
Abstract
Aza-sulfur compounds, such as sulfilimines, sulfoximines, etc, are increasingly recognized as essential contributors to advancements in drug development and asymmetric synthesis. Among them, the catalytic enantioselective synthesis of sulfinimidate esters remains an uncharted and formidable challenge. Herein, we unveil an efficient enantioselective oxidative esterification of sulfenamides via chiral sulfinimidoyl iodide intermediates. Using stereogenic-at-Co(III) complexes as catalysts, this approach enables the synthesis of an extensive array of enantioenriched sulfinimidate esters (>70 examples, up to 98.5:1.5 er), offering a versatile platform for the preparation of structurally diverse aza-sulfur compounds.
Similar content being viewed by others

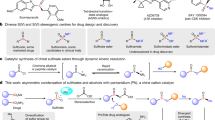
Introduction
Optically active S(IV) compounds have emerged as highly promising candidates, owing to their distinctive physicochemical and pharmacokinetic properties1,2,3,4,5,6,7. Furthermore, sulfur atoms’ Lewis basicity and chirality enable S(IV) compounds to play a pivotal role in asymmetric synthesis as novel chiral ligands, organocatalysts, and auxiliaries (Fig. 1a)8,9,10,11,12,13,14. Therefore, the catalytic synthesis of chiral S(IV) compounds has attracted significant interest and has become a long-standing goal for organic chemists over the past decades15,16,17,18,19. Owing to the stability of S=O bonds, numerous efforts were made to facilitate the catalytic synthesis of chiral sulfinyl compounds (Fig. 1b). For instance, catalytic synthesizing chiral sulfoxides were well studied since Kagan, et al., completed the first asymmetric oxidation of sulfides20,21,22,23,24. The significant advances in asymmetric catalysis have led to the development of several sulfoxides, including Esomeprazole and Armodafinil, as marketed drugs1,2,3,4,5,6,7. To access chiral sulfinate esters, catalytic nucleophilic substitutions of sulfoxide electrophiles (RS(O)LG) have been recognized as a feasible approach pioneered by Ellman, Toru, and others25,26,27,28,29,30,31. However, while replacing the oxygen atom with nitrogen is essential for effectively modulating physicochemical properties and introducing molecular diversity4,5, the catalytic approach to the asymmetric synthesis of sulfinimidoyl compounds featuring S=N bonds, such as sulfilimines, remains underexplored19,32,33,34,35,36,37,38,39. Notably, the efficient catalytic asymmetric synthesis of chiral sulfinimidate esters is still unknown40,41,42 (During our manuscript revision, Wu and coworkers present a novel catalytic sulfinamide activation protocol to access chiral sulfinimidate esters43). Moreover, the sulfimidation of S(II) species could be unsuitable for synthesizing sulfinimidate esters due to the inherent instability of the S(II)−O substrates44,45,46. The obtainment of chiral sulfinimidate esters mainly depended on the enantiospecific O-isopropylation of chiral sulfinamides, which seriously limited the variety and the bioactivity exploration of chiral sulfinimidate esters (Fig. 1c)47. Considering their great potential to access enantioenriched aza-sulfur compounds and in chiral medicine discovery, developing a catalytic asymmetric procedure for preparing chiral sulfinimidate esters is highly desired and urgently desired.

a Importance of stereogenic-at-sulfur compounds. b Representative example of S(IV) species and challenge to access chiral sulfinimidate esters. c Enantiospecific transformation to access chiral sulfinimidate esters. d Catalytic enantioselective synthesis of sulfinimidate esters (This work).
To address this challenging objective, halonium-promoted enantioselective oxidative esterification of sulfenamides could be a promising protocol41,48,49,50. However, the highly reactive sulfinimidoyl halide intermediates mean that the stereocontrol remains greatly challenging, as background reactions are almost inevitable. Moreover, the sulfinimidate esters could also undergo a Swern-type reaction during the reaction process, resulting in the oxidation of alcohols51. Recently, we unveiled the asymmetric oxidation of sulfenamides52,53,54 enabled by anionic stereogenic-at-cobalt(III) complexes55,56,57,58,59,60,61, leading to various enantioenriched sulfinamides in excellent yields62. DFT studies revealed that the iodination of sulfenamides was the enantio-determining step, delivering chiral sulfinimidoyl iodides, which subsequently underwent an enantiospecific SN2 substitution with water to release products.
Encouraged by these findings and the inherent difficulty of synthesizing chiral sulfinimidate esters catalytically, in this work, we disclose a catalytic synthesis of sulfinimidate esters via I+-promoted oxidative esterification of sulfenamides catalyzed by anionic stereogenic-at-cobalt(III) complexes (Fig. 1d). Various sulfenamides and alcohols are successfully incorporated into our reaction system, resulting in a broad of sulfinimidate esters with excellent enantioselectivities (>70 examples, up to 98.5:1.5 er). The synthetic application of chiral sulfinimidate esters is demonstrated by their versatile reactivity, allowing the preparation of various chiral aza-sulfur compounds under mild conditions.
Results
Investigation of reaction conditions
Our investigation commenced with the oxidative esterification reaction involving sulfenamide 2a and isobutanol (3a) by using sodium salt of anionic stereogenic-at-cobalt(III) complex Λ-(S, S)-1a (10 mol%) as the catalyst and THF as solvent. To our satisfaction, sulfenamide 2a was fully converted within 30 min at room temperature, affording chiral sulfinimidate ester 4a in 66% yield with 83.5:16.5 er (Table 1, entry 1, for details, see Table S1–S6 in SI). Employing acetone as the solvent enhanced the enantioselectivity of 4a to 86:14 er, whereas DCM or hexane provided poor stereocontrol (entries 2–4). Subsequently, we screened various X+ reagents. Notably, only a trace amount of 4a was produced when using NCS as the oxidant and no desired products could be detected with NBS (entries 5, 6). Substituting NIS with either I(0) or I(III)41 species also led to unsatisfactory results (entries 7, 8). Interestingly, introducing 5 Å molecular sieves (MS) in the reaction system reduced the enantioselectivity of 4a (entry 9 vs. entry 4), but both the yield and enantioselectivity of 4a were significantly improved by adding 5 Å MS when THF was used as the solvent (entry 10). The addition of water (5 μL) led to an increase in enantioselectivity (92:8 er), however, this was accompanied by a decrease in yield (50%) due to the formation of sulfenamides (entry 11). Next, we evaluated a series of anionic stereogenic-at-cobalt(III) complexes Λ-(S, S)-1b–1g. As exhibited in entries 12–17, the employment of the bulky catalyst Λ-(S, S)-1d furnished product 4a in 90% yield with 90:10 er (entry 14). The enantioselectivity of 4a was significantly increased at lower reaction temperatures (entries 18–20). The optimal result (94% yield, 96.5:3.5 er) was obtained when the reaction was carried out at –40 °C for 36 h (entry 19), and catalyst 1d performed less efficiently than 1a at lower temperatures (entry 20 vs. entry 19), albeit higher enantioselectivity was offered with 1d at room temperature. At last, we tested several classic chiral Brønsted acid and thiourea catalysts under the optimized condition, providing poor yields or enantioselectivities (for details, see Table S7 in SI).

Substrate scopes
With the optimized reaction condition in hand, we first explored the scope of alcohols suitable for the catalytic synthesis of chiral sulfinimidate esters (Fig. 2). A wide range of linear alcohols, including methanol and ethanol, were excellent nucleophiles, delivering chiral sulfinimidate esters 4b–4f with up to 97.5:2.5 er. Branched alcohols were also well tolerated in our reaction system with products 4g–4h exhibiting high-level enantioselectivities. The oxidative esterification reaction of sulfenamide 2a with various alicyclic alcohols worked smoothly (4i–4m), yielding corresponding products with up to 96:4 er. Subsequently, several phenyl-containing alcohols were subjected to asymmetric esterification reactions, delivering sulfinimidate esters 4n–4q with excellent enantioselectivities. Moreover, the introduction of substituents on the aromatic ring had little to no impact on either the yield or stereoselectivity (4r–4y). Encouragingly, the method proved compatible with alkyl alcohols bearing functional groups, such as CF3 (4z), F (4aa), Cl (4ab), and alkynyl groups (4ac–4ae), further demonstrating the versatility and broad substrate compatibility of this protocol. The oxidative esterification between sulfenamide 2a and dihydrocholesterols afforded chiral sulfinimidate ester 4af in 92% yield with >95:5 dr. However, using sugar derivatives, as nucleophiles could not provide desired chiral sulfinimidate esters.

Unless otherwise noted, the reaction was performed with sulfenamide 2a (0.10 mmol), 3 (0.20 mmol. 2.0 equiv), Λ-(S, S)−1a (0.01 mmol, 10 mol%), NIS (0.2 mmol, 2.0 equiv), and THF (1.0 mL) at –40 °C for 36 h. Isolated yields are reported, and er values were determined by chiral stationary HPLC. N. D. not detected.
Next, we investigated the scope of sulfenamides (Fig. 3). Electron-withdrawing aryl groups, such as halogen atoms, CF3, CN, NO2, and CO2Me, on the sulfur of sulfenamides were compatible with the oxidative esterification reactions, delivering desired sulfinimidate esters with good to excellent enantioselectivities (4ag–4am, up to 97.5:2.5 er). Sulfenamides containing electron-rich aryls could be regarded as competent substrates, leading to 81–97% yields with slightly decreased enantioselectivities (4an–4aq). This decrease may be attributed to their stronger electron-donating properties, which enhance reducibility and accelerate the reaction rate, thereby compromising stereoselectivity. The oxidative esterification reactions proceeded smoothly with sulfenamides bearing meta-substituents, giving chiral products with satisfactory outcomes (4ar–4av). The enantioselectivities could be also influenced by the electronic properties of the substituents. The challenging sterically hindered ortho-substitution was also tolerated, affording the corresponding sulfinimidate ester with moderate enantiocontrol (4aw). Our catalysis was well accommodated with sulfenamides bearing a multi-substituted aryl group or heteroaryl group, as evidenced by the obtainments of enantioenriched sulfinimidate esters 4ax and 4ay in good yields. In general, most S-aryl sulfenamides exhibited lower enantioselectivities compared to 2a, implying a possible steric interaction between the tert-butyl groups and the chiral catalysts, despite their spatial distance from the reaction site. Subjecting various S-alkyl sulfenamides to our reaction system gave chiral products in good yields with up to 90.5:9.5 er (4az–4bc). Subsequently, the N-acyl substitution of sulfenamides was examined. To our delight, the influence of para-substituted groups, including various functional groups (CF3, CN, NO2, CO2Me, and COPh) on stereocontrol was minimal, furnishing the corresponding sulfinimidate esters with excellent enantioselectivities (4bd–4bm, ranging from 95:5 er to 98:2 er). Outstanding stereocontrol was also achieved when the sulfenamides bearing meta-substituted aryl groups were subjected to the catalytic reactions (4bn–4bq). Importantly, the sulfenamides containing a multi-substituted aryl group, 2-naphthyl group, or heteroaryl groups, were competent substrates, affording enantioenriched sulfinimidate esters efficiently (4br–4bu). This methodology also worked well for the sulfenamides, assembled from alkyl amides (4bw–4bz), affording excellent stereocontrol in all cases. The oxidative esterification worked smoothly with the urea-derived sulfenamides, providing chiral product 4ca in good yield with moderate enantioselectivity.

Unless otherwise noted, the reaction was performed with sulfenamide 2 (0.10 mmol), 3a (0.20 mmol. 2.0 equiv), Λ-(S,S)−1a (0.01 mmol, 10 mol%), NIS (0.2 mmol, 2.0 equiv), and THF (1.0 mL) at –40 °C for 36 h. Isolated yields are reported, and er values were determined by chiral stationary HPLC. aDCM was used instead of THF (For details, see Table S8 in SI).
Synthetic applications and mechanistic studies
To showcase the synthetic potential of this oxidative esterification reaction, a gram-scale reaction was conducted, yielding enantioenriched sulfinimidate ester 4a without erosion of enantioselectivity (for details, see Section 5.1 in SI). To our delight, the chiral sulfinimidate ester could be a wonderful molecular platform for accessing structurally diverse chiral aza-sulfur compounds (Fig. 4a). For instance, the treatment of chiral sulfinimidate ester 4a (96:4 er) with PhMgBr, EtMgBr, and iPrMgBr individually resulted in the enantiospecific formation of chiral sulfilimines (5a–5c) within 5 min under mild conditions (room temperature, air atmosphere)47,63. Treating 4a with in-situ generated pyridin-3-ylmagnesium bromide enabled the access of S-heteroaryl sulfilimine 5d with 95.5:4.5 er64. The amide-ester exchange reaction also proceeded smoothly with the secondary amine (5e) or primary amine (5f) in the presence of strong bases, delivering chiral sulfinamidines in excellent yields and enantioselectivities27. Notably, chiral sulfinamidines’ general and concise synthesis remains extremely rare and challenging65. The absolute configuration of the enantioenriched 5f was determined via X-ray crystallographic analysis (CCDC 2418774). At last, the enantiospecific oxidation of sulfinimidate ester 4a with RuCl3/NaIO4 provided the sulfonimidate ester 5g in 85% yield43. These straightforward derivatizations underscore the significance of our catalytic system in enabling the efficient synthesis of chiral aza-sulfur compounds, paving the way for the development of novel stereogenic-at-sulfur medicines and pesticides.

a synthetic application of chiral sulfinimidate ester 4a. b Nonlinear effect (NLE) study. c Experimental evidence for chiral sulfinimidoyl iodide intermediates. d. Proposed catalytic cycle.
The mechanistic investigation of the asymmetric oxidative esterification reaction commenced with an NLE study66. As exhibited in Fig. 4b, a slight negative nonlinear effect elucidated that a high-order catalyst aggregate might promote the transformation in this reaction system (for details, see Section 6.1–6.3 in SI)67,68. Interestingly, when water (20 μL) was introduced in the oxidative esterification reactions catalyzed by 1a with different enantioselectivities, the ee values of sulfinamides (water as nucleophile) were always nearly identical to the sulfinimidate ester 4a (alcohol as nucleophile). This observation suggests that both products likely arise from the same chiral intermediate, and that the subsequent nucleophilic substitution proceeds via an enantiospecific transformation (Section 6.3 in SI). In our previous asymmetric oxidation reaction, we proposed that the key sulfinimidoyl iodide was a stable chiral species based on the DFT studies62. To further support this proposal, a (L)-menthol was employed as a chiral probe in the esterification reaction with catalyst Λ-(S, S)-1a, Δ-(R, R)-1a or without chiral catalysts, respectively (Fig. 4c). As a result, sulfinimidate ester (R, L)-6 was obtained with 82:18 dr by using Λ-(S, S)-1a while using Δ-(R, R)-1a afforded (S, L)-6 with high diastereoselectivity (84:16 dr). Notably, the background reaction enabled product 6 with low diastereoselectivity albeit at –40 °C (44:56 dr, R/S). These results can be considered experimental evidence supporting the existence of chiral sulfinimidoyl iodides. Otherwise, significantly higher diastereoselectivity would be expected in the catalyst-free reaction due to the chiral induction effect of menthol27,69,70,71. According to the previous studies and experimental investigation, we showcased a concise catalytic cycle in Fig. 4d. In the presence of the in-situ generated acidic chiral complex [Co*]–H+, sulfenamide 2a reacted with NIS stereoselectively, leading to chiral sulfinimidoyl iodide intermediate 7, whose existence was indirectly supported by HRMS (for the proposed stereocontrol model, see Section 6.5 in SI)62. With the assistance of another molecular NIS, an enantiospecific SN2 reaction happened between alcohol 3 and the Brønsted acid-activated intermediate 7, releasing chiral sulfinimidate ester 4 with the regeneration of [Co*]–H+.
Discussion
In conclusion, we have developed a groundbreaking catalytic approach for the synthesis of chiral sulfinimidate esters through iodonium-promoted enantioselective oxidative esterification of sulfenamides, utilizing an anionic stereogenic-at-Co(III) complex as the catalyst. This strategy enabled the efficient preparation of a broad range of chiral sulfinimidate esters, achieving excellent yields and enantioselectivities across more than 70 examples (up to 98.5:1.5 er). Importantly, the chiral sulfinimidate esters were showcased as versatile molecular platforms for synthesizing diverse chiral aza-sulfur(IV) compounds, under mild reaction conditions. By using (L)-menthol as a chiral probe, the mechanistic investigation afforded experimental evidence of chiral sulfinimidoyl iodide intermediates. Furthermore, the exploitation of chiral sulfinimidoyl electrophiles presents a robust and practical methodology for addressing challenging asymmetric issues in sulfur stereochemistry, paving the way for novel chiral sulfur-containing compounds with broad applications in pharmaceuticals, and agrochemicals.
Methods
General procedure for the synthesis of enantioenriched 4a–4ca
An oven-dried 10-mL tube equipped with a magnetic stirring bar was charged with sulfenamide 2 (0.10 mmol), catalyst Λ-(S,S)-1a (0.01 mmol, 10 mol%), alcohol 3 (0.20 mmol, 2.0 equiv), 5 Å MS (100 mg) and THF (1.0 mL). The mixture was cooled to –40 °C and stirred for 30 min. The NIS solid (0.20 mmol, 2.0 equiv) was poured into the tube all at once, and the reaction mixture was stirred at –40 °C for 36 h. The reaction was quenched by adding Na2S2O3 aq. and then separating the organic layer from the aqueous phase. The aqueous phase was further extracted with EtOAc (3 × 5.0 mL). The organic layer was dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using the petroleum ether/ethyl acetate (10/1) as an eluent to afford enantioenriched products 4a–4ca.
Data availability
Deposition Number 2418774 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruhe Access Structures service. These data can be obtained free of charge via www.ccdc.cam.ac.uk/ data_request/cif, by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 441223 336033. Data supporting the findings of this manuscript are also available from within the Supplementary Information and from the corresponding author upon request.
References
Lipinski, C. & Hopkins, A. Navigating chemical space for biology and medicine. Nature 432, 855–861 (2004).
Lücking, U. Sulfoximines: a neglected opportunity in medicinal chemistry. Angew. Chem. Int. Ed. 52, 9399–9408 (2013).
Bizet, V., Kowalczyk, R. & Bolm, C. A. Fluorinated sulfoximines: syntheses, properties and applications. Chem. Soc. Rev. 43, 2426–2438 (2014).
Lücking, U. Neglected sulfur(vi) pharmacophores in drug discovery: exploration of novel chemical space by the interplay of drug design and method development. Org. Chem. Front. 6, 1319–1324 (2019).
Mäder, P. & Kattner, L. A. Sulfoximines as rising stars in modern drug discovery? Current status and perspective on an emerging functional group in medicinal chemistry. J. Med. Chem. 63, 14243–14275 (2020).
Han, Y. et al. Application of sulfoximines in medicinal chemistry from 2013 to 2020. Eur. J. Med. Chem. 209, 112885 (2021).
Lücking, U. New opportunities for the utilization of the Sulfoximine group in medicinal chemistry from the drug designer’s perspective. Chem. Eur. J. 28, e202201993 (2022).
Mellah, M., Voituriez, A. & Schulz, E. Chiral sulfur ligands for asymmetric catalysis. Chem. Rev. 107, 5133–5209 (2007).
Robak, M. T., Herbage, M. A. & Ellman, J. A. Synthesis and applications of tert-Butanesulfinamide. Chem. Rev. 110, 3600–3740 (2010).
Sipos, G., Drinkel, E. E. & Dorta, R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 44, 3834–3860 (2015).
Zrost, B. M. & Rao, M. Development of chiral sulfoxide ligands for asymmetric catalysis. Angew. Chem. Int. Ed. 54, 5026–5043 (2015).
Otocka, S., Kwiatkowska, M., Madalińska, L. & Kiełbasiński, P. Chiral Organosulfur ligands/catalysts with a stereogenic sulfur atom: applications in asymmetric synthesis. Chem. Rev. 117, 4147–4181 (2017).
Jia, T., Wang, M. & Liao, J. Chiral sulfoxide ligands in asymmetric catalysis. Top. Curr. Chem. 377, 8 (2019).
Yang, M.-M., Wang, S. & Dong, Z.-B. Recent advances for chiral sulfoxides in asymmetric catalysis. Synthesis 54, 5168–5185 (2022).
Wojaczyńska, E. & Wojaczyński, J. Enantioselective synthesis of sulfoxides: 2000−2009. Chem. Rev. 110, 4303–4356 (2010).
Han, J., Soloshonok, V. A., Klika, K. D., Drabowicz, J. & Wzorek, A. Chiral sulfoxides: advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 47, 1307–1350 (2018).
Wojaczyńska, E. & Wojaczyński, J. Modern stereoselective synthesis of chiral sulfinyl compounds. Chem. Rev. 120, 4578–4611 (2020).
Zhang, X., Wang, F. & Tan, C.-H. Asymmetric synthesis of S(IV) and S(VI) stereogenic centers. JACS Au 3, 700–714 (2023).
Wei, M. K. & Wills, M. C. The catalytic synthesis of Aza-sulfur functional groups. Synthesis 57, 1429–1440 (2025).
Pitchen, P., Duñach, E., Deshmukh, M. N. & Kagan, H. B. An efficient asymmetric oxidation of sulfides to sulfoxides. J. Am. Chem. Soc. 106, 8188–8193 (1984).
Di Furia, F., Modena, G. & Seraglia, R. Synthesis 1984, 325 (1984).
Nemecek, C., Duñach, E. & Kagan, H. B. Asymmetric oxidation of some sulfur derivatives. N. J. Chem. 10, 761–764 (1986).
Zhang, Y.-Q., Hu, L., Yuwen, L., Lu, G. & Zhang, Q.-W. Nickel-catalysed enantioselective hydrosulfenation of alkynes. Nat. Catal. 6, 487–494 (2023).
Gao, L. et al. Angew. Chem. Int. Ed. 63, e202317626 (2024).
Evans, J. W., Fierman, M. B., Miller, S. J. & Ellman, J. A. Catalytic enantioselective synthesis of sulfinate esters through the dynamic resolution of tert-butanesulfinyl chloride. J. Am. Chem. Soc. 126, 8134–8135 (2004).
Shibata, N. et al. Cinchona alkaloid/sulfinyl chloride combinations: enantioselective sulfinylating agents of alcohols. J. Am. Chem. Soc. 127, 1374–1375 (2005).
Zhang, X., Ang, E. C. X., Yang, Z., Kee, C. W. & Tan, C.-H. Synthesis of chiral sulfinate esters by asymmetric condensation. Nature 604, 298–303 (2022).
Huang, S. et al. Organocatalytic asymmetric deoxygenation of sulfones to access chiral sulfinyl compounds. Nat. Chem. 15, 185–193 (2023).
Liao, M. et al. Enantioselective sulfinylation of alcohols and amines by condensation with sulfinates. Chem 10, 1541–1552 (2024).
Li, B. et al. Catalyst Control over S(IV)-stereogenicity via Carbene-derived Sulfinyl Azolium Intermediates. J. Am. Chem. Soc. 146, 25350–25360 (2024).
Wei, T., Wang, H.-L., Tian, Y., Xie, M.-S. & Guo, H.-M. Enantioselective construction of stereogenic-at-sulfur(IV) centres via catalytic acyl transfer sulfinylation. Nat. Chem. 16, 1301–1311 (2024).
Yoshitake, M., Hayashi, H. & Uchida, T. Ruthenium-catalyzed asymmetric N-acyl nitrene transfer reaction: imidation of sulfide. Org. Lett. 22, 4021–4025 (2020).
Liu, Z. et al. Iron-catalyzed asymmetric imidation of sulfides via sterically biased nitrene transfer. J. Am. Chem. Soc. 146, 18050–18060 (2024).
Greenwood, N. S., Champlin, A. T. & Ellman, J. A. Catalytic enantioselective sulfur alkylation of sulfenamides for the asymmetric synthesis of sulfoximines. J. Am. Chem. Soc. 144, 17808–17814 (2022).
Wang, F.-C. et al. Synthesis of chiral sulfilimines by organocatalytic enantioselective sulfur alkylation of sulfenamides. Sci. Adv. 10, eadq2768 (2024).
Champlin, A. T., Kwon, N. Y. & Ellman, J. A. Enantioselective S-alkylation of sulfenamides by phase-transfer catalysis. Angew. Chem. Int. Ed. 63, e202408820 (2024).
Yuan, Y. et al. Enantioselective arylation of sulfenamides to access sulfilimines enabled by Palladium Catalysis. Angew. Chem. Int. Ed. 63, e202409541 (2024).
Liang, Q.-J. et al. Enantioselective Chan–Lam S-arylation of sulfenamides. Nat. Catal. 7, 1010–1020 (2024).
Fang, W. et al. Asymmetric S-arylation of sulfenamides to access axially chiral sulfilimines enabled by anionic stereogenic-at-Cobalt(III) complexes. Angew. Chem. Int. Ed. 64, e202419596 (2025).
Andresini, M. et al. Synthesis of sulfinamidines and sulfinimidate esters by transfer of nitrogen to sulfenamides. Org. Lett. 22, 7129–7134 (2020).
Lu, X. et al. Hypervalent iodine-mediated synthesis of sulfinimidate esters from sulfenamides. Org. Lett. 25, 2151–2156 (2023).
Fan, L.-W. et al. Decoupling strategy-enabled radical generality via an asymmetric SH2 path. ChemRxiv, https://doi.org/10.26434/chemrxiv-2024-3n41t (2024).
Xiong, Q. et al. Asymmetric synthesis of S(IV)-stereogenic sulfinimidate esters by sulfinamide activation. Angew. Chem. Int. Ed. 64, e202500170 (2025).
Harpp, D. N., Steliou, K. & Chan, T. H. Organic Sulfur Chemistry. 26. Synthesis and reactions of some new sulfur transfer reagents. J. Am. Chem. Soc. 100, 1222–1228 (1978).
Rudzinski, D. M., McCourt, M. P. & Priefer, R. Thermolytic decomposition of Benzylic Dialkoxy Disulfides. Tetrahedron Lett. 50, 5520–5522 (2009).
Xue, J. & Jiang, X. Unsymmetrical polysulfidation via designed bilateral disulfurating reagents. Nat. Commun. 11, 4170 (2020).
Tsuzuki, S. & Kano, T. Asymmetric synthesis of chiral sulfimides through the O-alkylation of enantioenriched sulfinamides and addition of carbon nucleophiles. Angew. Chem. Int. Ed. 62, e202300637 (2023).
Yang, G. et al. One-pot tandem oxidative bromination and amination of sulfenamide for the synthesis of sulfinamidines. J. Org. Chem. 88, 4581–4591 (2023).
Huang, G. et al. Hypervalent iodine mediated synthesis of sulfinamidines from sulfenamides. J. Org. Chem. 88, 11728–11734 (2023). (b).
Wu, P., Demaerel, J., Statham, B. J. & Bolm, C. Azasulfur(IV) derivatives of sulfite and aulfinate esters by formal S–S bond insertion of Dichloramines. Chem. Sci. 15, 5333–5339 (2024).
Mukaiyama, T., Matsuo, J. & Yanagisawa, M. A new and efficient method for oxidation of various alcohols by using N-tert-Butyl Phenylsulfinimidoyl Chloride. Chem. Lett. 29, 1072–1073 (2000).
Ma, L. et al. Chiral Brønsted-acid-catalyzed asymmetric oxidation of sulfenamide by using H2O2: A versatile access to sulfinamide and sulfoxide with high enantioselectivity. ACS Catal. 9, 1525–1530 (2019).
Yang, G. et al. Synthesis of chiral sulfonimidoyl chloride via desymmetrizing enantioselective hydrolysis. J. Am. Chem. Soc. 145, 5439–5446 (2023).
Huang, H. et al. Enantioselective synthesis of chiral sulfonimidoyl fluorides facilitates stereospecific SuFEx Click chemistry. Angew. Chem. Int. Ed. 63, e202415873 (2024).
Cruchter, T. & Larionov, V. A. Asymmetric catalysis with octahedral stereogenic-at-metal complexes featuring chiral ligands. Coord. Chem. Rev. 376, 95–113 (2018).
Larionov, V. A., Feringa, B. L. & Belokon, Y. N. Enantioselective “Organocatalysis in Disguise” by the ligand sphere of chiral metal-templated complexes. Chem. Soc. Rev. 50, 9715–9740 (2021).
Yu, J., Jiang, H.-J., Zhou, Y., Luo, S.-W. & Gong, L.-Z. Sodium salts of anionic chiral Cobalt(III) complexes as catalysts of the enantioselective Povarov reaction. Angew. Chem. Int. Ed. 54, 11209–11213 (2015).
Jiang, H.-J., Liu, K., Yu, J., Zhang, L. & Gong, L.-Z. Switchable stereoselectivity in Bromoaminocyclization of Olefins: using Brønsted acids of anionic chiral Cobalt(III) complexes. Angew. Chem. Int. Ed. 56, 11931–11935 (2017).
Jiang, H.-J. et al. Assembling a hybrid Pd catalyst from a chiral anionic Co(III) complex and ligand for asymmetric C(sp3)–H functionalization. Angew. Chem. Int. Ed. 58, 1803–1807 (2019).
Zhang, X. et al. Atroposelective ring opening of cyclic diaryliodonium salts with bulky anilines controlled by a chiral Cobalt(III) Anion. Angew. Chem. Int. Ed. 59, 19899–19904 (2020).
Sun, B.-B. et al. Enantioselective Ugi and Ugi-Azide reactions catalyzed by anionic stereogenic-at-cobalt(III) complexes. Nat. Commun. 13, 7065 (2022).
Jiang, H.-J. et al. Unlocking Chiral Sulfinimidoyl electrophiles: asymmetric synthesis of sulfinamides catalyzed by anionic stereogenic-at-Cobalt(III) complexes. J. Am. Chem. Soc. 147, 2137–2147 (2025).
Andresini, M. et al. Sulfinimidate esters as an electrophilic sulfinimidoyl Motif Source: Synthesis of N-protected Sulfilimines from Grignard reagents. Org. Lett. 23, 6850–6854 (2021).
Zhang, Z.-X. & Willis, M. C. Sulfondiimidamides as new functional groups for synthetic and medicinal chemistry. Chem 8, 1137–1146 (2022).
Fimm, M. & Saito, F. Enantioselective synthesis of Sulfinamidines via asymmetric nitrogen transfer from N−H Oxaziridines to Sulfenamides. Angew. Chem. Int. Ed. 63, e202408380 (2024).
Satyanarayana, T., Abraham, S. & Kagan, H. B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 48, 456–494 (2009).
Liu, S., Chan, K. L., Lin, Z. & Sun, J. Asymmetric synthesis of remotely chiral naphthols and naphthylamines via naphthoquinone methides. J. Am. Chem. Soc. 145, 12802–12811 (2023).
Li, X.-Z., He, Y.-P. & Wu, H. Multicomponent Cyclizative 1,2-rearrangement enabled enantioselective construction of 2,2-Disubstituted Pyrrolinones. Angew. Chem. Int. Ed. 63, e202317182 (2024).
Hajipour, A. R., Mallakpour, S. E. & Afrousheh, A. One-pot and simple reaction for the synthesis of Alkyl p-Toluenesulfinate Esters under solid-phase conditions. Phosphorus, Sulfur Silicon 160, 67–75 (2000).
Hajipour, A. R., Falahati, A. R. & Ruoho, A. E. An efficient and novel method for the synthesis of sulfinate esters under solvent-free conditions. Tetrahedron Lett. 47, 2717–2719 (2006).
Ji, Y.-Z., Li, H.-J., Zhang, J.-Y. & Wu, Y.-C. Sodium Arenesulfinates-involved sulfinate synthesis revisited: improved synthesis and revised reaction mechanism. Eur. J. Org. Chem. 2019, 1846–1855 (2019).
Acknowledgements
We are grateful for the financial support from NSFC (Grants 92156022 and 22401004), Anhui Provincial Natural Science Funds (Grants 2308085MB43 and 2308085QB44), Shennong Scholar Program of Anhui Agricultural University, National Undergraduate Training Program for Innovation and Entrepreneurship.
Author information
Authors and Affiliations
Contributions
H.J.J. and J.Y. conceived the research and were responsible for the experimental design; X.Q.T., X.Y.K., and J.Y.W. performed the synthetic experiments, compound testing, and data analysis; Y.Y.L. and M.L.S. assisted in performing experiments and analyzing data. H.J.J. and J.Y. prepared the manuscript with assistance from C.Z.Y., Q.L., and M.L.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, HJ., Tu, XQ., Kong, XY. et al. Catalytic synthesis of chiral sulfinimidate esters via oxidative esterification of sulfenamides. Nat Commun 16, 6988 (2025). https://doi.org/10.1038/s41467-025-62197-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62197-9
