
Abstract
The heterodifunctionalization of alkenes is an efficient and straightforward method for the preparation of highly functionalized molecules. However, enantioselective introduction of two different carbon-based functional groups in a single step using readily accessible and inexpensive starting materials presents a significant challenge. Herein, we report an electrochemical copper-catalyzed protocol for the asymmetric cyanoesterification of vinylarenes using commercially available alkyl carbazates and trimethylsilyl cyanide (TMSCN) as the sources of ester and cyano groups, respectively. The desired products could be obtained with good yields and enantioselectivities under mild conditions without the need for stoichiometric oxidants, providing sustainable access to versatile synthetic intermediates that could be smoothly converted into a variety of useful chiral building blocks. Mechanistic data are consistent with electrochemical copper-catalyzed generation of alkoxycarbonyl radicals from alkyl carbazates and the copper catalyst is also responsible for the stereoselective C–CN bond formation. The potential synthetic utility of this new electrocatalytic protocol is demonstrated in the concise synthesis of pharmacologically active molecules.
Similar content being viewed by others
Introduction
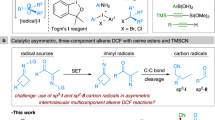
Given the ubiquity of alkenes in feedstock chemicals and natural products, the development of new catalytic strategies for difunctionalization of the double bonds has been an area of intense research in organic chemistry1,2,3,4,5,6,7,8. Specifically, the stereoselective heterodifunctionalization of alkenes is a powerful and widely employed tactic for the rapid construction of complex molecular architectures that are particularly valuable in synthetic and medicinal applications9,10,11,12. In this context, the asymmetric cyanoesterification of alkenes is highly desirable in the synthetic community, because two synthetically versatile functional groups—the ester and cyano groups are installed across the double bond in a single operational step. More importantly, such a methodology grants access to synthetic intermediates that are readily converted into a wide range of valuable chiral building blockings, including β-substituted-γ-aminobutyric acids (GABA) and pertinent structures in medicinally relevant compounds (Fig. 1A)13,14,15. Despite significant efforts in developing innovative methods for the practical synthesis of chiral GABA derivatives16,17,18,19,20,21,22,23,24,25, the direct utilization of more readily available alkenes as substrates for asymmetric cyanoesterification remains largely unexplored.

A Selected drug molecules derived from chiral β-cyano esters. B Synthesis of chiral β-cyano carbonyls with cyclopropanols. C Synthesis of chiral β-cyano carbonyls with styrenes. D This Work: electrochemical Cu-catalyzed asymmetric cyanoesterification of vinylarenes. SET single-electron transfer.
The incorporation of single-electron strategies into asymmetric transition metal catalysis has equipped chemists with innovative retrosynthetic approaches for solving challenging synthetic problems by harnessing highly reactive open-shell species26,27,28,29. As a particularly elegant example, copper-catalyzed asymmetric radical cyanation of carbon-centered radicals has achieved notable success in synthesizing enantiomerically enriched alkyl nitriles30,31,32,33,34. Directly related to the present work, the Liu group devised an efficient protocol for the asymmetric cyanation of cyclopropanols by copper-catalyzed radical relay processes, providing β-cyano esters with excellent enantioselectivity (Fig. 1B)35. In addition, carbonyl radicals including acyl, alkoxycarbonyl, and carbamoyl radicals, generated by single-electron activation or hydrogen atom abstraction of the aldehydic C(sp2)–H bonds, have been successfully applied in radical-based transformations36,37,38,39,40,41,42,43. For instance, Chen, Xiao, and co-workers reported the photoredox copper-catalyzed asymmetric acylcyanation of styrenes using redox-active oxime esters as the acyl radical precursors. Photoredox-mediated single-electron transfer (SET) of oxime esters produced the key nitrogen-centered iminyl radicals that triggered C–C bond cleavage to deliver the required acyl radicals for the reaction44. However, the use of pre-functionalized substrates for the generation of carbonyl radicals could reduce the practicality of the catalytic system (Fig. 1C).
Recently, we and others have demonstrated electrochemical transition metal catalysis as a viable and potentially general synthetic platform for the innovation of radical-based transformations that are often elusive or currently impossible using existing methods45,46,47,48,49,50,51,52. Enabled by the unique attributes of this strategy, redox reactions are frequently realized in a more sustainable, efficient, and chemo-selective manner53,54,55,56,57,58,59,60,61,62,63,64,65,66,67. Based on our previous investigation of the electrochemical iron-catalyzed azidoesterification of alkenes68, we envisioned that an enantioselective variant of cyanoesterification of vinyl arenes might be accessible via the synergistic merger of electrochemical iron catalysis and asymmetric copper catalysis69,70,71. In the proposed mechanistic pathways, electrochemical iron catalysis is designed to promote oxidative production of alkoxycarbonyl radical 1 from the commercially available carbazates68,72. After that, the radical addition to the vinylarene would generate a more thermodynamically stable benzylic radical 2. Taking the advantage of electrochemistry to accommodate multiple concurrent redox events at the same electrode, anodic oxidation could also lead to the formation of a Cu(II)–CN complex 3, which would intercept the newly generated benzylic radical. The resulting Cu(III)–CN adduct is expected to undergo stereoselective reductive elimination to forge the desired C–CN bond with the aid of an appropriate chiral ligand (Fig. 1D)73,74,75,76,77,78,79,80.
In this work, we report an electrochemical copper-catalyzed protocol for the asymmetric cyanoesterification of vinylarenes. Notable features of this electrocatalytic strategy are as follows: 1) direct use of readily accessible reagents for the preparation of synthetically versatile chiral β-cyano esters from vinylarenes; 2) excellent chemoselectivity, stereoselectivity, and good functional group tolerance without the need for stoichiometric chemical oxidants; 3) good scalability for synthetic applications and has been successfully demonstrated in the efficient synthesis of drug molecules.
Results and discussion
Optimization of reaction conditions
We set out to explore the feasibility of our reaction design in the asymmetric cyanoesterification of 2-vinylnaphthalene 4 with methyl carbazate 5 and TMSCN by the combinational use of iron phthalocyanine (PcFe) (5 mol%) and Cu(OTf)2 (1.0 mol%) together with a commercial bisoxazoline ligand 6 (3.0 mol%)81 in a simple and practical undivided cell under constant current conditions. We were delighted to find that the reaction indeed occurred to give the corresponding cyanoesterification product 7 in a promising yield with excellent enantioselectivity (Table 1, entry 1). However, further reaction conditions optimization and control experiments led to the discovery that the reaction proceeded smoothly without the need for iron catalysis, the desired cyanoesterification product could be obtained in an isolated yield of 80% with 92% ee when the reaction was performed at room temperature using Cu(OTf)2/6 as the catalyst (entry 2). Our investigation into the ratio of copper to the chiral ligand revealed that a 1:3 ratio is optimal for the reaction efficiency. Efforts to decrease the ligand loading proved unsuccessful, primarily due to the reduction of copper ion to plate out on the cathode, which was visibly observed upon reaction completion (entry 3). Replacing acetic acid (AcOH) with trifluoroethanol (TFE) seriously caused the undesired copper reduction issue; the alkene was fully consumed to form various side products (entry 4). We employed Pt as the cathode to ensure facile proton reduction to outcompete copper reduction. Therefore, as expected, the substitution of the Pt cathode with an iron resulted in an acceptable but reduced yield (entry 5). These results underscored the pivotal role of the proton source selection, the copper-to-ligand ratio, and the employment of Pt as the cathode for maximizing the electrocatalytic efficiency of the copper catalyst in the reaction. Modifying the current density or running the reaction with a constant cell potential led to suboptimal results but has no obvious effect on the stereoselectivity of the reaction (entries 6–8).
Substrate scope
With the optimized reaction conditions in hand, we next evaluated the scope of vinylarenes that can be employed in this new asymmetric cyanoesterification protocol. As illustrated in Fig. 2, a diverse array of vinylarenes successfully underwent the desired oxidative three-component coupling with good to excellent reaction efficiency (7–23, 48–80% yield, 81–98% ee), yielding chiral β-cyano esters featuring a broad spectrum of functional groups, including ethers (8, 13), halides (15–17, 20–23), and esters (19) that can be utilized for further diversification. In particular, vinylarenes with electron-rich groups such as the methoxy group that might generate the corresponding benzylic radicals with very low oxidation potentials for the formation of benzylic cation intermediates were competent substrates (13), demonstrating the mild nature of our electrocatalytic protocol. In addition, sulfide (14) that was supposed to be vulnerable to traditional chemical oxidation conditions was successfully preserved in the reaction. However, a notable amount of hydroesterification side product was observed in reactions involving styrenes with electron-withdrawing groups at the para position of the phenyl ring (18 and 19). We hypothesize that the corresponding benzylic radical intermediates are more susceptible to hydrogen atom transfer, leading to the formation of these side products. Styrenes featuring a substituent at the ortho position were found to be compatible substrates for the reaction; the ortho substituent appears to enhance stereocontrol but negatively impacts the reaction yield (20, 21). Notably, the benzyl chloride (23) that could potentially undergo nucleophilic substitution by AcO— or CN— in the reaction system, was also tolerated. Encouraged by the compatibility of the catalytic system to electron-rich substrates, we next examined the preparation of products 24 and 25, with the later as the key intermediate for the synthesis of an anti-depressant drug (R)-rolipram (vide infra); both of the desired products were obtained from the corresponding vinylarenes with good reaction results.

All yields are of isolated products. Unless otherwise noted, reaction conditions were as follows: alkene (0.15 mmol, 1.0 equiv), TMSCN (2.0 equiv), 5 (2.0 equiv), Cu(OTf)2/6 (1.0/3.0 mol %), TBAPF6 (2.0 equiv), AcOH (1.0 mmol), MeCN (3.0 mL), carbon felt as the anode, Pt as the cathode, under N2, in an undivided cell, 9.0 mA, for 4 h.
Next, we turned our attention to determining the tolerance of this electrocatalytic system to heteroaromatic rings that are commonly found in pharmaceutically relevant compounds. Remarkably, an array of electron-rich and electron-deficient heterocycles, including pyridine (26), benzothiophene (27, 28), benzofuran (29), and indole (30) were transformed to the corresponding products in good yields with good to excellent enantioselectivities (56–78% yield, 80–94% ee). In addition to styrenes, enynes also underwent chemoselective alkene cyanoesterification to yield the desired products with good enantioselectivities (31–33). Finally, we found that this electrocatalytic protocol could also be effectively applied to the functionalization of biologically active derivatives—styrenes derived from L-menthol (34), diacetonefructose (35), indometacin (36), gemfibrozil (37), and bezafibrate (38) proved to be viable substrates, affording the cyanoesterification adducts in synthetically useful yields with good stereocontrol.
Synthetic applications
For practical applications, we explored the scalability of this electrocatalytic asymmetric cyanoesterification protocol and found that it could be successfully scaled up to gram level under constant current conditions using a lower copper catalyst loading. The desired product was obtained with excellent stereoselectivity, albeit with a decreased yield (Fig. 3A). Attempts to improve the reaction yield through reactor design to address the possible mass transport issue are continuing in our laboratory. The synthesized product can be efficiently converted into the corresponding diacid compound 39, potentially useful in polymer synthesis82,83,84,85, without any loss of stereoselectivity. Moreover, the chiral β-cyano ester can be readily transformed into chiral γ-lactam 40 and γ-amino alcohol 41 with favorable reaction yields using well-established reduction methods. To further highlight the utility of our asymmetric electrocatalytic cyanoesterification method, we applied this protocol to the synthesis of pharmacologically active molecules. For example, when chiral β-cyano ester product 25 was subjected to the nickel-mediated reduction, the corresponding reductive cyclization product 42, an anti-depressant drug could be obtained with acceptable reaction efficiency. In the case of chiral β-cyano ester product 16, reductive cyclization followed by hydrolysis gave γ-amino acid product 43, which is an inhibitory neurotransmitter and is used to treat pain and certain types of spasticity (Fig. 3B).

A Large scale synthesis. B Product derivatization and application to the synthesis of bioactive molecules. For details, see the Supplementary Information.
Mechanistic studies
To elucidate the reaction mechanism, a series of experiments were designed and performed (Fig. 4). Cyclic voltammetry analysis indicated that the direct anodic oxidation of methyl carbazate 5 at the carbon electrode commenced with an onset potential of approximately 0.25 V (with reference to the ferrocenium ion/ferrocene redox couple in MeCN). The anodic oxidation of Cu(I)–CN to the corresponding Cu(II)–CN species in the presence of the chiral ligand, however, resulted in a feature at around 0 V (Fig. 4A). To ascertain whether the anodically generated Cu(II)–CN species could promote the oxidative fragmentation of methyl carbazate for the reaction, we conducted controlled anodic potential electrolysis at 0 V that is not sufficient to directly oxidize methyl carbazate 5, but we still observed the formation of product 7 in 15% yield with 92% ee (Fig. 4B). These data are consistent with a unique mechanistic scenario in which the copper catalyst plays a dual role: it is responsible not only for the stereoselective formation of the C–CN bond but also serves as a redox mediator that facilitates the oxidative fragmentation of methyl carbazate 5, leading to the generation of the methoxycarbonyl radical in the reaction86,87.

A Cyclic voltammetry studies. B Controlled potential electrolysis experiment. C Radical probe experiment. D Proposed catalytic cycles. For details, see the Supplementary Information.
In addition, a radical addition-trigged rearrangement experiment with cyclopropane-derived alkene 44 was performed to provide key support for the intermediacy of carbon-centered radical species in the reaction (Fig. 4C). Specifically, upon formation of the first C–C bond via homolysis of the double bond with alkoxycarbonyl radical, the resultant carbon-centered radical 45 would render ring rupture of the cyclopropyl group, leading to the generation of a more thermodynamically stable benzylic radical intermediate 46. This intermediate then enters the Cu-catalyzed asymmetric cyanation cycle to give the desired product via stereoselective reductive elimination from the putative Cu(III)–CN complex.
Taken together, we proposed an electrochemical copper-catalyzed mechanism for this novel asymmetric cyanoesterification reaction. As shown in Fig. 4D, the generation of methoxycarbonyl radical 48 from methyl carbazate was mediated by anodically generated L-Cu(II)–CN species in our catalytic system. Then, radical addition to vinylarene substrate produced the relatively more stable benzylic radical intermediate 49. Subsequently, radical oxidative addition to L-Cu(II)–CN to form the putative alkyl Cu(III)–CN species, which after reductive elimination forged the desired C–CN bond to furnish the final product along with the regeneration of L-Cu(I) for the next catalytic cycle.
In summary, we have established a sustainable and robust strategy for the direct and efficient preparation of chiral β-cyano esters from readily available vinylarenes and commercially available reagents with a low copper catalyst loading under electrochemical conditions. The electrochemical copper catalysis enabled the generation and utilization of the key alkoxycarbonyl radicals from alkyl carbazates in a controlled and sustainable manner and also imparted stereoselectivity control over the construction of C–CN bonds in the presence of a commercial chiral ligand. This electrocatalytic difunctionalization method exhibited excellent chemoselectivity, stereoselectivity, and high functional group tolerance, and can be successfully scaled up for preparative synthesis. We have demonstrated the utility of this protocol through the preparation of drug molecules. Given the generality and the synthetic potential of both the ester and cyano groups in the obtained products, we anticipate this new electrochemical copper-catalyzed cyanoesterification protocol will find broad applications in both synthetic and medicinal chemistry.
Methods
General procedure for cyanoesterification of vinylarenes
Solution A: Prior to use, Cu(OTf)2 (3.3 mg, 0.009 mmol) and ligand 6 (9.6 mg, 0.027 mmol) were combined stirred for 3 min in MeCN (6 mL) under a nitrogen atmosphere. Subsequently, TMSCN (96% wt, 186 mg, 1.8 mmol) was added, and the mixture was stirred continuously for an additional hour.
An oven-dried, 10 mL two-neck glass tube was equipped with a magnetic stir bar, a rubber septum, a threaded Teflon cap fitted with electrical feed-throughs, a carbon-felt anode (0.5 × 1.0 × 1.4 cm3) (connected to the electrical feedthrough via a 9 cm in length, 2 mm in diameter graphite rod), and a platinum plate cathode (0.5 × 1.0 cm2). To this reaction vessel was added methyl carbazate 5 (97% wt, 28 mg, 0.3 mmol, 2.0 equiv), tetrabutylammonium hexafluorophosphate (116 mg, 0.3 mmol). The cell was sealed and flushed with nitrogen gas for 5 min, followed by the sequential addition via syringe of solution A (1 mL), olefin substrate (0.15 mmol, 1.0 equiv), AcOH (60 mg, 1.0 mmol), and CH3CN (2 mL). The reaction mixture was then purged with nitrogen gas for another 5 min. A nitrogen-filled balloon was adapted through the septum to sustain a nitrogen atmosphere. Electrolysis was initiated at a constant current of 9 mA at room temperature. After 4 h, the tube cap was removed and electrodes were rinsed with ethyl acetate, which was combined with the crude mixture and the organic layers were concentrated in vacuo. The residue was subjected to flash column chromatography on silica gel (eluted with petroleum ether/EA) to yield the purified product.
Data availability
Materials and methods, optimization studies, experimental procedures, mechanistic studies, 1H NMR spectra, 13C NMR spectra and mass spectrometry data generated in this study are provided in the Supplementary Information. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
McDonald, R. I., Liu, G. & Stahl, S. S. Palladium(II)-catalyzed alkene functionalization via nucleopalladation: stereochemical pathways and enantioselective catalytic applications. Chem. Rev. 111, 2981–3019 (2011).
Yin, G., Mu, X. & Liu, G. Palladium(II)-catalyzed oxidative difunctionalization of alkenes: bond forming at a high-valent palladium center. Acc. Chem. Res. 49, 2413–2423 (2016).
Derosa, J., Tran, V. T., van der Puyl, V. A. & Engle, K. M. Carbon-Carbon π bonds as conjunctive reagents in cross-coupling. Aldrichim. Acta 51, 21–32 (2018).
Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287–4296 (2020).
Li, Y., Wu, D., Cheng, H.-G. & Yin, G. Difunctionalization of alkenes involving metal migration. Angew. Chem. Int. Ed. 59, 7990–8003 (2020).
Wickham, L. M. & Giri, R. Transition metal (Ni, Cu, Pd)-catalyzed alkene dicarbofunctionalization reactions. Acc. Chem. Res. 54, 3415–3437 (2021).
Jiang, H. & Studer, A. Intermolecular radical carboamination of alkenes. Chem. Soc. Rev. 49, 1790–1811 (2020).
Zhu, S., Zhao, X., Li, H. & Chu, L. Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel. Chem. Soc. Rev. 50, 10836–10856 (2021).
Coombs, J. R. & Morken, J. P. Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew. Chem. Int. Ed. 55, 2636–2649 (2016).
Li, Z.-L., Fang, G.-C., Gu, Q.-S. & Liu, X.-Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32–48 (2020).
Xiong, T. & Zhang, Q. Recent advances in the direct construction of enantioenriched stereocenters through addition of radicals to internal alkenes. Chem. Soc. Rev. 50, 8857–8873 (2021).
Han, J., He, R. & Wang, C. Transition metal-catalyzed asymmetric three-component dicarbofunctionalization of unactivated alkenes. Chem. Catal. 3, 100690 (2023).
Bowery, N. G. et al. (–)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature 283, 92–94 (1980).
Silverman, R. B. From basic science to blockbuster drug: the discovery of lyrica. Angew. Chem. Int. Ed. 47, 3500–3504 (2008).
Lee, G.-S. et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 (2012).
Sammis, G. M. & Jacobsen, E. N. Highly enantioselective, catalytic conjugate addition of cyanide to α,β-unsaturated imides. J. Am. Chem. Soc. 125, 4442–4443 (2003).
Sammis, G. M., Danjo, H. & Jacobsen, E. N. Cooperative dual catalysis: application to the highly enantioselective conjugate cyanation of unsaturated imides. J. Am. Chem. Soc. 126, 9928–9929 (2004).
Wiesner, M., Revell, J. D., Tonazzi, S. & Wennemers, H. Peptide catalyzed asymmetric conjugate addition reactions of aldehydes to nitroethylene—a convenient entry into γ2-amino acids. J. Am. Chem. Soc. 130, 5610–5611 (2008).
Tanaka, Y., Kanai, M. & Shibasaki, M. A Catalytic enantioselective conjugate addition of cyanide to enones. J. Am. Chem. Soc. 130, 6072–6073 (2008).
Mukherjee, H. & Martinez, C. A. Biocatalytic route to chiral precursors of β-substituted-γ-amino acids. ACS Catal. 1, 1010–1013 (2011).
Gómez, J. E., Guo, W., Gaspa, S. & Kleij, A. W. Copper-catalyzed synthesis of γ-amino acids featuring quaternary stereocenters. Angew. Chem. Int. Ed. 56, 15035–15038 (2017).
Li, X. et al. Rhodium-catalyzed asymmetric hydrogenation of β-cyanocinnamic esters with the assistance of a single hydrogen bond in a precise position. Chem. Sci. 9, 1919–1924 (2018).
Ma, J. et al. Synthesis of β-substituted γ-aminobutyric acid derivatives through enantioselective photoredox catalysis. Angew. Chem. Int. Ed. 57, 11193–11197 (2018).
Lahdenperä, A. S. K., Bacoş, P. D. & Phipps, R. J. Enantioselective giese additions of prochiral α-amino radicals. J. Am. Chem. Soc. 144, 22451–22457 (2022).
Xie, Y., Lai, Z.-M., Chan, A. S. C., Guo, J. & Lu, G. Nickel-catalyzed asymmetric synthesis of β- or β,γ-substituted GABA derivatives enabled by photoactive ternary electron donor–acceptor complex. ACS Catal. 14, 18734–18743 (2024).
Song, L. & Fu, N. Single-electron strategies in organometallic methods: photoredox, electrocatalysis, radical relay, and beyond, in comprehensive organometallic chemistry IV, G. Parkin, K. Meyer and D. O’Hare, Elsevier, 13, 339–403 (2022).
Levin, M. D., Kim, S. & Toste, F. D. Photoredox catalysis unlocks single-electron elementary steps in transition metal catalyzed cross-coupling. ACS Cent. Sci. 2, 293–301 (2016).
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692–700 (2017).
Liu, J., Lu, L., Wood, D. & Lin, S. New redox strategies in organic synthesis by means of electrochemistry and photochemistry. ACS Cent. Sci. 6, 1317–1340 (2020).
Wang, F., Chen, P. & Liu, G. Copper-catalyzed radical relay for asymmetric radical transformations. Acc. Chem. Res. 51, 2036–2046 (2018).
Wang, F., Chen, P. & Liu, G. Copper-catalysed asymmetric radical cyanation. Nat. Synth. 1, 107–116 (2022).
Zhang, Z., Chen, P. & Liu, G. Copper-catalyzed radical relay in C(sp3)–H functionalization. Chem. Soc. Rev. 51, 1640–1658 (2022).
Zhang, W. et al. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016).
Li, J. et al. Site-specific allylic C–H bond functionalization with a copper-bound N-centred radical. Nature 574, 516–521 (2019).
Wu, L., Wang, L., Chen, P., Guo, Y.-L. & Liu, G. Enantioselective copper-catalyzed radical ring-opening cyanation of cyclopropanols and cyclopropanone acetals. Adv. Synth. Catal. 362, 2189–2194 (2020).
Slutskyy, Y. & Overman, L. E. Generation of the methoxycarbonyl radical by visible-light photoredox catalysis and its conjugate addition with electron-deficient olefins. Org. Lett. 18, 2564–2567 (2016).
Zhang, X. & MacMillan, D. W. C. Direct aldehyde C–H arylation and alkylation via the combination of nickel, hydrogen atom transfer, and photoredox catalysis. J. Am. Chem. Soc. 139, 11353–11356 (2017).
Stache, E. E., Ertel, A. B., Rovis, T. & Doyle, A. G. Generation of phosphoranyl radicals via photoredox catalysis enables voltage–independent activation of strong C–O bonds. ACS Catal. 8, 11134–11139 (2018).
Zhang, M., Xie, J. & Zhu, C. A general deoxygenation approach for synthesis of ketones from aromatic carboxylic acids and alkenes. Nat. Commun. 9, 3517 (2018).
Li, Y., et al. Highly selective synthesis of all-carbon tetrasubstituted alkenes by deoxygenative alkenylation of carboxylic acids. Nat. Commun. 13, 10 (2022).
Fan, P., Lan, Y., Zhang, C. & Wang, C. Nickel/photo-cocatalyzed asymmetric acyl-carbamoylation of alkenes. J. Am. Chem. Soc. 142, 2180–2186 (2020).
Budai, B., Leclair, A., Wang, Q. & Zhu, J. Copper-catalyzed 1,2-methoxy methoxycarbonylation of alkenes with methyl formate. Angew. Chem. Int. Ed. 58, 10305–10309 (2019).
Li, W.-D., Jiang, Y.-Q., Li, Y.-L. & Xia, J.-B. Photoredox Ni-catalyzed selective coupling of organic halides and oxalates to esters via alkoxycarbonyl radical intermediates. CCS Chem. 4, 1326–1336 (2022).
Wang, P.-Z., et al. Asymmetric three-component olefin dicarbofunctionalization enabled by photoredox and copper dual catalysis. Nat. Commun. 12, 1815 (2021).
Lu, J., Wang, Y., McCallum, T. & Fu, N. Harnessing radical chemistry via electrochemical transition metal catalysis. iScience 23, 101796 (2020).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547–560 (2020).
Malapit, C. A. et al. Advances on the merger of electrochemistry and transition metal catalysis for organic synthesis. Chem. Rev. 122, 3180–3218 (2022).
Ackermann, L. Metalla-electrocatalyzed C–H activation by earth-abundant 3d metals and beyond. Acc. Chem. Res. 53, 84–104 (2020).
Gandeepan, P., Finger, L. H., Meyer, T. H. & Ackermann, L. 3d metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 49, 4254–4272 (2020).
Ma, C. et al. Transition metal-catalyzed organic reactions in undivided electrochemical cells. Chem. Sci. 12, 12866–12873 (2021).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300–310 (2020).
Ma, C., Guo, J.-F., Xu, S.-S. & Mei, T.-S. Recent advances in asymmetric organometallic electrochemical synthesis (AOES). Acc. Chem. Res. 58, 399–414 (2025).
Fu, N., Sauer, G. S., Saha, A., Loo, A. & Lin, S. Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579 (2017).
Yang, Q.-L. et al. Palladium-catalyzed C(sp3)–H oxygenation via electrochemical oxidation. J. Am. Chem. Soc. 139, 3293–3298 (2017).
Harwood, S. J. et al. Modular terpene synthesis enabled by mild electrochemical couplings. Science 375, 745–752 (2022).
Zhang, B. et al. Ni-electrocatalytic Csp3–Csp3 doubly decarboxylative coupling. Nature 606, 313–318 (2022).
Zhang, B. et al. Complex molecule synthesis by electrocatalytic decarboxylative cross-coupling. Nature 623, 745–751 (2023).
Yao, Q.-J., Huang, F.-R., Chen, J.-H., Zhong, M.-Y. & Shi, B. -f Enantio- and Regioselective Electrooxidative Cobalt-catalyzed C−H/N−H annulation with alkenes. Angew. Chem. Int. Ed. 62, e202218533 (2023).
Zhou, G., et al. Base-promoted electrochemical CoII-catalyzed enantioselective C−H oxygenation. Angew. Chem. Int. Ed. 62, e202302964 (2023).
Sauermann, N., Meyer, T. H., Tian, C. & Ackermann, L. Electrochemical cobalt-catalyzed C–H oxygenation at room temperature. J. Am. Chem. Soc. 139, 18452–18455 (2017).
Qiu, Y., Tian, C., Massignan, L., Rogge, T. & Ackermann, L. Electrooxidative ruthenium-catalyzed C−H/O−H annulation by weak o-coordination. Angew. Chem. Int. Ed. 57, 5818–5822 (2018).
von Münchow, T., Dana, S., Xu, Y., Yuan, B. & Ackermann, L. Enantioselective electrochemical cobalt-catalyzed aryl C–H activation reactions. Science 379, 1036–1042 (2023).
von Münchow, T. et al. Cobaltaelectro-catalyzed C–H activation for central and axial double enantio-induction. Angew. Chem. Int. Ed. 63, e202405423 (2024).
Liang, K., Zhang, Q. & Guo, C. Enantioselective nickel-catalysed electrochemical cross-dehydrogenative amination. Nat. Synth. 2, 1184–1193 (2023).
Lu, J., Yao, Y., Li, L. & Fu, N. Dual transition metal electrocatalysis: direct decarboxylative alkenylation of aliphatic carboxylic acids. J. Am. Chem. Soc. 145, 26774–26782 (2023).
Zhang, J., Zhu, W., Chen, Z., Zhang, Q. & Guo, C. Dual-catalyzed stereodivergent electrooxidative homocoupling of benzoxazolyl acetate. J. Am. Chem. Soc. 146, 1522–1531 (2024).
Huang, C., Tao, Y., Cao, X., Zhou, C. & Lu, Q. Asymmetric paired electrocatalysis: enantioselective olefin–sulfonylimine coupling. J. Am. Chem. Soc. 146, 1984–1991 (2024).
Zhou, K., Lv, C. & Fu, N. Electrochemical metal-catalyzed azidoesterification of alkenes. Eur. J. Org. Chem. 26, e202200977 (2023).
Rein, J., Zacate, S. B., Mao, K. & Lin, S. A tutorial on asymmetric electrocatalysis. Chem. Soc. Rev., 52, 8106–8125 (2023).
Lin, Q., Li, L. & Luo, S. Asymmetric electrochemical catalysis. Chem. Eur. J. 25, 10033–10044 (2019).
Chang, X., Zhang, Q. & Guo, C. Asymmetric electrochemical transformations. Angew. Chem. Int. Ed. 59, 12612–12622 (2020).
Taniguchi, T., Sugiura, Y., Zaimoku, H. & Ishibashi, H. Iron-catalyzed oxidative addition of alkoxycarbonyl radicals to alkenes with carbazates and air. Angew. Chem. Int. Ed. 49, 10154–10157 (2010).
Fu, N. et al. New Bisoxazoline Ligands Enable enantioselective electrocatalytic cyanofunctionalization of vinylarenes. J. Am. Chem. Soc. 141, 14480–14485 (2019).
Song, L. et al. Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes. Nat. Chem. 12, 747–754 (2020).
Cai, C.-Y. et al. Photoelectrochemical asymmetric catalysis enables site- and enantioselective cyanation of benzylic C–H bonds. Nat. Catal. 5, 943–951 (2022).
Fan, W. et al. Electrophotocatalytic decoupled radical relay enables highly efficient and enantioselective benzylic C–H functionalization. J. Am. Chem. Soc. 144, 21674–21682 (2022).
Lai, X.-L., Chen, M., Wang, Y., Song, J. & Xu, H.-C. Photoelectrochemical asymmetric catalysis enables direct and enantioselective decarboxylative cyanation. J. Am. Chem. Soc. 144, 20201–20206 (2022).
Yuan, Y., Yang, J. & Zhang, J. Cu-catalyzed enantioselective decarboxylative cyanation via the synergistic merger of photocatalysis and electrochemistry. Chem. Sci. 14, 705–710 (2023).
Lai, X.-L. & Xu, H.-C. Photoelectrochemical asymmetric catalysis enables enantioselective heteroarylcyanation of alkenes via C–H functionalization. J. Am. Chem. Soc. 145, 18753–18759 (2023).
Yang, K., Wang, Y., Luo, S. & Fu, N. Electrophotochemical metal-catalyzed enantioselective decarboxylative cyanation. Chem. Eur. J. 29, e202203962 (2023).
Hofstra, J. L., DeLano, T. J. & Reisman, S. E. Synthesis of chiral bisoxazoline ligands: (3aR,3a’R,8aS,8a’S)−2,2’-(cyclopropane-1,1-diyl)bis(3a,8a-dihydro-8H-indeno[1,2-d]oxazole). Org. Synth. 97, 172–188 (2020).
Liao, L.-L. et al. Electrochemical ring-opening dicarboxylation of strained carbon–carbon single bonds with CO2: facile synthesis of diacids and derivatization into polyesters. J. Am. Chem. Soc. 144, 2062–2068 (2022).
Zhang, W. et al. Electroreductive dicarboxylation of unactivated skipped dienes with CO2. Angew. Chem. Int. Ed. 62, e202301892 (2023).
Gui, Y.-Y. et al. Cu-catalyzed asymmetric dicarboxylation of 1,3-dienes with CO2. J. Am. Chem. Soc. 146, 2919–2927 (2024).
Sun, G.-Q., Liao, L.-L., Ran, C.-K., Ye, J.-H. & Yu, D.-G. Recent advances in electrochemical carboxylation with CO2. Acc. Chem. Res. 57, 2728–2745 (2024).
Gao, Y., Wu, Z., Yu, L., Wang, Y. & Pan, Y. Alkyl carbazates for electrochemical deoxygenative functionalization of heteroarenes. Angew. Chem. Int. Ed. 59, 10859–10863 (2020).
Wang, S.-N., Zhang, G.-Y., Shoberu, A. & Zou, J.-P. Copper-catalyzed coupling of amines with carbazates: an approach to carbamates. J. Org. Chem. 86, 9067–9075 (2021).
Acknowledgements
We thank the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0960300, N.F.), the National Natural Science Foundation of China (22071252, N.F. and 22471276, N.F.) and the Chinese Academy of Sciences for financial support.
Author information
Authors and Affiliations
Contributions
N.F. conceived and directed the project. K.Z. performed the experiments and collected the data. N.F. wrote the manuscript. Both authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chang Guo, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, K., Fu, N. Asymmetric cyanoesterification of vinylarenes by electrochemical copper catalysis. Nat Commun 16, 6767 (2025). https://doi.org/10.1038/s41467-025-62137-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62137-7
