
Abstract
The electrocatalysis of flue gas into CO in membrane electrode assembly (MEA) provides a sustainable route for realizing practical CO2 electrolysis technology but suffers from restricted CO2 mass transport due to thick gas boundary layer (GBL) and weak concentration gradient. Inspired by nutrient diffusion mechanism in plant, we introduce the concept of self-reinforced CO2 concentration gradient, which is realized via porous carbon nanosheets (PC) as soil for enriching CO2 and single-atomic Ni-doped carbon nanotubes (Ni-CNTs) as rhizome for electro-catalyzing CO2. A combined experimental and simulation study reveals optimal length of Ni-CNTs on PC reduces the GBL thickness and spontaneously enhances CO2 concentration gradient, synergistically breaking the limitation of CO2 transport. Consequently, the CO Faradaic efficiency attains >90% with varying CO2 concentration of 4-15 vol. % CO2 in MEA. Further through incorporation of an O2-adsorption packed column before MEA, we realize the stable and selective conversion of O2-containing flue gas into CO.
Similar content being viewed by others
Introduction
Electrochemical carbon dioxide reduction reaction (CO2RR) provides a sustainable technology to convert waste CO2 into valuable chemicals and fuel using renewable electricity1,2,3,4. Carbon monoxide (CO), as a product of the two-electron CO2RR, is suggested to be one of the most economically viable since it is an important carbon building block to synthesize long-chain industrial reagents5,6. In this respect, many efforts have been devoted to boosting the CO2RR-to-CO performance, including catalyst design and reactor upgrade7,8,9,10. In particular, the zero-gap membrane electrode assembly (MEA) electrolyzers based on gas diffusion electrode (GDE) enable the gaseous CO2 to directly reach the cathode catalyst layer (CL) through the microporous layer (MPL), which can circumvent the mass transport limited by the poor CO2 solubility and possess low ohmic loss10,11. Benefiting from these features, industrially relevant current density (>200 mA cm–2) with impressive CO Faradaic efficiency (>90%) has been attained in gaseous feedstock MEA by using CO-selective catalysts such as silver3,12 and metal single atoms8,13,14,15,16, pushing the CO2 electrolysis technology into the realm of industrial applications.
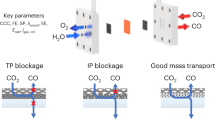
Despite the notable advances, a majority of studies have focused on concentrated CO2 streams, but the flue gas from combustion of fossil fuels typically contains 3–15% of CO2 (expressed as volume percentage unless specifically noted) primarily diluted by nitrogen (N2), meaning that it is necessary to first capture CO2 before flowing into electrolyzer as feed for CO2RR17. The mature technology for the production of pure CO2 from point sources requires absorption in amine/alkaline solution, followed by a regeneration process involving heating or pressure reduction, which are environmentally and economically unfavorable, sacrificing the viability of CO2RR technology18,19,20,21. In this context, it can be envisioned that integration of CO2 capture sites and CO2RR active sites into one single catalyst system for direct electrocatalysis of a dilute CO2 stream is more sustainable since it can avoid the cost caused by CO2 separation and enrichment. Nevertheless, the direct intake of low-concentration CO2 as gaseous feed suffers from serious limitations in mass transport due to the heavier CO2 molecule than N2, which causes slow mass transport speed based on the well-known Graham’s law of effusion22, thus leading to multiple challenges for low-concentration CO2RR. First, CO2 navigates through the cathode channel and approaches the catalyst surface via the gas boundary layer (GBL)23. At this point, the slow transport rate of CO2 in the bulk phase, coupled with the strong adhesion force of N2, jointly leads to a thicker GBL (Supplementary Note 1). This, in turn, results in a lower CO2 concentration on the gas-liquid-solid surface, which thermodynamically favors the competitive hydrogen evolution reaction (HER) over CO2RR (Fig. 1a). Second, according to Fick’s Law24,25,26, a decrease in the feeding concentration of CO2 weakens the concentration gradient effect within the CL, which directly incurs a weak CO2 flux to the catalytic sites, resulting in sluggish CO2RR kinetics and high overpotential (Fig. 1b and Supplementary Note 2).

a Dilute CO2 stream causes an increased thickness of gas boundary layer (δGBL), inversely proportional to Reynolds number (Re), thereby thermodynamically favors HER. b Dilute CO2 stream diminishes the CO2 concentration gradient (∂c/∂x) effect within catalyst layer (CL) that results in sluggish CO2RR kinetics. c Schematic of nutrient diffusion-inspired hierarchical electrocatalyst with ultramicropore-induced self-reinforced CO2 concentration gradient for low-concentration CO2RR.
To address the challenges in low-concentration CO2RR, limited attempts have been implemented on enriching CO2 in the vicinity of CO2RR active sites via the introduction of amino groups or physical confinement with optimal pore structures27,28,29,30,31. Beyond improving the CO2 adsorption ability, the gaseous CO2 transport behavior within the GBL and CL fundamentally determines the low-concentration CO2RR performance in MEA, which is seriously neglected. Additionally, how the CO2 capture sites and catalytic sites guide the CO2 mass transport behavior during the dilute CO2RR process remains unexplored; however, which is very critically important for CO2RR32, especially from industrial point sources. For these reasons, very few works on dilute gaseous CO2RR can realize the high conversion of CO2 into CO in MEA when CO2 concentration is below 10%, which cannot fully meet the requirement of CO2 from the iron and steel industry (CO2 concentration: 3–5%)33. Therefore, it is imperative to design an advanced electrocatalyst system that can simultaneously promote CO2 transport at the GBL and CL for improving dilute CO2RR performance.
Herein, mimicking natural nutrient diffusion that enables swift nutrient conveyance based on the difference in nutrient concentration between roots in soil and rhizome, we propose a hierarchical structure composed of nickel single atoms anchored on N-doped carbon nanotubes (Ni-CNTs) growing from porous carbon nanosheets (Ni-CNTs/PC), affording a self-reinforced CO2 concentration gradient effect, which facilitates CO2 transport to the Ni active sites for low-concentration CO2RR (Fig. 1c). The PC nanosheets featuring ultramicropores can capture and concentrate CO2 from CO2/N2 mixture, while the Ni single atoms can serve as active sites for CO2RR. Experimentally, Ni-CNTs/PC was synthesized through chemical vapor deposition (CVD) growth on swelled Ni2+-crosslinked aerogel, where the Ni-CNTs length can be simply controlled via the growth time. With optimal length of Ni-CNTs grown on PC NSs, the Ni-CNTs/PC is found to thin the GBL and establish a self-reinforced CO2 concentration gradient within CL in MEA electrolyzer, thereby significantly enhancing the CO2 transport ability. Such a nutrient diffusion-mimicking structure generates a CO Faradaic efficiency surpassing 90% with varying CO2 concentrations from 4 to 15%. Notably, the Ni-CNTs/PC enables the efficient conversion of dilute CO2RR containing 3% of CO2 with 87.7% of CO Faradaic efficiency. Further integrating an oxygen-adsorption packed column (OAPC) before the MEA reactor, the Ni-CNTs/PC enables the direct conversion of simulated flue gas containing 3–10% O2. This work provides a sustainable route for converting flue gas by introducing a self-reinforced concentration gradient, paving the way for the practical implementation of CO2 electrocatalysis technology.
Results
Material synthesis and characterization
The hierarchical structure of Ni-CNTs/PC was synthesized via CVD growth on the swelled Ni2+-crosslinked carboxymethylcellulose sodium (Ni-CMC) aerogel, as illustrated in Fig. 2a. Briefly, nickel chloride (NiCl2) was first added into the CMC solution for producing a green homogeneous colloidal solution based on the interaction between Ni2+ and OH–/–COO– groups in CMC (Supplementary Fig. 1). After molding on the frozen copper billet, the as-prepared solid was immersed in cold ethanol bath containing sodium hydroxide (NaOH) to form Ni-CMC hydrogel through H–bond interaction34 (Supplementary Fig. 1), followed by swelling treatment in deionized (DI) water and freeze-drying to obtain Ni-CMC aerogel (Fig. 2b), which presents porous networks comprising vertically arranged CMC layers with pore sizes of ~65 μm (Supplementary Fig. 2). Notably, the abundant porous structures rely on not only the Ni2+ crosslinking and NaOH activation, but also the employment of ice template (Supplementary Figs. 3, 4). Afterward, CVD growth was conducted on the well-ordered Ni-CMC aerogel with acetonitrile (CH3CN) as feedstock at 750 °C, where the Ni metal nanoparticles can catalyze the CH3CN decomposition into C–N fragments, forming Ni–N bonds and directly incorporating into the carbon lattice (Supplementary Fig. 5)6. Meanwhile, the Ni-CMC aerogel is converted into porous carbon nanosheets (PC NSs) during Ni, N co-doped CNTs growth (Supplementary Fig. 5 and Supplementary Table 1), forming Ni-CNTs/PC heterostructures, as evidenced by the scanning electron microscope (SEM) images in Fig. 2c and Supplementary Figs. 6, 7. Notably, the CNTs vertically grow on PC NSs due to a well-known tip-growth mode35, and thus the residual Ni nanoparticles can be easily removed by acid leaching (Supplementary Fig. 8). It is found that the Ni-CMC formation is the key factor for forming the well-defined Ni-CNTs/PC heterostructures (Supplementary Figs. 9, 10). Gram-scale purified product can be collected in one batch synthesis after etching in hydrochloric acid, followed by filtration (Supplementary Fig. 11), meeting the requirement of high mass loading for MEA devices.

a Schematic for the preparation of hierarchical Ni-CNTs/PC. b Schematic for synthesizing Ni-CMC hydrogel through the sol-gel strategy followed by swelling treatment. c Top-view SEM images and d HRTEM image of Ni-CNTs30/PC (the white circle in d refers to the location of the etched Ni particle). e Aberration-corrected HAADF-STEM image of Ni-CNTs. f EDS elemental mappings of C, N, and Ni. g Schematic for the tip-growth mode of Ni-CNTs. h Swelling ratio of Ni-CMC hydrogel and Ni nanoparticles (NPs) size versus swelling time. i The thickness of PC NSs and the length of Ni-CNTs in Ni-CNTs/PC versus growth time. Error bars represent the standard deviation of three independent measurements. Source data are provided as a Source Data file.
Transmittance electron microscope (TEM) images were carried out after 30 min of CVD growth and confirm the vertical growth behavior of Ni-CNTs from the CMC-derived PC NSs (Fig. 2d). Aberration-corrected high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) was employed to discern the distribution of Ni single atoms across the region of CNTs and PC NSs, respectively. As shown in Fig. 2e and Supplementary Fig. 12, the bright spots corresponding to Ni atoms are uniformly dispersed on the CNTs profile, without any observed clusters or nanoparticles. Conversely, extensive HAADF-STEM images reveal the absence of Ni species on the PC NSs (Supplementary Fig. 13). This contrast can be further supported by the energy-dispersive spectroscopy (EDS) mappings in Fig. 2f and Supplementary Fig. 14, where Ni species only homogeneously distribute on the CNTs skeleton. The observation can be explained by the tip-growth mode of CNTs36,37, where the Ni nanoparticles detach from the CMC-derived carbon substrate during CNT growth and can dope into the CNTs through Ni–N interaction7, as schematically shown in Fig. 2g. Since the Ni seeds are located at the ends of CNTs, they are susceptible to being etched away, leaving rare Ni nanoparticles or clusters on Ni-CNTs.
It is noted that the swelling treatment before free-drying of Ni-CMC hydrogel is very critical for growing CNTs arrays and avoiding the residual of Ni nanoparticles, possibly due to the increased interval of Ni species by water uptake that prevents the heavy Ni aggregation into large nanoparticles in the subsequent high-temperature growth (Fig. 2h, i and Supplementary Figs. 15–17)38. It is found that the swelling ratio reaches equilibrium after 48 h, where the Ni nanoparticles are the smallest with sizes of ~5.5 nm. Accordingly, well-defined CNTs with diameters of ~100 nm are successfully synthesized on PC NSs, which can effectively avoid the Ni aggregation as compared to other swelling times. Moreover, the length and coverage of CNTs can be easily adjusted by controlling the growth time while the thickness of PC NSs remains unchanged (Fig. 2i and Supplementary Fig. 18), which provides rich models to study the effect of CO2 transport distance and concentration gradient effect on dilute CO2RR. Specifically, CNTs with growth time of 10, 30, 60, 90, and 180 min, denoted as Ni-CNTs10/PC, Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, and Ni-CNTs180/PC, correspond to the length of ~100, ~500, ~1100, ~1300, and ~1800 nm, respectively. Besides, the inductively coupled plasma mass spectroscopy (ICP-MS) analyses reveal the Ni content in Ni-CNTs10/PC, Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, and Ni-CNTs180/PC was ~0.75, ~2.00, ~2.14, ~2.34, and 2.75 wt% (Supplementary Table 2), respectively, suggesting that the Ni content slightly increases as growth time was over 30 min. All these results demonstrate the successful construction of hierarchical structures composed of Ni, N co-doped CNTs, and PC nanosheets, where the Ni single atoms exclusively distribute on the CNTs and the length of CNTs can be readily adjusted.
The Ni-CNTs/PC samples were further analyzed by X-ray diffraction (XRD), all of which display a distinct peak at ~26° (Fig. 3a), assigned to the plane (002) of graphitic carbon. The (002) diffraction peak becomes sharper as growth time proceeds, ascribed to the increased CNTs coverage. The absence of characteristic Ni species collaborates with the isolated Ni single atoms, consistent with the HAADF-STEM results. X-ray photoelectron spectra (XPS) analyses were performed to reveal the chemical compositions and valence states of Ni-CNTs/PC. As shown in Fig. 3b, the Ni 2p peaks of the four samples are all higher than Ni foil, but lower than NiO, which suggests that the valence state of Ni species in Ni-CNTs/PC is situated between Ni0 and Ni2+. In addition, the high-resolution N 1s XPS spectra can be deconvoluted into three peaks at binding energies of 398.4, 399.4, and 401.0 eV (Fig. 3c), corresponding to pyridinic N, Ni–N, and graphitic N, respectively13,39, which confirms the formation of Ni–N bonds via a direct CVD approach. The N configurations and their ratios show an inapparent difference (Supplementary Table 3), which can exclude their effect on CO2 adsorption and conversion. Besides, pure PC NSs were also prepared without introducing CH3CN during annealing of the Ni-CMC aerogel, followed by acid etching, which shows negligible Ni and N signals (Supplementary Figs. 19, 20).

a XRD patterns of Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, and Ni-CNTs180/PC. Corresponding high-resolution of Ni 2p XPS spectra (b) and N 1s XPS spectra (c). d Ni K-edge XANES spectra. e k2-weighted Fourier transform of the EXAFS spectra at the Ni K-edge of Ni foil, Ni-CNTs30/PC, and NiO. f EXAFS fitting curve for Ni sites in Ni-CNTs30/PC. In situ ATR-FTIR spectra recorded at –0.75 V vs. RHE in a mixture of CO2/N2 (v/v = 15/85) on (g) Ni-CNTs30/PC and (h) PC NSs. i Schematic illustration of the CO2RR process on Ni-CNTs/PC, where the Ni–N3 on CNTs are the active sites for forming *COOH intermediate and low-concentration CO2RR. Source data are provided as a Source Data file.
To delve deeper into the coordination structures of Ni single atoms, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses were conducted. As illustrated in Fig. 3d, the Ni K-edge position of Ni-CNTs30/PC is located between that of NiO and Ni foil, suggesting a positive Ni valence state between 0 and +2. The EXAFS spectrum of Ni-CNTs30/PC displays a dominant peak at around 1.41 Å (Fig. 3e), which can be attributed to the Ni−N path scattering. Meanwhile, the coordination of Ni−Ni is negligible, evidencing that the Ni species mostly exist in the form of single atoms. Furthermore, quantitative EXAFS fitting was conducted to extract the structure parameter. The coordination number of N and C for Ni-CNTs30/PC in the first coordination shell was around 3 and 1, with the average scattering length of 1.85 and 1.86 Å, respectively, reflecting that the proposed local structure is Ni−N3C (Fig. 3f, Supplementary Fig. 21, and Supplementary Table 4).
Following the identification of Ni species, in situ electrochemical attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) was carried out to elucidate the contribution of Ni−N3 to CO2RR. The experiments were performed at a potential of −0.75 V vs. reversible hydrogen electrode (RHE) in 0.5 M potassium bicarbonate (KHCO3) using a home-made ATR-FTIR setup under dilute CO2 stream (CO2/N2 = 15/85), as shown in Supplementary Fig. 22a. A characteristic peak located at 1356.8 cm–1 appears in Ni-CNTs30/PC after 5 min of catalysis and increases gradually with reaction time (Fig. 3g). The characteristic peak can be ascribed to the vibration of C−O stretching of the *COOH, a widely recognized key intermediate for CO2RR to CO40. In sharp contrast, the *COOH intermediate cannot be detected on pure PC NSs without Ni-CNTs, indicating CO2RR barely happens (Fig. 3h). While for Ni-CNTs180/PC, the appearance of *COOH signal takes a longer time with a weaker intensity in comparison with Ni-CNTs30/PC (Supplementary Fig. 22b), manifesting that the high coverage of CNTs, even with a higher loading of Ni single atoms, has an adverse effect on CO2 activation, possibly due to insufficient CO2 supply. These findings imply that Ni single atoms in CNTs are the active sites for CO2RR rather than PC NSs, as shown in Fig. 3i.
CO2/N2 adsorption selectivity in Ni-CNTs/PC catalyst
Since the low-concentration CO2RR highly depends on the adsorption capability of CO2, we proceeded with a detailed study of the CO2 adsorption behavior in the catalysts composed of PC NSs with varying growth periods of Ni-CNTs from 30 to 180 min, namely Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, and Ni-CNTs180/PC. Furthermore, bare PC NSs and Ni-CNTs30 were also studied for comparison. The uptake capacity of CO2 was evaluated from adsorption isotherms at 298.15 K. As shown in Fig. 4a and Supplementary Table 5, PC NSs achieve a remarkable CO2 uptake of 1.36 mmol g–1 at 1.00 bar. When Ni-CNTs grow on the surface, the CO2 uptake shows a negative correlation to the CNTs coverage. Without PC NSs, the bare Ni-CNTs present a poor CO2 uptake capability (~0.11 mmol g–1) at 1.00 bar, which is close to that of Ni-CNTs180/PC. The differences in adsorption capacity among the six samples provide a strong hint that the PC NSs fundamentally determine the CO2 adsorption ability of Ni-CNTs/PC catalyst, while Ni-CNTs growth may block the pores in PC NSs, which affects the CO2 capture. Under conditions of low CO2 concentration (0.15 bar), the CO2 adsorbed amount is calculated to be 0.29, 0.07, 0.03, 0.02, and 0.02 mmol g–1 for Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, Ni-CNTs180/PC, and Ni-CNTs30, respectively. This observation aligns perfectly with the electrochemically active surface area (Supplementary Fig. 23). Combining with the ATR-FTIR results in Fig. 3g–i, it can be concluded that the PC NSs are responsible for CO2 adsorption in the dilute stream and facilitate the access of CO2 to Ni−N3 active sites, jointly realizing dilute CO2RR. Impressively, with an ultralow CO2 pressure of 0.03 bar, the Ni-CNTs30/PC still exhibits 0.094 mmol g–1 of CO2 uptake, which is expected to realize ultralow-concentration CO2RR.

a CO2 adsorption isotherm at 298.15 K on PC NSs, Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, Ni-CNTs180/PC, and Ni-CNTs30. b Corresponding CO2/N2 IAST selectivity at 298.15 K and 1.0 bar for the binary mixture of CO2/N2 (15/85). c Pore-size distribution curves for PC NSs, Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, Ni-CNTs180/PC, and bare Ni-CNTs30. d Schematic illustration of the column breakthrough experiments, analogous to the MEA cell experiments. e Experimental column breakthrough curves for dry condition and 60% of RH CO2/N2 (v/v = 15/85) separations of PC NSs, Ni-CNTs30/PC, and Ni-CNTs180/PC. f Schematic of a two-bed VSA process by Aspen adsorption. The Aspen simulation results of CO2 recovery and purity (g), CO2 velocity and corresponding h LDF coefficient, and the i total energy consumption for the CO2 separation process in Ni-CNTs30/PC, Ni-CNTs180/PC, and Ni-CNTs30. Source data are provided as a Source Data file.
To quantify the adsorption selectivity of CO2 and N2, we employed the ideal adsorbed solution theory (IAST) to predict the adsorption equilibrium for the CO2/N2 mixture with a volume ratio of 15/8541,42, based on a set of pure N2 and CO2 adsorption isotherms data which were described by the Quadratic and BET model (Supplementary Note 3 and Supplementary Figs. 24–29), respectively. As shown in Fig. 4b and Supplementary Table 5, the calculated IAST selectivity for CO2 in the six catalysts decreased in the order of PC NSs (40.36) > Ni-CNTs30/PC (16.17) > Ni-CNTs60/PC (10.28) > Ni-CNTs90/PC (7.73) > Ni-CNTs30 (6.94) > Ni-CNTs180/PC (6.34), evidencing that PC NSs are responsible for the physical adsorption of CO2 and dense Ni-CNTs coverage sacrifices the CO2 capture capability.
To make it clear, the specific surface area and pore distribution of the as-prepared catalysts were measured based on the CO2 gas adsorption–desorption isotherms at 273.15 K. As depicted in Fig. 4c and Supplementary Table 6, PC NSs possess an average pore size of 0.5 nm, which just falls into the region of ultramicropore (pore size <1 nm, especially those ranging from 0.3 to 0.6 nm)41, indicating the swelling-assisted Ni-CMC aerogel enables the formation of a well-defined ultramicroporous structure. Accordingly, the total ultramicropore volume (<1 nm) is calculated to be ~0.051 cm3 g–1, testifying to the presence of abundant ultramicropores in PC NSs responsible for CO2 adsorption. In comparison, the bare Ni-CNTs30 without PC NSs have almost no pore size distribution in the ultramicropore region, and the corresponding volume is quite small (only 0.002 cm3 g–1). When CNTs gradually grow on the PC NSs, there is an obvious decline in porosity. The total ultramicropore volume of Ni-CNTs30/PC is 0.039 cm3 g–1, which is higher than that of Ni-CNTs60/PC (~0.012 cm3 g–1) and Ni-CNTs90/PC (~0.008 cm3 g–1). While for Ni-CNTs180/PC, no porous structure can be observed, which is similar to bare Ni-CNTs, affirming our conclusion that the as-grown Ni-CNTs cover the ultramicropores of PC NSs. Noted that the gas adsorption–desorption isotherms of Ni-CNTs30/PC were also conducted in the N2 atmosphere, which presents a narrow pore size distribution over a wide range of 0.60 to 2.30 nm (Supplementary Fig. 30), alongside a larger fitting standard deviation as compared to that in the CO2 atmosphere. This can be explained by the larger kinetic diameter of N2 (3.64 Å) than that of CO2 (3.30 Å), which induces a steric effect41 only available for the smaller CO2 molecules diffusion into the ultramicropores in PC NSs, thus greatly contributing to the superior adsorption selectivity of CO2 in the dilute CO2 stream.
To simulate and reflect the CO2 diffusion ability in the cathode channel of MEA, column breakthrough experiments were further carried out to examine the feasibility of the ultramicroporous PC NSs for the separation of CO2/N2 (v/v = 15/85) with 60% of relative humidity (RH) at ambient temperature and pressure (Fig. 4d). As shown in Fig. 4e, the breakthrough time of N2 and CO2 on bare PC NSs is 0.11 and 3.26 min g–1, respectively, serving as direct evidence that the PC NSs are capable of separating N2 and CO2. When Ni-CNTs30 covers PC NSs, the breakthrough time decreases to 0.11 min g–1 for N2 and to 1.30 min g–1 for CO2, respectively, apparently retaining the separation ability, largely due to the partial exposure of ultramicropores. The longer breakthrough time of CO2 than N2 further corroborates that the PC NSs can physically adsorb CO2 and delay the CO2 from penetrating through. While for Ni-CNTs180/PC, the breakthrough times for CO2 (0.03 min g−1) and N2 (0.02 min g−1) are very close, ascribed to the almost complete concealing of ultramicropores by highly dense Ni-CNTs, in good agreement with the observation in Fig. 4a–c.
To quantitatively evaluate the CO2 transport rate on Ni-CNTs30/PC in MEA, a two-bed vacuum pressure swing adsorption (VSA) process was conducted through the Aspen Adsorption simulation43. 15% of CO2 balanced by N2 serves as the feedstock with a total feed rate of 1.45 kmol h–1 (Fig. 4f, Supplementary Note 4, Supplementary Figs. 31–36, and Supplementary Tables S7–11). During the adsorption process at 1.00 bar, the CO2 loading on Ni-CNTs30/PC is ~0.34 mol kg–1, approximately sixfold higher than that on Ni-CNTs180/PC (0.06 mol kg–1) and Ni-CNTs30 (0.05 mol kg–1) (Supplementary Table 12). On account of superior adsorption capability, Ni-CNTs30/PC achieves 94.8% of recovery and 79.5% of purity for CO2, both of which are higher than those of bare Ni-CNTs30 and Ni-CNTs180/PC (Fig. 4g). Besides, the Ni-CNTs30/PC attains a remarkable CO2 velocity of 0.19 kmol h–1 within the bed, corresponding to a high CO2 linear driving force (LDF) coefficient of 2.02 × 10–2 s–1 (Fig. 4h). By comparison, Ni-CNTs180/PC and bare Ni-CNTs30 exhibit an almost identical CO2 velocity (0.12 kmol h–1) with low LDF coefficient (3.24 × 10–3 s–1), much lower than those of Ni-CNTs30/PC. Benefiting from the high adsorption capacity and transport rate for CO2, Ni-CNTs30/PC costs a lower energy consumption of \(2.59\,{{{\rm{MJ}}}}\,{{{{{\rm{kg}}}}}_{({{{{\rm{CO}}}}}_{2})}}^{{-}1}\), which is only half that of Ni-CNTs180/PC (\(4.64\,{{{\rm{MJ}}}}\,{{{{{\rm{kg}}}}}_{({{{{\rm{CO}}}}}_{2})}}^{{-}1}\)) and Ni-CNTs30 (\(4.43\,{{{\rm{MJ}}}}\,{{{{{\rm{kg}}}}}_{({{{{\rm{CO}}}}}_{2})}}^{{-}1}\)). Furthermore, COMSOL Multiphysics simulations were employed to model the CO2 velocity within the MEA channel under a dilute CO2 stream (CO2/N2 = 15/85). As depicted in Supplementary Fig. 37, Ni-CNTs/PC achieves a velocity of 0.0045 m s–1 at the channel outlet, which is ~1.2 times higher than that observed in Ni-CNTs. Taken together, the Ni-CNTs30/PC with opportune Ni-CNTs coverage integrates the complementary functions of CO2 capture and activation as well as enhanced CO2 transport kinetics, ensuring feasibility for the dilute CO2 reduction in MEA.
Direct electrocatalysis of dilute CO2 in MEA
Based on the simulation results, the porous carbon nanosheets in Ni-CNTs30/PC can accelerate the outlet velocity of CO2 towards CL ascribed to the high uptake capacity for CO2, which is supposed to thin the GBL (Supplementary Note 1), as schematically illustrated in Fig. 5a. In this case, the local CO2 concentration in the vicinity of active sites can be enhanced, thus favoring CO2 reduction even at flue gas concentration. To this end, we evaluated the electrocatalytic performance of dilute CO2 with a volume ratio of 15% (balance gas: N2) with varying inlet CO2 flow rates at 100 mA cm–2 in MEA. The Ni-CNTs30/PC shows a consistently high CO Faradaic efficiency (FECO) of ~95%, irrespective of the dilute CO2 stream flow rate varying from 10 to 120 sccm (Fig. 5b and Supplementary Fig. 38), providing powerful experimental evidence that Ni-CNTs30/PC with self-accelerated outlet CO2 velocity can maximally thin the GBL, which is barely affected by CO2 feed rate. In this context, the accessible CO2 molecule to the Ni–N3 sites are markedly increased, thus favoring dilute CO2 reduction over competing HER. Conversely, the FECO on bare Ni-CNTs30 increases from 74.2 ± 4.5% to 85.9 ± 0.6% as the CO2 feed rate increases from 10 sccm to 120 sccm, assuring the dilute CO2 stream indeed forms a thick GBL, which can be narrowed by increasing CO2 flow rate, in good accordance with Supplementary Equations 1-2. On the other hand, the CO selectivity on Ni-CNTs30/PC is consistently higher than that of Ni-CNTs30 even at a high CO2 feed rate, strongly confirming that the porosity of the catalyst critically influences the thickness of GBL, which directly affects the dilute CO2RR selectivity. With the same CO2 concentration (15%) and flow rate (60 sccm), we further compared the FECO of CO2RR on Ni-CNTs30/PC and bare Ni-CNTs30 under various cell voltages. As shown in Fig. 5c and Supplementary Fig. 39, Ni-CNTs30/PC and Ni-CNTs30 exhibit similar FECO under low cell voltage (<3.0 V) that corresponds to the CO2RR reaction kinetics-limited region13, indicating comparable intrinsic CO2RR activity derived from their Ni–N3 sites. In the mass transport-limited region (>3.0 V), Ni-CNTs30/PC achieves the maximum FECO of 98.3 ± 0.6% at 3.5 V. In sharp contrast, the FECO over bare Ni-CNTs30 without PC NSs rapidly decreases to 79.9 ± 0.6% at 3.6 V, largely ascribed to the limited CO2 transport caused by the thick GBL. Meanwhile, the Ni-CNTs30/PC exhibits a consistently high partial current density of CO (jCO) over the entire cell voltage with a positively increasing trend as cell voltage (Fig. 5d). Specifically, the jCO over Ni-CNTs30/PC reaches 196.5 ± 2.2 mA cm–2 at 3.1 V, double that of bare Ni-CNTs30 (~83.8 ± 1.9 mA cm–2). The above comparison experimentally collaborates the ultramicropore structure in Ni-CNTs30/PC guarantees CO2 capture/enrichment and breaks the dilute CO2 mass transport limit, thus preferentially converting CO2 into CO even at high current densities. Besides, the calculated energy conversion current density (ECCD) of Ni-CNTs30/PC in Supplementary Fig. 40 is consistently higher than of Ni-CNTs30 over the measured cell voltage29, indicating the reinforced CO2 concentration gradient effect can lower the energy consumption, consistent with the Aspen simulation in Fig. 4i.

a Schematic to illustrate the dependence of GBL thickness on CO2 concentration. b The comparison of CO Faradaic efficiency for Ni-CNTs30 and Ni-CNTs30/PC at 100 mA cm−2 with dilute CO2 flow rates ranging from 10 sccm to 120 sccm. Cell voltage-dependent c CO Faradaic efficiency and d jCO on Ni-CNTs30 and Ni-CNTs30/PC. e Schematic to illustrate the effect of CL length and CO2 concentration in the concentration gradient. The comparison of CO Faradaic efficiency for f Ni-CNTs30/PC and g Ni-CNTs180/PC measured in MEA under different CO2 concentrations. h CO and H2 Faradaic efficiency for Ni-CNTs30/PC in a 5 cm2 MEA eletrolyzer. i The comparison of CO Faradaic efficiency over Ni-CNTs30, Ni-CNTs30/PC, and Ni-CNTs180/PC with the dilute CO2 feeding from 15 to 3% at a current density of 100 mA cm–2. j Comparison of the dilute CO2RR performance metrics (e.g., FECO, jtotal, cell voltage, CO2 concentration, and CO2 adsorption capacity) in this work with previous works on low-concentration CO2RR with gaseous feedstocks27,29,31. k Schematic illustration of introducing an O2-adsorption pack column before CO2RR MEA electrolyzer for direct electrocatalysis of O2-containing simulated flue gas (CO2/N2/O2 = 15/82/3). l, m The corresponding CO Faradaic efficiency with the OAPC system and the stability test of Ni-CNTs30/PC at 100 mA cm–2. Error bars correspond to the standard deviations based on triplicate electrochemical measurements. Source data are provided as a Source Data file.
According to Supplementary Note 1 and Supplementary Equation 21, the narrowed GBL accelerates CO2 molecular transport toward Ni–N3 sites. To validate this, we further measured the jCO and HER on Ni-CNTs30/PC and bare Ni-CNTs30 under a cell voltage of 3.2 V at varying temperatures from 298.15 to 338.15 K and calculated the activation energy (\({E}_{a}\)) for dilute CO2RR and HER by Arrhenius-fitting jCO and \({j}_{{H}_{2}}\) over 1/T, respectively28. As shown in Supplementary Fig. 41, Ni-CNTs30/PC displays a substantially lower \({E}_{a}\) for CO2 conversion into CO (3.50 kJ mol–1), as compared to that for HER (83.19 kJ mol–1). In contrast, for bare Ni-CNTs30 without the CO2 enrichment effect, the \({E}_{a}\) for CO2RR (11.61 kJ mol–1) and HER (20.59 kJ mol–1) differ little, highlighting the crucial role of PC NSs in enhancing CO2RR reaction kinetics through accelerating CO2 mass transfer velocity within GBL. Besides forming a thick GBL, the dilute CO2 feed causes a weak concentration gradient effect within the CL based on the well-known Fick’s law24 (Fig. 5e), which diminishes the CO2 transport ability and slows down the CO2RR kinetics. Since the CO2 concentration gradient shows a strong dependence on both the feeding concentration of CO2 and the length of CL, we compared the CO2RR performance using Ni-CNTs/PC with varying lengths of Ni-CNTs and different concentrations of CO2 with the cathode geometric area of 1 cm2. As shown in Fig. 5f and Supplementary Fig. 42, it is surprising to find that the FECO in Ni-CNTs30/PC shows an irrelative trend to CO2 concentration, all exceeding 90% with 15, 50, and 100% purity of CO2 as feedstock over a wide current density from 50 to 250 mA cm–2 in MEA. For Ni-CNTs180/PC, the FECO is consistently higher than 90% with pure CO2, indicating the high activity of Ni–N3 sites. However, when N2 was introduced to dilute the CO2, the FECO sharply decreases, especially at high current densities (Fig. 5g and Supplementary Fig. 42), which can be ascribed to the limited CO2 transport. The marked contrast aligns with our hypothesis that a higher length of Ni-CNTs would lead to a weak CO2 concentration gradient effect, which induces weak transport ability and low local CO2 concentration on the top of CNTs, particularly during the low-concentration CO2RR, resulting in sluggish CO2RR kinetics (Supplementary Figs. 43–45). While for Ni-CNTs30/PC, the presence of both porous carbon nanosheets for concentrating CO2 and optimal length of Ni-CNTs for converting CO2 synergistically contributes to the establishment of the self-reinforced CO2 concentration gradient effect. This effect is analogous to the nutrient diffusion process from soil to rhizome and leaves in plants, which can, in turn, promote CO2 diffusion and accelerate CO2RR kinetics.
Impressively, the FECO on Ni-CNTs30/PC reaches 95.4 ± 1.2% at 250 mA cm–2, far surpassing those on Ni-CNTs60/PC (73.8 ± 3.4%), Ni-CNTs90/PC (36.1 ± 1.5%), and Ni-CNTs180/PC (20.0 ± 0.9%) (Fig. 5f, g and Supplementary Fig. 46). The negative correlation of FECO to Ni-CNTs length further collaborates with the weakened CO2 concentration gradient with increasing the length of Ni-CNTs as CL. Accordingly, the Ni-CNTs30/PC requires the smallest cell voltage to drive the same magnitude of current density, which is also irrelevant to CO2 concentration (Supplementary Fig. 47). A similar changing trend was also observed in H-type cell, where the Ni-CNTs30/PC display the FECO higher than 90% over the potential ranging from –0.57 to –0.85 V, with the highest value of 97.0 ± 1.9% at –0.75 V, and shows independence on CO2 concentration (Supplementary Fig. 48). With respect to bare PC NSs, CO can be scarcely detected in either H-type cell (FECO <1%) or MEA device (FECO <1%), further validating the active sites for dilute CO2RR originate from Ni-CNTs rather than porous carbon nanosheets, which aligns well with ATR-FTIR results (Supplementary Figs. 49, 50).
Considering the variance in Ni content across different lengths of Ni-CNTs, the turnover frequency (TOF) was calculated to evaluate the pristine activity of Ni-CNTs/PC. Even the Ni-CNTs10/PC with the strongest CO2 concentration gradient demonstrates the highest TOF at different current densities, its FECO rapidly decreases to 44.3 ± 9.4% at 250 mA cm−2 (Supplementary Figs. 51, 52), largely due to the insufficient active sites. On the contrary, Ni-CNTs30/PC with suitable CO2 concentration gradient and active sites achieves a high TOF of 1.31 ± 0.02 × 105 h–1 at 250 mA cm–2 (Supplementary Figs. 52, 53 and Supplementary Table 13), which is slightly lower than that Ni-CNTs10/PC (1.62 ± 0.34 × 105 h–1) and around eightfold enhancement in comparison with Ni-CNTs180/PC (1.99 ± 0.09 × 104 h–1). These contrasts substantiate that the low-concentration CO2RR performance primarily depends on the CO2 concentration gradient, especially when the active site density reaches a certain level (i.e., 2 wt%). Meanwhile, the Ni-CNTs30/PC shows a negligible change in both FECO (~90%) and cell voltage (~3.3 V) during 40 h of continuous electrocatalysis under dilute CO2 stream (15%) at 100 mA cm–2 (Supplementary Fig. 54), demonstrating the superior CO2RR stability, consistent with the high recovery rate in Aspen simulation. This can be further supported by the postmortem analyses, where Ni-CNTs30/PC can well preserve its original morphology with uniform distribution of Ni and N throughout the CNTs profile (Supplementary Figs. 55, 56). In contrast, the FECO on bare Ni-CNTs30 and Ni-CNTs180/PC both rapidly decrease below 50% after only 1 h of CO2RR operation under the same conditions. Notably, the low CO2 concentration also significantly reduces the formation rate of (bi)-carbonate species, thereby extending the time to reach KHCO3 or K2CO3 saturation (Supplementary Figs. 57, 58).
To assess the scalability of Ni-CNTs30/PC for the CO2RR to CO under dilute conditions, we enlarged the cathode geometric area to 5 cm2, maintaining a constant dilute CO2 stream flow rate of 60 sccm (CO2/N2 = 15/85). As the total current incrementally rises from 0.25 to 1.00 A, the FECO consistently exceeds 90%. Even when the applied current reaches 1.25 A, the system sustains a FECO of 86.2 ± 3.7% with a CO partial current surpassing 1.00 A (Fig. 5h and Supplementary Fig. 59), demonstrating the great potential of the self-reinforced concentration gradient strategy for industrial application. Inspired by the high selectivity on Ni-CNTs30/PC for 15% purity of CO2, we further decreased the CO2 concentration and compared the selectivity of CO2-to-CO at 100 mA cm–2 in MEA. Remarkably, the Ni-CNTs30/PC catalyst achieves a FECO of 92.3% at 5% CO2 feed concentration and retains 87.7% even under an ultralow CO2 concentration of 3% (Fig. 5i and Supplementary Figs. 60), which realizes the ultralow concentration for studying CO2 electroreduction as reported so far9,27,28,31,44,45,46,47 (Supplementary Figs. 61 and Supplementary Table 14). Considering the key performance metrics, including FECO, jtotal, CO2 concentration, and CO2 adsorption capacity, this work explicitly stands out among the reported works27,29,31,48, (Fig. 5j), demonstrating the superiority of the self-reinforced CO2 concentration gradient effect under dilute CO2 conditions. By contrast, a rather low FECO of 26.5 and 48.2% was observed under 5% of CO2 on Ni-CNTs180/PC and Ni-CNTs30, respectively. The above phenomenon elucidates that the narrow GBL and self-reinforced CO2 concentration gradient in Ni-CNTs30/PC ensures both high CO2RR selectivity and good stability due to enhanced mass transport ability, which is expected to realize direct electrocatalytic conversion of CO2 from the ultra-dilute CO2 point sources.
Furthermore, we examined the feasibility of a self-reinforced concentration gradient strategy for converting O2-containing simulated flue gas (15% CO2 and 3–10% O2, balanced by N2) using different O2 removal systems49. By connecting the oxygen reduction reaction (ORR) and CO2RR MEA electrolyzers in series (Supplementary Fig. 62), the flue gas flows into the first reactor for ORR with oxidized CNTs (O-CNTs) as a catalyst (Supplementary Figs. 63, 64), followed by entering the second reactor for CO2RR. The tandem ORR reactor not only enables the on-site production and utilization of H2O2, but also mitigates the adverse effect of O2 on CO2RR performance by consuming the O2 in the flue gas (Supplementary Figs. 65–68). With a 5 cm2 cathode for ORR, the H2O2 yield reaches 3.34 ± 0.70 mol gcat–1 h–1 at 0.50 A, which not only rapidly removes the organic effluent in 20 min but also attains a FECO of 77.7 ± 10.7% at 200 mA cm−2 (Supplementary Fig. 69). To further improve the practical viability, we introduced a home-made O2-adsorption packed column (OAPC) containing sodium dithionite (Na2S2O4) and quartz sand before CO2RR MEA electrolyzer (Fig. 5k). The OAPC exhibits an ultra-high O2 removal efficiency (~99.4%), leading to a high FECO of 91.5 ± 2.0% at 200 mA cm−2 over the Ni-CNTs30/PC (Fig. 5l and Supplementary Figs. 70, 71). Inspiringly, the OAPC strategy sustains an O2 removal efficiency of 93% during 24 h of continuous operation, where the FECO consistently exceeds 90% with a stable cell voltage of ~3.1 V at 100 mA cm−2 (Fig. 5m and Supplementary Fig. 72). For the higher-concentration O2-containing flue gas (e.g., 5–10% typically from ore roasting and coal combustion processes)50, the OAPC treatment provides >99% of O2 removal efficiency along with a FECO above 90% (Supplementary Fig. 73). The coupling of OAPC with the self-reinforced CO2 concentration gradient strategy enables favorable economic viability (CO cost = 378.3 $ tonCO−1), which significantly outperforms the conventional amine-based flue gas capture methods (Supplementary Fig. 74 and Supplementary Note 6).
Fundamental principles and mechanisms of self-reinforced CO2 concentration gradient
To further understand the origin of self-reinforced CO2 concentration gradient within the CL as well as its role in boosting CO2 transport, we employed Ni-CNTs/PC with varying CNTs lengths to regulate the CO2 concentration gradient for comparing the local CO2 concentration and CO2RR kinetics. Notably, the CO2 concentration is fixed at 15% with N2 as a balance gas in the following study. The difference in hydrophobicity among PC NSs, Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, and Ni-CNTs180/PC was first excluded by contact angle measurements, all of which show hydrophobic character with contact angles higher than 130° (Supplementary Fig. 75). Since the acidic CO2 dissolution typically results in a pH decrease, a rotating ring disc electrode (RRDE) assembly was employed to determine the local pH change for further evaluating local CO2 concentration during dilute CO2RR51. As shown in Supplementary Fig. 76, the peak shift separation induced by pH (∆EpH-shifted), which is a linear response to the pH change, is the least noticeable in Ni-CNTs30/PC across a wide potential range. While for Ni-CNTs180/PC, the ∆EpH-shifted is obviously the largest and sharply increases at high potential range, mainly because of the limited CO2 transport. The above result reflects that the Ni-CNTs30/PC with a suitable length of CNTs can provide sufficient CO2 for electroreduction in the vicinity of the catalyst layer.
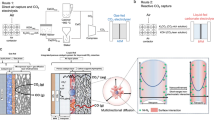
COMSOL Multiphysics simulations were further employed to visually comprehend the CO2 concentration distribution on bare PC NSs and Ni-CNTs, as well as Ni-CNTs/PC heterostructures in MEA under a cell voltage of 3.0 V. As shown in Fig. 6a, it is found that the abundant ultramicropores in PC NSs effectively concentrate CO2 near the CL, consistent with the conclusion from adsorption selectivity in Fig. 4. The admirable CO2 capture capacity of Ni-CNTs/PC enables the formation of a pronounced concentration gradient along with the Ni-CNTs spanning a length of ~800 nm. Beyond this length, the CO2 concentration stabilizes along the CNTs, and a significant concentration gradient ceases to exist. This well explains the strong dependence of dilute CO2RR selectivity on CNTs length. For Ni-CNTs30/PC, the CO2 concentration gradient distributes on the entire CNTs, which drives CO2 transport from porous carbon to Ni-CNTs and guarantees a high flux of CO2 accessible to the Ni–N3 sites based on Fick’s law, thus accelerating dilute CO2RR kinetics and rapid conversion. In contrast, the bare Ni-CNTs exhibit no discernible concentration gradient, which is responsible for the inferior CO2RR performance. Meanwhile, the Ni-CNTs/PC with self-reinforced CO2 concentration gradient induces a concentration gradient for CO product (Supplementary Fig. 77), which ensures prompt CO desorption and avoids the blocking of active sites by CO. For Ni-CNTs with a length of ~1000 nm, without the presence of PC NSs, results in a notably low CO2 concentration, which in turn aggravates the HER and leads to poor CO2RR selectivity, underscoring that the CO2 enrichment is the key premise for dilute CO2 reduction.

a Comparison of modeled CO2 concentration along the CL for PC NSs (left), Ni-CNTs/PC (middle), and bare Ni-CNTs (right). The color bar represents the concentration of CO2. DRT plots for b Ni-CNTs30/PC, c Ni-CNTs60/PC, and d Ni-CNTs180/PC in MEA. e Time-dependent local CO2 concentration during the low-concentration CO2RR on Ni-CNTs30/PC, Ni-CNTs60/PC, and Ni-CNTs180/PC. f Proposed mechanism in Ni-CNTs/PC for enhancing CO2 transport across GBL and CL in low-concentration CO2RR through a self-reinforced CO2 concentration gradient. Source data are provided as a Source Data file.
To assess the improved CO2 transport ability via the self-reinforced concentration gradient, the distributed relaxation time (DRT) was acquired through deconvolving the electrochemical impedance spectroscopy (EIS) during dilute CO2 electroreduction in MEA at a cell voltage from 2.8 to 3.6 V (Fig. 6b–d and Supplementary Figs. 78, 79). The DRT plots exhibit three distinct peaks over a timescale (τ) ranging from 10−5–10−3, 10−3–10−1, and 10−1–10, corresponding to contact resistance, charge transfer, and CO2 mass transport, respectively52,53. As Ni-CNTs length decreases, the CO2 mass transport process in the timescale region of 10−1 to 10 s appears high-frequency shifts along with decreased impedance, corroborating Ni-CNTs30/PC can ameliorate the problem associated with mass transport. Accordingly, the charge transfer process in the timescale of 10–3 to 10–1 is accelerated in Ni-CNTs30/PC as compared to Ni-CNTs60/PC and Ni-CNTs180/PC, which aligns well with the Tafel slopes (Supplementary Fig. 80), declaring the improved dilute CO2RR kinetics by the self-reinforced CO2 concentration gradient. A comparable trend was observed in the H-type cell (Supplementary Figs. 81, 82), where Ni-CNTs180/PC shows much higher resistance associated with both mass and charge transfer processes. This observation further demonstrates that a weak concentration gradient impedes CO2 mass transport, consequently resulting in diminished reaction kinetics.
To quantify the mass transport rate of CO2 within CL, the CO2 mass transport coefficient (k) was introduced, which can be calculated using the following equation54,55:
where n, F, [CO2]0, and [CO2]i represent the electron transfer number for CO2 to CO, Faradaic constant (96485 C mol–1), and the initial and instantaneous CO2 concentration near the CL during CO2RR, respectively. For the dilute CO2RR in this work (CO2/N2 = 15/85), [CO2]0 is calculated to be 6.12 mM. [CO2]i is dynamically monitored by in situ fluorescence electrochemical spectroscopy (FES) during the CO2RR process (Supplementary Fig. 83). As shown in Fig. 6e, Ni-CNTs30/PC shows a shorter relaxation time and higher equilibrium concentration of [CO2]i of 4.17 mM, as compared to Ni-CNTs60/PC (3.68 mM) and Ni-CNTs180/PC (3.24 mM). Accordingly, k was calculated to be 1.24, 0.98, and 0.59 cm s–1 for Ni-CNTs30/PC, Ni-CNTs60/PC, and Ni-CNTs180/PC, respectively. The results strongly support our conclusion that the self-reinforced CO2 concentration gradient accelerates the CO2 transport rate and elevates the CO2 concentration at the interface between the catalyst and the electrolyte.
Based on the above simulated and experimental results, we justifiably propose the mechanism of the self-reinforced CO2 concentration gradient for low-concentration CO2 to CO conversion in MEA, as schematically in Fig. 6f. In the case of dilute CO2 system, PC NSs features abundant ultramicroporous architectures that preferentially adsorb and concentrate CO2, which effectively reduces the thickness of GBL and facilitates the access of CO2 to active sites (Supplementary Fig. 25). Under these circumstances, the Lewis basic sites of Ni–N3 in Ni-CNTs chemically adsorb the accessible CO2 molecules (Supplementary Fig. 84), forming the self-reinforced CO2 concentration gradient along the CNTs profile, which weakens as the CNTs length increases. With a suitable Ni-CNTs growth time on porous carbon, the enhanced gradient concentration can continuously drive CO2 transport to the Ni–N3 active sites, which breaks the CO2 mass transport limit and enables rapid conversion.
Discussion
In summary, we constructed heterostructures composed of porous carbon nanosheets and Ni single atoms anchored in CNTs (Ni-CNTs/PC) to mimic the biological function of nutrient diffusion in plants. Specifically, the ultramicroporous feature of the PC NSs serves a role analogous to soil, enabling selective CO2 adsorption, while the rhizome-like Ni-CNTs incorporating Ni–N3 active sites efficiently transport and catalyze the captured CO2. This appealing architecture fosters a complementary effect between the PC and Ni-CNTs for dilute CO2RR, where the porous carbon nanosheets enrich CO2 and the Ni-CNTs catalyze CO2, allowing to establish a self-reinforced concentration gradient during the dilute CO2RR. The combined experimental and theoretical analyses reveal that the self-reinforced CO2 concentration gradient effect can narrow the gas boundary layer and strengthen the concentration gradient within the catalyst layer. This enhancement in CO2 mass transport accelerates dilute CO2RR kinetics and suppresses the HER. Consequently, the Ni-CNTs30/PC achieves a high CO Faradaic efficiency of 95.4 ± 1.2% under 15% CO2 at an industrial-level current density of 250 mA cm–2. When enlarging the MEA size, the CO partial current density reaches 1.08 ± 0.05 A. Notably, the integrated OAPC-CO2RR MEA electrolyzer achieves >90% of CO selectivity from simulated flue gas containing 3–10% of O2. This self-reinforced CO2 concentration gradient strategy offers a promising solution to overcome the challenges of CO2 mass transport in the conversion of dilute CO2 from industrial point sources, marking a substantial step toward practical application.
Methods
Synthesis of the Ni-CMC hydrogel
The swelled Ni2+-crosslinked carboxymethylcellulose sodium (Ni-CMC) hydrogel was fabricated through a combination of the sol-gel method and directional freeze-casting method. Typically, 1.0 g of CMC (Sigma-Aldrich, with a viscosity ranging from 1500–3000 mPa·s, USP grade) and 0.2 g of NiCl2 (Alfa Aesar, 98%) were dissolved in 50 mL of deionized water to form a green homogeneous colloidal solution. Then, the uniform sol was poured into a plastic mold (15 mm × 15 mm × 15 mm) and placed on a frozen copper billet (frozen in liquid nitrogen), enabling the upper sol surface to freeze solid. The as-prepared solid was immersed in an ethanol/NaOH (Sigma-Aldrich, 98%) bath (–30 °C) until the ice crystals completely dissolved in ethanol to form Ni-CMC hydrogel.
Synthesis of the Ni-CMC aerogel
The as-fabricated Ni-CMC hydrogel was soaked in deionized water (18.25 MΩ cm) for swelling, with the water being replaced every 2 h. After a 48-h swelling period, the same directional freeze-casting method was replicated. Subsequently, the sample underwent freeze-drying treatment at –60 °C for 48 h, and the Ni-CMC aerogel was ultimately obtained.
Synthesis of the Ni-CNTsX/PC
The Ni-CNTsX/PC was synthesized via the chemical vapor deposition method, where X represents the growth time. Specifically, for the Ni-CNTs30/PC sample, the Ni-CMC aerogel was placed in the center of a tube furnace and heated to 750 °C with a heating rate of 5 °C min–1 under an Ar atmosphere (Air Liquide Tianjin Co. Ltd, 99.9999%). After reaching this temperature, CH3CN (Sigma-Aldrich, 99.9%) was introduced by Ar to initiate the growth of Ni-CNTs for 30 min. Then the system was cooled to 25 °C. Finally, 1.0 M HCl (Gaofeng, AR) was used to etch the residual Ni species. For comparison, PC NSs were synthesized by the same process except for the introduction of CH3CN.
Characterization
Field emission SEM (FE-SEM) images were captured using JSM-7900F. FTIR spectroscopy was performed utilizing a Nicolet iS50 (Thermo Fisher Scientific). XPS spectra was recorded using Thermo Scientific K-Alpha with aluminum Kα source (1486.6 eV) and a collimator at 12 kV and 72 W. N2 adsorption–desorption isotherms were obtained at 77 K using a Micromeritics 3Flex system, with surface areas calculated via the Brunauer–Emmett–Teller (BET) method. The porosity distribution was analyzed using the density functional theory (DFT) method, employing CO2 adsorption isotherms measured on a Micromeritics 3Flex instrument at the temperature of 273.15 K. Prior to conducting the tests, all samples underwent a degassing process at 573.15 K for a duration of 8 h. TEM equipped with EDS mappings was carried out in the Talos F200X G2. HAADF-STEM images were collected by JEM-ARM200F. XRD was performed using SmartLab 9KW with 2θ ranging from 5˚ to 80˚, where Cu Kα radiation (λ = 1.54 Å) was operated at 40 kV and 150 mA. XAS measurements were conducted at the XAS Beamline of the Australian Synchrotron, located in Melbourne. The analysis of the Ni K-edge was performed using Si (111) crystal monochromators. The contact angles were measured at ambient temperature utilizing the Dataphysics OCA 25, employing a probe consisting of 10 μL of water. ATR-FTIR analyses were carried out using a Thermo Fisher Scientific Nicolet iS50 FTIR spectrometer. The electrolyte of 0.5 M KHCO3 was bubbled with a mixture gas (CO2/N2 = 15/85) for 30 min before the test. Electrochemical curves were recorded during 20 min of continuous electrolysis while maintaining a constant potential of –0.75 V versus RHE.
Electrochemical measurements and product quantification
The electrochemical measurements were conducted using an Ivium electrochemical workstation at room temperature (25 °C). In the case of the H-type cell, 5 mg of catalyst was sequentially dispersed in 500 μL of isopropyl alcohol (Sigma-Aldrich, 99.9%), 480 μL of DI water, and 20 μL of Nafion D520 binder solution (5 wt%, SCI Material Hub). Then, the mixture was sonicated for 30 min to achieve a homogenous ink. The resulting ink was dropped onto AvCarb P75T (20% PTFE treated, SCI Material Hub) to form a working electrode with a catalyst loading of ~0.5 mg cm2. SELEMION anion exchange membrane (size: 2 cm × 2 cm, thickness: 100 μm, SCI Material Hub) was used to separate the cathode and anode chambers, which was presoaked in deionized water for 24 h before being employed. Platinum (Pt) gauze (size: 15 mm × 15 mm, SCI Material Hub) served as the counter electrode. The gas flow controller was used to continuously supply various concentrations of a CO2/N2 mixture gas into electrolyte of 0.5 M KHCO3 with pH of 7.0 ± 0.2 (Sigma-Aldrich, 99.99%), including 15, 50, and 100% of CO2 with a total flow rate of 30 sccm (CO2 and N2, 99.9999%, Air Liquide Tianjin Co. Ltd). The potential, measured against the saturated calomel electrode (SCE, SCI Material Hub), was transformed into RHE values with an iR-correction using the following equation:
The iR compensation was implemented during the electrolysis experiments at each applied potential in the H-type cell. The ohmic resistance was determined through electrochemical impedance spectroscopy (EIS) measurements, which were conducted by applying an AC voltage with an amplitude of 5 mV across a frequency range spanning from 100 to 0.01 kHz.
During the electrochemical reaction, the gas products were analyzed utilizing a gas chromatograph (GC-2014), where H2 was detected by a thermal conductivity detector (TCD) and CO was detected by a flame ionization detector (FID). The Faradaic efficiency (FE) for gas production (H2 and CO) was calculated as follows:
where x represents the volume fraction of H2 or CO which determined by GC, v represents gas flow rate (mL min–1), F represents Faraday constant, P0 is 101.325 kPa, T represents temperature (K), R represents gas constant, and j represents total current density at a specific voltage. The corresponding partial current density was calculated by following:
The energy conversion current density (ECCD) for the electrochemical reduction of dilute CO2 to CO (ECCDCO) were determined according to the following equations28:
where EOER and ECO represent the thermodynamic equilibrium potential for OER and CO2RR, respectively.
Additionally, TOF (h–1) for CO was calculated by the following formula:
where mcat represents the weight of catalysts (g), ω represents the metal loading of Ni, which was detected by ICP-MS, MNi represents the atomic mass of Ni (58.69 g mol–1).
As for the dilute CO2RR test in MEA electrolyzer, the Sustainion X37-50 Grade FA anion exchange membrane (2 cm × 2 cm, thickness 50 μm, SCI Material Hub) served as the separator, which was soaked in 1.0 M KOH (Sigma-Aldrich, 95%) at least 24 h before being employed. In the case of the MEA cell, 20 mg of catalyst with 0.2 mg PTFE powder (~200 nm, Teflon, SCI Material Hub) was sequentially dispersed in 1 mL of isopropyl alcohol, 0.9 mL of deionized water, and 0.1 mL of Nafion D520 binder solution. Then, the mixture was sonicated for 30 min to achieve a homogenous ink. The resulting ink was air-spray coated onto the Sigracet 39BB gas diffusion layer electrode (size: 1.0 cm × 1.0 cm, thickness: 0.3 mm, SCI Material Hub) to form a working electrode with a catalyst loading of ~1.0 mg cm2. A Pt/Ti mesh (size: 1.5 cm × 1.5 cm, thickness: 0.3 mm, Pt coating thickness: 0.5 μm, SCI Material Hub) with the IrO2 (mass loading: ~2.0 mg cm−2) served as the anode catalyst. Detailly, 20 mg IrO2 (Sigma-Aldrich, 99.9%) was sequentially dispersed in 2 mL of isopropyl alcohol and 0.1 mL of Nafion D520 binder solution, followed by 30 min of sonication to achieve a homogenous ink. The resulting ink was air-spray coated onto the Pt/Ti mesh. The MEA measurements used a freshly prepared 1 M KOH electrolyte (pH = 13.6 ± 0.2), which was replaced daily to ensure consistency. In this work, all electrolytes are stored in a thermostat at 25 °C between experimental sessions. All cell voltage measurements were conducted without iR compensation.
Direct conversion of O2-containing simulated flue gas
The tandem ORR-CO2RR MEA electrolyzer system was designed to operate with simulated flue gas containing 15% CO2, 3% O2, and 82% N2 at a controlled total flow rate of 60 sccm. The 5 cm2 ORR-MEA electrolyzer utilized 0.5 M KHCO3 as the anode electrolyte with a pH of 7.1 ± 0.1 to minimize carbon loss while maintaining stable H2O2 production. The cathode was fabricated by casting a homogeneous slurry of O-CNTs with 1 wt% PTFE (Teflon, SCI Material Hub) onto a 5 cm2 reinforced 39BB GDL. IrO2/Pt/Ti mesh (area: 5 cm2) served as the anode catalyst. The cathode and anode chambers were separated by a Sustainion X37-50 Grade FA anion exchange membrane. The oxygen removal efficiency (ORE) of the ORR-MEA electrolyzer can be determined using the following expression:
where co2, in and co2, out represent the O2 volume fraction at the electrolyzer inlet and outlet, respectively, which can be detected by GC.
To enhance the residence time of simulated flue gas, the O2-adsorption packed column system (OAPC) are helical with inner and outer diameters of 15 and 100 mm, respectively. The Na2S2O4 (Sigma-Aldrich, AR, 90%) and quartz sand (Sigma-Aldrich, 40 mesh) are packed under an anaerobic environment with a volume ratio of 1:1. In this OAPC system, Na2S2O4 serves as the primary O2 adsorbent while the quartz sand provides dual functions of increasing the gas-solid interfacial contact area and preventing the agglomeration of Na2S2O4 powder. To reflect the O2 removal ability of the OAPC, the ORE can be calculated by Eq. (7).
Quantification of the accumulation rate of K salt
The quantification of accumulation K salt rate was performed using 2-h CO2RR measurements with Ni-CNTs30/PC as catalyst at 100 mA cm–2 under different CO2 concentrations (3, 15, 50, and 100%) in MEA using 1.0 M KOH as anolyte. When the tests finished, the cathode channels were thoroughly and repeatedly rinsed with DI water while remaining the MEA assembled. The resulting solution was analyzed via ICP-MS to quantify the formation rate of the accumulated salt.
Single-component adsorption experiments and IAST prediction
The adsorption isotherm curves of CO2 and N2 on 100 mg of the PC NSs, Ni-CNTs30/PC, Ni-CNTs60/PC, Ni-CNTs90/PC, Ni-CNTs180/PC, and bare Ni-CNTs30 were measured using the Micromeritics 3Flex equipment at 298.15 K. The BET adsorption isotherm model and the quadratic model was employed to fit the adsorption isotherms of CO2 and N2, respectively. Meanwhile, IAST was utilized to predict the mixture adsorption isotherm based on the pure CO2 and N2 isotherms41,42. The BET adsorption isotherm model was used to describe the adsorption behavior of CO2 with three parameters as follows:
where IP1, IP2, and IP3, represent the isotherm parameters of the BET model, Pi represents the corresponding equilibrium pressure (bar). The quadratic adsorption isotherm model was used to describe the adsorption behavior of N2 with three parameters as follows:
where IP1, IP2, and IP3 represent the isotherm parameters of the quadratic model, Pi represents the corresponding equilibrium pressure (bar). For the calculation of IAST, see Supplementary Note 3 for more details.
CO2 chemical adsorption experiments
The CO2 chemical adsorption behavior was studied through temperature programmed desorption (TPD) measurements, conducted using an AutoChem1 II 2920. For a detailed assessment of CO2-TPD, 100 mg of catalyst powder was placed in a reaction cube and subsequently heated to 523.15 K at the rate of 10 K min–1 and maintained for 1 h for pre-drying under a helium (He) atmosphere. Once cooling down to 323.15 K, the adsorption of CO2 was conducted in mixed gas (10% CO2 and 90% He) flow for 1 h. Following this, He flow (99.999%) was introduced to purge the residual CO2 from the catalyst surface for an additional hour. Finally, the reaction cube was gradually heated from 323.15 to 1073.15 K at a rate of 10 K min–1. The desorbed gas was detected using the TCD.
CO2/N2 breakthrough experiments
The CO2/N2 breakthrough experiments were conducted using a multicomponent competitive adsorption equipment (BSD-MAB) at 298.15 K. The stainless-steel column with an inner diameter of 10 mm and a length of 70 mm served as the fixed-bed reactor to minimize wall-flow effects and associated breakthrough curve tailing. The weight of the sample packed in the column was 500–800 mg. Prior to the breakthrough experiment, all samples were activated via purging He flow at 573.15 K for 2 h. Subsequently, the mixed gas containing 15% CO2 and 85% N2 was introduced at a flow rate of 10 sccm. The outlet gas from the column was analyzed using online mass spectrometry (INFICON). For humidified CO2 tests, the feed gas was conditioned to 60% relative humidity via water vapor saturation, accurately reflecting the humidified gas for MEA operation.
Local pH measurement
Local pH environments of various samples were measured using RRDE (RRDE-3A, ALS). This device comprises a catalyst disc electrode and a Pt ring electrode, with SCE and Pt foil functioning as the reference and counter electrode, respectively. Prior to testing, cyclic voltammetry (CV) was employed to purify the Pt ring under N2 atmosphere, sweeping from 0.45 to 1.15 V at a rate of 100 mV s–1. Subsequently, the same technique was utilized to activate the catalyst. Operating at a rotation speed of 1600 rpm, a constant potential ranging from –0.68 to −0.98 V was held on the catalyst disc to perform the CO2RR process Simultaneously, potential cycling from 0.17 to 1.36 V was applied to the Pt ring electrode for detecting the peak of CO oxidation. Consequently, the difference value between the concentration-dependent peak of CO (ECO) and the actual oxidation peak (Eobs) observed is designated as the pH-induced peak shift (∆EpH-shifted), a direct indicator of the local pH level51.
Operando EIS experiments
Operando EIS measurements were conducted in both an H-type cell and an MEA at 298.15 K utilizing the Ivium electrochemical workstation. The impedance data were galvanostatically recorded, employing an amplitude of 10 mV across a frequency range of 0.01 to 100 kHz. The cell voltages in the MEA were adjusted from 2.8 to 3.6 V in increments of 0.2 V, while the potentials in the H-type cell were adjusted from –0.68 to –0.98 V in increments of 50 mV. The EIS data were analyzed using the DRT method, which were discretized by employing radial basis functions of Gaussian type56. Fitting without inductance was adopted to address the inductive features:
where R∞ represents Ohmic resistance, γ(lnτ) represents the DRT function that describes the time relaxation characteristics of the electrochemical system studied, τ represents relaxation time and f represents frequency.
The regularization parameter λ utilized in Tikhonov regularization was optimally adjusted by employing the regularization method of mGCV.
In situ fluorescence electrochemical spectroscopy (FES)
A fluorescence spectrophotometer (QM/TM/NIR) connected with a CH Instruments 760E was utilized to monitor local CO2 concentrations. Initially, a pH-sensitive fluorescent probe was synthesized by adding 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS, ACMEC, 98%) to 1 mL of a 0.5 M tetraoctylammonium hydroxide solution (TH, Macklin, 20 wt% in methanol), followed by the incorporation of an additional 2.5 mL of methanol to create the fluorescent probe solution. Subsequently, 30 μL of this solution was applied to the reverse side of the gas diffusion layer. The excitation and emission wavelengths for HPTS were set at 485 and 520 nm, respectively. Experiments were conducted in a custom-built cell using Ni-CNTs30/PC or Ni-CNTs180/PC as the cathode, a Pt foil (SCI Material Hub) as the anode, and 1 M KOH as the electrolyte, at a current density of 50 mA cm–2.
Aspen adsorption simulation
A two-bed vacuum swing adsorption (VSA) was employed to evaluate the performance of Ni-CNTs30/PC, Ni-CNTs180/PC, and Ni-CNTs30 in separating CO2 from a dilute CO2 stream consisting of 85% N2 and 15% CO2 under 298.15 K41,43. Aspen Adsorption was utilized for simulating and calculating the above process. During the simulation process, CFD4 was employed for partial differential equations. Detailed information regarding the mass balance model, momentum balance model, dynamic hypotheses, adsorption equilibrium models, bed parameters, VSA simulation methodology, and performance evaluation metrics for the VSA process can be found in Supplementary Note 4.
COMSOL simulation
A multiphysics model, implemented using COMSOL 6.1, was developed to elucidate the distribution and CO2 transport dynamics within the local environment during the low-concentration CO2RR process. This model was constructed based on an MEA electrolyzer, comprising anode and cathode catalyst layers, an electrolyte chamber, and a gas channel. The cathode catalysts were modeled using three configurations: 200 nm-thick PC NSs, 1-μm length of Ni-CNTs with anchored Ni–N3 active sites, and a heterarchical structure of Ni-CNTs/PC integrated with PC NSs and Ni-CNTs. The total ultramicropores volume parameters of these model catalysts were derived from Supplementary Table 5. The Butler–Volmer equation was used to correlate the relationship between current density and applied electrode potential, and Faraday’s law was applied to convert the current density to the generation/consumption rates of chemical species in the system, which were used as the source/sink terms in the convection-diffusion-reaction equation. Detailed information can be found in Supplementary Note 5.
Data availability
All data that support the findings of this study are present in the paper and the Supplementary Information. Source data are provided with this paper.
References
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Zheng, T. et al. Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386–1393 (2021).
Yang, H. et al. Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities. Nat. Commun. 11, 593 (2020).
Liu, Y. C. et al. Electrosynthesis of pure urea from pretreated flue gas in a proton-limited environment established in a porous solid-state electrolyte electrolyser. Nat. Nanotechnol. https://doi.org/10.1038/s41565-025-01914-3 (2025).
Wong, H. H. et al. Neighboring effect in single-atom catalysts for the electrochemical carbon dioxide reduction reaction. eScience 4, 100140 (2024).
Ren, X. et al. In-situ spectroscopic probe of the intrinsic structure feature of single-atom center in electrochemical CO/CO2 reduction to methanol. Nat. Commun. 14, 3401–3411 (2023).
Wang, H. et al. Precursor-mediated in-situ growth of hierarchical N-doped graphene nanofibers confining nickel single atoms for CO2 electroreduction. Proc. Natl Acad. Sci. USA 120, e2219043120 (2023).
Wang, M. et al. Hydrophobized electrospun nanofibers of hierarchical porosity as the integral gas diffusion electrode for full-pH CO2 electroreduction in membrane electrode assemblies. Energy Environ. Sci. 16, 4423–4431 (2023).
Ma, Z. et al. P-block aluminum single-atom catalyst for electrocatalytic CO2 reduction with high intrinsic activity. J. Am. Chem. Soc. 146, 29140–29149 (2024).
Xiao, T. et al. Proton antagonist membrane towards exclusive CO2 reduction. Nano Res. 16, 4589–4595 (2022).
Lees, E. W., Mowbray, B. A. W., Parlane, F. G. L. & Berlinguette, C. P. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 7, 55–64 (2021).
Ko, Y. et al. Extrinsic hydrophobicity-controlled silver nanoparticles as efficient and stable catalysts for CO2 electrolysis. Nat. Commun. 15, 3356–3367 (2024).
Wu, Z. et al. A general synthesis of single atom catalysts with controllable atomic and mesoporous structures. Nat. Synth. 1, 658–667 (2022).
Zhang, W. et al. Exclusive Co–N4 sites confined in two-dimensional metal-organic layers enabling highly selective CO2 electroreduction at industrial-level current. Angew. Chem. Int. Ed. 62, e202219241 (2023).
Xia, C. et al. General synthesis of single-atom catalysts with high metal loading using graphene quantum dots. Nat. Chem. 13, 887–894 (2021).
Wang, B. et al. Nanocurvature-induced field effects enable control over the activity of single-atom electrocatalysts. Nat. Commun. 15, 1719–1731 (2024).
Spurgeon, J. M. & Kumar, B. A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products. Energy Environ. Sci. 11, 1536–1551 (2018).
Sullivan, I. et al. Coupling electrochemical CO2 conversion with CO2 capture. Nat. Catal. 4, 952–958 (2021).
Keith, D. W., Holmes, G., St. Angelo, D. & Heidel, K. A process for capturing CO2 from the atmosphere. Joule 2, 1573–1594 (2018).
Papangelakis, P. et al. Improving the SO2 tolerance of CO2 reduction electrocatalysts using a polymer/catalyst/ionomer heterojunction design. Nat. Energy 9, 1011–1020 (2024).
Xiong, J. et al. Synergetic energy coupled thermal catalytic systems for CO2 reduction. eScience 5, 100306 (2025).
Islam, M. A. & Ulbricht, M. A new interpretation of gas viscosity for flow through micro‐capillaries and pores. Small 19, 2205827 (2023).
Tan, Y. C., Lee, K. B., Song, H. & Oh, J. Modulating local CO2 concentration as a general strategy for enhancing C−C coupling in CO2 electroreduction. Joule 4, 1104–1120 (2020).
Guzmán-Lastra, F., Löwen, H. & Mathijssen, A. J. T. M. Active carpets drive non-equilibrium diffusion and enhanced molecular fluxes. Nat. Commun. 12, 1906–1921 (2021).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Weng, L. C., Bell, A. T. & Weber, A. Z. Towards membrane-electrode assembly systems for CO2 reduction: a modeling study. Energy Environ. Sci. 12, 1950–1968 (2019).
Liu, Z., Yan, T., Shi, H., Pan, H. & Kang, P. Grafting amine-functionalized ligand layer on catalyst for electrochemical CO2 capture and utilization. Appl. Catal. B Environ. 343, 123456 (2024).
Kim, D. et al. Electrocatalytic reduction of low concentrations of CO2 gas in a membrane electrode assembly electrolyzer. ACS Energy Lett. 6, 3488–3495 (2021).
Liu, Y. Y., Huang, J. R., Zhu, H. L., Liao, P. Q. & Chen, X. M. Simultaneous capture of CO2 boosting its electroreduction in the micropores of a metal–organic framework. Angew. Chem. Int. Ed. 62, e202311265 (2023).
Jiao, L. et al. Single‐atom electrocatalysts from multivariate metal–organic frameworks for highly selective reduction of CO2 at low pressures. Angew. Chem. Int. Ed. 59, 20589–20595 (2020).
Chen, B. et al. Molecular enhancement of direct electrolysis of dilute CO2. ACS Energy Lett. 9, 911–918 (2024).
Propagating progress in transport processes. Nat. Chem. Eng. 1, 553 (2024).
Lei, T. et al. Global iron and steel plant CO2 emissions and carbon-neutrality pathways. Nature 622, 514–520 (2023).
Fang, W. et al. Organic–inorganic covalent–ionic molecules for elastic ceramic plastic. Nature 619, 293–299 (2023).
Ding, L. P. et al. Why carbon nanotubes grow. J. Am. Chem. Soc. 144, 5606–5613 (2022).
He, M. et al. Growth kinetics of single-walled carbon nanotubes with a (2n, n) chirality selection. Sci. Adv. 5, eaav9668 (2019).
Wang, X. & Ding, F. How a solid catalyst determines the chirality of the single-wall carbon nanotube grown on it. J. Phys. Chem. Lett. 10, 735–741 (2019).
Graeber, G. et al. Extreme water uptake of hygroscopic hydrogels through maximized swelling‐induced salt loading. Adv. Mater. 36, 2211783 (2024).
Cho, J. H. et al. Crystallographically vacancy‐induced MOF nanosheet as rational single‐atom support for accelerating CO2 electroreduction to CO. Carbon Energy 6, e510 (2024).
Guo, S. T. et al. Stabilizing undercoordinated Zn active sites through confinement in CeO2 nanotubes for efficient electrochemical CO2 reduction. Angew. Chem. Int. Ed. 63, e202314099 (2023).
Zhou, Y. et al. Self-assembled iron-containing mordenite monolith for carbon dioxide sieving. Science 373, 315–320 (2021).
Goel, C., Bhunia, H. & Bajpai, P. K. Prediction of binary gas adsorption of CO2/N2 and thermodynamic studies on nitrogen enriched nanostructured carbon adsorbents. J. Chem. Eng. Data 62, 214–225 (2016).
Durán, I., Rubiera, F. & Pevida, C. Vacuum swing CO2 adsorption cycles in waste-to-energy plants. Chem. Eng. J. 382, 122841 (2020).
Kumagai, H. et al. Electrocatalytic reduction of low concentration CO2. Chem. Sci. 10, 1597–1606 (2019).
Sun, Q., Zhao, Y., Ren, W. & Zhao, C. Electroreduction of low concentration CO2 at atomically dispersed Ni-N-C catalysts with nanoconfined ionic liquids. Appl. Catal. B Environ. 304, 120963 (2022).
He, D. Y., Ma, X. H., Zhou, H., Zhang, Y. & Wu, Y. Continuous conversion of flue gas into syngas by a bipolar membrane-integrated single-cell cyclic system. Joule 9, 101806 (2025).
Zhang, W. et al. Electrolyte design using “porous water” for high-purity carbon monoxide electrosynthesis from dilute carbon dioxide. Angew. Chem. Int. Ed. 64, e202424104 (2025).
Hu, Y. et al. Carbon-based materials for low concentration CO2 capture and electrocatalytic reduction. Carbon 230, 119574 (2024).
Zhao, Z. H. et al. Efficient capture and electroreduction of dilute CO2 into highly pure and concentrated formic acid aqueous solution. J. Am. Chem. Soc. 146, 14349–14356 (2024).
Chen, J. C., Liu, Z. S. & Huang, J. S. Emission characteristics of coal combustion in different O2/N2, O2/CO2 and O2/RFG atmosphere. J. Hazard. Mater. 142, 266–271 (2007).
Zhang, F. & Co, A. C. Direct evidence of local pH change and the role of alkali cation during CO2 electroreduction in aqueous media. Angew. Chem. Int. Ed. 59, 1674–1681 (2019).
Hansen, K. U., Cherniack, L. H. & Jiao, F. Voltage loss diagnosis in CO2 electrolyzers using five-electrode technique. ACS Energy Lett. 7, 4504–4511 (2022).
Heinzmann, M., Weber, A. & Ivers-Tiffée, E. Advanced impedance study of polymer electrolyte membrane single cells by means of distribution of relaxation times. J. Power Sources 402, 24–33 (2018).
Shi, R. et al. Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces. Nat. Commun. 11, 3028–3038 (2020).
Lin, Y. et al. Tunable CO2 electroreduction to ethanol and ethylene with controllable interfacial wettability. Nat. Commun. 14, 3575–3587 (2023).
Liu, J. & Ciucci, F. The Gaussian process distribution of relaxation times: a machine learning tool for the analysis and prediction of electrochemical impedance spectroscopy data. Electrochim. Acta 331, 135316 (2020).
Acknowledgements
We acknowledge the support from the National Natural Science Foundation of China (No. 22375103 and 22105107), the Natural Science Foundation of Tianjin (No. 24JCZDJC00540), Beijing National Laboratory for Molecular Sciences (BNLMS202410), the “111 Center” (B25010) and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
H.W. and J.C. conceived the original idea. J.C. and H.W. designed all the experiments. J.C., T.L., X.L., R.L., Y.W., W.C., W.L., and L.C. carried out the experiments. J.C., T.L., S.C., Y.L., W.W., Z.C., Z.Z., W.S., S.Z., and H.W. analyzed the experimental data. J.C. performed the COMSOL simulation with the assistance of J.L. J.C. and H.W. co-wrote the paper. All authors were involved in the discussion of the experimental results and preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dunfeng Gao, Wenlei Zhu, Pei-Qin Liao, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, J., Lu, T., Liao, X. et al. Nutrient diffusion-inspired catalysts with self-reinforced concentration gradient for sustainable electroreduction of dilute CO2. Nat Commun 16, 7598 (2025). https://doi.org/10.1038/s41467-025-62240-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62240-9
