutathione agarose beads: 50 µL). Asterisk: GST-TRIM25. i Immunoblotting of HEK293T cells transfected with HA-Ub, pcDNA3.1, and Myc-LC3B (wildtype) or Myc-LC3B mutant plasmids (n = 3 independent experiments). j Immunoblotting of HEK293T cells transfected with Myc-LC3B and HA-Ub-wildtype or HA-Ub-K48R/K63R mutant plasmids (n = 3 independent experiments). k Recombinantly expressed human GST-TRIM25 was incubated with recombinant Ub, LC3B, UbcH5a/UbcH5c, and His-UBE1 in a reaction system containing MgATP at 37 °C for 4 h. Samples were analyzed by SDS-PAGE and immunoblotting with antibody to Ub epitope (n = 3 independent experiments). As shown in the result of GST-TRIM25, the polyubiquitination of LC3B (LC3B-Ub) is shown only when all the components are present in the mix. e, f, h–j The samples derived from the same experiment were run on parallel gels, with each gel probed for a different antibody. Statistics are provided in the source data. Source data are provided as a Source Data file.
We next investigated the mechanism by which LC3 is ubiquitinated. LC-MS/MS analysis of anti-LC3 immunoprecipitates identified two candidate E3 ubiquitin ligases: RNF213 and TRIM25 (Fig. 6b). TRIM25, but not RNF213, exhibited a negative correlation with LC3 protein levels based on immunohistochemistry (IHC) data from renal cancer tissues in the Human Protein Atlas (HPA) database38 (Supplementary Fig. 6b). TRIM25 was significantly upregulated in ccRCC, as shown by both TCGA-KIRC transcriptomic data and our IHC analysis (Fig. 6c, d), with no observed sex-specific differences (Supplementary Fig. 6c). Functionally, TRIM25 knockdown increased LC3 protein levels (Fig. 6e), while TRIM25 overexpression decreased LC3 levels (Fig. 6f), indicating that TRIM25 promotes LC3 degradation. Co-IP confirmed that TRIM25 interacts with LC3 (Fig. 6g), and this interaction was further validated by GST pulldown assays using recombinantly expressed human TRIM25 and LC3B (Fig. 6h).
To identify the ubiquitination site on LC3B, we individually mutated each of its ten lysine (K) residues to arginine (R), which cannot be ubiquitinated (Supplementary Fig. 6d). Among these, only the K51R mutation abolished ubiquitination, indicating that lysine 51 is the primary site of polyubiquitination on LC3B (Fig. 6i). Furthermore, LC3B ubiquitination was reduced by K63R, but not K48R mutant ubiquitin, suggesting that TRIM25 mediates K63-linked polyubiquitination of LC3B (Fig. 6j). To directly test whether TRIM25 catalyzes LC3B polyubiquitination, we performed an in vitro ubiquitination assay using recombinant LC3B, ubiquitin, E1 (His-UBE1), E2 (UbcH5a/UbcH5c)39,40, a source of ATP (MgATP), and GST-tagged human TRIM25 purified from E. coli. Immunoblot analysis of the complete reaction mixture with recombinant GST-TRIM25 revealed high-molecular-weight bands consistent with polyubiquitinated LC3B, as detected by anti-ubiquitin antibody (Fig. 6k). Together, these findings demonstrate that TRIM25 directly mediates K63-linked polyubiquitination of LC3B at lysine 51.
Functional states and clinical value of DSS1-driven cells in ccRCC
To evaluate the clinical significance of DSS1-driven cells in ccRCC, we analyzed single-cell RNA sequencing (scRNA-seq) data from 49 ccRCC samples across three independent datasets. After quality control, a total of 201,810 cells were retained and integrated (Fig. 7a). Following batch correction and cell-type annotation, we identified 30,662 epithelial cells (EPCAM and KRT8), 22,180 fibroblasts (ACTA2 and MYL9), 18,821 endothelial cells (ENG and CLEC14A), 2768 B cells (BANK1 and MS4A1), 933 plasma cells (MZB1 and TNFRSF17), 83,987 T/NK cells (CD2, CD3, NKG7, and GZMK), 39,758 myeloid cells (CD68 and FCGR3A), and 2701 mast cells (KIT and VWA5A, Fig. 7b, c). To identify malignant epithelial cells, we isolated the epithelial population for re-clustering and copy number variation (CNV) analysis using the inferCNV algorithm. Prior to CNV estimation, four clusters (5893 cells) expressing marker genes from multiple lineages (e.g., immune cells) were removed as likely doublets. The analysis identified 11,501 malignant epithelial cells (Fig. 7d, e). At single-cell resolution, DSS1 expression was significantly higher in malignant epithelial cells compared to normal kidney epithelial cells (Fig. 7f).

a Quality control for single-cell RNA-seq data from ccRCC tissues (n = 49 distinct samples). b, c Uniform Manifold Approximation and Projection (UMAP) of cells of ccRCC tissues before (b) and after batch effect correction (c). Right panel: annotation of major cell types. d, e Identification of malignant cells using inference of copy number variation (inferCNV, CNV score threshold: 0.04) of epithelial cells. Del, deletion; Amp amplification. f Comparison of DSS1 pseudo-bulk expression between malignant cells (tumor samples) and normal epithelial cells (normal samples, error bar: mean ± SD, n = 47 distinct samples with detected DSS1 RNA; two-tailed Welch’s t-test). g DSS1 amplification correlates with higher DSS1 expression (DESeq2 log2CPM) in tumors (n = 513). h Functional module analysis for malignant cells. Upper graph: hierarchical clustering dendrogram of genes; lower graph: gene module (x-axis, different genes). i UMAP showing DSS1-driven ccRCC cells and other malignant cells. j Comparison of the epithelial-mesenchymal transition (EMT)/autophagy module scores between DSS1-driven cells and other malignant cells. KEGG v113.0, Kyoto Encyclopedia of Genes and Genomes. k Proportion of DSS1-driven cells across different stages in treatment-naive patients (I-IV: n = 1, 6, 6, 3 patients, respectively). l DSS1-driven cell signature scores in patients with different grades and pathologic stages (n = 521). m Comparison of DSS1-driven cell signature scores across molecular subtypes defined by Clinical Proteomic Tumor Analysis Consortium (CPTAC) in TCGA-KIRC dataset (n = 495). n High DSS1-driven cell signature predicts poorer survival (Log-rank test, n = 506 and 509, respectively). HR, Hazard Ratio. o–q Differences in signature scores of DSS1-driven cells between treatment-naive/response and treatment/resistance groups (VEGFR inhibitors) in ccRCC patients or patient-derived xenograft (PDX) models (left to right, TCGA-KIRC, n = 430; GSE65615, n = 122; GSE64052, n = 28; GSE76068, n = 16). MUT, mutant; WT, wildtype. g, j, l, m, o–q Two-tailed Mann-Whitney U test. Box plots (Turkey style): Center line = median; box = 25th to 75th percentiles; whiskers = ±1.5×interquartile range; Violin plots: data distribution (minima to maxima) via kernel density estimation. Statistics are provided in the source data. Source data are provided as a Source Data file.
DSS1 is located on chromosome 7q21.3, and consistent with prior genomic studies41 gains of chromosome 7 were observed. To examine whether DSS1 expression is affected by CNV status, we analyzed matched transcriptomic and genomic profiles. Samples harboring DSS1 amplification showed significantly elevated DSS1 expression compared to those without amplification (Fig. 7g), with no significant sex-specific differences (Supplementary Fig. 6e). Notably, DSS1 amplification and its associated transcriptional upregulation were recurrent across 32 human cancer types (Supplementary Fig. 6f), underscoring its potential as a broadly relevant oncogenic driver.
Given the high degree of heterogeneity among malignant cells, we applied high-dimensional weighted gene coexpression network analysis (hdWGCNA) to characterize their functional states (Fig. 7h). This analysis revealed 14 coexpression modules associated with diverse biological processes, including cell metabolism (e.g., ALDHs, FABPs), EMT (e.g., LAMC2, VIM, FN1, COL1A1), and programmed cell death (e.g., LC3B) (Fig. 7h, Supplementary Fig. 7a). We identified 1,403 DSS1-driven malignant cells (referred to as DSS1hi) with elevated DSS1 expression—defined as at least 50% higher than the average expression in normal epithelial cells (Fig. 7i, Supplementary Fig. 7c). Compared to other malignant cells, DSS1hi cells exhibited significantly higher DSS1 levels (log₂ fold change = 2.08, P = 0, two-tailed Mann-Whitney U test). Functionally, DSS1hi cells showed higher EMT module scores and lower autophagy scores than other malignant cells (Fig. 7j). In patients with metastatic ccRCC (stage IV), DSS1hi cells had significantly elevated EMT and wound healing scores compared to those from non-metastatic cases (Fig. 7j, Supplementary Fig. 7b). Additionally, with disease progression, DSS1hi cells displayed increased cell cycle activity, modestly enhanced angiogenic signatures, and reduced programmed cell death compared to other malignant cells (Supplementary Fig. 7b). In treatment-naive patients, the frequency of DSS1hi cells was significantly higher in stage IV tumors than in earlier-stage tumors (Fig. 7k), further underscoring their potential role in tumor progression and metastasis.
Given the accessibility and clinical relevance of bulk RNA sequencing data, we investigated the significance of DSS1-driven cells across bulk ccRCC cohorts. Marker genes of DSS1hi cells were used to calculate signature scores for each bulk sample. Consistent with single-cell findings, higher signature scores were associated with poorer tumor differentiation (Grade), larger tumor size (T), and the presence of distant metastasis (M) (Fig. 7l). When stratified by immune subtype42, the signature score was highest in the CD8⁻ inflamed subtype, which was characterized by elevated endothelial, fibroblast, and macrophage infiltration, high EMT, oxidative phosphorylation, and angiogenesis, as well as frequent chromosome 7 gains. Scores were also elevated in the CD8⁺ inflamed subtype, which exhibits high immune checkpoint expression and interferon response. In contrast, scores were lowest in the metabolic/VEGF immune-desert subtype, marked by high MTORC1 signaling, HIF-1/glycolysis activity, and angiogenesis (Fig. 7m).
To assess the prognostic relevance, patients were classified into high- and low-score groups based on the median signature score. High-score patients had significantly worse survival outcomes (Fig. 7n). It is expected that signature scores were associated with chemotherapy, as DSS1 was demonstrated to be a critical protein of DNA repair, such as double-strand breaks43 (Fig. 7o). Signature scores were significantly higher in patients resistant to anti-VEGFR therapy than in those with progression-free disease, independent of Von Hippel-Lindau (VHL) mutation status (Fig. 7o). These results were validated in an independent cohort44, where patients treated with sunitinib exhibited higher DSS1 signature scores than treatment-naive patients, again independent of VHL mutation status (Fig. 7p). Consistently, RCC cell lines (with VHL mutations)45 treated with anti-VEGFR agents showed enrichment of DSS1-driven transcriptional signatures compared to untreated cells (Fig. 7q). Moreover, in patient-derived xenograft (PDX) models46 treated with sunitinib (40 mg/kg/day orally for 4 weeks), tumors from non-responders had significantly higher levels of DSS1-driven cells than those from responders (Fig. 7q). These findings collectively suggest that DSS1-driven cells are associated with aggressive clinical features, resistance to anti-VEGFR therapies, and adverse patient outcomes.
To identify potential therapeutic strategies targeting DSS1-driven cells, we applied two predictive frameworks: beyondcell47 and scTherapy48. Drug sensitivity analysis using beyondcell revealed candidate compounds targeting key downstream effectors of DSS1, such as SQSTM1 (e.g., glucocorticoid receptor agonist prednisolone), Bcl-2 (e.g., CCR8 agonist ZK-756326), and TWIST1 (e.g., GABA receptor agonist NCS-382). However, no single compound was predicted to simultaneously target all three effectors (Supplementary Fig. 7d). In contrast, scTherapy analysis based on DSS1-driven cell transcriptomes identified several compound classes with potential efficacy, including proteasome inhibitors, histone deacetylase (HDAC) inhibitors, and autophagy activators (Supplementary Fig. 7d). These findings suggest that a therapeutic combination involving proteasome inhibition and autophagy activation may represent a rational strategy for selectively targeting DSS1-driven tumor cells.
DSS1-driven cells communicate with microvascular endothelial cells through invasion-related ligand-receptor interactions
To elucidate the spatial distribution and microenvironmental context of DSS1-driven cells, we analyzed spatial transcriptomics data from 38 ccRCC specimens. Cell type deconvolution revealed the spatial landscape of DSS1-driven and tumor microenvironment (TME) cell populations across different tumor stages (Fig. 8a, Supplementary Fig. 8). Among the endothelial subsets, microvascular endothelial cells were most frequently co-localized with DSS1-driven cells within the same spatial spots. In contrast, hypoxic, lymphatic, and inflammatory endothelial cells showed less co-occurrence (Fig. 8b, c).

a Cell type deconvolution of DSS1-driven cells using spatial transcriptome data from ccRCC tissues. b Spearman correlations of proportions between DSS1-driven cells and tumor microenvironment (TME) cells in spatial spots. c Correlations between the proportion of DSS1-driven cells and signature scores (AddModuleScore) of endothelial subsets in spots. Scores were rescaled (to a 0–1 range), then adjusted by dividing by the summed weights of TME cell types within a spot. d Re-clustering (single-cell RNA-seq) and annotation of endothelial cell subsets. e Cell-cell communication analysis between DSS1-driven cells (n = 1403 cells) and microvascular cells (n = 7916 cells, single-cell RNA-seq). f Correlations between DSS1-driven cell weights and coexpression strength of predicted ligand-receptor pairs in spots (pT1/pT2: n = 8 distinct tumors, pT3: n = 27 [21 distinct tumors with 1 slide, and 3 distinct tumors with 2 slides each], pT4/IV: n = 2 distinct tumors available, error bar: mean ± standard error of mean). Ligand-receptor pair coexpression (log-normalized counts) strength: rescaled (to 0-1) product of ligand L and receptor R expression (R1 + R2, if two receptors). g SPP1-integrin colocalizations in late-stage sample slides. h Multiplex immunohistochemistry showing expression and colocalization of ligand SPP1 from DSS1-driven cells (DSS1high and LC3low, and pan-CK+) and receptor ITGB1 of microvascular cells (CD105+) (n = 4 distinct patients; boxes: zoom-in regions; scale bar: 20 μm). i Quantification of DSS1-driven cells and colocalized puncta in ccRCC patient samples (n = 4 distinct patients). j Proportions of SPP1-ITGB1 interaction within 25, 50, or 75 μm radius of CD105 puncta. b, c Boxplot: Center line: median; box: 25th to 75th percentiles; whiskers: ±1.5×interquartile range. a–c, f, g. n = 38 (32 distinct tumors with 1 section each, 3 distinct tumors with 2 sections each). Source data are provided as a Source Data file.
The microvascular cells were identified in single-cell RNA-Seq data by markers such as FLT1 (VEGFR1), NOTCH4, and ESM1, and were distinguished from the broader endothelial compartment (Fig. 8d). To investigate potential intercellular communication between DSS1-driven and microvascular cells, we performed ligand-receptor interaction analysis using single-cell RNA-seq data. Among the identified ligand-receptor pairs, VEGF-VEGFR1/R2, SPP1-ITGA5/ITGB1, ANGPTL4-ITGA5/ITGB1, and FN1/COL4A1/COL4A2-SDC4 were recurrently identified (Fig. 8e). Coexpression of these ligand-receptor pairs was enriched in late-stage lesions, whereas early-stage lesions exhibited weaker interactions in spots (Fig. 8f). Notably, SPP1-ITGA5/ITGB1 interactions showed a consistent positive correlation with DSS1-driven cell abundance in advanced ccRCC (Fig. 8f). The SPP1-ITGB1 interaction, recurrently co-occurring with VEGF-VEGFR1/R2 interactions, was enriched at the tumor-normal interface (Fig. 8g, Supplementary Fig. 9a, b). ITGB1, a central member of the integrin family, mediates cell-extracellular matrix adhesion and intracellular signaling. Compared to SPP1-ITGA5, SPP1-ITGB1 interaction demonstrated stronger co-localization with DSS1-driven cells and higher coexpression intensity at the tumor boundary (Fig. 8g). Multiplex immunohistochemistry of ccRCC specimens confirmed that DSS1-driven tumor cells (DSS1hi, pan-CK+) exhibited reduced autophagy (LC3lo) and were enriched within a vascularized invasive niche at the tumor-stromal interface. These cells were in spatial proximity to CD105+ microvascular endothelial cells, with evidence of SPP1-ITGB1 interactions (Fig. 8h, boxes 1 and 2). With disease progression, both the proportion of DSS1-driven cells and the frequency of SPP1-ITGB1 interactions increased (n = 4 patients, Fig. 8i).
Spatial transcriptomic data revealed that spots enriched with DSS1-driven cells exhibited higher EMT scores and lower autophagy scores (Supplementary Fig. 9c). Consistently, multiplex immunohistochemistry demonstrated that DSS1-driven cells with reduced E-cadherin and elevated Vimentin levels were frequently located near CD31-positive vascular endothelial cells at the tumor-stromal boundary. This spatial arrangement suggests that the interaction between DSS1-driven cells and vascular endothelial cells involves the induction of EMT via DSS1 (n = 4 patients, Fig. 9a).

a Multiplex immunohistochemistry showing spatial distribution of DSS1-driven cells with a high epithelial-mesenchymal transition (EMT) feature (E-cadlow, Vimentinhigh) around vascular cells (CD31+, arrows, n = 4 distinct patients). Scale bar: 40 μm. b SPP1 RNA and SPP1 protein levels in tissues of diverse cancer types (kidney cancer: RNA, n = 35 distinct cell lines, protein, n = 110 distinct samples) from the Human Protein Atlas (HPA, www.proteinatlas.org). c SPP1 Protein levels in normal tissues from the HPA database (kidney, high expression in proximal tubules, n = 3 slides). d Pseudo-bulk RNA levels of SPP1 in epithelial and immune cell types. AvgExp, Average expression. e Protein abundance of SPP1 between tumor and normal tissues in Clinical Proteomic Tumor Analysis Consortium (CPTAC)-ccRCC dataset (n = 194 distinct samples, two-tailed Mann-Whitney U test). Boxplot: Center line = median; box = 25th to 75th percentiles; whiskers = minima to maxima. f Spearman correlation between SPP1 and DSS1 mRNA expression (log2 Count Per Million, two-tailed spearman’s rank correlation test). g Immunoblotting showing SPP1 protein levels in ccRCC cells (shDSS1 vs. shNC, n = 3 independent experiments, error bar: mean ± standard deviation [SD], two-tailed Welch’s t-test). The samples derived from the same experiment were run on parallel gels, with each gel probed for a different antibody. h Spearman correlation between CD68 expression and coexpression of SPP1-ITGB1 in spots of sample sections from either tumor core or tumor-stromal interface (Li2022 spatial dataset, n = 13 sections from 10 distinct patients; error bar: mean ± SD). i Schematic diagram (by Figdraw.com) illustrates the proposed mechanistic model of DSS1-driven cells in ccRCC metastasis: DSS1 (oligomer) interacts with pro-LC3B or LC3B-I, promoting LC3B degradation via E3 ubiquitin ligase TRIM25-mediated Lys-63 (K63)-linked polyubiquitination at LC3B-K51, leading to impaired macroautophagic flux and p62 accumulation, TWIST1 stabilization and increased TWIST1 nuclear transport, promoting EMT activation. DSS1 highly expressed (DSS1hi) tumor cells were increased in late-stage tumors and linked to microvascular invasion within a vascularized invasive niche at the tumor-stromal interface, mediated by SPP1-ITGB1 interactions. Statistics are provided in the source data. Source data are provided as a Source Data file.
SPP1 is a secreted phosphoprotein that modulates diverse forms of cell-cell signaling. Although both normal and cancerous kidney tissues exhibit high levels of SPP1 mRNA and SPP1 protein (Fig. 9b, c), the protein level of SPP1 is slightly reduced in kidney cancer tissues compared to normal tissues (Fig. 9e), with no sex-specific differences observed in expression patterns (Supplementary Fig. 9d). Correlation analysis revealed that DSS1 expression was not significantly associated with SPP1 expression (Fig. 9f), a finding consistent with immunoblot results showing that silencing DSS1 did not alter SPP1 protein levels in ccRCC cells compared to the negative control (Fig. 9g). Notably, more than 50% of the positive SPP1-ITGB1 interactions were located within a 50 µm radius of CD105-positive microvascular puncta, and over 70% were within a 75 µm radius (Fig. 8h, box 3; Fig. 8j), indicating that SPP1-ITGB1 interactions are spatially enriched around microvascular cells, despite no overall increase in SPP1 expression at the bulk-tissue level (Fig. 9e).
SPP1 expression was higher in DSS1-driven cells than in other cell types, including SPP1-positive tumor-associated macrophages (Fig. 9d). This raises the question of whether SPP1-secreting macrophages contribute to DSS1-driven niches. Previous studies have shown that SPP1-positive macrophages are enriched in hypoxic and necrotic tumor regions18,19, which are spatially distinct from the areas enriched in DSS1-driven cells (Fig. 8c, h; Fig. 9a) and from regions where SPP1-ITGB1 colocalization occurs (Fig. 9h), both of which are associated with vascularized niches. Furthermore, SPP1 puncta were detected in close proximity to DSS1-driven cells, suggesting that in these vascularized niches, SPP1 is likely secreted primarily by DSS1-driven cells rather than macrophages (Fig. 8h). Taken together, these observations suggest that DSS1 enhances the invasive and migratory capabilities of ccRCC cells by promoting local SPP1 expression within vascularized niches and facilitating spatial interactions with microvascular endothelial cells, thereby contributing to microvascular metastasis. This supports a mechanistic model in which DSS1-driven invasive niches play a critical role in ccRCC metastasis (Fig. 9i).
Discussion
Through integrative analyses of transcriptomic datasets, patient-derived tissues, and animal models, we demonstrate that DSS1 is upregulated in ccRCC and promotes metastatic progression. Mechanistically, DSS1 binds to LC3 and facilitates its proteasomal degradation via TRIM25-mediated Lys63 (K63)-linked polyubiquitination at lysine 51 of LC3B. This degradation impairs macroautophagic flux, resulting in the accumulation of p62 and stabilization of TWIST1, thereby enhancing its nuclear translocation and activating EMT. We further identified a distinct subset of ccRCC cells characterized by high DSS1 expression. These DSS1-driven cells were enriched in advanced-stage and metastatic tumors, as shown by single-cell transcriptomic, bulk RNA-Seq, spatial transcriptomics, and multiplex immunohistochemistry analyses. Notably, the frequency of DSS1-driven cells was inversely correlated with patient survival, suggesting their potential as a prognostic biomarker. Spatial profiling revealed that DSS1-driven cells preferentially reside at the tumor-stroma interface, where they engage in cell-cell communication with microvascular endothelial cells. These interactions are mediated by invasion-associated ligand-receptor pairs, including VEGF-VEGFRs and SPP1-ITGB1/ITGA5. Such spatially restricted signaling likely facilitates microvascular invasion and dissemination of tumor cells. Together, our findings delineate a mechanistic model in which DSS1 suppresses autophagy and activates EMT, thereby promoting a pro-metastatic cellular state in ccRCC. This model not only deepens our understanding of ccRCC progression but also identifies DSS1-driven cells as potential therapeutic targets for mitigating metastasis in advanced disease.
Autophagy plays a critical role in maintaining cellular homeostasis in renal tubular epithelial cells by eliminating damaged organelles and senescent proteins49. In tumor cells, autophagy can theoretically support survival by degrading intracellular components to generate metabolic substrates. However, autophagy appears to be highly niche-dependent in ccRCC. The tumor suppressor VHL contributes to autophagic homeostasis by promoting the ubiquitin-mediated degradation of LC3 through direct binding50. Loss of VHL function occurs in approximately 60% to 80% of ccRCC cases51. In hypoxic niches, VHL deficiency enables transient receptor potential melastatin 3 (TRPM3) to activate autophagy by stimulating AMPK and ULK1 signaling, thereby facilitating phagophore formation and supporting tumor cell survival52. In contrast, within vascularized niches, ccRCC cells secrete succinate, which induces autophagy in PDGFR-β⁺ GPR91⁺ pericytes. These pericytes, in turn, release metabolites that nourish adjacent tumor cells53. Our findings suggest that DSS1 may function as an alternative effector in the context of VHL loss. By promoting the polyubiquitination and proteasomal degradation of LC3, DSS1 suppresses autophagy while concurrently activating EMT, thereby facilitating microvascular metastasis. The highly vascularized nature of ccRCC may also help explain the limited clinical efficacy of autophagy inhibitors. For instance, hydroxychloroquine demonstrated minimal therapeutic benefit in a phase II trial, with only 2 of 33 patients achieving partial responses54. These observations are consistent with our data and prior studies showing reduced autophagy activity in ccRCC tissues compared with matched adjacent non-tumorous tissues55. Furthermore, autophagy levels were found to be lower in advanced or metastatic ccRCC relative to localized disease55. This pattern correlates with the progressive upregulation of DSS1 and the relatively low-hypoxia phenotype of DSS1-driven cells. Thus, elucidating the molecular mechanisms underlying autophagy suppression in ccRCC is crucial for understanding disease progression and metastasis, and may inform the development of more effective therapeutic strategies.
Autophagy comprises a series of tightly regulated steps, including initiation, nucleation, elongation, maturation, and the autophagic lysosome reformation (ALR) cycle30. mTOR serves as a critical negative regulator by inhibiting autophagy initiation and promoting the ALR cycle. In our study, DSS1 knockdown did not alter total or phosphorylated mTOR (p-mTOR) protein levels. Instead, it attenuated the ubiquitin-mediated degradation of LC3, suggesting that DSS1 inhibits autophagic flux independently of mTOR signaling. Beclin 1, together with VPS15 and VPS34, mediates phagophore nucleation. However, its activity can be suppressed by Bcl-2 through direct interaction30. Our findings suggest that DSS1 may impair autophagy by promoting LC3 ubiquitination and degradation as well as upregulating Bcl-2 expression, although the mechanism underlying Bcl-2 regulation remains unclear. Therefore, DSS1-mediated autophagy suppression may represent a distinct regulatory pathway, operating independently of both mTOR and VHL signaling.
The loss of VHL function leads to the stabilization of hypoxia-inducible factors (HIFs), which activate downstream angiogenic programs51. This mechanism underpins the benefits of VEGFR inhibitors in treatment. However, resistance to anti-VEGFR therapy remains a major clinical challenge, and the molecular mechanisms underlying this resistance remain unclear56. Recent studies have identified ITGB1 as a key mediator of tumor angiogenesis and metastatic niche formation57,58. In our analysis, VEGF-VEGFR and SPP1-ITGB1 ligand-receptor interactions frequently co-occurred within tumor regions, particularly at invasive fronts. This spatial co-localization suggests that SPP1-ITGB1 signaling may compensate for suppressed VEGF signaling during anti-VEGFR treatment, thereby maintaining the invasive phenotype of tumor cells. Furthermore, the increased abundance of DSS1-driven cells in patients who had received anti-VEGFR therapy supports the hypothesis that these cells contribute to therapy resistance. Taken together, our data imply that DSS1-driven cells, through their engagement in alternative angiogenic signaling pathways such as SPP1-ITGB1, may play a critical role in the failure of anti-angiogenic therapies in advanced ccRCC.
DSS1 is an intrinsically disordered protein that lacks a stable tertiary structure under physiological conditions, posing substantial challenges for the development of small-molecule inhibitors. Upstream of DSS1, genomic copy number amplification is associated with its overexpression across various cancers, including ccRCC. Previous studies have also suggested that DSS1 expression may be linked to elevated levels of ROS34. In addition, DSS1 levels are reduced upon treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA, also known as PMA), a protein kinase C activator59, although the underlying regulatory mechanism remains unclear. A comprehensive investigation of the regulatory mechanisms driving DSS1 overexpression in ccRCC may facilitate the identification of upstream therapeutic targets. Downstream of DSS1, TWIST1, lacks enzymatic activity and possesses a flexible structure, rendering it undruggable using conventional approaches. Currently, no approved therapies directly target TWIST1. Interestingly, DSS1 interacts with structured proteins such as components of the proteasome, although proteasome complexes lacking DSS1 remain functional in yeast60,61. Given these limitations, targeting DSS1 or TWIST1 through proteolysis-targeting chimera (PROTAC) technology may offer a promising therapeutic strategy for ccRCC. PROTACs have recently demonstrated substantial advantages in degrading previously undruggable proteins62. Alternatively, combination strategies that exploit vulnerabilities downstream of DSS1 may also be beneficial. For example, proteasome inhibitors such as carfilzomib, when combined with autophagy agonists such as MLN0128, may synergistically suppress DSS1-driven tumor progression by counteracting the autophagy blockade induced by DSS1. However, the therapeutic efficacy and safety of such combinations require rigorous validation through preclinical models and clinical trials.
Methods
Ethical compliance
This study complied with all relevant ethical regulations. The human research protocol was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval no. 2021-465), and the animal research protocol was approved by the Ethics Committee of Chongqing Medical University.
Integrative analysis of DSS1 as a driver gene in ccRCC metastasis
-
i)
Genes with positive expression and differentially expressed in ccRCC
Differentially expressed genes were identified using data from TCGA. Specifically, the TCGA-KIRC dataset, comprising 523 tumor samples and 72 normal kidney tissue samples, was analyzed. The HTSeq-count expression matrix was downloaded from the Genomic Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/). Differential expression analysis was performed using the DESeq263 package (version 1.46.0) in R. Genes with a Benjamini-Hochberg adjusted P-value < 0.05 were considered statistically significant. Basal gene expression levels in ccRCC and normal kidney tissues were evaluated using both the TCGA-KIRC dataset and the HPA database (version 19.3; http://www.proteinatlas.org)38. For mRNA-level analysis, an average Fragments Per Kilobase of transcript per Million mapped reads (FPKM) value > 1 in either tumor or normal tissues was used to define gene expression abundance. Genes with low expression abundance (FPKM ≤ 1 in both tissue types) were excluded from further analysis. For protein-level evaluation, immunohistochemistry data from the HPA database were used. Only genes with protein expression detected in at least three independent samples were considered for downstream analyses.
-
ii)
Genes associated with patient survival and metastasis
First, univariate and multivariate Cox proportional hazards regression analyses were performed to identify genes associated with overall patient survival. Kaplan-Meier survival curves were generated, and genes with log-rank P values < 0.05 in univariate analysis were further evaluated by multivariate Cox regression. Clinical covariates, including age, sex, tumor stage, grade, and TNM classification, were included in the multivariate model to assess whether gene expression was an independent prognostic factor. Genes with P values < 0.05 after adjustment for these clinical variables were considered statistically significant. Second, a 154-gene signature related to cancer invasion and metastasis was curated as a subset of Catalogue of Somatic Mutations in Cancer (COSMIC) Cancer Gene Census (version 92, https://cancer.sanger.ac.uk/cosmic)64. Pearson correlation analysis was conducted between the expression of these 154 genes and candidate prognostic genes. Gene pairs with an absolute Pearson correlation coefficient (|r | ) > 0.4 and a false discovery rate (FDR) < 0.001 were considered significantly correlated. The COSMIC Cancer Gene Census was selected for this analysis because it not only catalogs genetic mutations but also provides expert-curated annotations linking genes to cancer hallmarks such as invasion and metastasis. These annotations are supported by strong experimental evidence, including knockdown, knockout, and overexpression studies, which elucidate the functional roles of these genes in cancer progression mechanisms.
Metastasis-related hub genes with high connectivity in the gene regulatory network were further selected based on their degree of interaction. Predicted regulatory interactions were considered valid if supported by at least one of nine computational tools: RNA22 v2.065, miRcode v1166, TargetScan v7.267, PITA v668, PicTar 200769, miRDB v6.070, mirTarBase v201971, miRanda v3.3a72, RNAhybrid v2.1.273. For putative interactions, only those with P-values < 0.05 (when applicable) were retained. Additionally, interactions directly supported by low-throughput experimental evidence were included. Genes with a degree of connectivity ≥ 5 were defined as high-degree hub genes.
-
iii)
Identification of candidate genes
To evaluate the concordance of gene differential expression in ccRCC at the protein level, data from the HPA database were analyzed. Only genes showing high concordance between mRNA and protein expression levels were retained. For example, if differential expression analysis identified a gene as upregulated at the mRNA level, the corresponding immunohistochemistry data from HPA tumor samples should show high or medium expression in at least two-thirds of cases, while normal kidney samples should show no or low expression in at least two-thirds of cases.
Subsequently, a comprehensive literature review was performed to identify genes previously reported in ccRCC or cancer metastasis. An independent gene signature analysis using Spearman correlation was conducted to confirm associations between candidate genes and metastasis-related signatures (CHANG_CORE_SERUM_RESPONSE_UP20, CSR_Activated_1570170021). A correlation coefficient |ρ | > 0.4 with P-value < 0.01 was considered statistically significant. Next, independent dataset analyses were performed to validate gene dysregulation in cancer metastasis. Specifically, GEO datasets GSE11320474 (n = 12) and GSE7230475 (n = 14) were analyzed using two-tailed Welch’s t-tests to assess differential expression of candidate genes.
Functional and pathway analyses
First, pathway enrichment analysis based on hypergeometric tests was performed using a DSS1-related gene set to identify candidate pathways. Pathways with a Benjamini-Hochberg adjusted P-value < 0.05 were considered statistically significant. Second, Gene Set Enrichment Analysis (GSEA, version 4.3.3)76 was conducted with default parameters to explore potential pathways associated with DSS1 expression. Samples were divided into high- and low-DSS1 expression groups based on the median expression value of DSS1, and DESeq2-normalized log2 count per million (CPM) values were used. Pathways were ranked by NES and nominal P values.
Identification of downstream transcription factors regulated by DSS1 through autophagy
The Consensus Scoring of Genes in Cascade (CSGC) algorithm was developed to identify candidate genes that potentially play a role in the DSS1-mediated cascade associated with autophagy and metastasis. EMT-inducing transcription factors (TF, i) ZEB1/2, SNAI1/2, TWIST1, CTNNB1, FOXC1/2, TCF3, and KLF8 were considered as candidates. First, these TFs should be upregulated in ccRCC or metastatic ccRCC because they are considered EMT inducers. Fold change (FC) was calculated based on DESeq2. Second, TFs should be correlated (Coef, Spearman) positively with DSS1 and EMT, while correlated negatively with autophagy to be logically consistent with the hypothesis that the TFs are regulated by DSS1-suppressed autophagy. Log2CPM expression (DESeq2, TCGA-KIRC) was used for calculation of pathway activities (ssGSEA77, MSigDB gene sets v2022.1 [e.g., autophagy, EMT]) and Spearman correlation. This was performed by introducing a logical variable (Sign, 1 or −1). Variable Sign was assigned to 1 if the expected correlation was shown, otherwise −1. A CSGC score of a candidate TFi was calculated as follows:
Where j represents the activities of elements (n; j₁, j₂, jn) of the DSS1-related cascade (DSS1, autophagy pathway, EMT process). Proteomics-based analysis can, in theory, offer more accurate insights than transcriptome-based analysis for identifying functional regulatory cascades. The Clinical Proteomic Tumor Analysis Consortium (CPTAC) project42 has quantified the protein levels of approximately 10,000 proteins in ccRCC tissues using trypsin-based mass spectrometry. However, several proteins interested in this analysis were not detected in the CPTAC-ccRCC cohort (trypsin-based mass spectrometric analysis), such as SNAI1/2, TWIST1, FOXC2, KLF8, and DSS1. While transcriptomic data do not fully account for post-transcriptional and post-translational regulation, the RNA expression levels of key EMT-related genes (e.g., TWIST178) have been shown to correlate with EMT activation42. Furthermore, it has been demonstrated that, in general, mRNA levels positively correlate with protein abundance across a broad range of genes in cells79. Therefore, despite inherent limitations, transcriptome data offer a high-throughput and broad-coverage platform that enables the quantitative estimation of pathway activities, such as autophagy80,81, and have been widely used for hypothesis generation and pathway modeling.
Analysis of mTOR and autophagy pathways
It is well established that the mTOR pathway negatively regulates autophagy. GSEA using the TCGA-KIRC dataset (comparing DSS1-high vs. DSS1-low, defined by the median expression level) showed negative NES for both mTOR signaling and autophagy-related pathways. However, the mTOR pathway exhibited a modestly negative NES and ranked relatively low among all tested pathways. To evaluate whether this inverse association between DSS1 expression and both mTOR/autophagy pathways is consistently observed, we performed additional GSEA (v4.3.3) analyses under the same settings (default parameters, DSS1-high vs. DSS1-low, median as cut-off) across three independent ccRCC datasets. The GSE3538 dataset24 includes 177 human ccRCC tumor samples across stage I–IV (n = 49, 29, 40, and 59, respectively). Log2-transformed expression data from different microarray platforms were merged and quantile-normalized. The GSE251905 dataset82 contains RNA-seq data from 54 samples (32 primary and 22 metastatic ccRCC tumors), and the GSE254566 dataset83 includes RNA-seq data from 91 ccRCC tumors. For RNA-seq datasets, expression profiles (log2CPM, DESeq2) were downloaded.
Single-cell transcriptome data
Single-cell RNA sequencing (10x Genomics platform) data from ccRCC tumor tissues and adjacent normal tissues were obtained from the Gene Expression Omnibus (GEO) database, including GSE159115 (n = 14)84, GSE178481 (n = 26)85, and GSE210038 (n = 9)86, comprising a total of 251,681 cells. These datasets were integrated using the merge function in the Seurat package (v4.3)87. High-quality cells were retained based on the following filtering criteria: nFeature_RNA > 500, nCount_RNA > 1000, and mitochondrial gene percentage (percent.mt) <20%. Cells with nFeature_RNA > 7000 were identified as potential doublets and excluded. Additionally, clusters containing cells expressing markers from multiple lineages were flagged as doublets and removed14. The raw count data were normalized, and 2000 highly variable genes were selected. Batch effects among datasets were corrected using the FastMNN algorithm from the SeuratWrappers package (v0.3.1, https://github.com/satijalab/seurat-wrappers). Dimensionality reduction and clustering were conducted using the standard Seurat pipeline, including RunUMAP and FindNeighbors, based on MNN-corrected principal components (top 30 PCs). Cell clusters were identified using the Louvain algorithm via the FindClusters function.
Cell type annotation and inferCNV analysis
Major cell types were annotated based on canonical marker genes. Specifically, B cells were identified using BANK1 and MS4A1, plasma cells by MZB1 and TNFRSF17, mast cells by KIT and VWA5A, myeloid cells by CD68 and FCGR3A, fibroblasts by ACTA2 and MYL9, endothelial cells by ENG and CLEC14A, epithelial cells by EPCAM and KRT8, and T/NK cells by CD2, CD3, NKG7, and GZMK. Endothelial cell subtypes were further delineated using distinct markers: inflammatory endothelial cells by HLA-DRA and ICAM1, microvascular endothelial cells by FLT1, NOTCH4, and ESM1, hypoxic endothelial cells by HIF1A and FGF2, and metabolically altered endothelial cells by LDHA and PKM. Pericytes were identified using RGS5 and ACTA2. To distinguish malignant epithelial cells, we applied inferCNV (v1.10.0)35 using normal epithelial cells and immune cells as reference controls. Copy number variation (CNV) scores were estimated, scaled, and subsequently rescaled to a range between −1 and 1. The mean squared value of rescaled CNV estimates across the genome was calculated as the CNV score. Cells with a CNV score > 0.04 were classified as malignant88.
High-dimensional weighted gene coexpression network analysis (hdWGCNA)
To investigate the functional programs within malignant cells, high-dimensional weighted gene coexpression network analysis (hdWGCNA, R package v0.3.00)89 was performed following the developer’s guidelines. Briefly, metacells were constructed by grouping every 10 cells, with the maximum number of shared cells between any two metacells set to 10. A soft-thresholding power was selected to construct a signed coexpression network. Functional annotations of each module were determined using Gene Ontology (GO, July 2024 release) enrichment analysis. Module signature scores for individual cells were calculated using the AddModuleScore function in Seurat, based on the expression of all genes within each module.
Identification of DSS1-driven cells and analysis of the clinical value
DSS1-highly expressed ccRCC cells (referred to as DSS1-driven cells) were defined as those exhibiting expression levels at least 50% higher than the mean expression of normal epithelial cells, comprising the top 12% of malignant cells. This threshold yielded an average log₂ fold change (log₂FC) of 2.08 between DSS1-driven cells and all other malignant cells, exceeding the empirical four-fold cutoff used by the HPA (v24.0)38 to classify elevated expression. To assess the clinical relevance of DSS1-driven cells using bulk RNA-Seq data, signature scores were computed as the mean scaled expression of marker genes identified via the FindMarkers function in Seurat. Comparisons of signature scores across tumor stages and molecular subtypes were performed using the two-tailed Mann-Whitney U test. The prognostic significance of the signature score in ccRCC patients was evaluated via survival analysis (log-rank test) based on the TCGA-KIRC cohort. Associations between DSS1-driven cell signature scores and anti-VEGFR treatment efficacy were analyzed using the Mann-Whitney U test across multiple datasets, including GSE6561544, GSE6405246, and GSE7606845.
Analysis of potential drugs or compounds targeting DSS1-driven cells
To identify potential drugs that may effectively inhibit ccRCC metastasis, we analyzed the single-cell RNA sequencing (scRNA-seq) data of DSS1-driven cells using two computational tools: scTherapy48 and beyondcell47. Both tools were employed to predict responses to FDA-approved drugs, preclinical candidates, and laboratory compounds. scTherapy48 (v1.0.0) is a methodological framework that integrates scRNA-seq data with therapeutic response prediction by identifying cell-type- or cell-population-specific drug targets and biomarkers. Differentially expressed genes between DSS1-driven cells and other malignant cells were identified using the FindMarkers function in Seurat. Genes with |avg_log₂FC | > 0.5 and adjusted P-values < 0.05 (Bonferroni correction) were used as input for drug response prediction. All other parameters were set to default. Beyondcell47 is a computational tool designed to predict drug sensitivity at the single-cell level by integrating scRNA-seq data with pharmacogenomic resources such as the NIH LINCS project. We applied beyondcell to DSS1-driven cells to compute a switch point score (ranging from 0 to 1) for each cell in response to each drug. A lower switch point indicates higher predicted drug sensitivity. Following the developer’s guidelines, drugs with switch point values < 0.1 were considered potentially effective against DSS1-driven cells.
Spatial transcriptome analysis
Spatial transcriptome data of ccRCC tumor tissues were obtained from three datasets: GSE175540 (n = 2)90, GSE210041 (original n = 24)86, and Li2022 dataset (original n = 16)91. Li2022 dataset is available on the CELLxGENE platform92. Samples lacking clinical staging information (n = 1 from GSE210041) or determined to be of low quality according to the original publication (n = 3 from Li2022) were excluded, resulting in a final dataset of 38 spatial transcriptomics samples. Cell type deconvolution was performed using the RCTD algorithm, implemented in the spacexr (v2.2.1) package93, with our annotated single-cell RNA-seq dataset as the reference. To analyze spatial relationships between DSS1-driven cells and other tumor microenvironment (TME) cell types, the raw weights assigned by RCTD were normalized: for each spot, the weight of a given TME cell type was divided by the sum of all TME cell weights within that spot. Similarly, to assess the correlation between DSS1-driven cell abundance and functional states of endothelial subsets, signature scores for endothelial subtypes were computed using AddModuleScore, rescaled to [0–1], and divided by the summed TME cell weights in each spot. Endothelial subtypes were defined by curated marker genes. Microvascular endothelial cells were defined by PECAM1, KDR, VCAM1, NOS3, PROCK, ANGPT2, TEK, ITGAL, ESM1, FLT1, NOTCH4, and TNFRSF1A. Lymphatic endothelial cells were defined by PROX1, LYNE1, FLT4, and PDPN. Hypoxia endothelial cells were defined by HIF1A. Inflammatory endothelial cells were defined by SELE, ICAM1, VCAM1, and IL6. To define tumor-normal boundaries, spots were first categorized as tumor-cell dominant if more than 50% of their total RCTD-derived cell weights were from malignant cells. All other spots were classified as non-tumor-cell dominant. To further identify boundary spots, Euclidean distances between spatial coordinates of each non-tumor-cell dominant spot and its nearest tumor-cell dominant spot were calculated. Non-tumor-cell dominant spots located within a distance of less than 2 units from a tumor-cell dominant spot were defined as tumor-normal boundary spots.
Cell-cell communication analysis
Cell-cell communication between DSS1-driven cells and microvascular endothelial cells was analyzed using CellChat (v2.1.2)94 based on ligand-receptor pairs curated in CellChatDB v294. The TriMean method was applied to compute communication probabilities, and interactions involving fewer than 10 cells were excluded to ensure robustness. To validate these ligand-receptor interactions in spatial transcriptomics data, a coexpression strength metric was calculated for each spot by computing the product of the ligand expression and the sum of receptor expressions, followed by rescaling to a 0–1 range. To assess the relationship between the abundance of DSS1-driven cells and the strength of ligand-receptor coexpression, Spearman correlation coefficients were calculated across spatial spots for each individual sample (slide).
Human tissues and ethics approval
Formalin-fixed and paraffin-embedded (FFPE) tissue samples from patients with ccRCC were randomly selected from the pathology archives (November 2019 to June 2022) of the First Affiliated Hospital of Chongqing Medical University using a random number generator. Eligibility criteria included: (a) histologically confirmed diagnosis of ccRCC; (b) complete clinical information, including sex (biological attribute), age, and pathological grading or staging; and (c) sufficient residual tissue from both tumor and adjacent normal regions. No subjective criteria were applied for sample exclusion. In total, FFPE sections from 74 patients (47 males and 27 females; median age: 63 years) were collected, including both tumor and paired adjacent normal kidney tissues (left/right kidney). Among these, 46 patients were diagnosed with World Health Organization/International Society of Urological Pathology (WHO/ISUP) grade 1 or 2, and 28 patients with grade 3 or 4. All tissue sections were stored at 4 °C prior to analysis. Written informed consent was obtained from all participants, and the study protocol was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval No. 2021-465). Patient identities were anonymized prior to any downstream analysis.
Immunohistochemistry (IHC) and H&E staining
FFPE human and murine tissue sections were processed following standard protocols. Briefly, sections were deparaffinized in xylene, rehydrated through graded ethanol series, and subjected to heat-induced epitope retrieval in 10 mM sodium citrate buffer (pH 6.0) using a microwave for 15 min. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide for 10 min at room temperature (RT), followed by blocking in 10% goat serum for 1 hour at RT. Sections were then incubated overnight at 4 °C with primary antibodies diluted in antibody diluent. The following antibodies were used: DSS1 (Proteintech, 13639-1-AP, 1:100), LC3 (Proteintech, 14600-1-AP, 1:100), SQSTM1/p62 (Proteintech, 18420-1-AP, 1:100), TWIST1 (Proteintech, 25465-1-AP, 1:100), TRIM25 (Proteintech, 12573-1-AP, 1:100), and CDH1/E-cadherin (Cell Signaling Technology, 3195, 1:100). After washing, sections were incubated with a biotinylated secondary antibody at 37 °C for 30 min and developed with DAB (Absin, abs9210) according to the manufacturer’s instructions. Nuclear counterstaining was performed using hematoxylin. The IHC assay was performed using the rabbit-enhanced polymer detection kit (ZSGB-BIO, PV-9001). IHC staining was visualized and imaged using a bright-field microscope (Nikon or equivalent). Tumor cell content and staining intensity were independently evaluated by two pathologists blinded to clinical information. DSS1 IHC was performed in 74 matched tumor and adjacent normal tissue pairs, and TRIM25 IHC was performed in 66 pairs with sufficient tissue availability. H&E staining was conducted using a standard hematoxylin and eosin kit (ServiceBio, G1076-500ML) for morphological assessment.
Multiplex immunohistochemistry (mIHC)
FFPE tissue sections from four patients with ccRCC were used for multiplex immunohistochemistry (mIHC) analysis. Slides were incubated overnight at 4 °C with primary antibodies against DSS1 (Proteintech, 13639-1-AP; 1:200), LC3 (Proteintech, 14600-1-AP; 1:200), SPP1 (Abcam, ab214050; 1:200), ITGB1 (Abcam, ab30394; 1:200), pan-Cytokeratin (pan-CK, Abcam, ab7753; 1:200), and CD105 (Abcam, ab231774; 1:200). To analyze the autophagic status of DSS1-driven cells in tissue, FFPE sections from an additional four ccRCC patients were processed using the same protocol, with additional primary antibodies against CD31 (Proteintech, 66065-2-Ig; 1:200), E-cadherin (Cell Signaling Technology, 3195; 1:200), and Vimentin (Cell Signaling Technology, 5741; 1:200). Signal amplification was performed using a tyramide signal amplification kit (RecordBio Co. Ltd, RC0086Plus-67RM) according to the manufacturer’s instructions.
Cell lines and cell culture
The 786-O (CRL-1932), ACHN (CRL-1611), Caki-1 (HTB-46), and HEK293T (CRL-11268) cells were obtained from the American Type Culture Collection (ATCC). Cell line authentication was performed by Short tandem repeat (STR) profiling at the time of purchase. Cells were passaged at ~80% confluency using 0.25% trypsin-EDTA (Gibco) and monitored daily for morphological consistency. Mycoplasma contamination was not detected in cells. The 786-O cells were cultured in Roswell Park Memorial Institute (RPMI)−1640 medium, ACHN cells in Minimum Essential Medium (MEM) medium, Caki-1 in McCoy’s 5 A medium, and HEK293T cells in Dulbecco’s Modified Eagle Medium (DMEM). All media were supplemented with 10% fetal bovine serum (FBS; ExCell, South America origin) and 1% penicillin-streptomycin (Gibco). Cells were maintained at 37 °C in a humidified incubator with 5% CO₂.
Cell morphology
Cell morphology was quantified by calculating the aspect ratio, defined as the ratio of the major axis length (longest diameter) to the minor axis length (shortest perpendicular diameter). Measurements were performed using the Fit Ellipse function in ImageJ software (v1.53t, NIH). Independent experiments were performed in triplicate.
Inhibitors
Chloroquine (CQ, HY-17589A, 10 μM), MG-132 (HY-13259, 20 μM), and Cycloheximide (CHX, 50 μg/mL) were purchased from MedChemExpress® and administered to cultured cells at the indicated concentrations following the manufacturer’s instructions.
RNA extraction and reverse transcription-quantitative (RT-q) PCR
Total RNA was extracted from cells using TRIzol® reagent (Invitrogen, Thermo Fisher Scientific) and 500 ng RNA was reverse transcribed into a 20 µl final volume of cDNA using Reverse Transcription kit (Takara, RR092A), according to the manufacturer’s instructions. RNA purity was assessed by A260/A280 ratio (Nanodrop 2000, Thermo Fisher), and integrity was confirmed via agarose gel electrophoresis (28S/18S ratio >1.8). MAP1LC3B and SQSTM1 RNA expression were measured using qPCR on the QuantStudio Real-Time PCR system (Thermo Fisher Scientific). qPCR was conducted using TB Green® Premix Ex Taq™ II (Takara, RR820A) and analyzed using the 2-ΔΔCt method. qPCR reaction conditions were as follows: Initial denaturation at 95 °C for 2 min, followed by 39 cycles at 95 °C for 15 s and 60 °C for 30 seconds. GAPDH was used as endogenous controls.
Additionally, GSEA analyses showed a consistent negative correlation between DSS1 and KEGG (v113.0) autophagy, with a big number of autophagy genes (Core Enrichment gene set) negatively correlated with DSS1 expression. To validate the mRNA-level expression correlation between DSS1 and autophagy-related genes, we conducted RT-qPCR to evaluate the mRNA expression of autophagy-related genes (Core Enrichment gene set) between DSS1-knockdown cells and control cells. Specifically, the genes identified as Core Enrichment genes by at least 3 of 4 datasets (TCGA-KIRC, GSE353824, GSE25190523, and GSE25456625) were obtained (37 genes) and were analyzed using RT-qPCR. Primer specificity was confirmed by a single peak in melt curve. This experiment was biologically replicated 3 times, with 3 technical replicates for each sample. Primer sequences were provided in Supplementary Table 1.
Western blot
Total proteins were extracted using RIPA lysis buffer supplemented with PMSF, phosphatase, and protein inhibitors, following the Whole Cell Lysis Assay protocol (KeyGEN Biotech, KGP2100). The protein concentration was determined using a BCA reagent kit (Beyotime, P0010S). The proteins were separated by SDS-PAGE and then transferred to PVDF membranes. After blocking in 5% bovine serum albumin or 5% nonfat milk at RT for 2 h, the membranes were incubated with primary antibodies against DSS1 (Proteintech, 13639-1-AP, 1:1000), CDH1/E-cadherin (Proteintech, 20874-1-AP, 1:1000), CDH2/N-cadherin (Proteintech, 22018-1-AP, 1:1000), VIM/Vimentin (Proteintech, 10366-1-AP, 1:1000), LC3 (Proteintech, 14600-1-AP, 1:1000), SQSTM1/p62 (Proteintech, 18420-1-AP, 1:1000), TWIST1 (Proteintech, 25465-1-AP, 1:1000), TRIM25 (Proteintech, 12573-1-AP, 1:1000), ubiquitin (Abcam, ab134953, 1:1000), Myc-tag (Proteintech, 16286-1-AP, 1:1000), HA-tag (Proteintech, 51064-2-AP, 1:1000), mTOR (Abways, CY5306, 1:1000), p-mTOR (Abways, CY5996, 1:1000), SPP1 (Abcam, ab214050, 1:1000), Bcl-2 (Abways, CY5032, 1:1000), Beclin 1 (HUABIO, HA721216, 1:1000), TGF-β (Immunoway, YM8257, 1:1000), ZEB1 (Cell Signaling Technology, 70512, 1:1000), ZEB2 (Cell Signaling Technology, 97885, 1:1000), SNAI1 (Proteintech, 13099-1-AP, 1:1000), SNAI2/SLUG (Cell Signaling Technology, 9585, 1:1000), TCF3 (Proteintech, 21242-1-AP, 1:1000), FOXC2 (Proteintech, 23066-1-AP, 1:1000), GST-tag (Proteintech, 66001-2-Ig, 1:1000) and ACTB/β-actin (Proteintech, 20536-1-AP, 1:1000) at 4 °C overnight. Subsequently, the membranes were incubated with an HRP-conjugated secondary antibody (Proteintech, SA00001; or Abbkine, IPKine™ A25022 light chain specific; 1:5000 dilution) for 1 h at RT and detected using the ECL system. Independent experiments were performed in triplicate.
Plasmids, small interfering RNA (siRNA), and cell transfection
Small interfering RNAs (siRNAs) targeting DSS1, TRIM25, and TWIST1 were purchased from GenePharma Co., Ltd. Plasmids encoding FLAG-DSS1 (N-terminal), HA-DSS1W27GW39GW43GF52A (N-terminal), DSS1, TRIM25, TWIST1, LC3B, Myc-LC3B (N-terminal), Myc-LC3B mutants (K5R-K8R, K30R-K39R, K42R-K48R, K51R, K65R, K103R, LC3B-K122R, all N-terminal), HA-Ub (wildtype), HA-Ub-K48R, HA-Ub-K63R (N-terminal) were constructed using the pcDNA3.1 vector. Empty vector (pcDNA3.1) was obtained from Tsingke Biotech Co., Ltd. For the Glutathione-S-Transferase (GST) pull-down assay, the full-length sequence of DSS1, DSS1W27GW39GW43GF52A, and TRIM25 were cloned into a pGEX-4T-1 vector (Tsingke) containing an N-terminal GST tag. The empty pGEX-4T-1 vector (Tsingke) was used as a control. Plasmid transfection was performed using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s protocol. siRNAs were transfected into cells using GP-Transfect-Mate (GenePharma, G04009). The sequences of the human siRNAs are as follows:
DSS1 siRNA: 5’- GACAAUGUAGAGGAUGACUUCUCUA-3’.
TWIST1 siRNA: 5’- CCUGAGCAACAGCGAGGAATT-3’.
TRIM25 siRNA-1: 5’- AUGGAUUUUCUCUAAGAGGAA-3’.
TRIM25 siRNA-2: 5’- UAUGGAUUUUCUCUAAGAGGA-3’.
Control siRNA (siNC): 5’- GCAGGCGAUUCAGAUCUGGUGCUUA-3’.
Lentivirus and short hairpin RNA (shRNA)
The lentiviral overexpression construct for DSS1 (vector GV492: Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin) and the lentiviral shRNA knockdown construct for DSS1 (vector GV493: hU6-MCS-CBh-gcGFP-IRES-puromycin) were generated by Genechem Co., Ltd. Lentivirus infection was conducted according to the manufacturer’s instructions. Seventy-two hours post-infection, 786-O, ACHN, and Caki-1 cells expressing green fluorescent protein (GFP) were selected. Stable DSS1-overexpressing or DSS1-knockdown cell lines were established by culturing in medium containing 2 μg/mL puromycin. The efficiency of DSS1 overexpression or knockdown was confirmed by western blot analysis. Target sequences for ShDSS1: ShDSS1 #1: 5’-GGTAGACTTAGGTCTGTTAGA-3’; ShDSS1 #2: 5’-GAGTTTGAAGAGTTCCCTGCC-3’; ShDSS1 #3: 5’-GACGACGAGTTTGAAGAGTTC-3’. Target sequence for shNC: 5’-TTCTCCGAACGTGTCACGT-3’.
Cell proliferation assay
Cell proliferation was assessed using the Cell Counting Kit-8 (CCK-8; Yeasen Biotechnology, 40203ES60). Briefly, 2000 cells were seeded per well in 96-well plates. At indicated time points, 10 μL of CCK-8 reagent was added to each well and incubated at 37 °C for 1 hour in the dark. Absorbance was measured at 450 nm using a microplate reader every 24 h. Additionally, proliferation was evaluated by 5-ethynyl-2′-deoxyuridine (EdU) incorporation using the BeyoClick™ EdU Cell Proliferation Kit (Beyotime, C0071S) according to the manufacturer’s protocol. Cells were stained with Alexa Fluor 488-conjugated dye at RT for 30 min in the dark and imaged by fluorescence microscopy (Nikon Eclipse Ts2R). All experiments were performed in biological quintuplicates (CCK-8) or triplicates (EdU assay). CCK-8 experiment was biologically replicated 5 times, with 3 technical replicates for each sample. EdU experiment was biologically repeated 3 times.
Transwell assay
8 μm-pore transwell chambers (BIOFIL, TCS020024) coated with Matrigel (BD Bioscience) were used for cell invasion assay, while those without any pre-treatment were for cell migration assay. 2 × 104 (migration) and 5 × 104 (invasion) suspended transfected cells in 200 μL FBS-free medium were loaded into the upper chamber of each 24-well transwell chamber, while 600 μL 10%-FBS medium was added into the lower chamber. After culture for 48 h at 37 °C with 5% CO2, the cells on the lower surface were fixed with methanol for 30 min and stained with a 0.5% crystal violet solution for 15 min after the non-adhering cells in the upper chamber were scraped. Images were captured using an inverted microscope and analyzed using ImageJ software. Independent experiments were performed in triplicate.
Co-immunoprecipitation (Co-IP)
The total proteins (5% of proteins for input) were incubated with primary antibodies against Myc-tag (Proteintech, 16286-1-AP, 1:50), FLAG-tag (Proteintech, 20543-1-AP, 1:50), LC3 (Proteintech, 14600-1-AP, 1:50), TWIST1 (Proteintech, 25465-1-AP, 1:50), TRIM25 (Proteintech, 12573-1-AP, 1:50), and IgG (Abcam, ab172730, 1:50), followed by precipitation with Protein A-Agarose beads (Santa Cruz, sc-2001). After washing the beads with wash buffer (Beyotime Biotechnology, P0013) and retrieving the proteins, they were subjected to western blot analysis. Independent experiments of co-IP were performed in triplicate.
GST pull-down
The recombinant plasmids of GST-DSS1, GST-DSS1W27GW39GW43GF52A, GST-TRIM25, and pGEX-4T-1 vector were transfected into E. coli BL21 (DE3), followed by the selection of monoclonal colonies for extended culture. Expression of recombinant plasmids were induced by addition of 0.5 mM IPTG and incubation at 25 °C for 6 h. 1 mg of fusion protein was immobilized in 50 µL of glutathione agarose and incubated at 4 °C for 4 h with gentle rocking. Purified human LC3B protein was purchased from MedChemExpress® (HY-P70909). 50 ng of LC3B fusion proteins was added to the immobilized GST-DSS1, GST-DSS1W27GW39GW43GF52A, GST-TRIM25, and GST-tag and then incubated overnight at 4 °C with gentle rotation. Following this, the complex of agarose and bound proteins was washed, and the bound proteins were eluted for immunoblotting. This experiment was conducted using a GST pull-down kit (FITGENE, FI88808). Independent experiments were performed in triplicate.
DSS1 adducts analysis
Cells were lysed using Cell Lysis Solution (Beyotime Biotechnology, P0013J) supplemented with Protease Inhibitor Cocktail (Beyotime Biotechnology, P1005), with or without 10 mM EDTA. Lysates were then heated at 95 °C for 5 min in a dry bath incubator prior to immunoblot analysis. The recombinant DSS1W27GW39GW43GF52A plasmid was also utilized to assess the formation of DSS1 adducts.
Immunofluorescence
Cells were transfected with siDSS1 or siNC and cultured for 48 h at 37 °C, then seeded onto glass slides in 24-well plates. After an additional 36 h of culture at 37 °C, cells were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100 for 20 min, and blocked with 10% goat serum for 1 hour at room temperature. Slides were incubated overnight at 4 °C with primary antibodies against LAMP1 (Abcam, ab25630, 1:100) and LC3 (Proteintech, 14600-1-AP, 1:100). Following washes, slides were incubated for 1 h at RT with DyLight 488-conjugated goat anti-mouse secondary antibody (for LAMP1) and DyLight 549-conjugated goat anti-rabbit secondary antibody (for LC3). Nuclei were counterstained with DAPI (BOSTER Biological Technology, AR1176) for 10 min in the dark at RT. For multiplex immunofluorescence, primary antibody against DSS1 (Proteintech, 13639-1-AP, 1:200), LC3 (ServiceBio, GB11124, 1:500), PSMD3 (Proteintech, 12054-1-AP, 1:200), Bcl-2 (ServiceBio, GB154830, 1:500), TWIST1 (Proteintech, 25465-1-AP, 1:200), and p62 (Proteintech, 18420-1-AP, 1:200) were applied. Signal amplification was performed using the Tyramide Signal Amplification Kit (RecordBio Co. Ltd, RC0086-34RM) according to the manufacturer’s instructions. Imaging was conducted on a Leica SP8 confocal microscope with LAS X software (v3.5.7) or scanned using an automated slide scanner (3DHISTECH, Pannoramic MIDI). Independent experiments of immunofluorescence were performed in triplicate.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
Protein samples (Caki-1) separated by SDS-PAGE were excised as gel bands and subjected to in-gel digestion. Gel pieces were destained with alternating washes of 50% acetonitrile (ACN) in 25 mM ammonium bicarbonate (NH4HCO3) until complete decolorization, followed by dehydration in 100% ACN. Proteins were reduced with 10 mM dithiothreitol (DTT) at 57 °C for 60 min and alkylated with 55 mM iodoacetamide (IAA) in darkness for 45 min. After sequential washes with NH4HCO3 and ACN, tryptic digestion was performed using sequencing-grade trypsin (Promega) at 37 °C overnight (enzyme-to-substrate ratio 1:50). Peptides were extracted using 50% ACN/0.1% formic acid (FA) with ultrasonic assistance, followed by vacuum concentration. Desalting was performed using Pierce C18 Spin Tips (Thermo Scientific) according to manufacturer’s protocol. Purified peptides were reconstituted in 0.1% FA and analyzed on a Q-Exactive HF mass spectrometer (Thermo Scientific) coupled with an EASY-nLC 1200 nanoflow HPLC system. Chromatographic separation was achieved using a C18 analytical column (75 μm × 15 cm, 1.9 μm particles) with a 300 nL/min gradient from 2% to 35% ACN in 0.1% FA over 60 min. MS acquisition employed data-dependent acquisition (DDA) mode. Raw data were processed using Proteome Discoverer 2.4 (Thermo Scientific) against the SwissProt Homo sapiens database (release 2023_03) with the following search parameters: tryptic digestion (max 2 missed cleavages), carbamidomethylation (C) as fixed modification, oxidation (M) and N-terminal acetylation as variable modifications, 10 ppm precursor mass tolerance, and 0.02 Da fragment mass tolerance. FDR was controlled at ≤ 0.01. Independent experiments were performed in triplicate.
Transmission electron microscope
The sample was pre-fixed with 3% glutaraldehyde at 4 °C overnight, avoiding light, and subsequently fixed with 1% osmium tetroxide. Following fixation, the samples underwent dehydration in a graded series of acetone solutions. Next, the samples were treated with a mixture of embedding agent (SPI-Pon™ 812) and acetone (at ratios of 1:3, 1:1, and 3:1 by volume, for 1 h each), followed by embedding with the embedding agent (SPI-Pon™ 812). Sectioning of the samples was conducted using a Leica UC7rt (at thicknesses of 60–90 nm). After staining with uranyl acetate for 15 min and lead citrate for 2 min, the samples on copper grids were imaged using a transmission electron microscope JEM-1400FLASH (JEOL, Japan). Two pathologists engaged in identifying and labeling cellular structures, such as autophagosomes. Independent experiments were performed in triplicate.
Ubiquitination assay
HEK293T cells were selected for ubiquitination assays due to their high transfection efficiency, low background ubiquitin-proteasome activity, and broad compatibility with established protocols. Cells were transfected with Myc-LC3B and HA-Ub, along with control siRNA (siNC) or DSS1-targeting siRNA (siDSS1), and cultured for 48 h at 37 °C. Prior to harvest, cells were treated with either 0.02% DMSO (vehicle control) or 20 μM MG-132 (MedChemExpress) for 6 h at 37 °C to inhibit proteasomal degradation. Cells expressing Myc-LC3B were lysed on ice for 20 min in Cell Complete Lysis Buffer (Beyotime, P0037) supplemented with a protease inhibitor cocktail (Beyotime, P1005). Lysates were clarified by centrifugation at 14,000 g for 15 min at 4 °C. The supernatant was incubated with anti-Myc magnetic beads at 4 °C for 12 h to immunoprecipitate Myc-LC3B. Beads were then washed, and bound proteins were eluted for immunoblot analysis. In vitro ubiquitination analysis was performed based on E2 Select Ubiquitin Conjugation Kit (Yeasen Biotechnology, 20440ES10) according to the manufacturer’s instructions. 20 µL reaction system (2 µL 10× MgATP, 4 µL 5× Ubiquitin, 2 µL 10× His-tagged E1 enzyme UBE1, 1 µL 20× E2 enzyme UbcH5a/UbcH5c, 0.5 µg E3 enzyme GST-TRIM25, 0.5 µg substrate LC3B [MedChemExpress, HY-P70909], 2 µL 10× Reaction Buffer, and ddH2O) was prepared and incubated for 4 h at 37 °C. Samples were analyzed by immunoblotting. Independent experiments were performed in triplicate.
Xenograft model
Male BALB/c-nu nude mice (4 weeks old) were obtained from HuaFuKang Bioscience (Beijing, China) and housed under pathogen-free conditions in individually ventilated cages (IVCs) with autoclaved corncob bedding. Mice were maintained on a 12 hour light/dark cycle with free access to food and water. Environmental conditions were controlled at 22 ± 1 °C temperature and 55 ± 5% relative humidity, with HEPA-filtered air ventilation providing ≥15 air changes per hour. Given the higher incidence of ccRCC in males and the absence of observed sex-related differences in DSS1 effects in patients, male mice were used exclusively. Inclusion criteria comprised all healthy animals, while exclusion criteria included procedural errors or unexpected mortality; no animals were excluded, and all completed the study. Body weights were recorded weekly.
For the experimental metastasis model, 1 × 106 Caki-1 or 786-O cells stably expressing lentiviral constructs (Lv-shDSS1, Lv-shNC, Lv-DSS1, or Lv-Vector) were injected into the lateral tail vein of mice (n = 6 per group; total n = 24), randomized prior to injection. Mice were monitored for up to seven weeks and euthanized by cervical dislocation unless humane endpoints were met earlier (> 20% body weight loss from baseline or signs of distress). Lung tissues were harvested for evaluation of metastatic foci and histopathological analyses.
For the subcutaneous tumor model, 1 × 106 Caki-1 cells stably transduced with Lv-shDSS1 or Lv-shNC (n = 6 per group; total n = 12) were injected into the axillary fossa. Tumor dimensions (length, width, height) were measured weekly using a vernier caliper, and tumor volume was calculated as π/6 × (L × W × H). Animals were euthanized four weeks post-inoculation or earlier if humane endpoints were reached (tumor volume ≥ 1500 mm³, ulceration, or > 20% body weight loss). No animals required early euthanasia.
All experimental data were analyzed without subjective exclusions. Investigators performing experiments and outcome assessments were blinded to group allocation, and data analysts remained blinded until completion of statistical analyses. One sample per group was excluded from immunohistochemical analysis due to tissue processing damage. All animal procedures were approved by the Ethics Committee of Chongqing Medical University. Throughout the study, tumor sizes remained within institutional ethical limits (maximum diameter 1.8 cm; volume 1800 mm³), ensuring compliance with all relevant ethical guidelines.
Statistical analysis
Omics data of ccRCC patients were analyzed using R 3.6.1 software (https://www.r-project.org/). Images were evaluated using ImageJ software (v1.53t, https://imagej.nih.gov/ij/). Experimental data were analyzed using Prism 10.4 software (GraphPad Prism Software Inc.). A two-tailed Welch’s t-test (effect size: Welch-corrected Cohen’s d, or Hedges’ g if n < 20) or Mann-Whitney U test P < 0.05 (effect size: Δ with 95% confidence interval) was considered statistically significant. Descriptive mean difference (MD) with 95% confidence interval was also calculated. Benjamini-Hochberg adjustment for P values was performed for multiple tests. Error bars (mean ± standard deviation) of data were shown in experiments. Detailed statistics for each experiment or analysis were shown in Source Data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Proteomics data of LC-MS/MS generated in this study have been deposited in PRoteomics IDEntifications Database (PRIDE) under accession code: PXD063023. The transcriptome and copy number data used in this study from TCGA project, including TCGA-KIRC cohort, are available in the Genomic Data Commons (GDC) portal [https://portal.gdc.cancer.gov/]. Human Protein Atlas (HPA, v19.3 and v24.0) [http://www.proteinatlas.org/] for RNA and protein expression data of DSS1 and SPP1 in cell lines and tissues. Proteomics data of ccRCC used in this publication were generated by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) [https://proteomic.datacommons.cancer.gov/pdc/]. Microarray and transcriptome data used in this study are available in the Gene Expression Omnibus (GEO) database under accession codes GSE72304, GSE113204, GSE3538, GSE251905, GSE254566, GSE65615, GSE64052, GSE76068. Single-cell transcriptome data are available under accession codes GSE159115, GSE178481, GSE210038. Spatial transcriptome data are available under accession codes GSE175540 and GSE210041. Spatial transcriptome data is available from CELLxGENE database [https://cellxgene.cziscience.com/] under Li2022 dataset [https://cellxgene.cziscience.com/collections/f7cecffa-00b4-4560-a29a-8ad626b8ee08]. Source data are provided with this paper.
Code availability
The script to run CSGC algorithm was deposited in GitHub with a DOI [https://zenodo.org/records/14768671] (Song, J. JingSonglab/CSGC-algorithm: CSGC v1.0. Zenodo. https://doi.org/10.5281/zenodo.15628528 (2025))95. Other analyses use public software with default parameters, otherwise parameters were described in this article.
References
Bukavina, L. et al. Epidemiology of Renal Cell Carcinoma: 2022 Update. Eur. Urol. 82, 529–542 (2022).
Rathmell, W. K. et al. Management of Metastatic Clear Cell Renal Cell Carcinoma: ASCO Guideline. J. Clin. Oncol. 40, 2957–2995 (2022).
Hsieh, J. J. et al. Renal cell carcinoma. Nat. Rev. Dis. Prim. 3, 17009 (2017).
Luan, F. L. et al. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 63, 917–926 (2003).
Kotecha, R. R., Motzer, R. J. & Voss, M. H. Towards individualized therapy for metastatic renal cell carcinoma. Nat. Rev. Clin. Oncol. 16, 621–633 (2019).
Motzer, R. J. et al. Nivolumab plus cabozantinib versus sunitinib in first-line treatment for advanced renal cell carcinoma (CheckMate 9ER): long-term follow-up results from an open-label, randomised, phase 3 trial. Lancet Oncol. 23, 888–898 (2022).
McDermott, D. F. et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 24, 749–757 (2018).
Ng, M. S. F. et al. Deterministic reprogramming of neutrophils within tumors. Science 383, eadf6493 (2024).
Yang, D. et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185, 1905–1923.e1925 (2022).
Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 12, 31–46 (2022).
Lengrand, J. et al. Pharmacological targeting of netrin-1 inhibits EMT in cancer. Nature 620, 402–408 (2023).
Shi, Q. et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell 40, 1207–1222.e1210 (2022).
Calon, A. et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 22, 571–584 (2012).
Barkley, D. et al. Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat. Genet. 54, 1192–1201 (2022).
Kragelund, B. B., Schenstrøm, S. M., Rebula, C. A., Panse, V. G. & Hartmann-Petersen, R. DSS1/Sem1, a Multifunctional and Intrinsically Disordered Protein. Trends biochemical Sci. 41, 446–459 (2016).
Mishra, A. P. et al. BRCA2-DSS1 interaction is dispensable for RAD51 recruitment at replication-induced and meiotic DNA double strand breaks. Nat. Commun. 13, 1751 (2022).
Li, C. et al. SEM1 promotes tumor progression of glioblastoma via activating the akt signaling pathway. Cancer Lett. 577, 216368 (2023).
Matusiak, M. et al. Spatially Segregated Macrophage Populations Predict Distinct Outcomes in Colon Cancer. Cancer Discov. 14, 1418–1439 (2024).
Bill, R. et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 381, 515–524 (2023).
Chang, H. Y. et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2, E7 (2004).
Chang, H. Y. et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc. Natl Acad. Sci. USA 102, 3738–3743 (2005).
Wang, C. et al. Blocking the autocrine regulatory loop of Gankyrin/STAT3/CCL24/CCR3 impairs the progression and pazopanib resistance of clear cell renal cell carcinoma. Cell death Dis. 11, 117 (2020).
Bezwada, D. et al. Mitochondrial complex I promotes kidney cancer metastasis. Nature 633, 923–931.
Zhao, H. et al. Gene expression profiling predicts survival in conventional renal cell carcinoma. PLoS Med. 3, e13 (2006).
Mehra, R. et al. Discovery and Validation of a 15-Gene Prognostic Signature for Clear C ell Renal Cell Carcinoma. JCO Precis. Oncol. 8, e2300565, https://doi.org/10.1200/PO.23.00565.
Suzuki, H. I., Kiyono, K. & Miyazono, K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy 6, 645–647 (2010).
Yin, L. et al. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. EBioMedicine 51, 102596 (2020).
Bertrand, M. et al. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle 14, 364–374 (2015).
Sun, T. et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma. Hepatol. (Baltim., Md.) 54, 1690–1706 (2011).
Kang, R., Zeh, H. J., Lotze, M. T. & Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell death Differ. 18, 571–580 (2011).
Rogov, V., Dötsch, V., Johansen, T. & Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. cell 53, 167–178 (2014).
Stefanovie, B. et al. DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic Acids Res 48, 694–708 (2020).
Wang, W., Chen, Z., Billiar, T. R., Stang, M. T. & Gao, W. The carboxyl-terminal amino acids render pro-human LC3B migration similar to lipidated LC3B in SDS-PAGE. PloS one 8, e74222 (2013).
Zhang, Y. et al. DSSylation, a novel protein modification targets proteins induced by oxidative stress, and facilitates their degradation in cells. Protein cell 5, 124–140 (2014).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Tiwari, P., Kaila, P. & Guptasarma, P. Understanding anomalous mobility of proteins on SDS-PAGE with special reference to the highly acidic extracellular domains of human E- and N-cadherins. Electrophoresis 40, 1273–1281 (2019).
Tomko, R. J. Jr. & Hochstrasser, M. The intrinsically disordered Sem1 protein functions as a molecular tether during proteasome lid biogenesis. Mol. cell 53, 433–443 (2014).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Mei, P. et al. E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis. Cell death Differ. 28, 2888–2899 (2021).
Lee, J. M. et al. The E3 ubiquitin ligase TRIM25 regulates adipocyte differentiation via proteasome-mediated degradation of PPARγ. Exp. Mol. Med. 50, 1–11 (2018).
Bui, T. O. et al. Genomics of Clear-cell Renal Cell Carcinoma: A Systematic Review and Meta-analysis. Eur. Urol. 81, 349–361 (2022).
Clark, D. J. et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e931 (2019).
Murakawa, Y. et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res. 67, 8536–8543 (2007).
Stewart, G. D. et al. Sunitinib Treatment Exacerbates Intratumoral Heterogeneity in Metastatic Renal Cancer. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 21, 4212–4223 (2015).
Diaz-Montero, C. M. et al. MEK inhibition abrogates sunitinib resistance in a renal cell carcinoma patient-derived xenograft model. Br. J. cancer 115, 920–928 (2016).
Zhang, L. et al. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal Cancer. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 21, 1925–1934 (2015).
Fustero-Torre, C. et al. Beyondcell: targeting cancer therapeutic heterogeneity in single-cell RNA-seq data. Genome Med. 13, 187 (2021).
Ianevski, A. et al. Single-cell transcriptomes identify patient-tailored therapies for selective co-inhibition of cancer clones. Nat. Commun. 15, 8579 (2024).
Liu, S. et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 8, 826–837 (2012).
Choi, M. E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 82, 297–322 (2020).
Schödel, J. et al. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur. Urol. 69, 646–657 (2016).
Hall, D. P. et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer cell 26, 738–753 (2014).
Zhang, C. et al. Methionine secreted by tumor-associated pericytes supports cancer stem cells in clear cell renal carcinoma. Cell Metab. https://doi.org/10.1016/j.cmet.2024.01.018 (2024).
Haas, N. B. et al. Autophagy Inhibition to Augment mTOR Inhibition: a Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 25, 2080–2087 (2019).
Deng, Q. et al. Lower mRNA and protein expression levels of LC3 and Beclin1, markers of autophagy, were correlated with progression of renal clear cell carcinoma. Jpn. J. Clin. Oncol. 43, 1261–1268 (2013).
Jin, J. et al. Sunitinib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Drug resistance updates: Rev. commentaries antimicrobial anticancer Chemother. 67, 100929 (2023).
Hoshino, A. et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015).
Ji, Q. et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat. Commun. 11, 1211 (2020).
Wei, S. J., Trempus, C. S., Cannon, R. E., Bortner, C. D. & Tennant, R. W. Identification of Dss1 as a 12-O-tetradecanoylphorbol-13-acetate-responsive gene expressed in keratinocyte progenitor cells, with possible involvement in early skin tumorigenesis. J. Biol. Chem. 278, 1758–1768 (2003).
Sone, T., Saeki, Y., Toh-e, A. & Yokosawa, H. Sem1p is a novel subunit of the 26 S proteasome from Saccharomyces cerevisiae. J. Biol. Chem. 279, 28807–28816 (2004).
Paraskevopoulos, K. et al. Dss1 is a 26S proteasome ubiquitin receptor. Mol. cell 56, 453–461 (2014).
Ding, Y., Xing, D., Fei, Y. & Lu, B. Emerging degrader technologies engaging lysosomal pathways. Chem. Soc. Rev. 51, 8832–8876 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Tate, J. G. et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Loher, P. & Rigoutsos, I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 28, 3322–3323 (2012).
Jeggari, A., Marks, D. S. & Larsson, E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 28, 2062–2063 (2012).
Agarwal, V., Bell, G. W., Nam, J. W. & Bartel, D. P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, https://doi.org/10.7554/eLife.05005 (2015).
Kertesz, M., Iovino, N., Unnerstall, U., Gaul, U. & Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 39, 1278–1284 (2007).
Chen, K. & Rajewsky, N. Natural selection on human microRNA binding sites inferred from SNP data. Nat. Genet. 38, 1452–1456 (2006).
Wong, N. & Wang, X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 43, D146–D152 (2015).
Huang, H. Y. et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res 48, D148–d154 (2020).
John, B. et al. Human MicroRNA targets. PLoS Biol. 2, e363 (2004).
Krüger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 34, W451–W454 (2006).
Dagher, J. et al. Multiple metastatic clones assessed by an integrative multiomics strategy in clear cell renal carcinoma: a case study. J. Clin. Pathol. 75, 426–430 (2022).
Iacobas, D. A. et al. Genomic Fabric Remodeling in Metastatic Clear Cell Renal Cell Carcinoma (ccRCC): A New Paradigm and Proposal for a Personalized Gene Therapy Approach. Cancers 12, https://doi.org/10.3390/cancers12123678 (2020).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009).
Yang, J. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 (2004).
Jiang, L. et al. A Quantitative Proteome Map of the Human Body. Cell 183, 269–283.e219 (2020).
Quarato, G. et al. Mitophagy restricts BAX/BAK-independent, Parkin-mediated apoptosis. Sci. Adv. 9, eadg8156 (2023).
Li, J. et al. PBMC transcriptomics identifies immune-metabolism disorder during the development of HBV-ACLF. Gut 71, 163–175 (2022).
Bezwada, D. et al. Mitochondrial complex I promotes kidney cancer metastasis. Nature 633, 923–931 (2024).
Mehra, R. et al. Discovery and Validation of a 15-Gene Prognostic Signature for Clear Cell Renal Cell Carcinoma. JCO Precis. Oncol. 8, e2300565 (2024).
Zhang, Y. et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA. 118, https://doi.org/10.1073/pnas.2103240118 (2021).
Alchahin, A. M. et al. A transcriptional metastatic signature predicts survival in clear cell renal cell carcinoma. Nat. Commun. 13, 5747 (2022).
Davidson, G. et al. Mesenchymal-like Tumor Cells and Myofibroblastic Cancer-Associated Fibroblasts Are Associated with Progression and Immunotherapy Response of Clear Cell Renal Cell Carcinoma. Cancer Res. 83, 2952–2969 (2023).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Morabito, S. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet. 53, 1143–1155 (2021).
Meylan, M. et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 55, 527–541.e525 (2022).
Li, R. et al. Mapping single-cell transcriptomes in the intra-tumoral and associated territories of kidney cancer. Cancer cell 40, 1583–1599.e1510 (2022).
Program, C. Z. I. S.-C. B. et al. CZ CELL×GENE Discover: A single-cell data platform for scalable exploration, analysis and modeling of aggregated data. bioRxiv, 2023.2010.2030.563174, https://doi.org/10.1101/2023.10.30.563174 (2023).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Song, J. JingSonglab/CSGC-algorithm: CSGC v1.0. Zenodo. https://doi.org/10.5281/zenodo.15628528 (2025).
Acknowledgements
This research is supported by Chongqing Natural Science Foundation (CSTB2024NSCQ-MSX0232 to Mingming Zhao), and CQMU Program for Youth Innovation in Future Medicine (W0144 to Mingming Zhao). We sincerely thank Professor Delin Wang of the First Affiliated Hospital of Chongqing Medical University for his assistance in submitting the ethical review.
Author information
Authors and Affiliations
Contributions
J.S. and X.C. designed the study. J.S. performed bioinformatic analyses. J.S. and X.C. performed the laboratory experiments. Q.L., M.Z., J.W., and P.Z. contributed samples or clinical data. J.S., X.C., Q.L., M.Z., J.W., and P.Z. interpreted the data. J.S. and M.Z. drafted the manuscript. J.S. and M.Z. supervised the study. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Liu, Q., Wu, J. et al. DSS1 inhibits autophagy to activate epithelial-mesenchymal transition in a pro-metastatic niche of renal cell carcinoma. Nat Commun 16, 6769 (2025). https://doi.org/10.1038/s41467-025-62135-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62135-9
