
Abstract
A substantial portion of lung cancer-associated genetic elements in East Asian populations remains unidentified, underscoring the need for large-scale genome-wide studies, particularly on non-coding regulation. We conducted a whole genome sequencing (WGS)-based genome-wide scan in 13,722 Chinese individuals to identify regulatory elements associated with lung cancer. We verified common-variant-based loci by meta-analysis across the available East Asian studies. Integrating a genome-transcriptome reference panel of lung tissue in 297 Chinese, we bridged the variant-lung cancer associations, highlighting genes including TP63 and DCBLD1. Implementing the STAAR pipeline for rare variant aggregate analysis, we identified and replicated novel genes, including PARPBP, PLA2G4C, and RITA1 in the context of non-coding regulation. Adapting a deep learning-based approach, potential upstream regulators such as TP53, MYC, ZEB1, and NFKB1 were revealed for the lung cancer-associated genes. These findings offered crucial insights into the non-coding regulation for the etiology of lung cancer, providing additional potential targets for intervention.
Similar content being viewed by others

Introduction
Lung cancer is the most frequently diagnosed cancer and a leading cause of death in China and worldwide1,2, posing a severe threat to population health. Germline variants play an important role in lung cancer development, with an estimated heritability of 18%3. Substantial progress has been made through large-scale genome-wide association studies (GWAS) that have uncovered over 40 lung cancer-associated loci4,5,6,7.
However, on the one hand, the molecular mechanisms underlying these associations remain largely inconclusive8. Given the fact that most of the trait-associated variants are located in non-coding regions8,9, gene expression regulation is considered as one of the major steps for filling the knowledge gap between genetic variants and complex traits8,10. The Genotype-Tissue Expression (GTEx) project provided a great multi-omics reference panel for genome and transcriptome data in ancestrally Europeans and Africans10, while there is limited data available for a multi-omics reference panel of a similar size in Asian populations. Comprehensive multi-omics-informed causal element mapping has proven to be a desirable and imperative tool to reveal the “missing biology” for variant-lung cancer associations8,11.
On the other hand, the genome-wide significant common variants can only account for a limited fraction of the narrow sense heritability of lung cancer4,5,6, leaving a substantial portion of “missing heritability” unexplained. Emerging evidence suggests that rare variants might play a nontrivial role in most complex traits12,13. Only a limited number of single-nucleotide variants (SNVs) have been discovered through chip-based and whole exome sequencing (WES)-based studies14,15. Whole genome sequencing (WGS)- based scans could capture the entire spectrum of variants, but the main focuses have often been placed on putative loss-of-function (pLoF) variants. The role of rare variants located in non-coding regions, which cover more than 97% of the genome, remained largely unexplored16. Hence, large-scale WGS-based studies are crucial to explore the influence of rare variant-based elements on lung cancer, especially in the Chinese population.
To address the above challenges, we performed an extensive WGS study in a total of 13,722 Chinese individuals with a particular emphasis on non-coding regulation. We validated several common variant-trait associations and further projected them to the transcript level by integrating a genome and transcriptome reference panel with 297 samples from normal lung tissues. Rare variant-based gene-centric and fixed/dynamic sliding window analyses were performed to identify potential predisposition genes for lung cancer using the STAAR pipeline16,17,18. Furthermore, a deep learning-based in silico fine-mapping approach was implemented to identify replicable lung cancer-associated elements and potential upstream regulators.
Results
Study overview
Figure 1 shows the overall study design. In the discovery stage, we sequenced the whole genome for 11,058 Chinese subjects and performed a full minor allele frequency (MAF) spectrum scan on lung cancer-associated genetic elements. We sequenced an additional 3055 Chinese subjects to verify the findings. We also measured the gene expression levels in 297 normal lung tissue samples from lung cancer patients and generated a multi-omics reference panel to understand the biology of lung cancer-associated genetic elements.

A genome-wide scan was performed to probe lung cancer-associated elements across the whole allele frequency spectrum. A genome-wide association study was carried out to identify trait-associated common variants. Informed by the SNP-expression regulation in lung tissues, the variant-level signal was projected to the gene level by a transcriptome-wide association study. Following the STAAR pipeline, lung cancer-associated genes were revealed by rare variant-based gene-centric and fixed/dynamic sliding window analyses. Fine-mapping and enrichment analyses were implemented to provide additional insights into the etiology and genetic architecture.
For common variants, variant-level analysis was performed to identify lung cancer-associated loci using a generalized linear mixed-effect model17. The GWAS results were then meta-analyzed in ancestrally East Asian populations18,19. Conditional and joint analysis was implemented to identify independent trait-associated variants20. A transcriptome-wide association study21 probed lung cancer-associated genes by assuming gene expression regulation as the mediator between genetic variants and lung cancer.
For rare variants, we followed the STAAR pipeline22,23,24 and grouped the variants into multiple annotation-informed coding and non-coding regions25. Gene-centric and sliding window analyses were implemented by pooling variants with the same annotation category to boost the statistical power. An omnibus test, STAAR-O, was used to aggregate multiple annotation-weighted burden test, SKAT, and ACAT-V in the STAAR framework23. Novel candidate genes were identified and replication. Leveraging the large-scale transcriptional factor (TF) footprint and multiple ChIP-seq data, we performed in sillico fine-mapping to identify potentially causal variants and related regulatory elements26,27. Enrichment analyses were conducted to reveal lung cancer-related cell types by incorporating scRNA-seq data28. Finally, we studied the architecture of lung cancer-associated molecular elements by integrating evidence across multiple omics29.
Variant calling, quality control, and a landscape of variants
Following the GATK pipeline30, the sequencing data were processed. The mean sequencing depth was 21.23×. Samples with high contamination rates, duplication rates, or low coverage were removed (details in “Methods”). Joint calling was performed by including an additional 50 CHB (Han Chinese in Beijing)/CHS (Han Chinese South) samples from 1000 genome project phase 331 (sequenced by the same platform) and 65 downsized samples. A total of 143,124,918 (Ts/Tv = 1.961) from 11,006 samples were identified after joint calling. Excess heterozygosity filtration and Variant Quality Score Recalibration (VQSR) were conducted, leaving 115,881,216 variants (Ts/Tv: 2.061). Then, we calculated the genotype posteriors, refined the genotype (GT), and updated the Genotype quality (GQ). Variants with imbalanced allele counts were removed. After setting GT with either low GQ or low DP (depth) to missing, variants with a call rate <90% were filtered out. Genotype refinement was performed for 94,261,918 biallelic variants (Ts/Tv: 2.092). Monomorphic variants and variants with Phwe < 1.0 × 10−6 were removed. Potential monozygotic (or duplicated samples) and heterozygosity outliers (± 6 standard deviations, SD) were removed. By manually reviewing the updated pathological diagnosis and self-reported questionnaires in the recent follow-up, 1104 lung cancer (non-small cell lung cancer, NSCLC) cases and 9635 cancer-free controls with 90,399,472 autosome biallelic variants (Ts/Tv: 2.096) were finally included as the discovery set. Detailed information can be found in “Methods” and Supplementary Fig. 1. A similar process was conducted in the replication stage (Supplementary Fig. 2), resulting in 1487 NSCLC cancer cases and 1496 cancer-free controls. The basic characteristics of the subjects are described in Table 1.
Taking the officially released genotype of the 50 CHB/CHS samples from the 1000 Genome Project31 as the ground truth, we evaluated the genotype concordance between the variants that we called and the truth (“Methods”). Consistent with previous studies32, the non-reference concordance rate of our study improved from 94.49% to 99.60% after genotype refinement, indicating the effectiveness of the refinement, especially for samples with low sequencing depth (Fig. 2a, b). As expected, we found that a higher sequencing depth greatly improved the ability to identify rare variants, as compared with common variants (Fig. 2c, d).

The blue and red dots denote the 50 CHB/CHS samples before and after refinement (a: SNP, b: INDEL). Refinement largely increased the variant concordance (y-axis) between the officially released genotype and the genotype called by the current pipeline, especially for samples with low sequencing depths (x-axis). c, d show the association (linear regression) between the number of variants (x-axis) for each sample and the sequencing depth (y-axis) for relatively common variants (P = 2.78 × 10−12) and rare variants (P = 1.00 × 10−599), respectively. Principal component analysis shows the genetic background of the study samples by pooling the participants from the 1000 genome projects phase 3 in (e). In (f), the PC1 and PC2 were visualized for lung cancer cases and controls in the discovery stage and the replication stage, respectively.
Among all variants identified in this study, 28.02 million SNPs and 2.86 million INDELs (insert and deletion) had not been reported in either the ChinaMap reference dataset or in the dbSNP build 154. Based on the results of functional annotation, 57.0% and 25.8% of variants were located in intronic and intergenic regions, respectively. Missense and synonymous variants accounted for 60.7% and 34.7% of the variants in coding regions, respectively. The subsequent categories were frameshift variants (1.8%), stop-gained variants (1.6%), and inframe insertion/deletion (0.9%). We also used SIFT33 and PolyPhen234 to predict the effect of the change on protein function. A large portion of nonsynonymous variants (41%) was annotated as deleterious variants by SIFT, and 27.1% of non-synonymous variants were predicted to be damaging (Table 1). These results expanded the diversity of low-frequency and rare genetic variants in the Chinese population.
To locate the genetic background of the study participants, principal component analysis (PCA) was performed using linkage disequilibrium (LD)-pruned common variants. Samples in the current study were projected to the region overlapping EAS (East Asia) samples from the 1000 genome project31 (Fig. 2e). The PC1 and PC2 were also visualized for lung cancer cases, and for controls in the discovery stage and the replication stage, respectively (Fig. 2f). No evidence of potential population stratification for the study subjects was observed (Fig. 2f).
Altogether, these results showed the effectiveness of refinement and quality control for variant calling and verified the genetic background of the study participants.
GWAS for NSCLC and meta-analysis
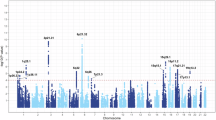
We performed a genome-wide association study for NSCLC using common variants (MAF ≥ 0.01) and a generalized linear mixed-effect model implemented in fastGWA17,35. As shown in the Manhattan plot (Supplementary Figs. 3 and 4 and Supplementary Data 1), 19 variants (involving two loci located near TERT [5p15] and TP63 [3q28]) were identified to be associated with NSCLC at the genome-wide level (P = 5.0 × 10−8). Among the 19 variants, 13 were successfully genotyped in the replication stage. The identified genetic variant-NSCLC associations were all validated in the replication dataset (P < 0.05/13) with a concordant direction of effect. Then, we combined our results with the GWAS results for lung cancer from Biobank Japan19 using fixed-effect meta-analysis. Except for the HLA region, we further verified four loci, namely 6q22.1, 10q25.2, 12q13.13, and 19q13.2, that were associated with lung cancer in 7035 cases and 185,413 controls (Fig. 3a). No evidence for inflation was found (lambda = 1.054, estimated by the median of Chi-squared, intercept estimated using ldsc = 1.033, Fig. 3b). Conditional and Joint Analysis (COJO) identified 14 independent trait-associated variants (Supplementary Data 2). For the genome-wide significant independent variants, we adjusted for NSCLC-associated variants reported in previous studies4,7,19. As we expected, we didn’t observe any signals from common variants that were independent of reported ones.

The meta-analyzed genome-wide association results (generalized linear mixed-effect model) were displayed in a Manhattan plot and a QQ plot in (a and b), respectively. The Manhattan plot for the transcriptome-wide association study (linear regression model) projected the SNP-level signal to the gene level by assuming gene expression regulation as the mediator. Each tested gene-tissue/cell type pairs were marked as a dot or a hollow diamond if the significance passed the multiple testing adjustment. Genes with top-ranked associations were labeled, followed by the source from where the gene expression model was trained (c). The raw p-values are presented in (a–c). For each gene, we only labeled the one with the lowest p-value among tissue or cell types. The correlation between predicted and observed gene expression of TP63 in lung tissue samples was visualized in (d). Each dot denotes a lung tissue sample.
Transcriptome-wide association study probes lung cancer-associated genes
A transcriptome-wide association study (TWAS) was performed to project the variant-level signal to the transcript/gene level36. TWAS is generally viewed as an in vivo knockdown for genes of interest. Specifically, a set of germline regulatory variants naturally “knocked down” the gene expression for certain subjects. i.e., subjects with lower Genetically Regulated eXpression (GReX), were compared with the ones with higher GReX21. We measured the gene expression level in 297 normal lung tissue samples from lung cancer patients and bridged the genetic variants to the risk of lung cancer. The RNA-seq data processing was detailed in the “Methods”. A total of 6388 genes were found to be predictable according to the commonly used definition (r > 0.1 and P < 0.05 for the association between predicted and observed expression in cross-validation). Supplementary Fig. 5 shows the distribution of r for prediction accuracy.
GWAS summary statistics-based TWAS projected the meta-analyzed SNP-level GWAS results to the gene level and identified eight genes associated with lung cancer (Fig. 3c). The GReX of TP63 and CLDN18 were found to be associated with a lower risk of lung cancer (P = 5.13 × 10−13 and P = 7.39 × 10−7, respectively). The TWAS analysis indicated FOXP4 as a potential oncogene (P = 1.63 × 10−9). Notably, genetic variants explained 4.8%, 24.8%, and 20.3% of the expression variance for TP63 (Fig. 3d), CLDN18, and FOXP4 in lung tissues, respectively (Supplementary Data 3).
We also trained the gene expression prediction models in immune cells from East Asian samples using cis-eQTL summary statistics37. We demonstrated the prediction accuracy by comparing individual-level data-based model training and PRS-CS-based38 model training approaches (Supplementary Fig. 6). The GReX of 26 gene-cell type pairs (13 unique genes) was found to be associated with the risk of lung cancer. Genes including DCBLD1 were identified in multiple immune cell types. We also observed a few genes (including P3H2 and RSPH4A) that only showed significant associations in Type 1T helper (Th1) cells and Type 2T helper (Th2) cells, respectively, indicating cell-type dependent regulation.
We further performed differential gene expression (DGE) analysis in 346 lung cancer tumor samples and 401 normal samples using a negative binomial generalized linear model implemented in edgeR39 (“Methods”). Genes with TPM > 1 in at least 20% samples were included. The Fold-change and P-values were reported in Supplementary Data 4. The DGE analysis supported the potential role of multiple genes, including DCBLD1 (tumor vs. normal logFC [log-fold change] = 0.63, P = 2.16 × 10−54), in lung cancer development.
Rare variant-based analysis
Following the STAAR pipeline22,23, we annotated the low-frequency and rare variants (MAF < 0.01) into five categories for coding regions and seven categories for non-coding regions25 (“Methods”). Gene-centric analysis was performed for each gene by pooling all the variants in each category. Genes with lower than 10 cumulative minor allele counts (cMAC) in the lung cancer group or with less than 10 variants were filtered out. In the discovery set, 147 genes were found to be associated with the risk of lung cancer (FDR < 0.1, stratified by categories, Fig. 4a, b)40. In the replication set, the associations for nine genes (ENO1, EFHD2, MACF1, LRRFIP2, PSMB9, C11orf21, RAD52, RITA1, and PLA2G4C) were replicated (nominal P < 0.05, Fig. 4a, b) in the matched category. i.e., in the replication stage, significant associations were observed in the same category compared with the category in the discovery stage (we merged the two promoter categories and the two enhancer categories). Among the nine replicated associations, the significance of ENO1 (enhancer category, Pdiscovery = 1.52 × 10−4, Preplication = 2.92 × 10−3) and PLA2G4C (promoter category, Pdiscovery = 5.67 × 10−5, Preplication = 7.66 × 10−4) passed the stratified FDR correction40 in the replication stage. It is worth noting that seven of the nine (77.8%) replicated genes showed significance in non-coding categories (Table 2 and Supplementary Data 5). We also observed replicable associations for 27 genes in the replication stage. i.e., nominally significant associations were observed in a different category in the replication stage. Among the 27 genes, nominally significant associations were detected in six genes using WES-based SKAT-O/burden test from the UK biobank samples12 (Table 3 and Supplementary Data 6). We note here that the UKB results were considered as a line of informative evidence, not as a formal replication, since no adjustment across categories was applied to the UKB dataset. The final significance was reported by combining the p-values using Fisher’s method (Supplementary Data 7).

a shows a flowchart for the rare variant-based analyses in the discovery and the replication stages. The -log(P) of the gene-centric STAAR-O test results in the discovery stage were visualized in (b) (the raw p-values are presented). Each dot denotes a gene-category pair. Hollow triangles denote genes that passed the stratified FDR correction in the discovery stage and remained nominally significant either in the replication stage or in the UK Biobank WES-based results. Categorically matched (between the discovery and the replication stages) replicable genes were labeled with gene names and their categories. c displays the enhancer enrichment analysis results for the lung cancer-associated segments and their related genes identified by fixed and dynamic window-based tests. Two segments near FRMD6 were merged since they share a large proportion of positions. The enrichment test was performed to evaluate the representative levels of a segment overlapping an enhancer, given the background signal in multiple lung and immune-related cell lines.
Additionally, we performed the analysis using the cross-category pairs of each finding by pooling variants with annotations from the discovery stage and the replication stage. As shown in Supplementary Data 8, the statistical significance is highly consistent with the results using only the annotation from the discovery stage.
In the context of up and down gene expression regulation in tumor and normal tissues, among the 31 genes identified by gene-centric analysis and TWAS, 30 of them (96.8%) were differentially expressed (FDR < 0.05) between tumor and normal tissues (Supplementary Data 4).
Our results also supported some genes identified by Wang et al. in the context of rare coding variants16. Wang et al. reported 13 NSCLC-associated genes (leading by BRCA2 [P = 1.52× 10−05] and FANCA [P = 8.13× 10−04]) based on pLoF variants (nominal P < 0.05). We extended the pLoF category to all the available coding categories to provide a more informative result. In addition, we implemented the STAAR-O approach, which incorporates the burden test while considering rare variant models consistent with the assumptions of the SKAT test. Among the 13 genes, we successfully replicated 4 (namely NBN, WRN, FANCA, and GEN1) of the 13 genes with nominal P < 0.05. The results are detailed in Supplementary Data 9.
We then conducted fixed and dynamic window analyses to identify lung cancer-associated segments. To identify potential regulatory segments located in the gene desert, we scanned the genome by moving a window with no awareness of the location of genes and didn’t skip any intergenic regions. In the discovery stage, 28 fixed windows (sliding window) and 15 dynamic windows (SCANG) were probed after controlling the genome-wise (family-wise) error rate (GWER/FWER) for window-based analysis at α = 0.05 level using Bonferroni correction. In the replication stage, consistent results were observed in 4 of the 43 segments (Table 4). The identified four segments were in the intron of CMIP, intron of PRKG1, downstream of CSTF2T, and intron/upstream of FRMD6. Interestingly, high enhancer enrichment scores41 were observed, especially for the segment in CMIP, in multiple lung and immune-related cell lines, indicating potential regulatory roles of the identified segments (Fig. 4c and Supplementary Data 10).
Stratified analysis was conducted to identify lung cancer-associated genes in smokers and non-smokers. Among the top-ranked genes identified in the discovery stage, marginally significant associations were observed for MYO5B (P = 0.061, promoter/enhancer) and AUTS2 (P = 0.089, synonymous) in the replication stage. The results are detailed in Supplementary Data 11. We also performed the analysis for lung adenocarcinoma. Four genes, namely HOXA2, LGR6, ULBP2, and EDN3, were identified in the discovery stage. However, significance was not observed in the replication stage (P > 0.05, Supplementary Data 12). Lung squamous cell carcinoma-specific analysis was not performed due to limited cases.
In silico fine-mapping identifies potential causal regulators
Considering the large proportion of non-coding genetic elements identified in the study, we performed a deep learning-based fine-mapping to probe causal variants and their related regulators by implementing the DeepSEA/Sei framework26,27. The posterior probability of a segment showing regulatory signal binds to TF was estimated. Genetic variants with large differences in posterior probability for reference allele and alternative allele were prioritized as potential causal variants. We filtered out the variants with a difference in posterior probability lower than 0.1 for TF in lung or immune-related cell lines. Among the rare variants in replicable genes or segments, we found that the binding profiles of TP53, ZEB1, MYC, and NFKB1 might be affected by SNVs located in EFHD2 with enhancer mask (Fig. 5a). The potential interplays between ENO1 and TFs including FOXA1, FOXA2, and MYC were revealed by assuming the variants with a promoter mask in the ENO1 region affected TF binding sequencing (Fig. 5b). Similar patterns were observed for PLA2G4C (Fig. 5c) and RITA1 (Fig. 5d), another two replicated genes in rare variant-based genetic analysis. For example, DNA segments with different alleles for a rare variant (MAF = 4.65 × 10−05) located at 19:48061591(GRCh38) showed a different binding affinity for both FOXA1 and FOXA2 in the lung cancer cell line A549 (Supplementary Data 13). Notably, this variant itself showed a promising association with lung cancer risk (P = 3.93 × 10−03). Additional TF-gene links were presented in Supplementary Fig. 7. Variants showing potential perturbations on TF binding were detailed in Supplementary Data 13 and 14. These analyses provided informative evidence for potential candidates as upstream regulators for the lung cancer-associated genes.

For lung cancer-associated genes that showed concordance signals in both the discovery and the replication stage, we performed causal variant mapping to identify potential upstream regulators. For each gene, transcriptional factors (TFs) whose binding motif may be affected by single-nucleotide variant (SNV) were linked to the gene with blue lines. The darker blue lines indicate more instances from different SNVs or from different lung/immune-related cell lines. a–d show the results for EFHD2, ENO1, PLA2G4C, and RITA1, respectively.
Cell type-specific gene expression enrichment analysis
To prioritize the cell types specifically linked to lung cancer-associated genes identified in this study, we carried out a scRNA-seq-informed enrichment analysis for genes identified by rare variant-based analysis. ScRNA-seq data from lung tissue samples were reanalyzed for cell-type-specific expression analyses28. Cell subpopulations were clustered based on the first 30 PCs generated from the top 2000 highly variable genes. The permutation test was performed to estimate the P-value for enrichment, which is defined by the probability of observing scores from 10,000 times random draws greater than or equal to a score generated by empirical data. Significant enrichment was observed in proliferating T cells (P = 0.005), featured by PARPBP and ENO1. PARPBP showed high cell type specificity in proliferating T cells (Fig. 6a). Informed by C11orf21 and EFHD2, significant enrichment was also observed for NK cells (P = 0.004). The enrichment analysis prioritized specific cell types for lung cancer-associated genes in a non-coding regulation framework.

a By incorporating scRNA-seq data, cell type-specific enrichment analysis was performed to prioritize lung cancer-related cell types given the identified genes. The top heatmap shows the enrichment score for each gene-cell type pair. Hierarchical clustering was performed. The bottom row shows the significance of the enrichment estimated by the one-sided permutation test (“Methods”). b Implementing the Gene Set Enrichment Analysis (GSEA), we revealed the convergences of lung cancer-associated genes identified by WGS-based study and studies in other design or using other omics data. The heatmap shows the significance (raw p-values) of enrichment between category-specific gene sets identified in the current WGS-based study and the genes reported from the COMIC CSC gene list (with the prefix “cosmic”), GWAS catalog (with the prefix “GWAS”), differential gene expression analysis between tumor and normal lung tissues (up denotes a higher expression level in tumor samples), TWAS from blood and lung tissues, and somatic copy number variations in lung cancer samples (b). The WES-based UK Biobank gene-centric results were also included.
The architecture of lung cancer-related genes
To address the question of to what extent the rare variant-informed lung cancer-associated genes were significantly enriched in the gene lists identified by other approaches, we tested the convergence of lung cancer-associated genes identified by different approaches or using different omics data. Under a case-control design, the results of GWAS, TWAS, WES/WGS-based gene-centric analysis were included (“Methods”). For tumor-normal tissue comparisons, we investigated the results of DGE, copy number variant analysis, and WES-based somatic mutation analysis (“Methods”). As shown in Fig. 6b, genes in the pLoF category were significantly enriched in the COSMIC Cancer Gene Census (CSC) gene list29, specifically for tumor suppressor genes (in line with biology). Genes identified by untranslated region (UTR) mask in the current study were significantly overrepresented in lung cancer-related genes from COSMIC. Enhancer-DHS mask-informed genes were significantly enriched in COSMIC pan-cancer categories29, especially for TSGs. As shown in GWAS columns, rare and common variants didn’t hit the same list of genes in the context of a case-control design. In addition, significant enrichment was observed between the COSMIC CSC gene list and the UK Biobank11 pLoF gene list identified by WES-based scan. Altogether, the cross-omics enrichment analysis provided a preliminary picture of the overlapping of molecular elements for the etiology of lung cancer.
Discussion
In this investigation aimed at enhancing our understanding of the etiology of lung cancer, our study has illuminated the multifaceted nature of lung cancer genetics, highlighting potential regulatory mechanisms, immune-related processes, and the convergence of established and novel candidate genes, particularly in the non-coding regions. We successfully verified the well-established lung cancer-associated loci, uncovered potential regulatory genes including TP63 and DCBLD1, and fleshed out the transcriptional regulation context underlying their associations. Subsequently, we conducted a rare variant-based genome-wide scan as a complementary approach to provide additional insight into the etiology of NSCLC. Both well-established genes (including ENO1, EFHD2, and RAD52) and novel genes (including PARPBP, PLA2G4C, and RITA1) were identified and replicated.
Our study primarily focused on filling the knowledge gap regarding non-coding regulation in lung cancer for both common and rare variants. On the one hand, by assuming gene expression regulation as a potential mediator between variants and disease, gene expression prediction models were trained in 297 normal lung tissue samples. Empirical results estimated from GTEx showed that increasing the sample size from ~120 (Wang et al.16) to ~300 (the current study) would result in at least 50% increase in the number of predictable genes42. On the other hand, the comprehensive WGS data provided us with an opportunity to study regulatory variants in non-coding regions, which would have otherwise been difficult to assess. The applications of fixed and dynamic window analyses offered a more flexible approach to probe trait-associated genomic segments. Assuming the regulatory role of the identified segments, promising results were found given the highly significant enrichment of enhancer signals in lung and immune-related cell lines. This enriched understanding of the genetic regulation of lung cancer further illuminates the complex landscape of this disease.
We discovered novel evidence for several genes, including PARPBP, PLA2G4C, and RITA1 for lung cancer utilizing rare variant-based analysis. PARPBP, for instance, encodes the PARP1 binding protein, which plays an essential role in multiple DNA repair pathways43. Recently, the DNA repair-independent roles of PARP1 in carcinogenesis and metastasis, particularly in lung cancer metastasis, have been revealed44. Although the precise role of PARPBP in the etiology of lung cancer remains unclear, our study provided valuable insight showing a replicable association between PARPBP and lung cancer based on genetic variants located in promoter and UTRs. In the case of PLA2G4C, direct evidence regarding its roles in lung cancer is relatively limited, but promising clues were found in previous functional studies. Nanashima and colleagues45 reported that the Hirosaki hairless rat, which was deficient in the Pla2g4c gene (the orthologous gene PLA2G4C in rats)45, displayed reduced susceptibility to mammary carcinogenesis after enhancement of NF-κB/Lcn2-induced apoptosis45. This observation was consistent with our replicable observations for the role of PLA2G4C in lung cancer by using rare variants in the promoter region of PLA2G4C. Our rare variant-based association test also supported the role of RITA1 in the susceptibility of lung cancer. Although the functional role of RITA1 is not clear in lung cancer, a recent study has demonstrated that aberrant upregulation of RITA1 is crucial for maintaining the growth of breast cancer cells by inhibiting Notch1 signaling46. In line with our observations, these functional studies provided preliminary evidence for the roles of PARPBP, PLA2G4C, and RITA1 in lung cancer. It is worth noting that all three of these genes were first identified in non-coding categories, suggesting the potential significance of rare and non-coding variants in shaping the genetic architecture of lung cancer.
Our study not only unveiled novel candidate genes but also provided additional genetic epidemiological evidence for some well-studied genes regarding their roles in lung cancer carcinogenesis through tissue or cell type-specific gene expression regulation. A typical example is RAD52, located at 12p13.33. RAD52 is involved in homologous recombination, an essential mechanism for DNA repair47. Both genetic and functional studies have underscored the importance of RAD52 in the development of lung cancer48. Experiments involving the depletion of RAD52 in mouse lung tumor cells revealed alterations in cell cycle distribution and increased DNA damage accumulation within lung tumor cells48. Our results showing an association between RAD52 and lung cancer using germline missense variants further verified the roles of this gene. Another essential gene verified in our study is TP63, which encodes a TP53 family member protein, playing a substantial role in cellular differentiation, stress response, and cancer development49. TP63 can induce and facilitate tumor cell apoptosis both in vivo and in vitro, significantly inhibiting tumor growth50. Evidence from the current study showing the association between genetically predicted expression of TP63 in lung tissue and the risk of lung cancer strengthened previous findings. DCBLD1 is located at 6q22.1, a region strongly linked to lung cancer51. Existing experiments involving the modulation of DCBLD1 in lung cancer cell lines have demonstrated its tumor-promoting effect. DCBLD1 knockdown in mice resulted in a notable reduction in xenograft cell proliferation and cell cycle progression52. In our study, we verified the oncogenic role of DCBLD1 and extended it in immune cell types. EFHD2 has been considered as a distinctive target with genetic alterations in lung cancer. Studies have unveiled its link to the promotion of epithelial-to-mesenchymal transition in lung cancer, a critical process enabling cancer cells to acquire invasive characteristics and metastasize53. Additionally, EFHD2 has been implicated in drug resistance in lung cancer through the activation of the NOX4-ROS-ABCC1 axis54. Our study verifies the role of EFHD2 in lung cancer, but from a non-coding regulation perspective. ENO1 has the capacity to activate the HGFR and WNT signaling pathways and encodes a key enzyme of glycolysis and choline kinase α (CHKα), which promotes tumor cell proliferation and survival in patients with early-stage non-small-cell lung cancer55,56. Based on rare variants located in non-coding regions, we confirmed the role of ENO1 in lung cancer carcinogenesis from a genetic epidemiological perspective. Altogether, we verified the role of several well-established genes and extended the context of the regulatory role of these genes in lung cancer.
Leveraging the high-throughput TF footprint data, we employed a deep learning framework to decode the binding sequences and mofit26,27. This approach allowed us to identify potential upstream regulators, typically TFs, for lung cancer-associated genes. By assuming that causal variants affect the binding sequence, the in silico examination identified carcinogenesis-related regulators, including TP53, FOXA1, FOXA2, ZEB1, NFKB1, and MYC as potential regulators, providing valuable insights into the mechanisms underlying these associations. To further corroborate the roles of potential regulatory variants, a natural follow-up would involve the utilization of Massively Parallel Reporter Assays57 to provide additional support for the role of potential regulatory variants. Our cell type enrichment analysis revealed the crucial roles of proliferating T cells and NK cells associated with the identified genes in the immune response against cancer, including lung cancer58,59. Their enrichment emphasizes the potential significance of immune-related processes in lung cancer etiology, warranting further investigation.
An intriguing discovery from our study was the overrepresentation of coding variant-derived genes from COSMIC and non-coding variant-derived genes in enhancer/UTR categories, indicating a potential convergence between the well-established genes and non-coding-based novel candidates. It underscores the interconnectedness of genetic factors contributing to lung cancer, regardless of their location within the genome. Notably, we didn’t observe significant enrichment between the common and rare variant-based genes. This observation might indicate distinct regulatory mechanisms governing these two categories of variants or might result from the relatively limited sample sizes, especially in the context of rare variant-based investigations. This underscored the need for well-powered studies to comprehensively understand the intricate genetic architecture of lung cancer and the interplay between common and rare variants. While providing valuable insights, our findings also underscored the necessity for larger-scale studies to unveil the full complexity of lung cancer genetics.
Our study has several strengths that underpin the reliability and significance of our findings. First, we conducted the most extensive WGS-based association test to date among the Chinese population. By scanning the entire genome, we ensured that our investigation was comprehensive and encompassed a wide spectrum of genetic variation. Second, we constructed the largest genome-transcriptome reference panel for lung tissue to date in the Chinese population. This reference panel provided valuable data for unraveling potential mechanisms governing gene expression regulation. Third, the STAAR framework, featuring a systematic integration of rare variants (particularly those located in non-coding regions), which significantly bolstered the statistical power of our association tests. This emphasis on rare variants filled a crucial gap in our understanding of lung cancer genetics.
While our study contributes to the field of lung cancer genetics, we also acknowledge several limitations. Although we observed significant associations in the replication stage, additional confirmations are needed to establish the robustness of our findings. Functional studies are needed to verify the roles of candidate genes and their potential regulators, including TFs. The cross-omics enrichment analysis may be underpowered to provide accurate estimates.
In conclusion, we performed a WGS-based genome-wide scan across the entire allele frequency spectrum to unravel the intricate genetic landscape of lung cancer, particularly in the context of non-coding regulation. We successfully verified the presence of well-established lung cancer-associated genes, including TP63, DCBLD1, ENO1, EFHD2, and RAD52, underscoring the robustness and reliability of our research. We identified and replicated novel lung cancer-associated genes, including PLA2G4C, PARPBP, and RITA1, expanding our understanding of the genetic factors contributing to this complex disease. Furthermore, this study shed light on potential upstream regulators and illuminated the roles of specific cell types in the context of lung cancer, adding valuable layers of insight to our understanding of the molecular etiology of carcinogenesis. These findings provided novel and critical insights into the genetic architecture of lung cancer within Asians. They served as a foundation upon which further research can build to uncover the molecular intricacies of this disease.
Methods
Sample collection and library preparing
We performed a case-control study to probe lung cancer-associated genetic elements in Chinese individuals. NSCLC cases were recruited from the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China. Cases were diagnosed by doctors and manually confirmed by pathological reports. Based on the Healthy Zhejiang One Million Cohort project, cancer-free controls were recruited from Lanxi, China. Participants with self-reported cancer diagnoses were excluded from the control group. The study protocol was approved by the Ethics Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine. All participants provided written informed consent.
Except for the lung cancer cases whose DNA samples were extracted from normal lung tissues using the AllPrep DNA/RNA/miRNA Universal Kit (#80224, Qiagen, Hilden, Germany), the DNA samples were extracted from peripheral blood samples using the QIAamp 96 DNA QIAcube HT Kit (#51331, Qiagen). The library was constructed using the MGIEasy FS DNA Library Prep Set (cat. 1000006987; MGI, China).
Whole genome sequencing, variant calling, and quality control
WGS was performed using the DIPSEQ platform (BGI, Shenzhen). We followed the GATK Best Practice pipeline for germline short variant discovery (SNPs + Indels) for alignment30. We then consolidated GVCF files using GATK CombineGVCFs. Joint calling was implemented using GATK GenotypeGVCFs30. In addition, 50 CHB/CHS samples (sequenced by the same platform) were also included in the joint calling step. Variants of excess heterozygosity (ExcessHet: 54.69) were filtered out. Based on quality scores including QD, MQRankSum, ReadPosRankSum, FS, SOR, and DP, VQSR was conducted to identify the low-quality variants with truth sensitivity levels at 99.6% and 99.0% for SNPs and INDELs, respectively. Then, we calculated the genotype posterior probability and updated the GQ using samples from the 1000 Genome Project as a prior31. Only biallelic variants were included in the following steps. Hard filtration was performed to keep variants in good quality based on empirical evidence. Variants with heterozygote allele balance <0.2 were excluded. SNPs and INDELs with SnpGap > 3 and IndelGap > 5 were removed, respectively. The GT was set as missing if GQ < 20 or DP < 8. After setting low-quality GT to missing, variants with a call rate <90% were removed.
To improve the genotyping quality, refinement was performed using Beagle 5.460,61. Monomorphic variants and variants with Phwe < 1.0 × 10−6 were filtered out after refinement. By comparing the genotypes estimated from the current study with the officially reported ones for the 50 samples from the 1000 Genome Project31, genotyping concordance was estimated. Based on the common variants (MAF ≥ 0.01), we estimated the sample relatedness using KING62. Potentially monozygotic twins (MZ), duplicated samples, and samples that failed in sex check were removed. Additional outliers were removed in heterozygosity filtration (> ± 6 SD). Variants were annotated using VEP63 with default settings. The technical details for variant calling and quality control in the discovery and replication stages are described in Supplementary Figs. 1 and 2.
Variant level association test, meta-analysis, and conditional analysis
The principal components were estimated using LD-pruned (r2 = 0.9) variants. All samples in this study were projected to a PC1 v.s. PC2 map of all 1000 genome phase 3 samples31 to confirm the genetic background.
A common variant-based (MAF ≥ 0.01) genome-wide association study was performed using a generalized linear mixed-effect model (GLMM-fastGWA)17. Age, Age2, Sex, and PC1-PC10 were adjusted. Fixed-effect meta-analysis was performed to combine the results in East Asian (EAS) populations18. Conditional and joint multiple-SNP analysis (COJO) was implemented to identify independent genetic variants20.
RNA sequencing, transcriptome-wide association study, and differential gene expression analysis
For lung cancer samples, the total RNA of tumor and paired normal tissue samples was extracted and sequenced (paired-end 100 bp RNA-seq). Samples with high ribosome RNA (> 30%) or low uniquely mapped reads (< 20 million) were removed. Samples that failed in Crosscheckfingerprints analysis (LOD < 5) were excluded. Following the hmftools pipeline (https://github.com/hartwigmedical/hmftools), the RNA-seq aligner STAR64, sambamba65, and Isofox were implemented to align reads to a reference, mark duplicate reads, and generate the counts for each gene. Transcripts per million (TPM) was calculated and log-transformed.
To project SNP-level signals to gene levels, we performed TWAS21 in lung cancer tissue and in immune cell types. TWAS integrates genetic variation with gene expression data to identify genes associated with complex traits and diseases. The primary objective of TWAS is to link genetic variants to phenotypes by examining the mediating role of gene expression. Usually, predictive models are constructed to estimate gene expression levels based on local genetic variants. These models are then applied to GWAS summary statistics to predict the association between gene expression and the trait of interest. We estimated the probabilistic estimation of expression residuals (PEER) factors66 to account for potential batch effects. Age, sex, PC1-10, and PEER factors were regressed out from the log-transformed TPM to generate the residual of TPM, which was used in the following analysis. The gene expression prediction model was trained by an elastic net model with SNPs in 1 Mb on both sides of gene body as features. The SNP-SNP covariance matrix was estimated for each gene. Predictable gene was defined by commonly used criteria (r > 0.1 and P < 0.05)21,42. To generate prediction models in immune cell types, PRS-CS38 was implemented using eQTL summary statistics from 17 immune-related cell types from the ImmuNexUT dataset37. With the data from GEUVADIS67, we demonstrated the accuracy of summary statistics-based model training. The association between GReX and lung cancer was estimated.
DGE analysis was performed using a negative binomial generalized linear model implemented in edgeR39. The potential batch effect was adjusted. Genes with TPM > 1 in at least 20% samples were included.
Rare variant-based association test
To efficiently identify NSCLC-associated genetic elements, we implemented the STAAR framework. STAAR is a scalable and powerful rare variant association test method that effectively incorporates both variant categories and multiple epigenetic annotations using a dynamic weighting scheme. Following the STAAR pipeline22,23,24, the genotype file in plink binary format was converted to Genomic Data Structure (GDS) file. Then, the GDS file was decorated by FAVORannotator25 to generate the annotated GDS (aGDS) file, which was used in the following analysis. A sparse genetic relatedness matrix (GRM) was generated to account for sample relatedness in a generalized linear mixed model (GLMM). We fit a null model using phenotype data, covariates (age, age2, sex, age*sex, age2*sex, and PC1-PC10), and the sparse GRM. For each protein-coding gene, gene-centric coding analysis was performed for five categories to aggregate coding rare variants: (1) putative loss of function (pLoF) (stop gain, stop loss, and splice), (2) missense, (3) disruptive missense (ds), (4) putative loss of function and disruptive missense (pLoF + ds), and (5) synonymous. Similarly, gene-centric non-coding analysis was performed for eight genetic categories of regulatory regions to aggregate non-coding rare variants: (1) promoter RVs overlaid with Cap Analysis of Gene Expression [CAGE] sites (promoter-CAGE), (2) promoter RVs overlaid with DNase hypersensitivity [DHS] sites (promoter-DHS), (3) enhancer RVs overlaid with CAGE sites (enhancer-CAGE), (4) enhancer RVs overlaid with DHS sites (enhancer-DHS), (5) UTR RVs, (6) upstream region RVs, (7) downstream region RVs, and (8) non-coding RNA RVs. Using the aggregated Cauchy association test (ACAT) method, an omnibus test STAAR-O was implemented to combine p-values by leveraging the advantages of different types of tests, including SKAT, burden, and ACAT-V22,23,24. Given the limited number of cases, genes with less than 10 cMAC in cases or with less than 10 variants were filtered out in both the discovery and the replication stages.
In the gene-centric analysis, we applied the stratified FDR correction40. i.e., a stratified false discovery control approach for studies in which a large number of hypotheses have some natural stratifications among them. A regular aggregated FDR correction assumes that genes from all categories have a fixed chance of being associated with the trait of interest (we denote it as the “fixed chance” assumption). If the “fixed chance” assumption is true, Sun et al. demonstrated that the regular aggregated FDR is a weighted average of the stratified FDRs. While if the “fixed chance” assumption does not hold, i.e., the true positive rate is different across different categories, then the stratified FDR provides a more flexible and efficient way to define the significance for different categories while accounting for the overall false discovery rate. In other words, if there are differences among stratums, the stratified FDR approach is superior to the aggregated one because the stratified FDR automatically incorporates auxiliary information. The potential advantages of FDR control by stratum have been demonstrated and illustrated in many empirical studies.
In practice, considering the potential difference in the proportion of true positive genes across the annotation categories (a natural stratification), we applied stratified FDR correction. i.e., for each category, we adjusted the p-value following the Benjamini-Hochberg method.
Deep learning-based in silico fine-mapping
To map causal variants of the identified genes and their potential upstream regulators, we implemented the DeepSEA-Sei framework26,27. Briefly, DeepSEA-Sei is an in silico fine-mapping approach for identifying potential causal variants by using deep learning to predict the functional impact of non-coding variants on various regulatory activities such as transcription factor binding, DNase I sensitivity, and histone modifications. DeepSEA-Sei breaks the genome into numerous 1000 bp segments and estimates the posterior probability of DNA segment to transcription factor binding signals. More than 20,000 regulatory annotation signals were fed to the deep convolutional neural network to estimate the posterior probability. The difference in posterior probability for segments carrying reference and alternative alleles was used to identify potential regulatory variants. An empirical cutoff (0.1) for the posterior probability was used. For each replicable lung cancer-associated gene, variants with regulatory annotations were evaluated by the posterior probability, showing different abilities to bind to TFs. Potential TFs and the gene of interest were visualized by lines in the circos plot68. We only showed the links identified in lung and immune-related cell lines.
Cell-type-specific gene expression enrichment analysis
A cell-type-specific enrichment analysis was performed for genes identified by rare variant-based analysis. ScRNA-seq data from lung tissue were used as described in a previous analysis28,69. Cells were grouped into cell types based on the top 2000 highly variable genes. The gene expression was normalized to values between 0 and 1, indicating low and high specificity levels, respectively. In each cell type, the normalized expression was ranked in ascending order. Then, the cell type-specific score was calculated by summing the ranks of a set of candidate genes. We performed permutation tests to calculate the P-value for enrichment, which was defined as the number of permutated scores greater than or equal to the score from the empirical data divided by the number of permutations.
Enrichment analysis across multiple omics data for lung cancer-related genes
To gain a better understanding of the architecture of genes identified by the rare variant-based gene-centric scan, we performed a series of enrichment analyses to see, to what extent, these genes overlapped with the genes identified by other methods or through other omics data including (1) lung cancer-associated genes projected by MAGMA70 from GWAS; (2) genes whose genetically predicted expression level associated with lung cancer identified by TWAS; (3) DEG by comparing measured gene expression between tumor and adjacent normal tissues of lung cancer patients71; (4) the genes cataloged by the COSMIC Cancer Gene Census (CGC)29 which considered both tier 1 level genes (possessing a documented activity relevant to cancer, along with evidence of mutations that change the activity of the gene product in a way that promotes oncogenic transformation) and tier 2 level genes (with strong indications of a role in cancer but with less extensive available evidence). We considered all CGC genes as pan-cancer genes. Genes with a “lung cancer” or a “NSCLC” label were considered as lung cancer-related genes in CGC; The recurrent somatic copy number alterations were obtained from a previous study by Campbell et al.72. In addition, we included lung cancer-associated genes identified in three masks (namely pLoF, missense, and synonymous) from the UK Biobank as a parallel comparison12. GSEA73 was implemented to evaluate the enrichment levels across different gene sets.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data (WGS and RNA-seq) from this study are available in the Genome Sequence Archive (GSA; https://ngdc.cncb.ac.cn/gsa-human/) under accession number HRA009288. The GWAS summary statistics for NSCLC have been deposited in GWAS Atlas (https://ngdc.cncb.ac.cn/gwas/) under accession GVP000060. The eQTL results are available via the OMIX platform (https://ngdc.cncb.ac.cn/omix/) under ID OMIX006687.
Code availability
The code for this paper is available on the GitHub (https://github.com/zdangm/lungca_WGS) with https://doi.org/10.5281/zenodo.13825007.
References
Xia, C. et al. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin. Med. J. 135, 584–590 (2022).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Mucci, L. A. et al. Familial risk and heritability of cancer among twins in Nordic countries. Jama 315, 68–76 (2016).
Dai, J. et al. Identification of risk loci and a polygenic risk score for lung cancer: a large-scale prospective cohort study in Chinese populations. Lancet Respir. Med. 7, 881–891 (2019).
Bossé, Y. & Amos, C. I. A decade of GWAS results in lung cancer. Cancer Epidemiol. Biomark. Prev. 27, 363–379 (2018).
Amos, C. I. et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25. 1. Nat. Genet. 40, 616–622 (2008).
Shi, J. et al. Genome-wide association study of lung adenocarcinoma in East Asia and comparison with a European population. Nat. Commun. 14, 3043 (2023).
Uffelmann, E. et al. Genome-wide association studies. Nat. Rev. Methods Prim. 1, 59 (2021).
Visscher, P. M. et al. 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Consortium, T. G. et al. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Aguet, F. et al. Molecular quantitative trait loci. Nat. Rev. Methods Prim. 3, 4 (2023).
Karczewski, K. J. et al. Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes. Cell Genomics 2, 100168 (2022).
Liu, Y. et al. Rare deleterious germline variants and risk of lung cancer. NPJ Precis. Oncol. 5, 12 (2021).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532 (2021).
Jin, G. et al. Low-frequency coding variants at 6p21. 33 and 20q11. 21 are associated with lung cancer risk in Chinese populations. Am. J. Hum. Genet. 96, 832–840 (2015).
Wang, C. et al. Analyses of rare predisposing variants of lung cancer in 6,004 whole genomes in Chinese. Cancer Cell 40, 1223–1239. e6 (2022).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 51, 1749–1755 (2019).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Li, Z. et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat. Methods 19, 1599–1611 (2022).
Li, X. et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet. 52, 969–983 (2020).
Li, X. et al. Powerful, scalable and resource-efficient meta-analysis of rare variant associations in large whole genome sequencing studies. Nat. Genet. 55, 154–164 (2023).
Zhou, H. et al. FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res. 51, D1300–D1311 (2023).
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931–934 (2015).
Chen, K. M., Wong, A. K., Troyanskaya, O. G. & Zhou, J. A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 54, 940–949 (2022).
Habermann, A. C. et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 6, eaba1972 (2020).
Sondka, Z. et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
Consortium, G. P. A global reference for human genetic variation. Nature 526, 68 (2015).
Cong, P.-K. et al. Genomic analyses of 10,376 individuals in the Westlake BioBank for Chinese (WBBC) pilot project. Nat. Commun. 13, 2939 (2022).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7.20. 1–7.20. 41 (2013).
Jiang, L., Zheng, Z., Fang, H. & Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 53, 1616–1621 (2021).
Wainberg, M. et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 51, 592–599 (2019).
Ota, M. et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 184, 3006–3021. e17 (2021).
Ge, T., Chen, C.-Y., Ni, Y., Feng, Y.-C. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Sun, L., Craiu, R. V., Paterson, A. D. & Bull, S. B. Stratified false discovery control for large-scale hypothesis testing with application to genome-wide association studies. Genet. Epidemiol.30, 519–530 (2006).
Gao, T. & Qian, J. EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 48, D58–d64 (2020).
Zhou, D. et al. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 52, 1239–1246 (2020).
Spiegel, J. O., Van Houten, B. & Durrant, J. D. PARP1: structural insights and pharmacological targets for inhibition. DNA Repair (Amst.) 103, 103125 (2021).
Choi, E. B. et al. PARP1 enhances lung adenocarcinoma metastasis by novel mechanisms independent of DNA repair. Oncogene 35, 4569–4579 (2016).
Nanashima, N., Yamada, T., Shimizu, T. & Tsuchida, S. Deletion of phospholipase A2 group IVc induces apoptosis in rat mammary tumour cells by the nuclear factor-κB/lipocalin 2 pathway. Biochem. J. 469, 315–324 (2015).
Tang, H. et al. RITA1 drives the growth of bladder cancer cells by recruiting TRIM25 to facilitate the proteasomal degradation of RBPJ. Cancer Sci. 113, 3071–3084 (2022).
Rojas, S. & Oviedo, N. J. Can neural signals override cellular decisions in the presence of DNA damage?. DNA Repair 103, 103127 (2021).
Lieberman, R. et al. Functional characterization of RAD52 as a lung cancer susceptibility gene in the 12p13. 33 locus. Mol. Carcinog. 55, 953–963 (2016).
Flores, E. R. The roles of p63 in cancer. Cell Cycle 6, 300–304 (2007).
Katoh, I., Aisaki, K. -I, Kurata, S. -I, Ikawa, S. & Ikawa, Y. p51A (TAp63γ), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene 19, 3126–3130 (2000).
Lan, Q. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 44, 1330–1335 (2012).
Long, E., Patel, H., Byun, J., Amos, C. I. & Choi, J. Functional studies of lung cancer GWAS beyond association. Hum. Mol. Genet. 31, R22–r36 (2022).
Fan, C. C. et al. EFHD2 promotes epithelial-to-mesenchymal transition and correlates with postsurgical recurrence of stage I lung adenocarcinoma. Sci. Rep. 7, 14617 (2017).
Fan, C.-C. et al. EFHD2 contributes to non-small cell lung cancer cisplatin resistance by the activation of NOX4-ROS-ABCC1 axis. Redox Biol. 34, 101571 (2020).
Li, H. J. et al. ENO1 promotes lung cancer metastasis via HGFR and WNT signaling-driven epithelial-to-mesenchymal transition. Cancer Res. 81, 4094–4109 (2021).
Ma, Q. et al. The moonlighting function of glycolytic enzyme enolase-1 promotes choline phospholipid metabolism and tumor cell proliferation. Proc. Natl. Acad. Sci. USA 120, e2209435120 (2023).
Klein, J. C. et al. A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods 17, 1083–1091 (2020).
Pockley, A. G., Vaupel, P. & Multhoff, G. NK cell-based therapeutics for lung cancer. Expert Opin. Biol. Ther. 20, 23–33 (2020).
Kamphorst, A. O. et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1–targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. 114, 4993–4998 (2017).
Browning, B. L., Tian, X., Zhou, Y. & Browning, S. R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 108, 1880–1890 (2021).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348 (2018).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507 (2012).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013).
Zhang, H., Meltzer, P. & Davis, S. RCircos: an R package for Circos 2D track plots. BMC Bioinforma. 14, 1–5 (2013).
Liu, Q. et al. The genetic landscape of familial pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 207, 1345–1357 (2023).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
This work was supported by the Healthy Zhejiang One Million People Cohort (K-20230085), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2019R01007), the Key Laboratory of Intelligent Preventive Medicine of Zhejiang Province (2020E10004), the Key Research and Development Program of Zhejiang Province(2020C03002), and the Guangdong Provincial Key Laboratory of Human Disease Genomics (2020B1212070028).
Author information
Authors and Affiliations
Contributions
X.W. developed the study concept and design. M.W., L.S., Y.T., W.Z., X.X., H.T., W.L., and M.Y. performed investigation and data collection. Y.Z. and K.W. performed lab experiments. K.H. and F.H. performed data management. Q.T., D.Z., H.Z., T.L., F.Y., F.L., and X.J. undertook all analyses. D.Z., Q.T., W.L., and X.W. undertook the writing of the first draft of the manuscript. All authors were responsible for data interpretation and approved final manuscript submission. X.W. supervised the study and obtained funding for the work undertaken.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zilin Li, Jianxin Shi, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, D., Wu, M., Tan, Q. et al. Non-coding genetic elements of lung cancer identified using whole genome sequencing in 13,722 Chinese. Nat Commun 16, 7365 (2025). https://doi.org/10.1038/s41467-025-62459-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62459-6
