
Abstract
Recent technological advances have facilitated the assembly of telomere-to-telomere (T2T) genomes. The current T2T CHM13 showcases the complete architecture of the human genome, yet its use in functional experiments is limited by discrepancies with the actual genome of the specific biological system under study. Access to reference assemblies for experimentally relevant cell lines is therefore essential in advancing sequencing-based analyses and precise manipulation, particularly in highly variable regions such as centromeres. Here, we present RPE1v1.1, the near-complete diploid genome assembly of the hTERT RPE-1 cell line, a non-cancerous human retinal epithelial model with a stable karyotype. Using high-coverage Pacific Biosciences and Oxford Nanopore Technologies long-read sequencing, we generate a high-quality de novo assembly, validate it through multiple methods, and phase it by integrating high-throughput chromosome conformation capture (Hi-C) data. Our assembly includes chromosome-level scaffolds that span centromeres for all chromosomes. Comparing both haplotypes with the CHM13 genome, we detect haplotype-specific genomic variations, including the translocation between chromosome 10 and chromosome X t(X;10)(Xq28;10q21.2) characteristic of RPE-1 cells, and divergence peaking at centromeres. Altogether, the RPE1v1.1 genome provides a reference-quality diploid assembly of a widely used cell line, supporting high-precision genetic and epigenetic studies in this model system.
Similar content being viewed by others

Introduction
Over the past three decades, the field of genomics has seen rapid advancements. A pivotal moment was the launch of the Human Genome Project in 1990, which sequenced DNA from 20 anonymous individuals using Sanger and shotgun sequencing methods. The final assembly, released in 2003, represented a composite genome derived from this group1,2. Despite this progress, the reference from the Human Genome Project still lacks ~8% of total DNA pertaining to the repetitive regions of the human genome. Advances in sequencing technologies over the last 20 years, particularly those generating long reads, have enabled the assembly of more complete human genomes. This progress culminated in 2022 with the assembly of a high-quality, fully complete human genome derived from the CHM13 cell line, which originated from a hydatidiform mole resulting from an ectopic pregnancy3. This assembly was a remarkable achievement, as it represented the first complete human genome without gaps; however, the CHM13 cell line is essentially haploid, containing only the paternal haplotype duplicated within an enucleated oocyte. In contrast, phased diploid genomes, where both haplotypes are individually resolved, enable the study of maternal and paternal genetic diversity and gene expression imprinting, which can lead to haplotype-specific diseases. They also allow for the investigation of the functional consequences of allele combinations that co-segregate on the same haplotype. Recent years have seen notable progress in sequencing and assembling diploid genomes. These include the high-quality assemblies of HG002, an immortalized lymphoblastoid cell line derived from B lymphocytes of a male individual4, and CN1, a healthy male Chinese individual from Hubei5, as well as the T2T reference genome with a Y chromosome for the Han population (T2T-YAO)6. The Human Pangenome Reference Consortium (HPRC) represents the next major step in capturing human diversity. This initiative has released a draft that includes genomes from 47 individuals, aiming to better represent global genomic diversity7. Having representative genomes from diverse populations is essential for addressing biological questions and for a deeper understanding of variation both within and between individuals. However, the genomes of diploid cell lines commonly used in laboratories worldwide—serving as the basis for numerous in vitro experiments and biological observations—are still largely unexplored. The absence of reference-quality genome assemblies for these cell lines limits our ability to generalize experimental findings and fully understand the genetic variability and biological implications of laboratory observations.
Here, we present RPE1v1.1, the near-complete reference-quality de novo genome assembly of the hTERT RPE-1 cell line, derived from a noncancerous human retinal pigment epithelial (RPE) cell line, RPE-340, which was immortalized by the human telomerase reverse transcriptase (hTERT)8,9. The hTERT RPE-1 cell line features a diploid karyotype with 46 chromosomes and lacks transformed phenotypes, making it a valuable model for studying normal physiological functions of human chromosomes. It is among the top three most used cell lines in cellular and molecular biology laboratories worldwide, and RPE-1 cells have been utilized in thousands of studies10. The near-complete genome of hTERT RPE-1 was generated using Hi-C technology to support phasing, as trio-based phasing11 is not applicable to cell lines, achieving a final Quality Value (QV) higher than 61 for both haplotypes, with 17 chromosomes reaching a QV of 67—corresponding to an estimated base accuracy above 99.9999%. This human diploid reference serves as a key resource for the broader scientific community and is a turning point, enabling high-resolution phased genetic and epigenetic analyses across a wide range of studies involving this important cell line.
Results
Diploid genome assembly and gaps curation
We aimed to construct a haplotype-resolved reference genome for the immortalized cell line RPE-1. This cell line is characterized by a stable diploid karyotype of 46 chromosomes, including a previously reported unique chromosomal rearrangement10,12,13,14,15,16. Specifically, this rearrangement involves the duplication of the long arm of chromosome 10, which is translocated to the telomeric q-arm of chromosome X, denoted as t(X;10)(Xq28;10q21.2), resulting in the 46,X,der(X)(Xpter→Xq28::10q21.2→10qter) karyotype (Supplementary Fig. 1a–d). We employed state-of-the-art practices to achieve a near-complete diploid genome assembly using long-read technologies (“Methods”). We generated Pacific Biosciences (PacBio) High-Fidelity (HiFi) reads at 46× coverage and Oxford Nanopore Technologies (ONT) reads at 125× coverage, with 30× coverage for reads over 100 kb (ultra-long reads) (Supplementary Fig. 2a). Additionally, we used Hi-C data at 30× coverage to phase the haplotypes, leveraging its ability to capture long-range chromosomal interactions which enables the separation of the two haplotypes even in the absence of parental trio information17. The ONT sequencing data were produced exclusively using R10.4 chemistry, achieving a final basecalling accuracy of 99% (Supplementary Fig. 2b).
The sequencing data were used as input for the automated genome assembly pipeline Verkko v1.418. The draft assembly produced by the tool was composed of a total of 42 and 54 scaffolds per haplotype (Fig. 1a and Supplementary Table 1). The number of gaps was 49 for Hap1 and 25 for Hap2, with a total gap length of 734,056 bp for Hap1 and 994,785 bp for Hap2 (Supplementary Table 1). Chromosomes 10 and X, which are the two involved in the karyotypically stable rearrangement for this cell line, accounted for 76% of total gap length in Hap1. This is primarily due to the duplication of chromosome 10 q-arm, creating a triplex structure in this region that deviates from the expected diploid state and leads to local assembly collapse (Supplementary Fig. 3 inset). Chromosomes 1, 13, 14, 16, 18, 22, and X (Hap1), and chromosomes 5, 14, 18, and X (Hap2) were represented with more than one scaffold, suggesting regions of insufficient read coverage, misjoined or chimeric reads that were preventing the complete scaffolding of these chromosomes (Supplementary Table 2). We also tested a more recent version of the assembler, Verkko v2.019. However, the draft assembly generated with this version (Supplementary Fig. 2c) contained a greater number of gaps (70 for Hap1 and 26 for Hap2). While it yielded fewer unassigned contigs, their total base length was comparable to that of the Verkko v1.4 draft assembly (Supplementary Fig. 4a). When mapped against the CHM13 reference genome, the unassigned sequences produced by the two Verkko versions showed similar patterns, with most of them aligning to chromosome 10 or to ribosomal DNA regions (Supplementary Fig. 4b). These results prompted us to use the draft assembly produced by Verkko v1.4.

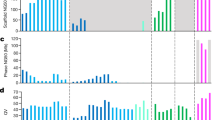
a Assembly graph of the RPE-1 diploid genome generated by Verkko v1.4, used to generate the final RPE1v1.1 reference, visualized using Bandage. The phased blocks are colored in red (Hap1) and blue (Hap2). The short arms of chromosomes 13, 14, 15, 21, and 22, which contain rDNA arrays, are unphased and appear as tangled regions. b Merqury assembly spectrum plot for the evaluation of RPE1v1.1 genome quality based on the comparison of k-mers from HiFi and k-mers from the assembly. The plot displays the multiplicity distribution of four categories of read-derived k-mers: k-mers found only in the reads (read-only), those unique to each haplotype assembly, and those shared between the two haplotypes (shared). c Circos plot showing Clip-based Regional Errors (CREs, blue) and the heterozygous sites (green) identified by CRAQ at specific genomic regions for each chromosome of RPE1v1.1 Hap1 and Hap2. d Bar plot showing, for each RPE1v1.1 chromosome, the amount of sequence (in kb), classified by Flagger as erroneous (orange), collapsed (blue) or haploid (structurally correct, green).
Next, we utilized Hi-C data mapped back to the draft assembly to achieve chromosome-level scaffolding and correct phasing errors. The Hi-C contact map, visualized using PretextView (see “Methods”), enabled us to identify misjoined points and to merge segments of the same chromosome into a single scaffold20 (Supplementary Fig. 5a). A subsequent round of dual manual curation yielded 23 chromosome-level scaffolds for each haplotype.
We then focused on the gaps present in the assembly after dual manual curation (Supplementary Fig. 5b and Supplementary Table 2). Apart from chromosomes 10 and X, gaps were found in chromosomes 1, 2, 3, 5, 12, 13, 16, 17, 18, and 21, including some centromeric regions (Supplementary Fig. 5b). To address these remaining gaps, we applied two complementary gap-closing strategies (see “Methods”). The first involved mapping ultra-long ONT reads (>100 kb) to the assembly (Supplementary Fig. 6a). Gaps spanned by ONT reads with at least 40 kb of mapped sequence on both sides were initially patched with the interleaving read segments (Supplementary Fig. 6b). To minimize errors introduced by ONT reads, we subsequently aligned PacBio HiFi reads to the patched genome and used these high-accuracy alignments to reconstruct a consensus sequence across the filled regions. This approach allowed to close gaps on chromosomes 2, 16, and 17 of Hap1, and on chromosomes 3, 5, 12, and 16 of Hap2 (Supplementary Table 2). In cases where no ultra-long reads were found with an extensive alignment on both side of a gap, we leveraged information from the assembly graph generated by Verkko (Supplementary Fig. 6c), as done for chromosome 3 of Hap1. In this case, we identified a bubble spanning positions 61.4–62.3 Mb, containing multiple alternative paths between the two ends. To resolve it, we mapped ONT reads to the graph and found alignments that unambiguously supported a single path through the unitigs. Using this graph-based approach, we were also able to close gaps on both haplotypes of chromosome 18 (Supplementary Table 2).
Finally, we did not curate two 3.5 Mb and 2.5 Mb contigs with sequence identity to centromere and pericentromere of CHM13 chromosome 18, and a 3 Mb-long contig that aligned only partially—171 kb—to the centromere of CHM13 chromosome 3, while also showing additional alignments to the short arms of acrocentric chromosomes (SAACs). As a result, these contig were excluded from further analysis and remain classified as unassigned. The telomeres were successfully assigned to all the chromosomes, excluding the unplaced SAACs and q-arm of chromosome 10, due to the aforementioned translocation (Supplementary Fig. 7). The p-arm telomeres of chromosomes 16 and X and the q-arm telomere of chromosome 4 were identified among the unassigned sequences generated by Verkko and subsequently recovered through manual curation. The total size of the final RPE1v1.1 assembly reached 3.06 Gb for Hap1 and 2.99 Gb for Hap2, closely aligning with CHM13v2.0 and HG002, except for ~43 Mb of unresolved rDNA from acrocentric chromosomes (Supplementary Fig. 8). A total of 15 and 16 chromosomes were assembled T2T without gaps for Hap1 and Hap2, respectively (Supplementary Table 2). The genome exhibited good contiguity (contig N50 equal to 136 Mb for Hap1 and 135 Mb for Hap2), just short of that of T2T assemblies CHM13v2.021 and T2T-YAO6 (Supplementary Table 3). To our knowledge, our final RPE1v1.1 represents the first assembly of a human diploid laboratory cell line at near-complete, reference-quality, and fully phased resolution.
Quality control of the RPE-1 genome
The RPE1v1.1 assembly was evaluated using a variety of quality control (QC) tools and strategies. We first assessed the alignment of PacBio HiFi reads across the entire genome (Supplementary Fig. 3). The coverage plot revealed a homogeneous distribution for the majority of chromosomes and for both haplotypes. As expected, chromosomes 10 and X exhibited distinct patterns in coverage. Specifically, we observed a dip in coverage within the translocated telomere on chromosome X and an increase within the duplicated long arm of chromosome 10, reflecting the rearrangement and consistent with the copy number gain (Supplementary Fig. 3). The chromosome 14, 15, 21, and 22 SAACs showed an increase in coverage (Supplementary Fig. 3), due to the unassigned repetitive rDNA. To further validate the assembly, HiFi read alignments were filtered to retain only the primary ones, and the frequencies of the most common (primary) and the second most common (secondary) base in the aligned reads at each position were calculated and displayed with a NucFreq plot22 (Supplementary Fig. 9). The assembly displayed a higher level of secondary bases in a few specific genomic locations, indicating potential misassemblies or errors: within human satellite (Hsat) regions of chromosomes 3, 4, and 9 (approximately 300 kb in total, including ~100 kb within pericentromeric satellites), centromere of chromosome 18, and chromosome 15 and 22 SAACs (Supplementary Fig. 9). The remaining chromosomes showed consistently low levels of secondary bases across both haplotypes, underscoring the high quality and reliability of the assembly (Supplementary Fig. 9).
We also employed the Merqury k-mer-based, reference-free evaluation pipeline23 using HiFi reads to determine genome quality and completeness20 (see “Methods”). We obtained a QV of 64.1 for Hap1 and 61.8 for Hap2, within the range observed for other high-quality human assemblies (Supplementary Table 3), and 99.8% completeness for both haplotypes. For the regions where gaps were resolved, the QV scores exceeded 65 (an error every 8.5 Mb), and no outstanding errors were reported. Merqury was also employed to compute and plot k-mer spectra, which revealed a multiplicity profile consistent with a near-complete, haplotype-resolved assembly (Fig. 1b). To further evaluate assembly quality, we analyzed HiFi read alignment patterns using the CRAQ24 and Flagger25 tools (see “Methods”). CRAQ uses read clipping information to identify regional and structural errors, and provides metrics expressing the regional and overall assembly quality (AQI). In the final RPE1v1.1 assembly, CRAQ detected only ~2 Mb of potential assembly errors, primarily corresponding to regions with unresolved gaps and specific telomeric sequences (Fig. 1c). The AQI score exceeded 98, underscoring the quality of the assembly. An AQI score greater than 90 is generally considered indicative of a reference-quality assembly24. Flagger enabled us to differentiate between assembly errors, collapsed regions and reliable sequences. Of the 6.05 Gb total assembly, 5.99 Gb were classified as error-free (Fig. 1d), with 2.4 Mb identified as assembly errors and 55 Mb as collapsed regions, primarily corresponding to the chromosome 10 duplicated region (Fig. 1d). Notably, the centromere of chromosome 18, which also showed high frequency of secondary bases (Supplementary Fig. 9), was flagged as erroneous in its terminal region (Supplementary Fig. 10). This suggests a potential assembly issue in this region, likely because of its being part of an extended run of homozygosity in the assembly graph, which may have led to unassigned contigs (Fig. 1a). As Flagger identified the largest number and extent of regions affected by gaps, errors, or collapses compared to other QC methods, we used the coordinates of these regions to define a low-confidence annotation track in the final assembly. Finally, we evaluated the presence of conserved single-copy ortholog genes using compleasm26. The analysis identified 99.71% complete genes (with 3.28% duplicated) in Hap1, and 99.73% (with 0.7% duplicated) in Hap2. Of the 452 duplicated genes in Hap1, 375 are located on the duplicated long arm of chromosome 10, indicating that the elevated duplication rate is largely driven by the presence of three gene copies in this triploid region. The missing genes were 0.21% and 0.09% (Supplementary Tables 1 and 4), while the fragmented genes were 0.08% and 0.09% for Hap1 and Hap2, respectively.
In addition to validating the completeness and correctness of the assembly, we tested whether errors were present when phasing the two haplotypes using Hi-C information. First, we used SecPhase25 (see “Methods”) to identify HiFi reads with incorrect haplotype alignment. No read needed to be relocalized, indicating that Hi-C data were sufficient to phase the assembly into two haplotype blocks. To further verify the phasing accuracy of the RPE1v1.1 genome, we analyzed Strand-seq data generated from 80 RPE-1 cells by Sanders and colleagues27. Strand-seq is a single-cell DNA sequencing technique that selectively targets parental template strands in daughter cells, enabling the identification of structural variants, chromosome misorientations, and the phasing of haplotypes across entire chromosomes28. Reads from each RPE-1 cell were aligned to the RPE1v.1.1 genome, and the breakpointR R package29 was used to detect strand state changes (see “Methods”). For each chromosome, the longest informative region—when available—presented reads mapping to both Watson and Crick strands in most cells. This pattern is indicative of proper chromosome-scale phasing in the RPE1v1.1 assembly (Supplementary Fig. 11).
To confirm that RPE-1 is karyotypically stable, we cytogenetically analyzed potential variations across the cell population and in different batches. We did not observe karyotypic changes at the cytogenetic level, with stable presence of the t(X;10)(Xq28;10q21.2) marker chromosome in all batches in this study (Supplementary Fig. 1d). RPE-1 cells have a diploid karyotype (Supplementary Fig. 1a–c). Indeed, we detected 46 chromosomes for the majority of metaphases, with only a subset showing 44 or 45 chromosomes, likely due to chromosome sliding during metaphase preparation (see “Methods”). We did not observe evidence of aneuploid, polyploid, tetraploid or pseudo-tetraploid clones in any of the samples analyzed (Supplementary Fig. 1a–c). Furthermore, no gross chromosomal rearrangements or other cytologically visible chromosomal abnormalities were observed comparing batches of RPE-1 acquired from different sources, implying that RPE-1 cells have a remarkably stable diploid asset and the assembly represents a universal reference for functional analysis of RPE-1 experiments and data across batches.
Altogether, the RPE1v1.1 represents a reference-quality, near-complete assembly to support genome-wide functional analysis with precision.
Translocation of q-arm of chromosome 10 on q-arm of chromosome X and hTERT plasmid insertion
Cytological and sequencing analyses described in the literature have shown that the RPE-1 cell line exhibits a stable marker chromosome10,12,13,14,15,16. This marker involves a duplication of the long arm of chromosome 10, which is thus present in three copies in RPE-1 cells, two copies in the intact chromosome 10 and one extra copy translocated to the telomeric region of chromosome X (Fig. 2a, b and Supplementary Fig. 1). The duplicated translocation was detected in the assembly graph generated by Verkko v2.0, where the interlinked triplex bandage of chromosome X q-arm is contiguous to chromosome 10 q-arm (Fig. 2a). Karyotype analysis performed in our laboratory using the same RPE-1 cell samples that were used for the sequencing experiments confirmed the 46,X,der(X)(Xpter→Xq28::10q21.2→10qter) karyotype with the presence of the marker chromosome t(X;10)(Xq28;10q21.2) (Fig. 2b and Supplementary Fig. 1). The chromosome contacts between chromosome 10 and chromosome X was also visible on the dual Hi-C contact map for both haplotypes (Fig. 2c, d). To accurately reflect this translocation in the final genome assembly, we mapped the precise breakpoint at base-pair resolution and curated the assembly accordingly. A multi-step approach was used to manually curate this large structural variant, starting from the phased haplotype assemblies (Fig. 2e and Supplementary Fig. 12). HiFi and ONT reads were aligned to the diploid genome, revealing chimeric alignments indicative of a 73.18 Mb segmental duplication of the q-arm of chromosome 10 fused to the telomeric region of chromosome X. This fusion was accompanied by a 3603 bp microdeletion at the X chromosome breakpoint. The presence of contiguous read alignments spanning the reconstructed junction further confirmed the precise location of the breakpoint. Read alignment scores suggest that the translocation involves chromosome 10 Hap2 and chromosome X Hap1, resulting in a 227.21 Mb rearranged chromosome. However, since Hi-C-based phasing assigns haplotype labels independently for each chromosome17, it is not possible to determine whether the translocation occurred between chromosomes of the same haplotype—as previously reported16—or between different haplotypes. Resolving this ambiguity would require further validation or complementary phasing approaches. Altogether, our assembly recapitulated some characteristic features of the RPE-1 genome, such as the presence of the t(X;10)(Xq28;10q21.2) translocation. We were also able to locate the complete hTERT plasmid sequence (Supplementary Fig. 13). The original cell line RPE-340 was immortalized using a construct (pGRN145 plasmid) to introduce the hTERT, resulting in hTERT RPE-1 cells8,9. To identify the genomic location of the insertion, the hTERT sequence was aligned to the RPE1v1.1 assembly using both NUCmer30 and Minimap231,32. Both tools consistently mapped the insertion to the p-arm of chromosome 20 in Hap2 (Supplementary Fig. 13a). The full 15 kb plasmid sequence was reconstructed using SnapGene (Supplementary Fig. 13b). The plasmid sequence was masked in the RPE1v1.1 assembly according to NCBI request during genome submission.

a Bandage visualization showing how chromosome 10 and chromosome X are joined in a single connected component in the assembly graph produced by Verkko v2.0, as a result of the RPE-1 specific t(X;10)(Xq28;10q21.2) rearrangement. b Representative G-banded karyogram of RPE-1 chromosomes obtained using GIEMSA staining. Scale bar: 10 μm. The normal and rearranged X chromosomes are shown (zoom in). This experiment was independently repeated ten times with similar results. c Hi-C contact maps, visualized using PretextView, showing the two haplotypes at chromosome-level resolution. d Zoom in of the Hi-C contact map of the diploid genome highlighting chromatin interaction between chromosome 10 and X due to the t(X;10)(Xq28;10q21.2) rearrangement. e Schematic representation of the multi-step approach followed to curate the breakpoint between chromosome 10 and X at base-pair resolution.
Genomic variation between the human RPE-1 haplotypes and CHM13 genome
Having generated a near-complete human genome, we reasoned that RPE1v1.1 is one of a few available chromosome-level assemblies that can be analyzed to understand diversity between human cell lines and across individuals at whole-genome level. A prior study conducted using the HG002 phased diploid genome indicated that differences between the two haplotypes of the same individual affect nearly half of all coding regions, with the highest levels of polymorphism concentrated in the centromeric regions4. To explore inter-haplotype variation within the RPE1v1.1 diploid genome, we first measured sequence identity and alignment length using NUCmer and dnadiff. On average, haplotypes aligned over segments of 471 kb with a sequence identity of 99.83%. 393 Mb of Hap1 did not align to Hap2, and 323 Mb of Hap2 did not align to Hap1, reflecting the different lengths of the two haplotypes. An additional comparison between the two haplotypes performed using SyRI33 identified 3,119,795 SNPs, 65 inversions, 1400 translocations, 1.4 Mb of insertions and deletions (30–500 bp), and 256 Mb of large structural variants (>100 kb) between the haplotypes (Supplementary Fig. 14 and Supplementary Table 5). Notably, many structural rearrangements were concentrated in centromeric regions (Supplementary Fig. 14). To investigate regional variation in more detail, we quantified SNP density in nonoverlapping 10 kb windows across the genome (see “Methods”). While the average SNP density across chromosome arms was 0.1% (one heterozygous marker every 1000 bp), it increased to 0.6% within centromeric regions (active α-satellite higher-order arrays), with the highest peaks in the centromere of chromosome 19 and in the pericentromeric region of chromosome 9, where it reached up to 7.1% and 7.3%, respectively (Fig. 3). We speculate that centromere divergence is primarily due to crossover suppression34,35, which limits meiotic recombination and preserves centromere blocks into intact parental haplogroups with high genetic diversity.

Karyoplot showing SNP density (blue line) across the RPE1v1.1 Hap1 genome, calculated in non-overlapping 10 kb windows, with SNPs identified by comparing Hap1 (reference) to Hap2 (query) with SyRI. Highly diverged regions (HDRs) detected by SyRI are shown as grey bars, and the positions of live higher-order repeats (HORs) are marked with underlying black boxes.
To expand our analysis beyond intra-genomic comparisons, we aligned each RPE-1 haplotype to the CHM13 assembly and assessed differences using SyRI (Fig. 4 and Supplementary Table 5). When restricting the analysis to autosomes, CHM13 was 95.64% syntenic with RPE-1 Hap1 and 95.85% with Hap2. The X chromosome was excluded due to the translocation and additional sequence length in RPE-1, which could confound alignment accuracy. Both haplotypes displayed a comparable number of variants relative to CHM13: Hap1 showed 3,064,100 SNPs, 54 inversions, and 796 translocations, while Hap2 had 3,221,002 SNPs, 77 inversions, and 1441 translocations. In both comparisons, approximately 445 Mb of sequence remained unaligned, and the average alignment length was 384 kb.

Visualization of synteny and structural rearrangements between CHM13 (reference) and RPE1v1.1 Hap1 (left panel) and Hap2 (right panel), as detected by SyRI. CHM13 live higher-order repeats (HORs) are marked with black boxes.
SyRI also identifies highly diverged regions (HDR)—segments within aligned syntenic or translocated regions that exhibit dense clusters of SNPs or structural variants, often associated with copy number variations or rearrangements. We identified 1653 HDRs comparing CHM13 vs. Hap1, 1623 comparing CHM13 vs. Hap2, and 1617 comparing Hap1 vs. Hap2. As expected, HDRs were enriched in centromeric regions, consistent with their elevated SNP density. We also found that the Major Histocompatibility Complex (MHC) locus, known for being one of the most gene-dense and polymorphic stretches of human DNA36, was classified as an HDR element in all comparisons (Supplementary Fig. 15).
Historically, incomplete human reference genomes, largely lacking linear sequences for centromeres and other HDRs, have obscured the true extent of variation in these regions, leading to the perception that humans are 99.9% identical at the sequence level. However, near-complete or complete diploid references now enable comprehensive assessment of genome-wide variation, both between haplotypes and across individuals. Our findings from the RPE-1 genome underscore a lower-than-expected sequence identity between haplotypes, with centromeres emerging as the most divergent regions of the human genome.
Comparing the RPE-1 genome to individuals from the human pangenome reference
Currently available human assemblies are often obtained by sequencing freshly isolated lymphocytes from individuals. Our RPE-1 genome provides an interesting opportunity to understand how representative commonly used laboratory cell lines are of the broader human population, and to characterize the differences—if any—between a laboratory cell line isolated from human tissue over 20 years ago and a human subject. To this end, we applied a pangenome analysis (see “Methods”) comparing the genomic landscape of the RPE-1 cell line to individual genomes from the HPRC25. To place these comparisons in a broader reference context, we also included the CHM133 and the GRCh3837 assemblies, enabling us to assess both inter-individual variation and differences among existing human genome references within a unified pangenomic framework (Fig. 5a). As expected, whole-genome analysis showed that the haplotype 2 of RPE-1 genome clusters within the broader group of HPRC genomes (Fig. 5a), confirming overall genomic similarity between the cell line and typical human genomes. RPE-1 haplotype 1, on the other hand, branched into a distinct cluster, indicating some degree of divergence from the HPRC individuals likely due presence of rearranged chromosome t(X;10)(Xq28;10q21.2) in this haplotype. This divergence was further supported by the Principal Component Analysis (PCA) of whole-genome data (Fig. 5b). To gain a more nuanced understanding of this genomic landscape, we conducted chromosome-specific analyses (Supplementary Fig. 16–19). These analyses confirmed that the distinctive positioning of RPE-1 Hap2 in whole-genome comparisons is primarily driven by the translocated chromosome X (Supplementary Fig. 19a). This translocation exemplifies the specific genomic alterations that can occur in established cell lines, and our analysis shows the impact of deviation from expected ploidy in the resulting clustering. Importantly, for all other chromosomes, our chromosome-specific pangenome analyses showed a similar pattern to individuals. For these non-X chromosomes, both haplotypes of RPE1v1.1 consistently clustered together with HPRC samples, converging also based on the expected population of origin (Supplementary Fig. 16–18). This finding indicates that the majority of the RPE-1 genome maintains close similarity to typical human genomes, with the exception of the altered chromosome X. PCA analysis of a cell line-specific locus such as the MHC further confirmed the trend for both haplotypes (Supplementary Fig. 19b). On the other hand, GRCh38 reference is the assembly that most deviates from human subjects, likely due to both being a composite of many individuals and to the remaining gaps within the assembly. The positioning of RPE-1 within the human genomic landscape indicates that it maintains core human genomic structures for the majority of its genome but has evolved distinct genomic features on chromosome X, which may reflect adaptations to in vitro culture conditions or its immortalized state (Supplementary Fig. 16–19). Altogether, the genomic stability of RPE-1—along with its close similarity to HPRC genomes in both whole-genome and chromosome-specific analyses, and its experimental tractability in culture—underscores its continued value as a model for studying human cellular processes and functional genomics. By capturing the specific alterations on chromosome X, the RPE1v1.1 assembly provides a crucial reference for studies involving X-linked genes and haplotype-resolved chromosome biology of this widely used cell line.

a Cartoon of comparison and dendrogram of the RPE-1 genome and HPRC samples based on whole-genome (all chromosomes) pangenome analysis. The tree shows the genomic relationships between RPE-1 haplotypes (green), individual HPRC genomes (red), CHM13 (blue), and GRCh38 (purple). Created in BioRender. Giunta, S. (2025) https://BioRender.com/nozlc3z. b Principal Component Analysis (PCA) of the whole pangenome. The plot shows the first two principal components, with the percentage of explained variance indicated on each axis. HPRC samples are represented by red dots, RPE-1 by green triangles, CHM13 by a blue square and GRCh38 by a purple cross. Given that the centromeric sequences of GRCh38 are masked, we interpret the first principal component (PC1) as representing the centromeric diversity of the assemblies. Furthermore, the distinct positioning of RPE-1 haplotype 1 (RPE-1#1) on PC2 highlights its divergence in chromosome X due to the t(X;10)(Xq28;10q21.2) translocation. AFR African, AMR American, EAS East Asian, SAS South Asian.
Discussion
Here, we present the near-complete and fully phased human diploid reference genome for the experimentally amenable laboratory cell line RPE-1. To our knowledge, this is the first reference-quality de novo assembly of a laboratory cell line. The RPE1v1.1 assembly presents a high base-level accuracy (QV > 61 for both haplotypes), complete phasing of haplotypes in the absence of trio-binning, and extensive completeness across all chromosomal regions, including hard-to-assemble ones like centromeres. Importantly, this assembly serves as a key resource for functional genomic studies. Unlike other human assemblies typically used to benchmark population-level diversity, the RPE-1 genome has the potential to enable high-precision, haplotype-resolved analysis of sequencing data generated from this cell line. By using a reference that matches the cell line under study (isogenomic reference), it is possible to mitigate reference bias, enhance multi-omics data processing, and improve downstream analyses—particularly within polymorphic regions most affected by sequence variation, such as centromeres.
The RPE1v1.1 assembly represents a bona fide reference because RPE-1 cells have a stable karyotype, as tested using different batches of RPE-1 cells, even from different laboratories. Of note, the genome we sequenced is able to capture a faithful representation of human diversity in line with the pangenome data. Our analysis suggests that, while the RPE-1 genome retains fundamental human genomic characteristics, it has acquired unique features during its establishment and maintenance as a cell line, likely due to adaptations to in vitro culture conditions, its immortalized state, and above all, the specific chromosomal rearrangements pertaining to the t(X;10)(Xq28;10q21.2) translocation. Despite these changes, cytogenetic analyses confirm that RPE-1 maintains a remarkably stable diploid karyotype across passages and batches, with no evidence of polyploidy or other extensive chromosomal rearrangements. This genomic stability, combined with the overall proximity of RPE-1 to the HPRC genomes in whole-genome analyses, supports its continued relevance as a model for human cellular processes.
As we enter the era of personalized reference genomes, our work underscores the value of extending genome assembly initiatives to include experimentally-relevant cell lines. This proof of concept serves as the foundation for a call for a broader effort to generate high-quality assemblies of cell lines widely used across the scientific community, enabling more accurate and informative functional genomics research. Work is underway to generate reference assemblies for embryonic stem cells, allowing improved monitoring of their genomic and epigenomic transitions from pluripotency to specialized cell types. Looking ahead, we envision the expansion of cell line reference genomes as a catalyst for advancing organoid research and precision genome engineering, ultimately paving the way for the creation of comprehensive cell line pangenomes.
To our knowledge, this is one of the first genomes assembled end-to-end within a single laboratory—starting from cell culture and pellet preparation, through sequencing, to the final assembly curation. However, there is an urgent need for more streamlined and optimized protocols and tools to handle the analysis of such assemblies, including read alignment and downstream processing, which remain particularly challenging due to their diploid nature and potential genome rearrangements. New standardized protocols for production criteria, assembly pipelines, and manual curation are essential to improve the fidelity of reference-quality genome generation and to make it more accessible and automated. Among current human reference-quality assemblies, the RPE-1 reference genome is, as far as we are aware, the first one completed outside of the United States or China, highlighting the importance of increasing international efforts and collaboration. This is particularly important, as we believe that near-complete genome assemblies for every cell line will be necessary to enhance multi-omics analyses and enable epigenomic and genomic manipulations at a truly whole-genome scale across diverse experiments.
A current limitation in working with cell lines is that, while Hi-C–based haplotype phasing can resolve haplotypes across entire chromosomes, it assigns haplotype labels (Hap1 and Hap2) arbitrarily for each chromosome in the absence of parental sequence data17. As a result, we were not able to determine the maternal or parental origin of the chromosomes in RPE1v1.1. To address this, we are actively investigating epigenetic and allelic signatures—such as telomere length and methylation patterns at imprinted loci—to reduce the arbitrariness of haplotype labeling. We are confident that these approaches will ultimately enable a consistent assignment of chromosomes to their respective haplotypes and allow for maternal and paternal distinction in cell lines.
Since the RPE1v1.1 assembly still contains some gaps, low-confidence regions, and unassigned rDNA, we provide a dedicated low-confidence annotation track based on Flagger output that highlights collapsed and uncertain regions to guide users. We are actively working to enhance the genome’s overall quality through additional sequencing experiments and the implementation of advanced computational methods aimed at achieving T2T completeness for each chromosome and validating assembly accuracy, especially at centromeres38.
Despite these limitations, RPE1v1.1 offers a much-needed, reference-quality genome that supports high-precision read alignment, due to the matching of reads and reference for isogenomic mapping, and minimizes reference bias. This marks the beginning of a new era in functional genomics, where matched reference genomes tailored to experimental systems enable haplotype-resolved, whole-genome analyses with unprecedented accuracy. We anticipate that additional diploid human cell lines of interest will soon be assembled in the same way, expanding the availability of high-quality matched references for functional genomics research.
Methods
Ethics declarations
The research study has been approved by the Comitato Etico Territoriale Lazio 1 and the University of Rome “La Sapienza” Institutional Review Board (prot.3/2024 of 08.01.2024), including consent for publication of research data by Geron Corporation that holds the license for the RPE-1 cell line (ATCC-CRL-4000).
Cell lines
The hTERT-immortalized RPE (hTERT RPE-1) cells used to generate the RPE1v1.1 assembly and for karyotype analysis were validated by MSKCC; a second batch of cells, used only for cytogenetic characterization, was purchased from ATCC (CRL4000). RPE-1 cells are near-diploid human cells of female origin with 46 chromosomes. Cells were cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum (Gibco 10270-106), 100 μg/ml streptomycin, 100 U/ml penicillin (Euroclone ECB3001D) and 2 mM glutamine (Euroclone ECB3000D). Cells were grown at 37 °C in a humidified atmosphere of 5% CO2, and tested negative for mycoplasma contamination.
Metaphase spread preparation
hTERT RPE-1 cells at 70% confluence were harvested by trypsinization after 2-h treatment with colcemid (100 ng/ml, Roche 10295892001) to arrest cells in mitosis, washed with PBS and incubated with a pre-warmed hypotonic solution (0.07 M KCl) for 30 min at 37 °C. After centrifugation, swollen cells were fixed with a solution of methanol-acetic acid (3:1) and washed twice with the same fixative solution. Fixed cells were dropped onto clean, wet slides and air dried overnight. All centrifugation steps were performed at 320 × g for 5 min at room temperature (RT).
Karyotype analysis
To confirm the chromosomal structure of the assembly, a karyotype for hTERT RPE-1 cells was generated. To obtain the G-banding staining, metaphase spreads from hTERT RPE-1 cells were treated with trypsin (0.5%, Thermo Scientific 15090046), washed with 1X PBS and stained with GIEMSA (10%, Thermo Scientific 10092013). For the R-banding, metaphase spreads were washed with a phosphate buffer (0.07 M NaH2PO4, 0.07 M Na2HPO4, 1 mM NaCl, pH 6.8) followed by 2-h incubation with Chromomycin A3 (0.6 mg/ml, Sigma-Aldrich C2659) at RT in a dark, moist chamber. Slides were then washed with NaCl-HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (0.15 M NaCl, 5 mM HEPES), stained for 15 min in a methyl green solution (0.1 mM, Sigma-Aldrich) and washed twice with NaCl-HEPES. The antifading (Vectashield H-1000; Vector Laboratories) isopropilgallate 1:300 was added to the slides before storing them at 4 °C for 3 days in a dark, moist chamber. Images were acquired using a Thunder fluorescent widefield microscope (Leica) at ×100 magnification.
DNA extraction
hTERT RPE-1 cells at 70–80% confluence were harvested by trypsinization, washed with PBS and centrifuged at 1000 rpm for 5 min at RT. Cell pellets were prepared in individual aliquots of 1.5 × 106 cells, frozen in dry ice and stored at −80 °C until further use. High molecular weight (HMW) DNA and ultra-high molecular weight DNA (UHMW) was extracted from hTERT RPE-1 dry cell pellets using the Monarch HMW DNA extraction kit for cells and blood (New England Biolabs, NEB T3050L) and for tissue (NEB T3060L), following manufacturer’s instructions and introducing few technical improvements. HMW DNA was quantified by Nanodrop and Qubit with a broad range kit (Thermo Scientific Q32850). Native DNA size distribution was assessed using Femto Pulse with the Genomic DNA 165 kb kit (Agilent FP-1002-0275).
Library preparation and sequencing
Sequencing data were obtained using two complementary long-read sequencing technologies for the assembly of hTERT RPE-1 cells: PacBio HiFi long reads and ONT long reads. Additionally, Arima Genomics Hi-C and Illumina sequencing technologies were used. HMW DNA from 1.5 × 106 cells was used to generate PacBio HiFi libraries with the SMRTbell express template prep kit 2.0 (PacBio 100-938-900). Size selection was performed with Megaruptor 2 (Diagenode) with default settings, and fraction sized 15–20 kb (as determined by Femto Pulse) was sequenced on the Sequel IIe platform with SMRT Cells 8 M (PacBio 101-389-001), using the chemistry 2.0 with 2-h pre-extension, 2-h adaptive loading and 30-h movie collection time, to reach a coverage of 46× in PacBio HiFi reads. Circular Consensus Sequencing reads were obtained using SMRT-Link (https://github.com/WenchaoLin/SMRT-Link) with default parameters. UHMW DNA from 6 × 106 cells was used to generate UL ONT libraries with the UL DNA sequencing kit (ONT SQK-ULK114) following manufacturer’s instructions. 90 μl of library was loaded in a R10.4.1 (FLO-PRO114M) flow cell and sequenced on the PromethION 24, with two nucleases washes and reloads after 24 and 48 h of sequencing resulting in a total coverage of 85× from reads with an average read length of 70 kb, and of ~30× from >100 kb reads with an average Phred Q score >20. These ONT data were basecalled with Guppy v6.3.9. In parallel, HMW DNA from 6 × 106 cells was used to generate additional UL ONT libraries with the same UL DNA sequencing kit and protocol. 90 μl of library was loaded in a R10.4.1 (FLO-PRO114M) flow cell and sequenced on the PromethION 2 Solo, with two nucleases washes and reloads after 24 and 48 h of sequencing to reach a total coverage of 10× (average 19 kb), with an average Phred Q score >35. These ONT data were basecalled with Guppy v6.5.7. HMW DNA from 1.5 × 106 cells was used to generate Illumina libraries with the Nextera XT DNA Library Preparation kit (Illumina FC-131-1024) and Illumina DNA PCR-Free Library Prep, Tagmentation Library Preparation kit (Illumina 20041795) following manufacturer’s instructions. HMW DNA from 2 × 106 cells was used to generate Hi-C libraries with the Arima High Coverage Hi-C+ kit (Arima Genomics A101030) and the Arima Library Prep Module kit v2 (Arima Genomics A303011) according to the manufacturer’s protocols. Hi-C libraries were sequenced on the Illumina Nova-Seq 6000 to reach a coverage of ~30× in Hi-C reads. Raw data were processed with Cutadapt v4.0 (https://github.com/marcelm/cutadapt)39 to trim the adapters, with the option -u 5.
Whole genome assembly and manual curation
Before assembling the genome of hTERT RPE-1 cells, global genomic features such as heterozygosity, repeat content and size were evaluated with GenomeScope v2.0 (https://github.com/tbenavi1/genomescope2.0)40 from unassembled HiFi raw sequencing reads. A first pseudo-phased assembly was generated using Hifiasm v0.19.8-r603 (https://github.com/chhylp123/hifiasm) with HiFi data41,42. To integrate the long-range information from ultra-long ONT reads for scaffolding, leverage Hi-C data for phasing, and enable dual manual curation, we selected Verkko (https://github.com/marbl/verkko)18 as the assembler for the final diploid genome reconstruction. Both Verkko v1.4 and v2.0 were employed in the assembly process, with the full set of HiFi, ONT, and Hi-C reads. The Bandage v0.8.1 tool (https://github.com/rrwick/Bandage)43 was used to visualize the graphs44. The number of gaps found in the draft genomes generated by both Verkko versions was calculated with Gfastats v1.3.6 (https://github.com/vgl-hub/gfastats)45. Unassigned contigs were aligned to the CHM13 v2.0 genome using MashMap v3.1.3 (https://github.com/marbl/MashMap)46 with parameters --pi 90 -s 1000 -f map to infer their most likely chromosomal origin. Contigs aligning to rDNA arrays, as defined in the CenSat annotation (https://github.com/marbl/CHM13), were classified as rDNA sequences. Based on the results of these analyses, we decided to use the draft assembly generated by Verkko v1.4.
Following the VGP pipeline17, Hi-C reads were independently mapped to haplotype 1 and 2, as well as to the diploid assembly (including unassigned contigs, used only to obtain a comprehensive alignment), using the BWA-MEM v0.7.17-r1188 aligner47 with default parameters. The final alignment file was converted into a contact map using PretextMap v0.1.9 (https://github.com/wtsihpag/PretextMap). PretextView v2.0 (https://github.com/wtsi-hpag/PretextView) was used to visualize the contact map and to manually curate the assemblies by resolving misjoins and integrating contigs larger than 300 kb into chromosome-scale scaffolds. Following the scaffold path assignment, the final contact map was converted into an AGP (A Golden Path) file, which describes the assembly scaffold structure. This AGP file served as input for the subsequent dual manual curation phase, aimed at separating the two haplotypes. The curation was performed using the Rapid-curation-2.0 pipeline (https://github.com/Nadolina/Rapid-curation-2.0), resulting in 23 chromosome-level scaffolds for each haplotype. MashMap46 was then used with parameters -f one-to-one --pi 95 -s 10000 to perform a genome-to-genome alignment, with the CHM13 assembly used as a reference; chromosome identity was assigned based on alignments longer than 500 kb with a sequence identity greater than 99%. Chromosome orientation and structural accuracy were then assessed by generating dot plots between RPE-1 and CHM13 chromosomes using the NUCmer tool from the MUMmer v4.4 suite (https://github.com/mummer4/mummer)30 with parameters --mum -c 100 -b 500 -l 50 and mummerplot. The ideogram showing the positions of gaps after dual manual curation was generated using the RIdeogram v0.2.2 R package48.
Gaps curation
To address the gaps remaining after dual manual curation, ONT reads >100 kb were mapped back to the assembly using minimap2 v2.17-r941 (https://github.com/lh3/minimap2)31,32 with the option -ax map-ont. After removing reads with MAPQ <20 with the view utility of SAMtools v1.17 (https://github.com/samtools/samtools)49, the alignment was visualized using IGV50 and, for each gap spanned by reads aligning for at least 40 kb on both flanking regions, the longest read with the lowest edit distance was selected to replace the corresponding stretch of N bases, thus generating a temporary gap-filled assembly. Given the lower base-level accuracy of ONT reads compared to HiFi data, we subsequently realigned HiFi reads to this assembly using minimap2 with the option -ax map-hifi. The resulting alignment file was given as input to DeepVariant v1.6.1 (https://github.com/google/deepvariant)51, setting the model type to PACBIO, to call variants in gap-filled regions. Variants were then filtered to retain only those with a Variant Allele Frequency of 1 with the view tool from the BCFtools v1.10.2 suite (https://github.com/samtools/bcftools)52. The filtered VCF file was used to construct the consensus sequence using bcftools consensus.
For gaps that remained unfilled after the initial approach–particularly those with multiple possible paths in the assembly graph—we employed an alternative strategy. First, gfalign (https://github.com/vgl-hub/gfalign)45 was used to map ONT reads to the assembly graph generated by Verkko. When alignments supported a single unambiguous path through the graph bubble, the corresponding sequence of unitigs was extracted and used to fill the gap.
Evaluation of de novo genome assembly
An initial assessment of the RPE1v1.1 assembly quality was performed by aligning HiFi reads to the diploid genome using minimap2 with the option -ax map-hifi, followed by coverage visualization with the kpBars function from the KaryoploteR v1.28.0 R package (https://github.com/bernatgel/karyoploteR)53. Primary alignments were extracted using samtools view with the option -F 2308 and used to generate a NucFreq plot with the NucPlot.py script from the NucFreq v0.1 toolkit (https://github.com/mrvollger/NucFreq)22. The HetDetection.R script54 was used to identify possible errors in the assembly.
Merqury v1.3 (https://github.com/marbl/merqury)23 was used with default parameters to evaluate the assembly using 31-mer counts calculated from the HiFi reads with Meryl v1.4.1 (https://github.com/marbl/meryl/)55.
Genome completeness was evaluated using compleasm v0.2.6 (https://github.com/huangnengCSU/compleasm)26 with the primates lineage. Additional assembly metrics were computed with Gfastats v1.3.6 (https://github.com/vgl-hub/gfastats)45 and QUAST v5.2.0 (https://github.com/ablab/quast)56. Assembly errors were also evaluated using CRAQ v1.0.9 (https://github.com/JiaoLaboratory/CRAQ)24 with parameters -mgs 1000 -q 10 -avgl 45 -avgs 60 -pl T -b T, which mapped HiFi reads to the diploid genome and calculated reads coverage and clipping information to compute AQI scores and generate a circos plot with heterozigous regions and errors. The read-based pipeline Flagger v0.3.3 (https://github.com/mobinasri/flagger)25 was used to detect errors, collapsed and reliable regions within the diploid assembly. The alignment file required by the pipeline was created by mapping HiFi reads to the diploid genome using Winnowmap v2.03 (https://github.com/marbl/Winnowmap)57 with parameters -ax map-pb -Y -L --eqx --cs -I8g and repetitive 15-mer counts generated from the assembly by using meryl count with the option k = 15, followed by meryl print with the option greater-than distinct = 0.9998.
SecPhase v0.4.3 (https://github.com/mobinasri/secphase)25 was used to assess the quality of haplotype phasing based on the alignment of HiFi reads to the diploid assembly produced by minimap2. To further validate haplotype phasing, we retrieved publicly available raw reads from Strand-seq libraries relative to 80 RPE-1 cells27 and aligned them to the RPE1v1.1 diploid genome using BWA-MEM v0.7.17-r1188 with default parameters. Then, strand state changes were identified using the breakpointr function from the breakpointR v1.20.0 R package29, setting the window size of 1,000,000. This analysis was performed on two sets of alignments: the complete set, which enabled identification of informative regions—defined as genomic intervals where reads mapped to both strands in a single cell—and a filtered subset containing only reads with a minimum MAPQ score of 10, indicative of uniquely mapping reads with haplotype-specific alignment. The latter allowed us to pinpoint informative regions that support correct phasing, based on the presence of reads mapping to opposite strands on the two haplotypes. For each chromosome in each cell, the longest informative region longer than 10 Mb was selected, if present; the duplicated region on the q-arm of chromosome 10 was excluded from analysis. These results were then summarized in a heatmap indicating, for each chromosome and cell, whether Strand-seq data supported phasing (reads mapping on opposite strands on the two haplotypes), did not support phasing (same strand on both haplotypes), showed mixed behavior, or if there was no sufficiently long informative region.
Telomere presence and length were evaluated using the find_telomere.sh script from the VGP assembly pipeline (https://github.com/VGP/vgp-assembly/blob/master/pipeline/telomere/find_telomere.sh).
Multi-step identification and curation of breakpoint in phased haplotypes
To map the exact t(X;10)(Xq28;10q21.2) translocation breakpoint characteristic of hTERT RPE-1 cells10, we first examined read coverage by independently aligning HiFi and ONT reads to each haplotype of the RPE-1 genome using minimap2 with the -x parameter set to map-hifi and map-ont, respectively. The reads were then aligned against the diploid genome to identify the haplotype with the translocated chromosome X. Chimeric alignments involving sequences from both chromosomes 10 and X were used to pinpoint the breakpoint location. Based on this evidence, the marker X chromosome containing the t(X;10)(Xq28;10q21.2) translocation was manually reconstructed. To validate this curation, reads were realigned to the resulting FASTA file, confirming the presence of contiguous breakpoint-spanning alignments with >99% identity.
pGRN145 plasmid insertion
The sequence of the pGRN145 plasmid containing the hTERT was retrieved from the ATCC website10. The hTERT sequence was aligned against the RPE1v1.1 genome with NUCmer with parameters --mum -l 50 and minimap2 with default parameters; alignment results were visualized using mummerplot and IGV, respectively. For both alignments, the only match between the hTERT sequence and the genome was within chromosome 20 Hap2. When realigning reads originally mapping to Hap1 onto Hap2 with minimap2, a gap in read alignment was found within a 15 kb region surrounding the insertion site. Based on this information, a 15 kb region from chromosome 20 Hap2 was extracted, and the complete plasmid sequence was reconstructed using SnapGene software (www.snapgene.com). During the submission of the genome to NCBI, we were asked to mask the plasmid sequence (genomic coordinates chr20_hap2: 3946958-3962318) due to it being flagged as non-human DNA.
Identification of structural variants and genome annotation
The genome-to-genome alignment files required to run SyRI were produced using minimap2 with the parameter -x asm5 for genomes with low sequence divergence. SyRI v1.6.3 (https://github.com/schneebergerlab/SyRI)33 was used with default parameters to compare Hap1 (reference) versus Hap2 (query), CHM13 versus Hap1, and CHM13 versus Hap2. SyRI outputs the complete information about structural variants and genomic rearrangements between two genomes, such as syntenic regions, copy number variation and HDRs, as well as SNPs and indels. Final visualization was obtained with Plotsr v1.1.1 (https://github.com/schneebergerlab/plotsr)58. The positions of SNPs between Hap1 and Hap2 were extracted from the SyRI TSV output file and used to calculate SNP densities across 10 kb genomic bins. These densities were visualized along HDR coordinates using KaryoploteR. The average SNP density in chromosome arms and live HORs was calculated using only bins with more than 50% overlap with syntenic, inverted or translocated regions identified by SyRI.
Whole-genome alignments were also generated using the NUCmer tool from the MUMmer v4.4 suite with parameters --maxmatch -c 100 -b 500 -l 50. The resulting alignments were filtered using delta-filter with options -m -i 99 -l 100000. Summary statistics were then computed with the dnadiff utility.
Liftoff v1.6.3 (https://github.com/agshumate/Liftoff)59 was used to map genes from the human transcriptome annotation of ENSEMBL 112 (GRCh38.p14 genome) to the RPE1v1.1 genome. Centromeres were annotated using the HumAS-HMMER_for_AnVIL tool (https://github.com/fedorrik/HumAS-HMMER_for_AnVIL), which outputs a BED file containing the locations of α-satellite higher-order repeat (HOR) arrays. From this file, we extracted the coordinates of live HOR arrays, marked by the character L.
Pangenome comparative analysis
We combined assemblies from the draft human pangenome reference25 with the RPEv1.1 assembly and then built chromosome-specific pangenome graphs with PGGB60 (commit: a15e350). We performed pairwise alignments with the wfmash sequence aligner (https://github.com/waveygang/wfmash; commit: 251f4e1), requiring homologous regions at least 50 kb long and nucleotide identity of at least 98%. We used the alignment between all assemblies to build unbiased pangenome graphs with the seqwish variation graph inducer61 (commit: 75e807c), ignoring alignment matches shorter than 23 bp (to remove possible spurious relationships caused by short repeated homologies). We used the ODGI toolkit62 (commit: 679e65e) to generate a comprehensive graph of all chromosomes and compute graph-based Jaccard distances between haplotypes. Data visualization and analysis, including the creation of graph-based dendrograms and PCA were performed with the R development environment (version 4.4.1), equipped with the following packages: tidyverse (version 1.3.1), ggplot2 (version 3.5.1), ggtree (version 3.12.0), ggrepel (version 0.9.5), and ape (version 5.8). For the all-chromosomes and chrX-specific analyses, we removed 17 HPRC haplotypes that behaved as strong outliers in chromosome X: HG00621#1, HG00673#1, HG01106#1, HG01109#1, HG01243#1, HG01258#1, HG01358#1, HG01928#1, HG01952#1, HG02055#1, HG02145#1, HG02486#1, HG02572#1, HG02717#1, HG03098#1, HG03492#1, and HG03579#1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The CHM13 v2.0 and GRCh38.p14 genome assemblies used in this study are available at NCBI under the GenBank assembly accession GCA_009914755.4 [https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_009914755.1] and GCA_000001405.29 [https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.40/], respectively. The HPRC pangenomes used in this study were downloaded from the human pangenomics S3 bucket (https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=pangenomes/freeze/freeze1/pggb/chroms/). The RPE1v1.1 genome generated in this study has been deposited in NCBI GenBank under the accession numbers JBJQNK000000000 (Hap1) and JBJQNL000000000 (Hap2), with links to BioProject accession numbers PRJNA1193286 (Hap1) and PRJNA1193302 (Hap2), both under the umbrella BioProject accession number PRJNA1195024. The versions described in this paper are JBJQNK000000000.1 and JBJQNL000000000.1. The genome can be visualized in the UCSC genome browser at https://genome.ucsc.edu/h/GCA_050656315.1 (Hap1) and https://genome.ucsc.edu/h/GCA_050656345.1 (Hap2). Release of the RPE-1v1.1 reference genome has been approved for public access by Geron Corporation. The HiFi, ONT and Hi-C sequencing data used to generate the genome have been deposited in SRA under the accession numbers SRR33464826, SRR33464827, SRR33464828, and SRR33464829 (HiFi), SRR33464817, SRR33464818, SRR33464819, SRR33464820, SRR33464821, SRR33464822, SRR33464823, SRR33464824, SRR33464830, and SRR33464831 (ONT), and SRR33464825 (Hi-C) with links to BioProject accession number PRJNA1193286. Annotation tracks for the RPE1v1.1 genome, including the low confidence RPE1v1.1 regions, and additional processed data are available and documented at the GitHub repository https://github.com/GiuntaLab/RPE1 and at the linked Zenodo repository https://doi.org/10.5281/zenodo.1578991363.
Code availability
Custom scripts and tools used to generate the figures of this manuscript are available and documented at the GitHub repository https://github.com/GiuntaLab/RPE1 and at the linked Zenodo repository https://doi.org/10.5281/zenodo.1578991363.
References
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Jarvis, E. D. et al. Semi-automated assembly of high-quality diploid human reference genomes. Nature 611, 519–531 (2022).
Yang, C. et al. The complete and fully-phased diploid genome of a male Han Chinese. Cell Res. 33, 745–761 (2023).
He, Y. et al. T2T-YAO: a telomere-to-telomere assembled diploid reference genome for Han Chinese. Genom. Proteom. Bioinform. 21, 1085–1100 (2023).
Wang, T. et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022).
Bodnar, A. G. et al. Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352 (1998).
Jiang, X. R. et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet. 21, 111–114 (1999).
hTERT RPE-1. ATCC Genome Portal: https://www.atcc.org/products/crl-4000.
Koren, S. et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 36, 1174–1182 (2018).
Janssen, A., van der Burg, M., Szuhai, K., Kops, G. J. P. L. & Medema, R. H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333, 1895–1898 (2011).
Soto, M. et al. p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep. 19, 2423–2431 (2017).
Giunta, S. & Funabiki, H. Integrity of the human centromere DNA repeats is protected by CENP-A, CENP-C, and CENP-T. Proc. Natl. Acad. Sci. USA 114, 1928–1933 (2017).
Giunta, S. et al. CENP-A chromatin prevents replication stress at centromeres to avoid structural aneuploidy. Proc. Natl. Acad. Sci. USA 118, e2015634118 (2021).
Tourdot, R. W., Brunette, G. J., Pinto, R. A. & Zhang, C.-Z. Determination of complete chromosomal haplotypes by bulk DNA sequencing. Genome Biol. 22, 139 (2021).
Larivière, D. et al. Scalable, accessible and reproducible reference genome assembly and evaluation in Galaxy. Nat. Biotechnol. 42, 367–370 (2024).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 41, 1474–1482 (2023).
Antipov, D. et al. Verkko2 integrates proximity-ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding. Genome Res. 35, 1583–1594 (2025).
Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021).
Rhie, A. et al. The complete sequence of a human Y chromosome. Nature 621, 344–354 (2023).
Vollger, M. R. et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94 (2019).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat. Commun. 14, 6556 (2023).
Liao, W. W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39, btad595 (2023).
Sanders, A. D. et al. Single-cell analysis of structural variations and complex rearrangements with tri-channel-processing. Nat. Biotechnol. 38, 343–354 (2020).
Falconer, E. et al. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 9, 1107–1112 (2012).
Porubsky, D. et al. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics 36, 1260–1261 (2020).
Marçais, G. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277 (2019).
Choo, K. H. Why is the centromere so cold? Genome Res. 8, 81–82 (1998).
Balzano, E. & Giunta, S. Centromeres under pressure: evolutionary innovation in conflict with conserved function. Genes 11, 912 (2020).
Trowsdale, J. & Knight, J. C. Major histocompatibility complex genomics and human disease. Annu. Rev. Genom. Hum. Genet. 14, 301–323 (2013).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Corda, L. & Giunta, S. Chromosome-specific centromeric patterns define the centeny map of the human genome. Science 389, eads3484 (2025).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Limasset, A., Cazaux, B., Rivals, E. & Peterlongo, P. Read mapping on de Bruijn graphs. BMC Bioinform. 17, 237 (2016).
Formenti, G. et al. Gfastats: conversion, evaluation and manipulation of genome sequences using assembly graphs. Bioinformatics 38, 4214–4216 (2022).
Jain, C., Koren, S., Dilthey, A., Phillippy, A. M. & Aluru, S. A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 34, i748–i756 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, e251 (2020).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Gel, B. & Serra, E. karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090 (2017).
Mc Cartney, A. M. et al. Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. Nat. Methods 19, 687–695 (2022).
Miller, J. R. et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24, 2818–2824 (2008).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710 (2022).
Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 2922–2926 (2022).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
Garrison, E. et al. Building pangenome graphs. Nat. Methods 21, 2008–2012 (2024).
Garrison, E. & Guarracino, A. Unbiased pangenome graphs. Bioinformatics 39, btac743 (2023).
Guarracino, A., Heumos, S., Nahnsen, S., Prins, P. & Garrison, E. ODGI: understanding pangenome graphs. Bioinformatics 38, 3319–3326 (2022).
Volpe, E., Colantoni, A., Corda, L., Guarracino, A. & Giunta, S. The reference genome of the human diploid cell line RPE-1. GiuntaLab/RPE1: v1.0. Zenodo. https://doi.org/10.5281/zenodo.15789913 (2025).
Acknowledgements
We thank Valentina Liguori and all members of the Giunta lab, as well as Alessandro Paiardini, Beniamino Trombetta, Fulvio Cruciani at University of Rome Sapienza, for helpful discussions and insights. We are thankful to T2T and HPRC members for the kind advice, especially Sergey Koren with Verkko, Nadolina Brajuka and members of Erich Jarvis lab for Hi-C data integration. We are grateful to Nicolas Altemose and Altemose lab members, as well as Riccardo Paone, Isabella Baldini, Juan Caceres and Noemi Di Sabatino at Dante Labs, and Margherita de Gaspari at Area Science Park, for the generous sharing of insight, space and resources. We thank Alessandra Bonito Oliva at Rockefeller University, as well as Sai Swaroop Chittoor, Giulio Marano, Veronica Marselli and Alessia Spurio, for critical reading of the manuscript and providing feedback. All computing was possible thanks to CINECA, and the Terastat2 HPC, with important support from Umberto Ferraro Petrillo and Edoardo Bompiani of the Statistics Department, University of Rome Sapienza. This work in the Giunta Lab was possible thanks to the Italian Foundation for Cancer Research (AIRC Start-Up Grant 2020 ID. # 25189), the ERC CENTROFUN Starting Grant #101078838, RICERCA_SAPIENZA CUP B83C24001270005-2024 and B83C23007030005-2023, and the Rita Levi-Montalcini program from the Italian Ministry of University and Research (MIUR) to S.G. E.V. was supported by the REACT-EU PON Ph.D. Fellowship through Italian Ministerial Decree no. 1061/2021. G.F. is supported by NIH NHGRI Grant U01HG010971.
Author information
Authors and Affiliations
Contributions
S.G. conceived the idea underlying this work; S.G. and E.V. designed the study with key input from A.C. E.V. and D.L. generated all sequencing data, with supervision from R.O. and M.C. for PacBio data. E.V. and A.C. performed all computational analyses, with support from L.C. and A.G. E.D.T., S.G., and F.P. designed and performed cytogenetic experiments. S.G. supervised experiments and analyses with key support from E.T., L.F., and G.F. S.G. and A.C. wrote the manuscript together with contributions from E.T., L.C., E.V., and A.G. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Volpe, E., Colantoni, A., Corda, L. et al. The reference genome of the human diploid cell line RPE-1. Nat Commun 16, 7751 (2025). https://doi.org/10.1038/s41467-025-62428-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62428-z
This article is cited by
-
A genomic and epigenomic view of human centromeres
Nature Reviews Genetics (2026)
-
Heterochromatin boundaries maintain centromere position, size and number
Nature Structural & Molecular Biology (2026)
-
Cell line-matched reference enables high-precision functional genomics
Nature Communications (2025)
-
40 years of CENP-A: the foundation of a new era of centromere biology
Chromosoma (2025)
-
40 years of CENP-A: the foundation of a new era of centromere biology
Chromosome Research (2025)
