
Abstract
Accurate prediction of the functional impact of missense variants is important for disease gene discovery, clinical genetic diagnostics, therapeutic strategies, and protein engineering. Previous efforts have focused on predicting a binary pathogenicity classification, but the functional impact of missense variants is multi-dimensional. Pathogenic missense variants in the same gene may act through different modes of action (i.e., gain/loss-of-function) by affecting different aspects of protein function. They may result in distinct clinical conditions that require different treatments. We develop a new method, PreMode, to perform gene-specific mode-of-action predictions. PreMode models effects of coding sequence variants using SE(3)-equivariant graph neural networks on protein sequences and structures. Using the largest-to-date set of missense variants with known modes of action, we show that PreMode reaches state-of-the-art performance in multiple types of mode-of-action predictions by efficient transfer-learning. Additionally, PreMode’s prediction of G/LoF variants in a kinase is consistent with inactive-active conformation transition energy changes. Finally, we show that PreMode enables efficient study design of deep mutational scans and can be expanded to fitness optimization of non-human proteins with active learning.
Similar content being viewed by others

Introduction
Accurate and comprehensive prediction of variant effects has been a long-standing fundamental problem in genetics and protein biology. Single amino acid (missense) variants are the most common type of coding variants that contribute to many human diseases and conditions1,2,3,4,5. The functional impact of most missense variants remains uncertain. At the molecular level, missense variants in only ~80 human genes have been screened in saturated mutagenesis experiments (as of Nov. 2024)6,7. At the genetic level, only about 2% of clinically observed missense variants are classified as pathogenic or benign, while the majority remain of uncertain clinical significance8. Such limitations make it challenging for accurate clinical diagnosis and timely clinical interventions. Furthermore, understanding the functional impact of mutations is important to protein engineering, especially in directed evolution methods, where proteins are iteratively mutated to optimize function or fitness9. Such efforts were often limited by the high cost and explosion of sequence space. It remains a challenge to understand and predict the fitness landscape of mutants to reduce the search space and improve the efficiency of engineering10,11.
In the past decade, many computational methods have been developed12,13,14,15,16,17,18,19,20,21,22,23 to predict variant effects in a binary manner aiming at distinguishing pathogenic and benign variants. These methods showed that pathogenicity can be predicted by manually encoded or self-learned features based on sequence conservation, protein structures, and population allele frequency. Moreover, recently developed methods based on protein language models, leveraging Transformer architectures and self-supervised training on billions of protein sequences from UniProt24, have demonstrated their capability to serve as versatile predictors of various protein features25,26,27. The embeddings from these models can offer zero-shot predictive potential for variant pathogenicity28,29. While those methods are helpful in genetic analyses, pathogenicity does not capture the complexity of functional and genetic effects of variants. For example, gain of function variants in SCN2A lead to infantile epileptic encephalopathy30,31 while loss of function variants in the same gene lead to autism or intellectual disability30,31. Such limitation reduced the utility of the methods in genetic analysis and clinical applications.
We use “mode-of-action” as a generic term to encapsulate the multi-dimensional molecular and genetic mechanisms through which pathogenic variants impact protein functionality and increase the risk of diseases, respectively. More specifically, at molecular level, pathogenic variants can change the biochemical properties of a protein in different ways. For example, decreasing/maintaining protein stability32,33, enzymatic activity33,34, regulatory functions, and interaction35,36. At genetics level, variants are often categorized into two major types, gain or loss of function (G/LoF). GoF variants encompass alterations that perturb the protein from its normal functions via increased or novel activities37,38. GoF variants are often found to be driver mutations in oncogenes39. LoF variants damage protein function via decreased activities, which are often found in tumor suppressors in cancer40 and other genetic diseases41. Gain and loss of function variants usually result in markedly different clinical phenotypes37,42,43,44,45, necessitating entirely distinct therapeutic approaches30,36,37,46.
While numerous methods have demonstrated the potential to predict pathogenicity on a genome-wide scale, the effort in G/LoF prediction has been limited. Stein et al. attempted to predict genome-wide G/LoF variants via assembly of human curated features47. However, we note that mode-of-action centers around how a variant disrupts the normal function of a protein. Given the inherent diversity of protein functions, attempting to define a universally applicable predictive task for all G/LoF variants across all proteins could lead to conceptual ambiguity. Therefore, we propose that such predictive tasks should be defined within the context of individual proteins or protein families that share similar functions. The main challenge is the limited availability of data for most genes and protein families.
We developed a new method, PreMode (Pretrained Model for Predicting Mode-of-action), to address these challenges with deep learning models through genome-wide pretrain and protein-specific transfer learning. PreMode is designed to capture the variant impact on protein function with regard to its structural properties and evolutional information. We built PreMode with SE(3)-equivariant graph attention transformers, utilizing protein language model embeddings26 and protein structures25 as inputs. We curated the largest-to-date labeled missense variants with mode-of-action annotations from clinical databases, genetic inference, and experimental assays. We applied PreMode to mode-of-action predictions of 17 genes. PreMode reached state-of-the-art performance at mode-of-action predictions compared to existing models. We further demonstrate PreMode’s practical utility in both improving data analysis in deep mutational scan (DMS) experiments and assisting protein engineering by significantly reducing the size of mutants for screening via active learning.
Results
Overview
We proposed a framework for predicting the mode-of-action at the molecular level and genetic level. Molecularly, the effect of a missense variant is about the change in biochemical properties of a protein, such as enzyme activity, stability, and the regulatory processes upon protein-protein interactions (Fig. 1a). These changes can be measured by DMS experiments (Fig. 1a). Genetically, the overall outcome of molecular effects results in different types of missense variants. One common categorization is “loss of function” (LoF) and “gain of function” (GoF) variants (Fig. 1a). To conceptualize this framework to variant effect prediction models, we introduced two parameters: “distance from wild type” (denoted as ‘\(r\)’) and the “direction of change” (notated as ‘\(\theta\)’). The distance parameter distinguishes between pathogenic and benign variants (Fig. 1a) and is shared across all genes. The direction parameter takes on different meanings both molecularly and genetically within various genes. Therefore, we proposed that a mode-of-action predictor would make separate predictions of \(r\) and \(\theta\) utilizing different datasets. It would first learn \(r\) prediction using labeled pathogenic and benign variants for all genes, just like conventional variant effect predictors, then learn \(\theta\) prediction using protein or protein family specific datasets via transfer learning (Fig. 1b).

a Mode-of-action definition at molecular level and genetic levels. b A proposed mode-of-action predictor framework. Blue arrows show the data flow at pretrain stage for pathogenicity prediction, green arrows show transfer-learning stage for mode-of-action prediction.
Curation and characterization of mode-of-action labeled missense variants
We curated the largest-to-date mode-of-action labeled missense variants datasets annotated at both molecular and genetic levels, including 41,081 missense variants in eight genes with multi-dimensional measurements of different biochemical properties by DMS experiments curated from MAVEDB6, 2043 gain- and 7889 loss-of-function missense variants in ~1300 genes. The gain and loss of function-labeled variants were collected from literature searches37,45,48,49, cancer hotspots50,51,52, and published databases53 (“Methods: Training and testing datasets”).
We first investigated global properties of GoF and LoF variants using the curated data set. While both GoF and LoF variants are enriched in high AlphaFold2 prediction confidence region (pLDDT ≥ 90) and protein core (rsa < 25%), GoF variants are relatively more likely to be located in regions with lower pLDDT (50 < pLDDT < 90) than LoF variants, and are relatively more likely to be on protein surfaces (rsa ≥ 25%) (Fig. 2a, Supplementary Fig. 1). In contrast, LoF variants have an overall bigger impact on protein folding energy than GoF variants (Fig. 2a), although both types of variants confer a greater folding energy change to protein folding than benign variants (Fig. 2a). As expected, both LoF and GoF variants are more likely to be in conserved regions than non-conserved regions, as shown in distribution of conservation represented by entropy of amino acid frequencies across species in multiple sequence alignments (MSA) (Fig. 2a), while benign variants are mostly located in non-conserved regions (Fig. 2a). Additionally, GoF variants in general are more likely to be located in disordered regions without specific secondary structures defined by DSSP54 than LoF variants (Fig. 2b). However, we note this pattern is different across protein families. For example, in Na+/Ca2+ channel genes, GoF variants are more enriched in alpha helixes that are critical for ion transport and selectivity domains than LoF variants (Fig. 2b), but we didn’t observe that trend in non-Na+/Ca2+ channel genes. Finally, the number of GoF and LoF variants are not evenly distributed across genes, with only a few of the genes having more than 15 GoF and 12 LoF variants (Fig. 2c). Overall, those results showed that protein structure, energy, and evolutionary features could help predict G/LoF variants while underscoring the necessity for the development of protein- and protein family-specific predictive models using limited data.

a Genome wide comparison of gain- and loss-of-function variant sites’ pLDDT (AlphaFold2 prediction confidence), protein folding energy change, relevant solvent accessibility, and conservation in 2043 GoF, 7889 LoF variants across 1300 genes and 13704 benign variants from same set of genes, p values were calculated by two-sided Mann-Whitney test. b Protein family wise comparison of gain- and loss-of-function variants enriched secondary structures, statistical tests were performed with two-sided binomial test followed by Benjamin-Hochberg procedure to control the FDR, “*” indicates statistically significant. c Gene wise comparison of gain- and loss-of-function missense variants numbers. Red colored and labeled genes have more than 15 GoF and 12 LoF variants.
A deep learning model for mode-of-action predictions
We developed PreMode, a model pre-trained on pathogenicity prediction task and optimized for transfer learning to mode-of-action prediction tasks. PreMode takes input features derived from amino acid biochemical properties, protein contexts, and cross-species conservation. PreMode models protein 3D context structure with SE(3)-equivariant graph neural networks (Fig. 3). PreMode was designed to not only capture the relative importance between residues by taking both backbone torsion angles and side chain directions into consideration, but also maintaining awareness of geometric equivariance so that rotation of the atom coordinates does not affect the predictions. PreMode’s SE(3)-equivariant learning ability was achieved by using a graph representation of protein 3D structures, where each residue was represented as nodes with features that explicitly represent local biochemical properties and evolutionary conservation including secondary structures54,55, pLDDT25, amino acid frequencies in MSA56, and relative coordinates of all atoms in sidechain with respect to alpha carbons (Methods: Input features). We also included protein sequence language model (ESM2) embeddings26 into node embeddings, which implicitly capture similar structural and evolution information. Such implicit representation could serve as a compensation of possible missing information limited by secondary structure annotation or MSA generation algorithms. For each edge connecting two residues, the features include Euclidean vector of two corresponding beta-carbons (for glycine we use alpha-carbon instead) to encapsulate static contacts. Additionally, we incorporate co-evolutional strength calculated from MSA and contact strength predictions from MSA transformer57 to represent potential dynamic contacts58 or inter-homopolymer contacts that are missed in a static structure (Methods: Input features). PreMode applies SE(3)-equivariant mechanisms on edge features and node features first through a star-graph that connects variant site with all other residues to capture the direct impacts of amino acid alterations on other residues, then through the k-nearest neighbor (KNN)-graph that connects each residue with its closest neighbor residues to capture the second order impacts, finally through a star-graph module to aggregate the impacts (Supplementary Fig. 2).

PreMode takes amino acid changes, protein language model embeddings, alphafold2 predicted structures, multiple sequence alignments as inputs and outputs two parameters. PreMode first predicts pathogenicity (\(r\)) for all genes during pretrain, next predicts the mode-of-action parameters (\(\theta\)) via transfer learning.
We pretrained PreMode using labeled pathogenicity data to let the model learn general representation of the variant effects. We collected 83,844 benign variants from ClinVar59 and PrimateAI18 in 13,748 genes, and 64,480 pathogenic variants from ClinVar59 and HGMD41 in 3703 genes (“Methods: Training and testing datasets”). We randomly selected 5% of the variants as a validation dataset and trained 20 epochs on the rest of the training data until validation loss stopped dropping (Supplementary Fig. 3a). Although predicting pathogenicity is not an objective of PreMode, we can still use pathogenicity prediction performance as a sanity check for the pretraining process and to investigate the contributions of various components of the model. As PreMode was pre-trained on human-curated ClinVar data, using variants from the same resource as testing data can result in inflated performances. Instead, we used independent testing data for which the pathogenicity label was entirely based on statistical evidence, that is, 533 pathogenic missense variants in cancer hotspots from cBioportal50,52,60, and same number of benign variants in the same genes randomly selected from common variants in primates18.
PreMode reached similar levels of performance as existing methods including AlphaMissense21 and gMVP20 on the testing dataset with AUROC (area under recall receiver operating characteristic curve) of 0.932 (Supplementary Fig. 3b). We performed ablation analysis to assess the contribution of language model embeddings, structural information, and MSA information to the prediction. Replacing ESM2 embeddings with one-hot encodings of amino acids resulted in a slight drop of AUROC to 0.922. Similarly, removing the MSA will drop the AUROC to 0.91, while removing the genes in training data that are paralogous to the testing (from Ensembl61 2.50.0) reduced the AUROC to 0.873 (Supplementary Fig. 3c). Removing the structure module will drop the performance slightly to 0.913 (Supplementary Fig. 3c). This showed that ESM2 embeddings, MSA module and SE(3)-equivariant module on AlphaFold2 predicted protein structures together provided complementary, non-redundant information for pathogenicity prediction, with PreMode learning joint representations from these components during pretrain.
PreMode reaches state-of-the-art in molecular mode-of-action predictions and facilitates interpretation of deep mutational scan experiments
We first investigated the utility of PreMode in predicting modes of action at the molecular level. We obtained DMS data on eight genes (PTEN, SNCA, CCR5, CXCR4, NUDT15, CYP2C9, GCK, ASPA) from MAVEDB6 with multiple assays of different biochemical properties. These assays broadly target two aspects of protein function, the stability and enzymatic activity. These two types of functional readout are moderately correlated as reduced protein stability often directly or indirectly affects function (Supplementary Fig. 4).
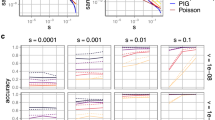
We split the data of each gene into 80% of training and 20% of testing data five times under different seeds and ran PreMode on each of them via transfer learning. While most other methods for pathogenicity prediction does not provide model weights for us to do transfer learning, we compared PreMode against four models (Augmented ESM1b, Augmented EVmutation, Augmented Unirep, Augmented EVE, “Methods: Curation of prediction scores from other methods”) with top transfer learning performances described in Hsu et al.62, and a baseline model utilizing ESM2 embeddings and a single layer perceptron (SLP, “Methods: Baseline Methods and ablation analysis”) as approximated transfer learning with ESM2. PreMode outperformed all methods with higher Spearman correlation (Fig. 4a) on all the assays of 8 genes. Overall, after transfer learning, PreMode is able to predict both the multi-dimensional protein stability and functional fitness with a Spearman correlation of 0.6 with experimental results, better than all other methods (Fig. 4b). The conclusion holds in larger benchmarking datasets like ProteinGym (as of Nov. 2024)7 (Supplementary Fig. 5), where PreMode is better than other methods in human proteins. Furthermore, we investigated the multi-dimensional transfer learning ability of PreMode under smaller sample sizes, where we randomly subsampled the training data and compared the performances in same testing dataset (Methods: Subsample of datasets). We found that PreMode is better than all other methods in transfer learning with ≥20% training data, while all methods had similar performance under smaller data sizes (Fig. 4c, Supplementary Fig. 6). Overall, PreMode is able to accurately predict variant effects of all missense variants with around 40% (~2000) of variants measured inside one gene, after which increasing the number of data points will have minimal improvement on the performance in the testing dataset (Fig. 4c).

a Spearman correlations of PreMode in multiplexed deep mutational scan experiments of 8 genes compared to other methods, mean and standard error were calculated from five random training/testing splits. b Comparison of overall Spearman correlations of PreMode compared to other methods, mean and standard error were calculated by weighted average of mean and standard error in 8 experiments based on dataset sizes. c Comparison of overall Spearman correlations of PreMode compared to other methods under sub-sampled data, mean and standard error were calculated by weighted average of mean and standard error in 8 experiments based on dataset sizes.
We further investigated the utility of PreMode to improve the analysis of experimental readouts in two applications. First, we hypothesized that PreMode scores could indicate abnormal measurements in each experiment by transfer learning as it had implicitly modeled the fitness of variants in all proteins during pretraining. We used the stability DMS experiment32 of PTEN as an example and trained PreMode on one of the eight biological replicates. We then compared the differences between PreMode’s predictions and the experimental readouts. We showed that this difference value is highly correlated to the difference between the readout of the single biological replicate and average readouts in all experiments (Supplementary Fig. 7a). The experimental readouts with large deviation from PreMode’s predictions are more likely to be abnormal measurements (Supplementary Fig. 7b). Next, we hypothesized that models trained on stability in a subset of genes are generalizable to other genes. We applied PreMode to the largest stability measurement experiments in MAVEDB across >30 genes. We trained PreMode on 80% of the data and tested on the other 20% of variants in completely different genes from training. PreMode outperformed all other methods (Supplementary Fig. 8).
PreMode is state-of-the-art in genetic mode-of-action predictions
We grouped the gain/loss of function variants dataset by genes and only kept those with ≥15 G/LoF variants (Fig. 2c, “Methods: Training and testing datasets”). We performed transfer learning on the selected genes using the pretrained model parameters as initial weights (Methods: Model training and testing). We note there are two reasons to perform transfer learning in individual genes rather than training a general model across all genes. First, G/LoF mechanisms are intrinsically different across genes, as they have different functions. A general model across all genes will have a poor generalizability and will perform random or nearly reversed predictions for unseen genes (Supplementary Fig. 9a). Second, the number of G/LoF variants are extremely unbalanced across genes. A deep neural network model with transfer-learning across genes will potentially reach a local minimum where gene-properties dominate its predictions that are better at predicting likely G/LoF genes but do not distinguish G/LoF variants in the same gene (Supplementary Fig. 9b).
For each gene, we randomly split the gain and loss of function variants into training and testing. The total amount of data for training and testing in each gene is shown in Supplementary Data 1. There were nine genes (ABCC8, BRAF, CACNA1A, FGFR2, KCNJ11, RET, SCN2A, SCN5A, TP53; Fig. 2c) in total. First, we compared PreMode against several baseline methods trained and tested on the same training/testing datasets. Overall, PreMode performed better in all the genes than baseline methods. It reached average AUC of 0.8–0.9 in genes RET, KCNJ11, CACNA1A and BRAF, average AUCs of 0.7–0.8 in SCN5A, SCN2A, TP53, and ABCC8 (Fig. 5a). PreMode is better than low-capacity models such as random forest using manually curated biochemical features and conservation information as input (Fig. 5a, “Methods: Baseline Methods and ablation analysis”). Pretrained PreMode is better than non-pretrained model for all genes except FGFR2, where all three models have average AUC lower than 0.6 (Fig. 5a). We compared PreMode’s performance when fine-tuned on G/LoF variants across genes and observed a performance drop in all genes compared to the per-gene fine-tuning strategy (Fig. 5a), with the most significant decreases seen in genes RET and ABCC8 (Fig. 5a). Next, we compared our model to LoGoFunc47, a method trained on G/LoF variants across genes. As the training and testing split information were not available for LoGoFunc, we re-split the training and testing data and completely removed the data curated from LoGoFunc in testing datasets for fair comparison. PreMode has similar performance as LoGoFunc in CACNA1A but better in all other genes (Fig. 5b). The trend still holds when we change the training/testing partition scheme by stratifying variants by residue position or even remove the data of their paralogous proteins from pretrain (Supplementary Fig. 10). Furthermore, we compared our model to funNCion37, a method trained only on voltage-gated sodium and calcium channels (encoded by SCNxA and CACNA1x family genes) with manually curated sequence and structural features. We found that PreMode is better than funNCion in all three genes SCN2A, SCN5A, CACNA1A, and all ion channel genes when using the same training testing data split (Fig. 5c). Overall, this showed PreMode reached state-of-the-art performances at gene specific genetic mode-of-action predictions.

a Performances of PreMode and baseline methods on gain- and loss-of-function predictions in 9 genes, mean and standard error were calculated from five random training/testing splits. b Performances of PreMode and LoGoFunc in 9 genes, training/testing data was split by presence in LoGoFunc data or not, mean and standard error were calculated from five random splits. c Comparison of PreMode and funNCion in three ion channel genes, training/testing data was split randomly or same as in their paper, mean and standard error were calculated from five random splits. d Ablation analysis of PreMode on gain- and loss-of-function predictions in 9 genes, for each model, mean and standard error were calculated by weighted average of mean and standard error in 9 genes based on dataset sizes. e Few-shot transfer learning of PreMode and random forest method on subsampled training data, mean and standard error were calculated by weighted average of mean and standard error in 9 genes based on dataset sizes. f PreMode performances when trained with G/LoF variants from the same gene (marked as “Gene Only”) or G/LoF data from same domain across itself and other genes (marked as “Protein Family”) or G/LoF data from same domain in other genes (marked as “Protein Family, exclude gene”) and tested on the same testing dataset, mean and standard error were calculated by weighted average of five random splits.
We next did ablation analysis on PreMode to identify the important features for Gain/Loss of function predictions. Removing ESM, homologous proteins in pretrain or protein structure information decreased PreMode’s overall performances in 9 genes (Fig. 5d, “Methods: Baseline Methods and ablation analysis”). Removing structural input decreased the performance in all genes except for FGFR2 (Supplementary Fig. 11a). We noticed that both the LoF variants and GoF variants in that gene were located in slightly lower pLDDT region (Supplementary Fig. 11c). Adding post-translational-modification (PTM) information (predicted by PhosphoSitePlus63) into the model input can slightly improve the predictions in TP53, SCN5A, and FGFR2. (Supplementary Fig. 11b). In these genes, the LoF variants are closer to the PTM sites than GoF variants while the trend is not significant in the other genes (Supplementary Fig. 11d). Overall, PreMode reached highest performance in the weighted sum of AUC across genes, followed by ablation models and baseline methods (Fig. 5d).
To be effective in genetic analysis, a machine learning method should be able to perform transfer learning with limited amount of data, as in most of the genes, there are fewer than 10 known G/LoF variants (Fig. 2c). We therefore examined PreMode’s performance with subsampled training data and compared with baseline methods like random forest. We found PreMode’s overall performances in 9 genes were better than random forest even if we down sample the training data to 10% of whole dataset (Fig. 5e). Furthermore, random forest’s overall performance stopped increasing after we increase the training data size to 40% of whole dataset, while PreMode’s performance is not saturated (Fig. 5e).
Next, we hypothesized that the gain and loss of function mechanisms are similar in the same domain across genes, and we can use this to further improve the GoF/LoF predictions in each gene. We split the data in each gene by domains and only selected the domains with ≥15 G/LoF variants for evaluation, including ion transport domains, tyrosine protein kinase domains and growth factor receptor domains. We performed PreMode transfer learning within the domain using either gene-specific data or data across genes while tested on the same data within one gene. We observed increased performance in all of the domains when using data across genes rather than using the gene alone (Fig. 5f). Furthermore, we performed PreMode transfer learning using data from other genes with the same domain and tested in unseen gene. We observed similar performances using this strategy compared to gene-specific transfer learning (Fig. 5f).
Overall, those result suggested that PreMode is the state-of-art method in G/LoF prediction even with limited data. For genes with small sizes of labeled G/LoF variants data, PreMode could utilize the information from G/LoF variants data in same protein family to further increase performances.
PreMode predicted mode-of-action landscapes in individual proteins through in silico saturated mutagenesis
We applied PreMode to infer the mode-of-action of all possible variants in BRAF, TP53, PTEN, RET, and KCNJ11. We averaged the prediction scores of all five models that trained on different training/testing splits and applied to all the other variants in corresponding gene.
In BRAF, PreMode identified two regions enriched with GoF variants. One is on a N-terminal phorbol-ester/DAG-type zinc-finger domain and the other is on the kinase domain which includes the well-known V600 GoF position64,65 (Fig. 6a). The variants on the two domains act through different gain/loss of function mechanisms. The phorbol-ester/DAG-type zinc-finger domain auto-inhibits BRAF activity through binding with 14-3-366 while also cooperating with Ras-binding domain to bind with Ras and activate BRAF activity67. To interpret PreMode’s predictions, we obtained B-Raf/14-3-3 and B-Raf/K-Ras binding structures from PDB (7MFD) and AlphaFold2 (Colabfold68 implementation) predictions, respectively (Supplementary Fig. 12a). We then calculated the energy change on both structures upon mutations using FoldX69. We found that the GoF variants predicted by PreMode only destabilize B-Raf binding to 14-3-3 and maintain the binding stability to K-Ras at similar level as benign variants (Fig. 6b). The ddG landscape suggested that the GoF variants in this region mostly act by abolishing its inhibitory regulation70. We further investigated the G/LoF variants predicted by PreMode in the kinase domain. The kinase domain has both active (PDB: 4MNE) and inactive (PDB: 4EHE) conformations with large and small enzyme pocket sizes, respectively71 (Supplementary Fig. 12b). The FoldX ddG results were consistent with previous findings that GoF variants V600E/D can destabilize the inactive state and stabilize the active state72, while the predicted LoF variants destabilize both conformations (Fig. 6c). Similarly, we found that predicted LoF variants can destabilize the complex of BRAF-MEK1 while GoF variants maintains the stability of BRAF-MEK1 complex at similar level of benign variants (Fig. 6d, Supplementary Fig. 12c).

a PreMode predictions visualized with BRAF protein domains, x-axis are annotations and amino acid changes, y-axis are amino acid positions. The first few columns indicate Alphafold2 pLDDT, relevant solvent accessibility, secondary structures, protein domains, respectively. The left panel showed PreMode’s pathogenicity predictions and labels used in training, pink indicates predicted pathogenic, light blue indicates predicted benign, red indicates pathogenic, and blue indicates benign in training data, white indicates reference amino acid. The right panel showed PreMode’s G/LoF predictions and labels used in training, pink indicates predicted GoF, light blue indicates predicted LoF, white indicates predicted benign. Red indicates GoF and blue indicates LoF in training data. b Folding energy change upon mutations of Braf phorbol-ester/DAG-type zinc finger domain on the inactive state (Braf-14-3-3 binding, y-axis) versus active state (Braf-Kras binding, x-axis), colored by PreMode predictions, a few known cancer driver mutations were labeled. c Folding energy change upon mutations of Braf kinase domain on the inactive state (y-axis) versus active state (x-axis), colored by PreMode predictions, a few known cancer driver mutations were labeled. d. Folding energy change upon mutations of BRAF-MEK1 complex. X-axis showed the ddG on BRAF-MEK1 complex, y-axis showed the ddG on BRAF active conformation alone (left panel, PDB: 4MNE), colored by PreMode predictions.
TP53 is a tumor suppressor gene, and most of the pathogenic variants act through LoF. However, there are a few regions enriched with GoF variants identified by PreMode, all on the DNA binding domain of the p53 protein (Supplementary Fig. 13a). Among those regions, sites 291 and 292 are essential post-translational modification sites for p53 ubiquitination and subsequent degradation73. A previous study showed the variants at sites 121 and 290-292 increased the ability to induce apoptosis in cultured cells74 (Supplementary Fig. 13a).
PreMode also identified several GoF enriched regions in RET and KCNJ11. In RET, PreMode identified GoF enriched regions both in the kinase domain and regulatory signaling domain near the extracellular binding sites (Supplementary Fig. 13b). In KCNJ11, the regions are located at transmembrane domains and cytoplasmic domains (48–54, 158–182, 200–210, 225–231, 320–331, PDB: 6C3O) (Supplementary Fig. 13c). The region spanning positions 160-179 forms the core part of potassium channel in the tetramer (Supplementary Fig. 14), especially residues L164 and F168 that form the inner helix gate75, and the regions spanning position 48–54, 179–182, 328–331 forms the ATP/ADP binding pocket (Supplementary Fig. 14).
PTEN is a tumor suppressor gene and an essential gene in fetal development. Loss of function in PTEN can lead to multiple syndromes. As shown by DMS experiments34,76, some variants in PTEN can have different impacts on stability and enzyme activities. We applied PreMode on 80% of the data from both functional assays to predict the effect of all possible variants in PTEN. Although most of variants decrease both the protein stability and enzyme function, PreMode identified variants that only disrupt stability but not enzyme function (Supplementary Fig. 15a, blue), and such variants are located all over the protein. Similarly, PreMode identified variants that only disrupt enzyme function but not stability, all located in the phosphatase domain (Supplementary Fig. 15a, red). These variants may have dominant negative effects. In fact, three known dominant negative variants in PTEN (C124S, G129E, and R130G)76,77 were successfully predicted by PreMode to maintain stability while causing loss of enzyme function, among which G129E was not in the training data. PreMode identified 4 regions enriched for such variants (Supplementary Fig. 15a, pink). Further analysis showed those regions are spatially close to each other and form the enzyme pocket around the phosphatase site (Supplementary Fig. 15b). Notably, PreMode can identify similar dominant-negative variants enriched in a region with only 20% (398 points) of the dataset (Supplementary Fig. 16).
PreMode trained with active learning allows efficient few-shot transfer
DMS and directed evolution-based protein design experiments often incur expensive time costs upon scaling up. We asked if PreMode can be used to optimize experimental design by active learning78. In an active learning framework, PreMode was iteratively trained on a set of experimental data and prioritized the rest of the variants with largest predicted variance to be measured in the next rounds of experiments (Methods: Active Learning experiments). We applied this framework to the protein design of green fluorescence protein (GFP) on fluorescence strength. PreMode was able to predict the fitness landscape of GFP with Spearman correlation above state-of-the-art performance (0.69)79,80,81 to the experimental data using only 40% of the training data by adaptive learning, which is much more efficient than randomly subsampling data (Supplementary Fig. 17).
Discussion
Previous methods for predicting the effect of missense variant have been focused on pathogenicity, which is a binary label. However, different pathogenic variants in the same gene can have different modes of action, i.e., change the protein function in different or sometimes opposite ways. It is challenging to predict mode-of-action because molecular and functional impact is gene specific that largely variable across different genes depending on the functions of encoded proteins36,82. And there is very limited amount of data in individual genes. In this study, we addressed this issue with a new deep learning method, PreMode, that enables pretraining on large pathogenic datasets across genes and then transfer learning in specific genes that have small number of variants with known mode-of-action.
To generate the training data for PreMode as well as understand the biochemical and evolutionary differences of gain/loss-of-function variants, we curated and characterized the largest-to-date missense variants that are known to act through different modes. Although both GoF and LoF variants are more enriched in high pLDDT region (pLDDT ≥ 90) and protein core (rsa < 25%) than the benign variants, GoF variants are relatively more enriched in low pLDDT regions (50 < pLDDT < 90) and surfaces (rsa ≥ 25%) in Alphafold2 predicted structures than LoF variants. Those regions are likely to be intrinsically disordered83, which implies conformational heterogeneity and dynamics that could be associated with protein binding and regulatory activities. On the contrary, loss of function variants tend to increase protein folding energy, suggesting that some of these variants destabilize protein structures, while gain-of-function variants tend to preserve protein folding integrity36. Overall, these findings showed how variant effects are associated with the protein structures and functions qualitatively. Gain-of-function variants often act through preserving overall protein structure while operating through diverse mechanisms by targeting specific structural domains, whereas LoF variants often destabilize protein structures and tend to be distributed across structured regions. However, this overall trend does not apply to some protein families, for examples GoF variants are more enriched in alpha helixes that forms the transmembrane domain in the ion channel proteins. This highlights the need for protein-specific rather than genome-wide Mode-of-Action predictions. Additionally, available data are heavily uneven across genes, making deep learning algorithms easily stuck at local minimum and gene properties to distinguish genes that tend to act through G/LoF mechanisms while hard to distinguish G/LoF variants within same gene.
To predict protein-specific mode-of-action from the sequence and structural context, we designed a deep learning method, PreMode. PreMode encodes the variant effects with implicit representation of biochemical properties and evolutionary information from protein language model embeddings, and explicit representations of protein structures via SE(3)-equivariant neural networks. PreMode performs gene-specific mode-of-action predictions through a genome-wide pretrain stage and a gene-specific transfer learning stage based on the hypothesis that the sequence and structural context that are informative for pathogenicity should also be informative for mode-of-action predictions. We selected 17 genes with sufficient deep mutation scan data or labeled gain/loss-of-function missense variants for model development and evaluation. At the molecular level, PreMode can simultaneously predict the multi-dimensional biochemical impact of single missense variants, which can reveal potential dominant negative variants that reduce protein function but maintain stability. We showed that PreMode is efficient at transfer learning and that it can capture the fitness landscape of all possible variants within one protein using around 40% of mutagenesis data. At genetic level, PreMode can efficiently utilize a small amount labeled data (a few dozens) to accurately distinguish G/LoF variants with an AUC of around 0.80 in most proteins. Additionally, we showed PreMode’s utility in DMS experiments and protein engineering. First, PreMode can improve efficiency of DMS experiments by detecting noisy data points in single measurements. Second, PreMode can be applied to unmeasured genes by fine tuning on the stability deep mutational experiments. Finally, PreMode can facilitate mutagenesis-based protein directed evolution through adaptive learning by efficiently lowering library sizes.
PreMode currently predicts a binary label as G/LoF. This is a limitation as it does not capture the complexity of protein functional changes. For instance, ion channel proteins undergo complex conformation changes and regulation to perform normal physiological functions30. Large-scale functional studies of these genes may provide additional data that enable training of improved models. Furthermore, gain/loss of function could be further divided into amorph, hypomorph, hypermorph, antimorph, and neomorph according to Muller’s morphs84. Accordingly, additional labeled data may facilitate the training of more accurate and comprehensive models.
PreMode can be potentially improved by considering protein dynamics, as ablation experiments showed it has lower performance for genes where both G/LoF variants are located in regions with relatively low pLDDT values. Currently, PreMode includes features representing a static protein structure, which is not sufficient to model the variant effects in those regions.
Methods
Training and testing datasets
To create a robust dataset for pathogenicity and gain/loss of function (GoF/LoF) prediction tasks, we curated labeled pathogenic/benign variants, GoF/LoF variants, and DMS experimental data from public databases and published studies.
Gain/Loss of Function Prediction Task
We compiled missense variants labeled as GoF or LoF from multiple sources for gain/loss of function prediction tasks, including 765 gain and 4,571 loss of function missense variants from Bayrak, et al.45; 669 gain and 1,232 loss of function variants from Clinical Knowledge Base53; 199 gain and 1,506 loss of function variants from cBioportal cancer hotspots50; 56 gain and 57 loss of function variants in ABCC8, GCK, KCNJ11 from Flanagan, et al.48; 45 gain and 7 loss of function variants in STAT1 from Kagawa, et al.49; 309 gain and 516 loss of function variants from Heyne, et al.37 (Table 1). For cBioportal, we identified cancer hotspots using established algorithms60. We annotated 199 variants in hotspots of 27 oncogenic genes as GoF and 1506 variants in 248 tumor suppressor genes as LoF based on COSMIC database51. Genes with multiple cancer roles were excluded.
As the gain and loss of function variants were extremely biased across genes, we split our data in a protein-wise manner with the following steps:
1. For each gene, 20% of GoF and 20% of LoF variants were randomly selected as testing data, with the remaining 80% designated for training. This split was repeated five times.
2. Only genes with a minimum of 15 GoF and 12 LoF variants were retained for model evaluation.
The final, detailed number of training and testing data for each gene is available in (Supplementary Data 1).
For comparisons with LoGoFunc and funNCion, we performed tailored splits:
1. For LoGoFunc, we selected a test set not present in LoGoFunc’s data, while the remainder served as the training set. The split was conducted five times.
2. For funNCion, the training and testing data were divided based on funNCion’s defined split criteria.
Pathogenicity Prediction Task
We collected a total of 148,324 missense variants for training, including 51,494 benign variants from PrimateAI18 and 32,350 non-overlapping benign variants from ClinVar59 with at least one-star non-conflict submits that labeled as “benign” or “likely-benign”; 64,480 “pathogenic” and “likely-pathogenic” variants from ClinVar database with at least one-star non-conflict submits8 and non-overlapping variants from HGMD41 (Table 2).
For testing, we included 1066 missense variants, including 533 pathogenic missense variants from somatic missense hotspots in 153 cancer driver genes that not annotated in the training data; and 533 benign variants from the same genes, randomly selected from ClinVar and PrimateAI that not overlapping with training dataset (Table 2).
Deep mutational scan assays
We collected multi-dimensional mutation scan datasets of PTEN, SNCA, CCR5, CXCR4, NUDT15, CYP2C9, GCK, ASPA from MAVEDB6 as of December 2023 (Supplementary Data 2).
Input features
For each missense variant, PreMode examines a 250 amino acid window flanking the variant position, treating each amino acid residue as a node in two distinct graph structures. The first graph \({{{\mathcal{G}}}}_{1}\) is a non-directed star graph where only the variant node connects to all other nodes. The second graph \({{{\mathcal{G}}}}_{2}\) is a non-directed KNN graph in which nodes are connected based on the 3D Euclidean distance of their alpha carbon atoms.
Each node contains both invariant and structural features.
Invariant features:
-
1.
Embeddings from the last layer of ESM2 (650 M)26 (d = 1280)
-
2.
Dssp54,55 annotated secondary structure and torsion angles from the AlphaFold2 predicted protein structures (d = 12)
-
3.
pLDDT25 for the AlphaFold2 prediction
-
4.
Amino acid frequencies from MSA of 199 species that reflects evolutionary conservation (d = 20).
-
5.
For the variant node specifically, an additional invariant feature includes the embedding for the alternative amino acid.
Structural features:
-
1.
Euclidean vectors from the alpha carbon to all other non-hydrogen side chain atoms (d = 3 × 35). If an atom does not exist in the side chain, then it is set as 0.
Each edge in the graph also has invariant features and structure features.
Invariant features:
-
1.
A weighted covariance matrix between the amino acid frequencies of two residue sites in MSA of 199 species20 (Box 1)
-
2.
Euclidean distances of beta carbons between two residues (For glycine we use alpha carbon), transformed by exponential smearing functions (Box 2)
-
3.
The contact strength predictions from MSA transformer57.
Structural features:
-
1.
Euclidean vector between beta carbons of connected residues.
Model architectures
PreMode is an SE(3)-equivariant graph attention neural network with 4 layers.
First layer is a feature embedding layer that encodes the input features into latent dimensions. This layer was designed separately for node invariant features, node structure features, and edge invariant features.
Node invariant features: This layer uses the GeLU85 as the activation function and a linear transformation with a 512-dimensional output, utilizing weights \({{{\bf{W}}}}_{{{\bf{i}}}}\) and bias\(\,{{{\bf{B}}}}_{{{\bf{i}}}}\). Additionally, the variant node has a unique embedding layer, with weights \({{{\bf{W}}}}_{{{\bf{c}}}}\) and bias\(\,{{{\bf{B}}}}_{{{\bf{c}}}}\), to integrate embeddings of the alternate amino acid (Box 3).
Node structure features: To maintain SE(3)-equivariance, the structural feature embedding uses a linear layer with weights \({{{\boldsymbol{W}}}}_{{{\bf{s}}}}\) but no bias. The latent dimension is set to 32 (Supplementary Fig. 1).
Edge invariant features: For each edge, this layer encodes the pairwise MSA, Euclidean distances, contact predictions, relative positions into a 444-dim invariant feature vector.
Second and third layers are equivariant graph attention layers designed to model interactions between the connected nodes in star or KNN graphs defined in the features section. Attention values are computed using a modified SE(3)-equivariant attention mechanism inspired from torchmd-net86 and gMVP20 that takes both co-evolutionary and structural features into consideration. The invariant and structure features were updated separately but share information across each other based on the attention values to maintain SE(3)-equivariance (Box 4). The second layer operates on the star graph \({{{\mathcal{G}}}}_{1}\), focusing primarily on the first-order effects of the amino acid change on all other residues. The third layer operates on the KNN graph \({{{\mathcal{G}}}}_{2}\), modeling the second-order, indirect consequences of the variant.
The last layer is a graph attention layer designed to summarize the overall impact of the variant on the protein and perform pathogenicity or mode-of-action prediction. This layer calculates attention values based solely on the invariant embeddings produced by the third layer, ignoring structural features. It computes attention between the center node (the variant node) to other nodes and update the center node representations. After this, a SLP will transform the updated representations into pathogenicity or mode-of-action predictions.
Model training and testing
We trained PreMode with Adam87 algorithm. For pretraining on the pathogenicity dataset, we set the initial learning rate to 1e-4 with a batch size of 256 and trained the model for 20 epochs. We randomly selected 5% of training data as validation and calculated the validation loss every 250 steps. The training schedule included a 500-step of warmup phase, where the learning rate increased linearly to the maximum rate of 1e-4. This was followed by a learning rate decay phase, where the learning rate was reduced by a factor of 0.8 if a plateau was observed on the validation loss, until the minimum learning rate 1e-6. After training, we selected the model with minimum validation loss for testing. The training process took about 50 h under 4 NVIDIA A40 GPUs.
For transfer learning on molecular mode-of-action tasks, we set a batch size of 8 and train the model for 40 epochs. The training schedule included a 200-step warmup phase, followed by a learning rate decay phase with a minimum rate of 1e-7. Validation loss was evaluated every 400 steps, and the model checkpoint with the lowest validation loss was selected for testing. The training process took about 4–6 h on a single NVIDIA A40 GPU, depending on the dataset size.
For transfer learning on genetic mode-of-action tasks, we set everything same as the molecular mode-of-action tasks, except that we trained the model with 20 epochs and evaluate validation loss every 5 steps in the learning rate decay stage. To create a validation dataset, we randomly split training data into 4 parts and did 4-fold cross-validation. After training, we selected the models with lowest validation loss from each fold and ensemble them by averaging their predictions. We trained two ensemble models with window size of 251 and 1251 following the same procedure, and chose the ensemble model with best overall AUC on complete training data for evaluation on testing data. The training process took about 20 min–1 h under one single NVIDIA A40 GPU depending on the dataset size.
Model evaluation
To evaluate the model performance in pretraining and transfer learning on genetic mode-of-action tasks, we calculated the AUROC (area under recall receiver operating characteristic curve) on testing data to assess the performances of models. For pretrain task, we reported a single AUROC value. For transfer learning tasks, we calculated the AUROC across five random training/testing splits and reported the mean and standard error. For molecular mode-of-action tasks, we assessed performance by calculating the Spearman correlation between model prediction and experimental measurement on testing data. We calculated Spearman correlation for all five random training/testing splits and reported the mean and standard error.
Baseline methods and ablation analysis
For both molecular and genetics level mode-of-action predictions, we created several baseline methods. First, we developed a SLP model on top of the ESM embeddings (ESM + SLP). This model took all invariant features (ESM2 embeddings, amino acid changes, conservation in MSA) that same as PreMode as inputs, followed by one linear layer and GeLU activation layer. Next, we conducted several ablation analyses on PreMode. For structure feature ablation, we replaced all structural features with zeros, and constructed the star and KNN graphs based on the 1D-distance on the sequences. For ESM embedding feature ablation, we replaced the ESM embeddings with one-hot encodings of 20 amino acids. For MSA feature ablation, we removed all MSA inputs and the MSA attention module. All the models above were trained using same configurations as PreMode during both the pretraining and transfer learning stages.
Additionally, we implemented low-capacity machine learning models that used only biochemical properties as inputs, including conservation, secondary structure, reference, and alternate amino acid identities, pLDDT and ddG. For molecular-level mode-of-action predictions, we implemented it with an elastic net linear regression model while for genetics-level mode-of-action predictions, we implemented it with a random forest classification model.
To compare overall model performances, we calculated a weighted average of the Spearman correlation for each gene based on their dataset sizes for molecular mode-of-action predictions. For genetics mode-of-action predictions, we first determined the effective dataset sizes as the harmonic average of gain and loss of function variants for each gene, then computed a weighted average of the AUC across genes.
Curation of prediction scores from other methods
In the pathogenicity prediction comparison, we directly obtained the prediction scores for PrimateAI, EVE, REVEL from dbNSFP (v4.4a)88. For gMVP, AlphaMissense, ESM1b, we obtained the prediction scores from their original publications20,21,29.
In the molecular mode-of-action prediction comparison, we selected three models with top performances reported in Hsu, et al.62 (https://github.com/chloechsu/combining-evolutionary-and-assay-labelled-data), Augmented ESM1b, Augmented EVmutation and Augmented Unirep. We didn’t compare with Augmented DeepSequence due to errors in the publicly available codes. Instead, we trained the augmented model using evolutionary density score from EVE17, as both models were based on variational autoencoders on MSA. We trained and tested those models using the same MSA and protein sequence inputs as well as same training and testing data as PreMode. In the benchmarking tasks on ProteinGym (https://github.com/OATML-Markslab/ProteinGym), we directly take the reported Spearman correlation using random training/testing split, filtered to human proteins then perform weighted average across proteins based on the dataset sizes.
In the genetic mode-of-action prediction comparison, we obtained LoGoFunc prediction scores from https://itanlab.shinyapps.io/goflof/. For funNCion, we obtained their codes and training/testing split information from their GitHub page (https://github.com/heyhen/funNCion). We compared PreMode and their model’s performance using their training/testing split as well as ours. For funNCion, their original implementation of gradient boosting machine learning method in R “caret” package will raise errors under small sample sizes, we reimplemented the gradient boosting method in python “sklearn” package and reported both AUCs.
Subsample of datasets
For DMS datasets, we subsampled the data to investigate how many points were required for sufficient adaptation to the tasks in transfer learning. For each of the multi-dimensional assays, we randomly subsampled training data to 10%, 20%, 40%, 60%, 80% of whole datasets 5 times with different random seeds and tested on the same 20% of whole datasets. We performed PreMode on each of the training data and evaluated the performance.
In silico saturated mutagenesis experiments
We did in silico saturated mutagenesis experiments for BRAF, TP53, KCNJ11, RET, and PTEN. For each gene, we calculate two predictions \(r\) (pathogenicity score) and \(\theta\) (Gain/Loss of function score) with pretrained model and corresponding transfer-learning models, respectively. For \(\theta\), there are 5 predicted scores from models trained on 5 training/testing splits under different random seeds. We selected the models with minimum validation loss under each training/testing split and ensembled the predictions by voting with a simple random forest model.
Active Learning experiments
We split the DMS experiment datasets to ~40% of training data, ~10% of validation data and ~50% of testing data the same way as previous methods79,80,81. Within the training data, we first performed PreMode transfer learning on 10% of the randomly selected data, then evaluated PreMode on the rest of 90% of training data. PreMode will output both predicted values and model confidence values. Then we selected the top 10% of data among the rest of training data where PreMode was most unconfident and added them to the next round of training. We repeated this procedure until all the training data were used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data used in model training and analysis have been deposited in GitHub: https://github.com/ShenLab/PreMode and Zenodo89: https://doi.org/10.5281/zenodo.15825903.
The data used for training in this study are available at: AlphaFold Protein Structure Database25 (https://alphafold.ebi.ac.uk); ESM-1b, ESM-226 (https://github.com/facebookresearch/esm); Bayrak et al.45 (https://itanlab.shinyapps.io/goflof/); Clinical Knowledge Base53 (https://ckb.genomenon.com); MAVEDB6 (https://mavedb.org) (URN numbers available in Supplementary Dataset 2).
Variant prediction results for analysis are collected: PrimateAI, EVE, REVEL: from dbNSFP v4.488 (https://www.dbnsfp.org); AlphaMissense21 (https://doi.org/10.5281/zenodo.8360242); gMVP20 (https://www.dropbox.com/s/nce1jhg3i7jw1hx/gMVP.2021-02-28.csv.gz?dl=0); ProteinGym7 (https://github.com/OATML-Markslab/ProteinGym); LoGoFunc47 (https://itanlab.shinyapps.io/goflof/); funNCion37 (https://github.com/heyhen/funNCion).
Code availability
All the codes for machine learning model, model training and data analysis are available at GitHub: https://github.com/ShenLab/PreMode and Zenodo89 (https://doi.org/10.5281/zenodo.15825903). The machine learning model is built under pytorch v2.0.0, and statistical analysis is performed by R 4.4.1. We also provided all the model weights and all mode-of-action predictions for genes PTEN, BRAF, KCNJ11, TP53 and RET in the repository.
References
Homsy, J. et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015).
Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601 (2017).
Huang, K. L. et al. Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355–370 e314 (2018).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584 e523 (2020).
Zhou, X. et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 54, 1305–1319 (2022).
Esposito, D. et al. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 20, 223 (2019).
Notin, P. et al. ProteinGym: Large-Scale Benchmarks for Protein Design and Fitness Prediction. bioRxiv. https://doi.org/10.1101/2023.12.07.570727 (2023).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Romero, P. A. & Arnold, F. H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009).
Yang, K. K., Wu, Z. & Arnold, F. H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019).
Wang, Y. et al. Directed evolution: methodologies and applications. Chem. Rev. 121, 12384–12444 (2021).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Samocha, K. E. et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv, 148353 (2017).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95 (2021).
Sundaram, L. et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 50, 1161–1170 (2018).
Gao, H. et al. The landscape of tolerated genetic variation in humans and primates. Science 380, eabn8153 (2023).
Zhang, H., Xu, M. S., Fan, X., Chung, W. K. & Shen, Y. Predicting functional effect of missense variants using graph attention neural networks. Nat. Mach. Intell. 4, 1017–1028 (2022).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Feng, B. J. PERCH: a unified framework for disease gene prioritization. Hum. Mutat. 38, 243–251 (2017).
Jagadeesh, K. A. et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48, 1581–1586 (2016).
UniProt, C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Brandes, N., Ofer, D., Peleg, Y., Rappoport, N. & Linial, M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 38, 2102–2110 (2022).
Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. bioRxiv. https://doi.org/10.1101/2021.07.09.450648 (2021).
Brandes, N., Goldman, G., Wang, C. H., Ye, C. J. & Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 55, 1512–1522 (2023).
Sanders, S. J. et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 41, 442–456 (2018).
Wolff, M., Brunklaus, A. & Zuberi, S. M. Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 60(Suppl 3), S59–S67 (2019).
Matreyek, K. A. et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet. 50, 874–882 (2018).
Suiter, C. C. et al. Massively parallel variant characterization identifies NUDT15 alleles associated with thiopurine toxicity. Proc. Natl. Acad. Sci. USA 117, 5394–5401 (2020).
Mighell, T. L., Evans-Dutson, S. & O’Roak, B. J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet. 102, 943–955 (2018).
Lage, K. Protein-protein interactions and genetic diseases: the interactome. Biochim. Biophys. Acta 1842, 1971–1980 (2014).
Gerasimavicius, L., Livesey, B. J. & Marsh, J. A. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat. Commun. 13, 3895 (2022).
Heyne, H. O. et al. Predicting functional effects of missense variants in voltage-gated sodium and calcium channels. Sci. Transl. Med. 12, https://doi.org/10.1126/scitranslmed.aay6848 (2020).
Lester, H. A. & Karschin, A. Gain of function mutants: ion channels and G protein-coupled receptors. Annu Rev. Neurosci. 23, 89–125 (2000).
Li, Y., Zhang, Y., Li, X., Yi, S. & Xu, J. Gain-of-function mutations: an emerging advantage for cancer biology. Trends Biochem Sci. 44, 659–674 (2019).
Wang, L. H., Wu, C. F., Rajasekaran, N. & Shin, Y. K. Loss of tumor suppressor gene function in human cancer: an overview. Cell Physiol. Biochem 51, 2647–2693 (2018).
Stenson, P. D. et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677 (2017).
Swan, H. et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ. Cardiovasc. Genet. 7, 771–781 (2014).
Blanchard, M. G. et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J. Med. Genet. 52, 330–337 (2015).
McDonell, L. M., Kernohan, K. D., Boycott, K. M. & Sawyer, S. L. Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Hum. Mol. Genet. 24, R60–R66 (2015).
Sevim Bayrak, C. et al. Identification of discriminative gene-level and protein-level features associated with pathogenic gain-of-function and loss-of-function variants. Am. J. Hum. Genet. 108, 2301–2318 (2021).
Zhang, C. et al. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 12, 674–687 (2020).
Stein, D. et al. Genome-wide prediction of pathogenic gain- and loss-of-function variants from ensemble learning of a diverse feature set. Genome Med. 15, 103 (2023).
Flanagan, S. E., Patch, A. M. & Ellard, S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet. Test. Mol. Biomark. 14, 533–537 (2010).
Kagawa, R. et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J. Allergy Clin. Immunol. 140, 232–241 (2017).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Chang, M. T. et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 8, 174–183 (2018).
Patterson, S. E., Statz, C. M., Yin, T. & Mockus, S. M. Utility of the JAX Clinical Knowledgebase in capture and assessment of complex genomic cancer data. NPJ Precis Oncol. 3, 2 (2019).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Joosten, R. P. et al. A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–D419 (2011).
Valdar, W. S. Scoring residue conservation. Proteins 48, 227–241 (2002).
Rao, R. M. et al. In Proc. 38th International Conference on Machine Learning Vol. 139 (eds Meila Marina & Zhang Tong) 8844–8856 (PMLR, 2021).
Fung, A., Koehl, A., Jagota, M. & Song, Y. S. The impact of protein dynamics on residue-residue coevolution and contact prediction. bioRxiv. https://doi.org/10.1101/2022.10.16.512436 (2022).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Chang, M. T. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016).
Harrison, P. W. et al. Ensembl 2024. Nucleic Acids Res. 52, D891–D899 (2024).
Hsu, C., Nisonoff, H., Fannjiang, C. & Listgarten, J. Learning protein fitness models from evolutionary and assay-labeled data. Nat. Biotechnol. 40, 1114–1122 (2022).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
Wan, P. T. et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 (2004).
Ng, P. K. et al. Systematic functional annotation of somatic mutations in cancer. Cancer Cell 33, 450–462 e410 (2018).
Martinez Fiesco, J. A., Durrant, D. E., Morrison, D. K. & Zhang, P. Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding. Nat. Commun. 13, 486 (2022).
Tran, T. H. et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 12, 1176 (2021).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Lu, H. et al. Engineering and functional characterization of fusion genes identifies novel oncogenic drivers of cancer. Cancer Res. 77, 3502–3512 (2017).
Kiel, C., Benisty, H., Llorens-Rico, V. & Serrano, L. The yin-yang of kinase activation and unfolding explains the peculiarity of Val600 in the activation segment of BRAF. Elife 5, e12814 (2016).
Maloney, R. C., Zhang, M., Jang, H. & Nussinov, R. The mechanism of activation of monomeric B-Raf V600E. Comput Struct. Biotechnol. J. 19, 3349–3363 (2021).
Lee, E. W. et al. Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis. EMBO J. 28, 2100–2113 (2009).
Kakudo, Y., Shibata, H., Otsuka, K., Kato, S. & Ishioka, C. Lack of correlation between p53-dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res. 65, 2108–2114 (2005).
Zhao, C. & MacKinnon, R. Molecular structure of an open human K(ATP) channel. Proc. Natl. Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2112267118 (2021).
Matreyek, K. A., Stephany, J. J., Ahler, E. & Fowler, D. M. Integrating thousands of PTEN variant activity and abundance measurements reveals variant subgroups and new dominant negatives in cancers. Genome Med. 13, 165 (2021).
Papa, A. et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595–610 (2014).
Ren, P. et al. A survey of deep active learning. arXiv. https://ui.adsabs.harvard.edu/abs/2020arXiv200900236R (2020).
Rao, R. et al. Evaluating protein transfer learning with TAPE. Adv. Neural Inf. Process. Syst. 32, 9689–9701 (2019).
Shanehsazzadeh, A., Belanger, D. & Dohan, D. Is Transfer Learning Necessary for Protein Landscape Prediction? arXiv. https://ui.adsabs.harvard.edu/abs/2020arXiv201103443S (2020).
Xu, M. et al. PEER: a comprehensive and multi-task benchmark for protein sequence understanding. arXiv. https://ui.adsabs.harvard.edu/abs/2022arXiv220602096X (2022).
Iqbal, S. et al. Comprehensive characterization of amino acid positions in protein structures reveals molecular effect of missense variants. Proc. Natl. Acad. Sci. USA 117, 28201–28211 (2020).
Ruff, K. M. & Pappu, R. V. AlphaFold and implications for intrinsically disordered proteins. J. Mol. Biol. 433, 167208 (2021).
Muller, H. J. Further studies on the nature and causes of gene mutations. In Proc. Sixth International Congress on Genetics 215–255 (1932). http://www.esp.org/books/6th-congress/facsimile/contents/6th-cong-p213-muller.pdf
Hendrycks, D. & Gimpel, K. Gaussian Error Linear Units (GELUs). arXiv:1606.08415 (2016). https://ui.adsabs.harvard.edu/abs/2016arXiv160608415H.
Thölke, P. & De Fabritiis, G. TorchMD-NET: equivariant transformers for neural network based molecular potentials. arXiv. https://ui.adsabs.harvard.edu/abs/2022arXiv220202541T (2022).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. arXiv. https://ui.adsabs.harvard.edu/abs/2014arXiv1412.6980K (2014).
Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12, 103 (2020).
Zhong, G. & Shen, Y. PreMode predicts mode-of-action of missense variants by deep graph representation learning of protein sequence and structural context (v1.0.0). Zenodo. https://doi.org/10.5281/zenodo.15825903 (2025).
Zhong, G. Mode-of-action definition and PreMode framework. BioRender. https://BioRender.com/irb5u6w (2025).
Acknowledgements
This work was supported by NIH grants: R35GM149527 (Y.S.), P50HD109879 (W.K.C. and Y.S.), Simons Foundation: SFARI #1019623 (Y.S.). We thank Dr. Mohammed AlQuraishi, Dr. David Knowles, Dr. Haicang Zhang, Dr. Haiqing Zhao, Xi Fu, Dr. Xiao Fan, Dr. Na Zhu, and Dr. Chao Hou for helpful discussions and suggestions.
Author information
Authors and Affiliations
Contributions
Y.S. conceptualized the study. G.Z. and Y.Z. curated the data. G.Z. and Y.S. developed the methodology. G.Z. and D.Z. conducted the formal analysis. G.Z. and Y.S. carried out the investigation. G.Z. and Y.S. wrote the original draft. G.Z., Y.Z., D.Z., W.K.C., and Y.S. reviewed and edited the manuscript. W.K.C. and Y.S. acquired funding for the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhong, G., Zhao, Y., Zhuang, D. et al. PreMode predicts mode-of-action of missense variants by deep graph representation learning of protein sequence and structural context. Nat Commun 16, 7189 (2025). https://doi.org/10.1038/s41467-025-62318-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62318-4
This article is cited by
-
Low frequency and rare coding variants affect susceptibility and progression of childhood acute lymphoblastic leukemia
Cancer Cell International (2026)
