
Abstract
Immunotherapy is a breakthrough in the treatment of triple-negative breast cancer (TNBC), although it is only effective in a portion of patients. Our clinical studies find that pathological elevated level of reactive oxygen species (ROS) and lipid homeostasis imbalance are closely associated with dysfunction of dendritic cells (DCs) in the immunosuppressive lymph nodes (LNs) microenvironment of TNBC patients following immunotherapy, which greatly affect the immunotherapeutic efficacy. Building on this, we introduce a chimeric exosomes-derived immunomodulator involving the polysulfide bond-bridged mesoporous silica as both the ROS scavenger and responsive carrier nucleus, loading with the lipid modulator toyocamycin and being coated with chimeric exosomes comprising DCs-derived exosomes and Salmonella outer membrane vesicles. This multifaceted immunomodulator can significantly enhance LNs’ homing through homologous targeting and chemokine-guided navigation, enabling ROS-responsive drug release, thereby restoring functions of DCs and LNs immuno-microenvironment. As expected, the immunomodulator significantly improves the responsiveness of TNBC to immunotherapy, exerting potent inhibition on both the primary tumor and metastases, while promoting a substantial increase in central memory T cells within LNs for sustained antitumor immunity. Our study provides a potent strategy for translational immunotherapy through optimizing the LNs microenvironment in TNBC.
Similar content being viewed by others


Introduction
Triple-negative breast cancer (TNBC) is a highly aggressive subtype of breast cancer, characterized by poor prognosis and limited treatment options1. Immunotherapy has emerged as a breakthrough in the therapeutic landscape of TNBC by leveraging the patient’s immune system to improve survival outcomes. However, its clinical success remains limited, with only a subset of patients experiencing significant benefits2.
Lymph nodes (LNs) play a crucial role in the immune response, facilitating antigen presentation and promoting tumor-specific immunity3,4,5,6. Tumor invasion, however, disrupts this immune function, causing immune cells within LNs to undergo metabolic reprogramming. This reprogramming leads to an immunotolerant environment that hampers effective antitumor immunity7,8,9,10. Our clinical studies have suggested that in TNBC, metastatic LNs (mLNs) exhibit lipid metabolic reprogramming in dendritic cells (DCs), driven by abnormal endoplasmic reticulum (ER) stress. The activation of the ER stress sensor inositol-requiring transmembrane kinase/endonuclease 1 (IRE1α) and the subsequent upregulation of X-box binding protein-1 (XBP1s) are highly associated with this dysfunction11,12,13. Furthermore, research has also demonstrated that dysregulated lipid metabolism in DCs can impair their functions and affect immune responses, linking lipid imbalance to reduced DC activity13,14. In addition, oxidative stress-induced reactive oxygen species (ROS) perpetuate ER stress, exacerbating lipid dysregulation and impairing DC function, resulting in downregulated MHC expression and diminished T-cell immunity15. These findings highlight a critical target for therapeutic intervention in TNBC, emphasizing the importance of revitalizing the immunosuppressive LNs microenvironment to enhance immunotherapy efficacy and improve patient prognosis.
Optimizing LNs immuno-microenvironment and addressing DCs’ dysfunction necessitates efficient internalization of DCs and their homing to LNs, targeting the underlying ER and oxidative stress pathways to restore lipid homeostasis. However, the complex structure of LNs and challenges posed by the mononuclear phagocyte system (MPS) hinder effective drug delivery16. In this context, biomimetic strategies leveraging extracellular vesicles (EVs), especially dendritic cell-derived exosomes (DEX), have emerged as a promising solution for targeted delivery to LNs17,18,19. DEX possess natural homing capabilities, excellent biocompatibility, minimal immunogenicity and efficient lymphatic navigation, making them ideal for enhancing antigen presentation and stimulating immune responses18,20. Further improving this approach, chimeric exosomes, which integrate functional proteins from various biological sources, offer a cutting-edge platform for drug delivery21. These chimeric exosomes provide targeted delivery with high specificity and an extended functional repertoire, critical for effective drug delivery and immunomodulation. However, challenges remain in their production, including intricate ex vivo processes and the sensitivity of biomembrane components22. Therefore, developing chimeric exosomes that overcome these limitations is highly desirable.
This study unveils an endogenous-production approach that uses bacteria-derived outer membrane vesicles (OMVs) to amplify DCs’ uptake and exocytosis and thereby facilitate efficient production of chimeric exosomes. Based on our clinical findings, we design a chimeric exosomes-derived immunomodulator, EMVs@SS-Toy, which optimizes the assembly of innate immunoregulators in DCs. The system features ROS-responsive polysulfide bonds-contained mesoporous silica (SS-MSN) that encapsulates toyocamycin (Toy) to target the IRE1α-XBP1 pathway. Its surface is modified with chimeric exosomes derived from DEX and Salmonella-derived OMVs. EMVs@SS-Toy exhibits potent LNs’ homing, revitalizes functions of DCs and regulates lipid homeostasis, remodeling the LNs immune microenvironment. In vivo, the immunomodulator significantly potentiates the immune response in TNBC, effectively suppresses both the primary tumor and metastases, and promotes sustained antitumor immunity. Such a chimeric exosome-derived immunomodulatory strategy may provide insights into addressing certain challenges in LNs-targeted immunotherapy for TNBC, although further studies are still required to fully evaluate its clinical feasibility and translational potential.
Results
Clinical data from TNBC patients reveal the link between lipid homeostasis imbalance of DCs and the immunosuppressive LNs microenvrionment
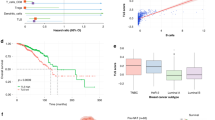
We initiated our investigation by analyzing a nationwide pharmaco-epidemiologic database to explore the correlation between LNs’ status and the efficacy of neoadjuvant therapy in TNBC patients (Fig. 1a)23. An analysis of 2849 patients from the SEER database identified LNs’ staging as an independent predictor of disease progression in TNBC (Table S1). Furthermore, we observed a notable association between LNs’ heterogeneity and overall survival rates, highlighting the critical role of LNs’ status in determining patient prognosis and its impact on treatment outcomes (Fig. 1b and Supplementary Fig. 1).

a Illustrative representation of nLNs and mLNs from TNBC patients. b Kaplan-Meier plots of overall survival (OS) categorizing TNBC patients by LNs’ stage (Log-rank P value < 0.0001). N0 samples (n = 1536), N1 samples (n = 945), N2 samples (n = 190), N3 samples (n = 178). c MRI images depicting the primary tumor and mLNs in TNBC patients, with red arrows highlighting the affected areas. d Histopathological comparison between the nLNs and mLNs using H&E staining. e Immunohistochemistry (IHC) analysis of Foxp3 in the nLNs versus mLNs. f CLSM showcasing the co-localization of CD11c (red) with BODIPY (green) in the nLNs versus mLNs. g Multiplex IHC for immune profiling in the nLNs and mLNs from the TNBC patient (n = 4 independent experiments). Scale bar=100 μm. h Clustering analysis of immune cells in matched nLNs and mLNs’ samples from the TNBC patient. i Gene Ontology (GO) enrichment analysis of gene expression variations in DCs within the mLNs vs nLNs. P values are derived from one-sided Fisher’s Exact Test for pathway and GO enrichment. Source data are provided as a Source Data file.
Moreover, we observed divergent therapeutic response rates between primary tumors and mLNs in patients receiving neoadjuvant immunotherapy through MRI. As indicated in Fig. 1c, the primary breast tumor demonstrated a reduction in maximum diameter (from 1.64 cm to 1.15 cm), while axillary mLNs resolved completely, transitioning from a baseline diameter of 2.62 cm to undetectable on post-treatment imaging. In contrast, extra-axillary mLNs in the outer quadrant exhibited only a mild regression (from 1.41 cm to 1.01 cm), with residual enhancing lesions indicative of persistent tumor burden. This regional disparity in the therapeutic response of LNs prompted an investigation into the molecular and pathological factors underlying this heterogeneity.
Pathological examination of mLNs biopsies from TNBC patients revealed a high density of metastatic tumor cells (Fig. 1d). Immunohistochemistry (IHC) further showed significantly elevated levels of Foxp3+ regulatory T cells (Tregs) in mLNs, compared to non-metastatic lymph nodes (nLNs), indicating a pronounced immunosuppressive microenvironment (Fig. 1e and Supplementary Fig. 2). Additionally, co-localization analysis revealed an abnormal lipid accumulation in DCs within mLNs (Fig. 1f and Supplementary Fig. 3a), as demonstrated by the increased overlap of CD11c and BODIPY co-localization from Pearson’s correlation coefficient (Supplementary Fig. 3b).
Further analysis using multiplex IHC identified that DCs in mLNs exhibited a significant upregulation of XBP1s expression and reduction in major histocompatibility complex class I (MHC I) expression (Fig. 1g and Supplementary Fig. 4, 5a-b), which may suggest impaired DCs functionality. Moreover, an analysis Pearson’s correlation coefficient was analyzed for the co-localization of XBP1s and CD11c (Supplementary Fig. 6). The results indicated a stronger spatial co-localization of XBP1s and CD11c in mLNs, which was consistent with the previous co-localization results of CD11c and BODIPY. Additionally, PD1 expression was significantly elevated in mLNs compared to nLNs, further exacerbating the immunosuppressive microenvironment and potentially explaining the inconsistent therapeutic responses observed with immunotherapy (Fig. 1g and Supplementary Fig. 5c).
Single-cell transcriptome sequencing of matched mLNs and nLNs from the same TNBC patients provided further confirmation of a reduced DCs population in mLNs (Fig. 1h and Supplementary Fig. 7). The transcriptomic analysis also demonstrated a significantly higher expression of XBP1s in DCs within mLNs compared to nLNs (Supplementary Fig. 8). Moreover, Gene Ontology (GO) enrichment analysis revealed a significant dysregulation of pathways essential for antigen processing and presentation of endogenous peptide antigen via MHC class I and antigen processing and presentation of endogenous peptide antigen via MHC class Ib (adjusted p < 0.05) in mLNs compared to nLNs (Fig. 1i and Supplementary Fig. 9). This transcriptional evidence aligns with the functional impairment of DCs observed in immunohistochemical and imaging analyses. Taken together, these findings suggest that DCs in mLNs may exhibit impaired antigen processing and presentation capabilities, which may also disrupt T cell activation and contribute to diminished responses to immune therapies in metastatic settings.
These findings elucidate valuable insight into the mechanisms driving DCs’ dysfunction within mLNs, suggesting that targeting lipid dysregulation, specifically by reducing XBP1s expression in DCs, may restore their antigen-presenting functionality and help reverse the immunosuppressive envrionment in mLNs. Such an approach holds promise for improving immunotherapeutic outcomes in TNBC patients.
Fabrication and characterization of EMVs@SS-Toy for multi-level lipid modulation of DCs
Herein, leveraging DCs’ biological processes, the self-assembled, chimeric exosomes-derived biomimetic immunomodulator was prepared. First, the ROS-responsive polysulfide bonds-contained mesoporous silica (SS-MSN) was synthesized as the core of the biomimetic immunomodulator through a sol-gel method for drug loading (Fig. 2a and Supplementary Fig. 10). This core structure not only provided a stable framework for the immunomodulator but also acted as a ROS scavenger, with multiple drug loading sites for responsive release. The successful preparation of SS-MSN was confirmed through XPS and TGA analysis, which showed characteristic S2p peaks and increased weight loss, indicating the incorporation of polysulfide bonds (Supplementary Figs. 11a–b). Nitrogen adsorption/desorption data showed a reduction in pore volume and average pore size of SS-MSN ( ~ 90 nm), confirming that polysulfide bonds altered the mesoporous structure (Supplementary Fig. 11c). FTIR analysis further verified the incorporation of polysulfide bonds, with distinct C-S, C-Si, and S-S stretching bands around 698, 1245 and 500-540 cm-1, respectively. Additionally, an asymmetric stretching vibration of methylene (-CH2CH2-) was observed at 2931 cm-1 in SS-MSN (Supplementary Fig. 11d).

a Stepwise schematic of EMVs@SS-Toy production. b TEM images of SS-Toy, OMVs, DEX, OMVs@SS-Toy and EMVs@SS-Toy. c Particle size distribution profiles for SS-Toy, OMVs, DEX, OMVs@SS-Toy and EMVs@SS-Toy. d CLSM showcasing the co-localization of SS-MSNFITC (green) with Dil (red) in OMVs@SS-Toy, and with APC-CD63 (red) in EMVs@SS-Toy. e Energy-dispersive X-ray spectroscopy (EDS) mapping displaying the composition of EMVs@SS-Toy with white arrow indicating the zoomed-in area, red represents carbon (C), blue represents oxygen (O), orange represents sulfur (S), purple represents silicon (Si), green represents nitrogen (N), and yellow represents phosphorus (P). f Differential protein expression in DCs following different treatments. The cutoff is defined by requiring the fold change (FC) to be greater than 1.5, with adjusted P < 0.05 (Benjamini-Hochberg-adjusted two-sided Welch’s t test). g Heatmaps representing gene expression involved in immune activation and antigen presentation in DCs following different treatments (n = 3 independent experiments). h Western blot probing for exosomal markers TSG101, CD9, and CD81 proteins. i SDS-PAGE analysis revealing the protein profiles of OMVs, OMVs@SS-Toy, DEX, and EMVs@SS-Toy. j CLSM images of cell uptake of SS-Toy and OMVs@SS-Toy in DCs, highlighting Nuclei (Hoechst 33342, blue) and SS-MSN (FITC, green). k Flow cytometric quantification of fluorescent uptake in DCs treated with Si-Toy, SS-Toy, OMVs@Si-Toy, and OMVs@SS-Toy over time (n = 3 independent experiments). **P < 0.01, ***P < 0.001 and ****P < 0.0001 by two-tailed Student’s t-test. l Kinetics of Si-Toy and SS-Toy release in a 10 mM H2O2 environment at pH 7.4 (n = 3 independent experiments). **P < 0.01 by two-tailed Student’s t-test. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Source data are provided as a Source Data file.
Next, Toy was encapsulated into SS-MSN using the solvent deposition method, resulting in SS-Toy, with an encapsulation efficiency of 13.96% and a drug loading content of 6.96% (Supplementary Fig. 12). The SS-Toy was then coated with OMVs by extrusion to form OMVs@SS-Toy, which was co-incubated with DCs to induce phagocytosis and trigger DEX release. Ex vivo cytotoxicity assays revealed that SS-MSN, SS-Toy, OMVs and OMVs@SS-Toy exhibited negligible cytotoxicity to DCs at concentrations ranging from 0 to 1 μg/mL (Supplementary Fig. 13). To quantify the purity of EMVs@SS-Toy, a dual-labeling approach was used in which SS-Toy was labeled with FITC and EVs were labeled with the exosomal marker APC-CD63. Flow cytometry analysis showed that approximately 70% of secreted EVs displayed both FITC and APC-CD63 labeling (Supplementary Fig. 14), validating the high purity of EMVs@SS-Toy within the total population of secreted EVs.
After coating with OMVs, the zeta potential of SS-Toy increased from −20 mV to −15 mV (Supplementary Fig. 15). TEM imaging revealed a distinct core-shell structure for both OMVs@SS-Toy and EMVs@SS-Toy, respectively, with a slightly larger particle size than SS-Toy (Fig. 2b, c). Co-localization of SS-MSNFITC with DiI-labeled OMVs, observed by confocal laser scanning microscopy (CLSM), indicated the successful preparation of OMVs@SS-Toy (Fig. 2d). Similarly, co-localization of SS-MSNFITC with CD63-labeled EMVs confirmed the successful implementation of the endogenous production (Fig. 2d). Energy-dispersive X-ray spectroscopy (EDX) revealed the presence of silicon (Si), carbon (C), nitrogen (N), phosphorus (P), and sulfur elements (S) in EMVs, verifying the composition of EMVs@SS-Toy (Fig. 2e). During the biogenesis of DEX, DCs generated chimeric exosomes and released EMVs@SS-Toy, which featured SS-Toy as the inner core and EMVs as the outer membrane (Supplementary Fig. 16). To further confirm that EMVs@SS-Toy secretion relies on the exosome biogenesis pathway, we used the pharmacological inhibitor GW4869, which specifically blocks endosomal invagination and multivesicular body (MVB) formation24. As shown in Supplementary Fig. 17, treating DCs with GW4869 led to a progressive decrease in extracellular green fluorescence at 24 and 36 h, concomitantly with a corresponding increase in intracellular fluorescence signals. These results collectively demonstrate that DCs may internalize OMVs@SS-Toy and then package the drug-loaded nanoparticles into exosomes, secreting them as EMVs@SS-Toy through the exosome biogenesis pathway.
Proteomic analysis of EMVs@SS-Toy revealed 38 upregulated and 58 downregulated proteins compared to OMVs@SS-Toy (Fig. 2f). Key proteins from OMVs, including ompA, ompX, tolB and tolC, were still preserved in EMVs@SS-Toy, which have been corroborated to boost DCs and T cell immune responses (Fig. 2g). Western blot confirmed the presence of hallmark exosomal proteins, such as TSG101, CD9, and CD81, in both EMVs@SS-Toy and DEX (Fig. 2h), while the SDS-PAGE further corroborated the protein profiles of both DEX and OMVs in EMVs@SS-Toy (Fig. 2i).
Compared to Toy-loaded mesoporous silica (Si-Toy) and SS-Toy, OMVs@Si-Toy and OMVs@SS-Toy displayed significantly increased cell uptake, as evidenced by strong green fluorescence (Fig. 2j, k and Supplementary Fig. 18). OMVs coating greatly augmented DCs’ uptake, with a 5.27- and 6.29-fold increase at 6 h post-treatment, respectively. Building on preliminary proteomic findings, we hypothesized that the pathogen-associated molecular patterns (PAMPs) in OMVs stimulated the enhanced uptake of DCs. Based on this premise, we examined whether OMVs could enhance the endogenous production efficiency of EMVs@SS-Toy through exocytosis. Inductively coupled plasma mass spectrometry (ICP-MS) showed that the silicon content in the OMVs@SS-Toy group was 1.45 times higher than that in the SS-Toy group at 36 h (Supplementary Fig. 19), indicating that the OMVs obviously enhanced DCs phagocytosis and secretion, resulting in a higher yield of EMVs@SS-Toy. Additionally, we measured the uptake of EMVs@SS-Toy in DCs and found a significant time-dependent effect (Supplementary Fig. 20).
We subsequently evaluated the ex vivo release profile of SS-Toy, Si-Toy, EMVs@Si-Toy and EMVs@SS-Toy, respectively, which demonstrated responsive release in the presence of H2O2 and DTT (Fig. 2l and Supplementary Figs. 21–22), confirming the redox-sensitive characteristics of the immunomodulator. Stability experiments also showed that EMVs@SS-Toy remained stable in serum (Supplementary Fig. 23).
Ex vivo reinvigoration of DCs via dual-stress pathway immunomodulatory
Tumor invasion disrupts the microenvironment of LNs, altering the distribution of oxygen, nutrient, and metabolites. This disturbance triggers immune cells reprogramming towards an immune-tolerant state, thereby reducing antitumor immunity. In particular, DCs within mLNs exhibited increased lipid accumulation accompanied by upregulation of XBP1s (Supplementary Figs. 3 and 5b), a phenotype that may be associated with compromised antigen-presenting function. To explore the immunomodulatory effects of EMVs@SS-Toy, we co-cultured 4T1 breast tumor cells with bone marrow-derived dendritic cells (BMDCs) in Transwell chambers, creating a tumor-associated DCs (TADCs) model (Supplementary Fig. 24). Qualitative CLSM and quantitative flow cytometry analysis showed a time-dependent increase in ROS and lipid levels, with 24 h co-cultured TADCs selected for subsequent studies (Supplementary Fig. 25).
Compared to mesoporous silica (Si-MSN), both SS-Toy and EMVs@SS-Toy significantly reduced ROS levels in TADCs in vitro, due to the conversion of polysulfide bonds into hydrophilic sulfoxide under high ROS conditions25, thus promoting pathological ROS scavenging (Fig. 3a, c and Supplementary Fig. 26). Moreover, EMVs@SS-Toy effectively reduced lipid accumulation in TADCs, compared to free Toy (Fig. 3b, d and Supplementary Fig. 27). This outcome leveraged the homotypic targeting properties and immunogenic proteins of EMVs, enhancing the uptake of EMVs@SS-Toy by TADCs and thereby amplifying Toy’s therapeutic impact. To better characterize the cellular tropism of EMVs@SS-Toy, we compared their uptake by TADCs and NIH/3T3 fibroblasts, a model for normal, non-immune somatic cells (Supplementary Fig. 28). Over a 2 to 6 h incubation period, TADCs internalized EMVs@SS-Toy significantly more efficiently than NIH/3T3 cells, as quantified by a markedly strong fluorescence signal. These findings suggest that EMVs@SS-Toy exhibited preferential uptake by DCs, rather than by normal somatic cells.

a CLSM image displaying ROS levels in TADCs 6 h post-treatment of PBS, Si-MSN, SS-Toy and EMVs@SS-Toy. b CLSM images displaying BODIPY levels in TADCs 6 h post-treatment of Toy and EMVs@SS-Toy. Nuclei stained with Hoechst 33342 (blue), ROS and lipids detected by DCFH-DA and BODIPY 493/503 probes (green), respectively. c The ROS level assessment in the TADCs by flow cytometry. d The BODIPY level assessment in the TADCs by flow cytometry. e. The differential gene expression between the samples treated with EMVs@SS-Toy and PBS. f Gene Ontology (GO) enriched pathways of the upregulated genes in the samples treated with EMVs@SS-Toy, showing immune-related terms. P values are derived from one-sided Fisher’s Exact Test for pathway and GO enrichment. g Gene set enrichment analysis (GSEA) of the term positive regulation of immune system process. P values for GSEA were calculated using two-sided permutation test, and adjusted using Benjamini-Hochberg methods. h Cytokine profiling in the TADCs culture using ELISA. i, j Flow cytometry analysis of CD80 and MHC I H-2 Kb expression in BMDCs (n = 3 independent experiments). *P < 0.05 and **P < 0.01 by two-tailed Student’s t-test. k, l Flow cytometry analysis of CD8+ expression in CD3+ T cells (n = 3 independent experiments). *P < 0.05 and **P < 0.01 by two-tailed Student’s t-test. Data are presented as mean ± SD. Source data are provided as a Source Data file.
To further explore the immunostimulatory potential of EMVs@SS-Toy, we performed GO enrichment analysis using transcriptomic data from DCs treated with the agent. Compared to PBS-treated controls, transcriptomic analysis of in vitro-cultured DCs treated with EMVs@SS-Toy revealed substantial upregulation of genes associated with innate immune activation pathways (Fig. 3e, f). Specifically, EMVs@SS-Toy treatment led to increased expression of genes involved in co-stimulatory signaling (CD80, CD86), antigen processing and presentation (Calr3, Rfx5, Psme2b, Atg7, Lamp1, Tap2), cytokine production (IL6, C3, Tnfaip3), and chemokine signaling (Cxcl10, Ccr7). These findings suggest that chimeric exosomes may synergistically enhance DCs maturation and antigen presentation capacity, thereby contributing to a more robust antitumor immune response. Additionally, gene set enrichment analysis (GSEA) of DCs treated with EMVs@SS-Toy revealed significant enrichment in pathways associated with the term of positive regulation of immune system processes, activation of immune response, immune response-activating singaling pathway and positive regulation of immune response (Fig. 3g and Supplementary Fig. 29).
To assess the EMVs@SS-Toy’s ability to restore DCs antigen-presenting function, we introduced the model antigen OVA into the TADCs model. Flow cytometry results demonstrated that EMVs@SS-Toy markedly upregulated the expression of CD80 and MHC class I molecules on DCs, with 90.2% of DCs expressing both CD80 and MHC I (H-2Kb+) (Fig. 3i, j). Notably, compared to EMVs@SS without Toy loading, EMVs@SS-Toy significantly enhanced antigen presentation by DCs, confirming that dual stress inhibition could effectively reverse the dysfunction of TADCs. In addition, following this reactivation, we co-cultured the TADCs with CD3+ T cells to assess T cell proliferation. As shown in Fig. 3k, l, EMVs@SS-Toy treatment significantly increased the proportion of CD8+ T cells. Furthermore, EMVs@SS-Toy treatment led to a notable increase in the secretion of pro-inflammatory cytokines, including IFN-γ, IL-6, IL-12, TNF-α, and IL-1β, all of which play critical roles in immune modulation and reflect enhanced cytotoxic T cell activity (Fig. 3h). These findings provided compelling evidence that EMVs@SS-Toy could effectively restore DCs’ functionality within an immunosuppressive environment, thereby enhancing CD8+ T cell activation.
In vivo LNs’ homing effect and immunomodulatory effects on DCs
LNs play a central role in DCs-mediated antigen presentation and are crutial targets for antitumor immunotherapies26. Enhancing drug accumulation in LNs is essential for improving immunotherapy efficacy27,28. We hypothesized that EMVs encapsulation could offer the dual benefits of low immunogenicity and homotypic targeting, enabling efficient LNs-specific delivery while evading mononuclear phagocyte system (MPS) clearance (Fig. 4a).

a Illustrative representation of EMVs@SS-Toy’s lymph node-targeting in mice. b Ex vivo IVIS fluorescence images showing the biodistribution of fluorescence in the heart, liver, spleen, lungs, kidneys and tumor (n = 3 mice). c Quantitative analysis results of fluorescent intensity in tumor regions (n = 3 mice). d LNs histopathology with H&E staining. e Quantification of silicon uptake in LNs via ICP-MS at 24 h post-administration (n = 3 mice). f, g LNs-specific ex vivo IVIS fluorescence images and quantitative analysis of signal intensity (n = 3 mice). h CLSM images of LNs’ sections in LNs 24 h post i.v. administration of OMVs@SS-Toy and EMVs@SS-Toy, highlighting Nuclei (Hoechst 33342, blue), DCs (CD11c, red), and SSFITC-Toy (FITC, green) with co-localization (n = 3 independent experiments). i Quantitative results for co-localization in LNs’ sections post i.v. administration. j ROS detection in LNs 24 h post i.v. administration of PBS, OMVs@SS-Toy, and EMVs@SS-Toy (n = 3 independent experiments). Data are presented as mean ± SD. *P < 0.05 and **P < 0.01 by two-tailed Student’s t-test. Source data are provided as a Source Data file.
Our in vivo biodistribution studies revealed that EMVs@SS-Toy achieved a significantly higher accumulation in tumor tissue compared to OMVs@SS-Toy, as evidenced by increased fluorescence intensity (Fig. 4b). However, no significant differences in distribution were observed in other tissues and organs between two formulations (Supplementary Fig. 30). Quantitative analysis of organ samples ex vivo confirmed that EMVs@SS-Toy accumulated 1.85, 1.51, and 1.53 times more in tumor tissues at 6, 12 and 24 h post-administration, respectively, compared to OMVs@SS-Toy, indicating superior retention at tumor sites (Fig. 4c). To address the concern regarding the enhanced LNs targeting property of EMVs@SS-Toy, we performed pharmacokinetic studies using ultra high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) to quantify the Toy concentration of serum and LNs over time. As shown in Supplementary Fig. 31, following the i.v. administration, EMVs@SS-Toy group, similar with SS-Toy group, displayed prolonged circulation time compared to Toy group which was rapidly cleared from blood. These results indicated that EMVs@SS-Toy consistently exhibited significantly higher LNs’ concentrations than both free Toy and SS-Toy.
Next, we obtained LNs tissues through surgical procedures and performed H&E staining to confirm accurate sampling for subsequent analyses (Fig. 4d). ICP-MS analysis revealed significantly greater silicon retention in the EMVs@SS-Toy group, with a 1.61-fold increase compared to OMVs@SS-Toy (Fig. 4e). To evaluate the targeting efficiency of EMVs@SS-Toy to LNs, ex vivo imaging was performed, showing significantly higher fluorescence intensities in LNs at various time points post-administration (Fig. 4f). Quantitative analysis confirmed that the fluorescence intensity of EMVs@SS-Toy was 1.59, 1.71, and 1.88 times higher than that of OMVs@SS-Toy at 6, 12, and 24 h, respectively (Fig. 4g). Remarkably, at 24 h post-injection, EMVs@SS-Toy maintained high accumulation in LNs. This differential accumulation may be attributed to the immune system’s recognition of OMVs, which interacted with plasma proteins, leading to aggregation or instability29. In contrast, EMVs@SS-Toy, coated with chimeric exosomes, displayed lower immunogenicity, enabling more stable and prolonged systematic circulation. Meanwhile, we quantified the content of Toy in LNs via UHPLC-MS/MS to more precisely elucidate the in vivo targeting efficiency brought by the coating of EMVs. Compared to Toy and SS-Toy, treatment of EMVs@SS-Toy could remarkably enhance the distribution of Toy in LNs, which was in line with the ICP-MS findings (Supplementary Fig. 32). These findings underscored the superior LNs-targeting capability of EMVs@SS-Toy, suggesting that EMVs were more likely to localize in LNs over time.
To further investigate the targeting specificity of EMVs@SS-Toy, we labeled DCs with CD11c in LNs. CLSM analysis of LNs’ sections confirmed that, compared to OMVs@SS-Toy, EMVs@SS-Toy (labeled with FITC) exhibited significantly deep infiltration into the the core of LNs and showed a high degree of co-localization with DCs, demonstrating a notable enhancement in the targeting specificity towards DCs in LNs (Fig. 4h, i and Supplementary Fig. 33). The results demonstrated that the coating of EMVs significantly enhanced infiltration into LNs and increased targeting of DCs. This may be attributed to the presence of homologous targeting proteins30 and the elevated levels of chemokines associated with DCs present in the chimeric exosomes, as confirmed by our previous transcriptomics findings (Fig. 3e). Furthermore, EMVs@SS-Toy demonstrated the capacity to reduce intracellular oxidative stress, evidenced by a significant decrease in ROS levels within LNs (Fig. 4j and Supplementary Fig. 34) and DCs (Supplementary Fig. 35), confirming its potential as a ROS scavenger. These results underlined the complex interaction between EMVs@SS-Toy and LNs, highlighting the potential of EMVs@SS-Toy to leverage cellular trafficking pathways for enhanced therapeutic targeting.
Sensitizing TNBC immunotherapy through multi-level lipid modulation
After confirming that EMVs@SS-Toy preferentially accumulated and remained in LNs, we evaluated its potential to promote antitumor effects in combination with anthracycline-based chemoimmunotherapy using the 4T1 orthotopic breast tumor model (Fig. 5a). First, we evaluated tumor suppression across the following groups (PBS, Toy, DOX+Toy, and DOX+EMVs@SS-Toy) to elucidate whether EMVs@SS-Toy combined with DOX could enhance tumor suppression. Notably, the DOX+EMVs@SS-Toy group demonstrated a significantly enhanced anti-tumor response, underscoring the synergistic benefit of EMVs@SS-Toy in potentiating the therapeutic effect of chemotherapy (Supplementary Fig. 36). This may be due to the fact that EMVs@SS-Toy could better actively target LNs and regulate abnormal lipid metabolism and ROS disorder, synergizing with anti-tumor therapy. Second, we tested the combination strategy with aPD1. Pharmacodynamic evaluations indicated that both DOX+EMVs@SS-Toy and DOX+aPD1+EMVs@SS-Toy significantly inhibited tumor growth in mice (Fig. 5b–d and Supplementary Fig. 37a). None of the treatments affect body weights, confirming the safety of each treatment regimen (Supplementary Fig. 37b).

a Schematic illustration of the assessment protocol evaluating antitumor efficiency. b Visual representation of tumor progression in treated mice, with tumors outlined by a yellow dashed line. c, d Comparison of tumor progression and weight post-treatment (n = 5 mice). e H&E and Ki67 immunohistochemistry in tumor sections, red arrows: regions of necrotic tumor cells, blue arrows: viable tumor cells. n = 5 mice. f, g Flow cytometry analysis of activated DCs (CD80+CD86+) in LNs and tumors (n = 5 independent experiments). h Flow cytometry analysis of CD3+CD8+ T cells in LNs (n = 5 independent experiments). i Flow cytometry analysis of CD8+ T cells within CD45+ cells from tumors (n = 5 independent experiments). j, k Flow cytometry analysis of Tregs (CD3+CD4+Foxp3+) in LNs and tumors (n = 5 independent experiments). CLSM imaging of (l) XBP1s and (m) BODIPY in LNs (n = 3 independent experiments). G1: PBS, G2: DOX, G3: DOX + SS-Toy, G4: DOX+OMVs@SS-Toy, G5: DOX+EMVs@SS-Toy, G6: DOX+aPD1+EMVs@SS-Toy. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 by two-tailed Student’s t-test. Source data are provided as a Source Data file.
IHC analysis of Ki67 expression indicated that the DOX+aPD1+EMVs@SS-Toy group led to substantial suppression of tumor cell proliferation (Fig. 5e and Supplementary Fig. 38). H&E staining of tumor section revealed varying degrees of necrosis, with the highest level tumor necrosis observed in the DOX+aPD1+EMVs@SS-Toy group (Supplementary Fig. 39).
To further investigate the mechanisms driving the antitumor immune response, we analyzed immune cell phenotypes, quantities, and cytokine secretion induced by the different treatment regimens. Flow cytometry analysis revealed that both DOX+EMVs@SS-Toy and DOX+aPD1+EMVs@SS-Toy significantly upregulated the expression of co-stimulatory markers CD80 and CD86 on DCs in both tumors and LNs (Fig. 5f, g and Supplementary Figs. 40–41). Notably, DOX+aPD1+EMVs@SS-Toy treatment increased the proportion of CD80+CD86+ DCs in LNs to 54.5%, a 5.1-fold increase compared to the PBS-treated group. These treatments also resulted in a marked increase in CD8+ T cell infiltration in both tumors and LNs, which is crucial for inducing effective tumor-specific immune responses (Fig. 5h, i, and Supplementary Figs. 42–44). The increased presence of activated DCs and CD8+ T cells was indicative of a potent immune response, emphasizing the importance of the combined therapeutic strategy in reshaping the immunological landscape within LNs.
Moreover, the DOX+aPD1+EMVs@SS-Toy combination treatment significantly reduced Foxp3 expression in both tumors and LNs, demonstrating that the treatment inhibited Treg recruitment and effectively reversed the immunosuppressive microenvironment (Fig. 5j, k and Supplementary Figs. 45–46). An important observation was a substantial increase in CD62L+CD44+ central memory T cells (TCM) within LNs (Supplementary Fig. 47), suggesting that EMVs@SS-Toy may facilitate antigen-specific recall responses crucial for sustained antitumor immunity. These results demonstrated that EMVs@SS-Toy could efficiently sensitize immunotherapy and enhance its therapeutic effect.
Further analysis of lipid homeostasis within LNs revealed significant reductions in both XBP1s expression and BODIPY levels after treatment with DOX+EMVs@SS-Toy and DOX+aPD1+EMVs@SS-Toy (Fig. 5l, m and Supplementary Figs. 48–49). Safety assessments, including histopathological examination of vital organs, revealed no substantial morphological changes across all groups (Supplementary Fig. 50a). Biochemical tests also indicated no significant deviations in AST, ALT, and Crea levels, implying the absence of significant hepatotoxic or nephrotoxicity (Supplementary Fig. 50b). These findings underscore the potential of integrating EMVs@SS-Toy with immunotherapy to leverage immune pathways for improved therapeutic outcomes. The synergistic approach remarkably suppressed tumor growth by reducing oxidative stress and lipid accumulation in LNs, restoring efficient antigen presentation and refining the LNs immuno-microenvironment. This approach enhanced the response to PD1 blockade and induced robust antitumor immune responses.
Reshaping LNs microenvironment to inhibit tumor metastasis
Metastasis remains a major challenge in the treatment of TNBC, with lung metastasis being a leading cause of mortality. To assess the potential of EMVs@SS-Toy in inhibiting metastasis, we developed a mouse model of lung metastasis using 4T1-Luc tumor cells and monitored tumor progression through in vivo luciferase imaging (Fig. 6a). The imaging results revealed that treatment with DOX+EMVs@SS-Toy significantly reduced pulmonary fluorescence intensity compared to PBS, DOX, and DOX+aPD1 groups, indicating a substantial decrease in metastatic tumor growth. Notably, the addition of aPD1 further enhanced this effect, resulting in nearly complete inhibition of metastasis, as evidenced by negligible fluorescence (Fig. 6b). Ex vivo fluorescence imaging of excised lung tissues and measurements of radiant flux confirmed these findings, with the DOX+aPD1+EMVs@SS-Toy group showing the lowest fluorescence intensity and radiance (Fig. 6c, d and Supplementary Fig. 51). Additionally, histological evaluations revealed a marked reduction in metastatic tumor nodules, with no detectable metastasis in the lungs following DOX+aPD1+EMVs@SS-Toy (Fig. 6f, g). Correspondingly, the combined treatment significantly prolonged the survival of the mice, as indicated by the survival curves (Fig. 6h).

a Schematic illustration of the administration strategy in a metastatic breast cancer model. b In vivo bioluminescence imaging of mice with 4T1-Luc metastatic tumors over time (n = 6 mice). c Ex vivo IVIS fluorescence imaging of excised lung tissues (n = 5 mice). d Quantification of fluorescence intensity in lung tissues (n = 5 mice). e Serum cytokines profiling for IL-6, IL-1β, IFN-γ, IL-12, TNF-α, and IL-10 post-treatment. f Representative images of lung morphology post-treatment. g H&E staining of lung sections for histophathological analysis. h Survival assessment after different treatment regimens (n = 9 mice). i Flow cytometry analysis of central memory T cells (CD3+CD8+CD62L+CD44+) in peripheral blood from treated groups (n = 5 independent experiments). j Quantitative analysis of central memory T cells (CD3+CD8+CD62L+CD44+) in peripheral blood from treated groups (n = 5 independent experiments). k H&E staining of hilar LNs after different treatments (n = 3 independent experiments). l Immunofluorescence characterization of CD8, CD11c, MHC I, and Foxp3 in hilar LNs (n = 3 independent experiments). G1: PBS, G2: DOX, G3: DOX+aPD1, G4: DOX+EMVs@SS-Toy, G5: DOX+aPD1+EMVs@SS-Toy. Data are presented as mean ± SD. **P < 0.01 and ****P < 0.0001 by two-tailed Student’s t-test. Source data are provided as a Source Data file.
In addition to inhibiting metastatic growth, we observed significant changes in the immune profile of treated mice. Serum levels of pro-inflammatory cytokines, such as IL-6, IL-1β, IFN-γ, IL-12, TNF-α, were notably elevated in both the DOX+EMVs@SS-Toy and DOX+aPD1+EMVs@SS-Toy groups, while levels of the anti-inflammatory cytokine IL-10 were reduced (Fig. 6e). Furthermore, a marked increase in CD62L+CD44+ TCM was detected in the peripheral blood of mice treated with DOX+aPD1+EMVs@SS-Toy (Fig. 6i, j and Supplementary Fig. 52). These results underscore the potential of EMVs@SS-Toy to strengthen the immune response and improve the prognosis of TNBC patients by enhancing immune memory and controlling both primary and metastatic tumors.
Given the substantial reduction in lung metastasis observed with DOX+aPD1+EMVs@SS-Toy treatment, we further examined its effects on the immune landscape within the hilar LNs, which play a key role in tumor metastasis. Pathological and immunofluorescent analyses of hilar LNs revealed significant differences between the PBS and treatment groups. In the PBS-treated mice, notable tumor presence was observed in the hilar LNs, whereas LNs from the DOX+EMVs@SS-Toy+aPD1 group showed virtually no detectable tumor cells (Fig. 6k and Supplementary Fig. 53). Further immunofluorescent co-localization studies of DCs in the hilar LNs demonstrated that DOX+aPD1+EMVs@SS-Toy treatment significantly upregulated CD8+ T cells, CD11c, and MHC I expression, while reducing Foxp3+ Tregs (Fig. 6l). This indicates that EMVs@SS-Toy modulated the immune environment of hilar LNs, facilitating a robust activation of systemic immune responses and effectively targeting metastatic growth. By counteracting the immunosuppressive milieu in mLNs, this treatment regimen demonstrated a pronounced ability to suppress pulmonary metastases. These insights are of significant clinical relevance for the management and treatment of lung metastasis in TNBC.
Discussion
Although immunotherapy represents progress in treating TNBC, its efficacy is constrained by an immunosuppressive microenvironment in the LNs that impairs DCs’ activation capabilities. In this study, we developed a chimeric exosomes-derived immunomodulator that efficiently leveraged OMVs-induced stimulation of DCs to generate EMVs@SS-Toy. This immunomodulator restored DCs’ lipid homeostasis and immunological functions by directly intervening in the ER and oxidative stress pathways in DCs, thereby enhancing antigen presentation and tumor surveillance in LNs’ immuno-microenvironment. This biomimetic immunomodulator improved the efficacy of TNBC immunotherapy, demonstrating inhibition of primary tumor progression and lung metastasis while enhancing overall survival rates. Our findings provide a paradigm for TNBC immunotherapy, offering scientific and clinical insights to improve treatment response rates and patient outcomes.
Methods
Ethical statement
The study complies with all relevant ethical regulations. Experiments involving biopsy samples and MRI scans of TNBC patients used in this study were obtained from the Ethics Committee of the first hospital of China Medical University (Protocol Number: [2022] 436). All participants provided written informed consent to donate their biological samples and MRI images.
All the animal protocols were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committee of China Medical University (Protocol Number: CMUKT2022255). Humane endpoints were established as follows: tumor burden exceeding 10% of average body weight, weight loss greater than 20%, ulceration at the tumor site, and persistent self-mutilation. These endpoints received approval by the Certification and Accreditation Administration of the People’s Republic of China (CNCA).
Animals
Female BALB/c mice (species: Mus musculus, strain: BALB/c, genetic background: inbred BALB/cAnN), aged 8–10 weeks, were used in accordance the Guidelines for the Care and Use of Laboratory Animals.
Cell lines
The 4T1 cell line was purchased from the Cell Bank of Typical Culture Conservation Committee of the Chinese Academy of Sciences (SCSP-5056). DC2.4 cell line was purchased from the BeNa Culture Collection (BNCC351939). 4T1 and DC2.4 cells were cultured in RPMI 1640 with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in 5% CO2. 4T1 cells were transfected with a luciferase-expressing lentivirus (4T1-Luc) to enable bioluminescence imaging. All cell lines were validated using short tandem repeat (STR) markers and were tested negative for mycoplasma contamination.
Bone marrow-derived dendritic cells (BMDCs) were harvested from BALB/c mice and cultured in RPMI 1640 complete medium supplemented with 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and 10 ng/mL interleukin-4 (IL-4). Antibodies used in this study are listed in Table S2.
Software and code
The software used for data collection in this study includes Malvern: Zetasizer Software 7.01, Microplate reader: SkanIt 2.4.3.37 and Confocal laser scanning microscopy: NIS 4.13. The software used for data analysis in this study includes GraphPad prism 8.0, Microsoft Office 2019, ImageJ 1.8.0, FlowJo_V10, Trim-galore (version 0.6.7); HISAT2 (version 2.2.1), featureCounts (version 2.0.1), DESeq2 (version 1.32.0) and R package clusterProfiler, ABI Prism 3130 XL Genetic Analyzer, Gene Mapper ID 3.2 software (Applied Biosystems), Zetasizer Software 7.01, SPSS version 26.0, ZEN 3.3 software.
Obtaining clinical data and statistical analysis
This study utilized the Surveillance, Epidemiology, and End Results (SEER) database, with data accessed via SEER Stat software version 8.3.6. We selected female patients diagnosed with TNBC based on criteria including breast disease location, diagnosis between 2010 and 2020, age at diagnosis of 18 or older, confirmed pathological diagnosis, and HR-/HER2- breast subtypes. Initially, 20,959 breast cancer patients were identified, from which 2849 patients were selected for having complete records on age, race, tumor grade, TNM staging, primary tumor site, and treatment details in the final analysis. Baseline characteristics were analyzed using chi-square tests. A univariate Cox proportional hazards model assessed potential risk or protective factors, considering variables like age, race, tumor grade, TNM stage, primary tumor site, and neoadjuvant therapy, with a significance threshold of P < 0.05. Survival differences among subgroups were explored using Kaplan-Meier curves. The SEER database’s use did not require explicit patient consent or ethical approval from review committees due to the nature of the data.
Samples from TNBC patients
Patients with TNBC underwent Magnetic Resonance Imaging (MRI) before and after preoperative immunoadjuvant therapy to assess the impact of LNs’ status on immunotherapeutic response. Subsequently, nLNs and mLNs’ tissues were collected and subjected to Hematoxylin and Eosin (H&E) staining to observe pathological changes within the LNs. Immunohistochemistry (IHC) was utilized to detect Foxp3 expression. Fluorescence probe BODIPY 493/503 (5 μM, 20 min, 37 °C) was used for lipid detection, and CLSM was employed to observe the BODIPY intensity. Additionally, LNs were evaluated for PD1, XBP1s, MHC I, CD8, and CD11c expression using the AlphaTSA 7-color fluorescence staining kit (AXT37100041, AlphaX Biotech, China). Multiplex IHC staining was conducted using the AlphaPainter X30 system, with the ZEISS Axioscan7 for whole slide imaging. Image analysis was performed using ZEN 3.3 software.
Single-cell transcriptome sequencing
A breast cancer single-cell sequencing dataset (GSE158399) was obtained from the GEO database, including single-cell transcriptome data from matched primary breast cancer, mLNs, and negative nLNs from the same patient. Data from mLNs and nLNs were selected for further analysis. The data were analyzed using Seurat and SingleR packages. First, cells and genes included in the study were filtered based on the following criteria: 1) cells expressing fewer than 200 genes were removed; 2) genes expressed in fewer than 3 cells were removed; 3) cells with gene expression fluctuating between 200 and 7000 expressed genes were retained; 4) cells with mitochondrial gene percentage below 10% were retained. The SCTransform function was used to eliminate the influence of the cell cycle on subsequent results. The SCT normalization method was applied to integrate different samples and eliminate batch effects. To identify different cell clusters, the FindAllMarkers function in Seurat was used with default non-parametric Wilcoxon rank-sum test and Bonferroni correction to identify differentially expressed genes (DEGs) between clusters. The clusterProfilter package was used for Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the DEGs in different clusters.
Synthesis and characterization of SS-MSN
Polysulfide-bridged MSN (SS-MSN) was synthesized as follows: 0.6 g CTAT, 0.15 g triethanolamine (TEAH3), and 40 mL deionized water were stirred at 80 °C for 30 min. Then, a solution of 4.0 g TEOS and 1.0 g bis[3-(triethoxysilyl)propyl]tetrasulfide (BTESPT) was added dropwise to the surfactant solution. The resulting mixture was stirred at 80 °C for an additional 4 h at a stirring speed of 200 g. The products were collected by centrifugation, washed three times with ethanol, and refluxed in a 1% (w/v) NH4NO3 for 12 h. Finally, the as-synthesized SS-MSN was collected, washed, and dried for subsequent experiments. X-ray photoelectron spectroscopy (XPS, transparent, Netherlands) was used to verify the composition of SS-MSN. The pore characteristics of nanomedicines were obtained by the nitrogen adsorption/desorption tests with a nitrogen adsorption analyzer (VSorb 2800 P, app-one, China). Fourier transform infrared spectroscopy (FTIR) confirmed the introduction of functional groups and polysulfide bonds. TGA was carried out via a microcomputer differential thermal analysis instrument (HCT-1, Beijing, China) using air as a carrier gas from 25 to 700 °C.
Bacteria culture and OMVs collection
Attenuated Salmonella YS1646 were purchased from Beina Biological Co., LTD. Salmonella typhimurium VNP20009 was grown overnight in LB medium (37 °C). The following day, 500 mL of bacterial culture was centrifuged at 4000 g for 30 minutes to remove the bacteria, and the supernatant was filtered through a 0.45 μm polyethersulfone membrane filter (Millipore, SHLP033RB). The filtrate was then concentrated by centrifugation at 3000 g for 20 minutes at 4 °C using a 100 kDa ultrafiltration tube (Millipore, UFC810008). The concentrated solution was transferred into ultracentrifuge tubes and subjected to ultracentrifugation (Himac, 10PC THICK-WALLED TUBE ASSY, 338455 A) at 200,000 g for 2 h at 4 °C. After discarding the supernatant, the pellet was resuspended in PBS, washed twice, and filtered again through a 0.45 μm membrane to obtain OMVs. The protein concentration of OMVs was measured using a BCA Protein Assay Kit (Meilunbio, MA0082).
Cytotoxicity assay
DCs (7 × 104) were cultured in 96-well plates at 37 °C until reaching approximately 80% confluence. Subsequently, cells were treated with MSN and SS-MSN for 24 h. The original medium was removed, and the culture plate was incubated with 200 µL of MTT at 37 °C for 4 h. Next, 150 µL DMSO was added. A microplate reader (SkanIt 2.4.3.37) was utilized to determine the optical density (490 nm) of each well.
Preparation and characterization of EMVs@SS-Toy
The SS-MSN (1 mg/mL) solution was sonicated to achieve a homogeneous dispersion. Toy (2.5 mg/mL) was then added to the SS-MSN solution and stirred for 24 h at room temperature. To remove free Toy, the mixture was dialyzed against water for one day (MWCO = 3500 Da), yielding SS-Toy. Similarly, Si-Toy was prepared from Si-MSN (mesoporous silica) and Toy under identical conditions.
For the chimeric exosome coating, SS-Toy (1 mg/mL) was first mixed with OMVs at the same concentration, and the mixture was successively filtered through polycarbonate membranes of descending pore sizes (1 μm, 0.8 μm, 0.4 μm, and 0.2 μm), extruded at least 10 times. The extrudate was centrifuged (10,000 g, 30 min, 4 °C), the supernatant was removed, and the pellet was washed three times in PBS, yielding OMVs@SS-Toy. Then OMVs@SS-Toy (1 μg/mL) were incubated with DCs (5 × 106) for 6 h, followed by 36 h in a serum-free medium. The culture supernatant was centrifuged at increasing speeds (300 g for 10 min, 2000 g for 10 min, and 10,000 g for 30 min) and then ultracentrifuged (120,000 g, 2 h, 4 °C). The pellet was washed with cold, sterile PBS and resuspended in 200 μL to yield EMVs@SS-Toy. In addition, EMVs@SS without Toy loading were prepared in the same manner as a reference formulation. To label the formulations with fluorescence, SS-MSNFITC was synthesized by adding FITC to a 10% (w/w) APTES ethanol solution, stirring in darkness to form FITC-APS, and reacted with SS-MSN.
Particle size and zeta potential were measured using Malvern (Zetasizer Software 7.01). The morphology and elemental composition were examined using Transmission electron microscope (TEM, Thermo Scientific, Talos F200X, USA) and SEM (Thermo Scientific, Verios 5UC, USA). The distribution of elements on EMVs@SS-Toy was acquired by EDS mapping.
Drug loading efficiency and purity
To evaluate the drug loading efficiency of Toy within EMVs@SS-Toy, OMVs@SS-Toy was incubated with DC2.4 cells for 6 h. After incubation, the culture medium was replaced with serum-free medium, and the cells were further cultured for 12, 24, and 36 h, allowing collection EMVs@SS-Toy at each time point. The drug loading efficiency of EMVs@SS-Toy was assessed two steps: 1) Protein quantification: protein concentration was determined using a BCA protein assay kit with absorbance measured at 562 nm, providing an estimate of EMVs’ protein content. 2) Toy concentration quantification: Toy concentration in EMVs@SS-Toy was measured using a high-throughput 96-well plate format on a multimode microplate reader by reading absorbance at 279 nm, enabling the calculation of the drug loading yield and efficiency of EMVs@SS-Toy at each time point.
The purity of EMVs@SS-Toy was evaluated by flow cytometry (BD FACSCelesta, BD Bioscience). FITC-labeled SS-Toy was used to construct OMVs@SS-Toy (OMVs@SSFITC-Toy). OMVs@SSFITC-Toy were incubated with DC2.4 cells for 6 h. After incubation, the medium was replaced with fresh culture medium, and the DC 2.4 cells were cultured for an additional 36 h. EMVs@SS-Toy were then collected and incubated with APC-labeled anti-CD63 antibody (BioLegend, 143905, 1:200) for 30 min at room temperature. Flow cytometry was subsequently performed to assess the labeling.
The fate of OMVs@SS-Toy within DCs
DC2.4 cells were incubated with FITC-labeled OMVs@SS-Toy (OMVs@SSFITC-Toy) for 6 h. After incubation, the cell culture medium was replaced, and the cells were further cultured for 12, 24, and 36 h. The Lyso-Tracker Red (LTR) working solution was prepared by diluting the LTR lysosomal fluorescent probe with complete medium at a ratio of 1:15000. The cell culture medium was discarded, and 1 mL of the LTR working solution was added to each well, followed by incubation at 37 °C for 30 min. After removing the working solution, 1 mL of 4% paraformaldehyde was added to fix the cells for 10 min, followed by three washes with PBS. The Nuclei were then stained with Hoechst 33342 for 10 min, followed by three additional washes of PBS. The fate of OMVs@SS-Toy within DCs was observed using CLSM. Related data was collected using Confocal laser scanning microscopy (NIS 4.13).
FITC-labeled OMVs@SS-Toy (OMVs@SSFITC-Toy) were added to a culture plate containing DC2.4 cells and incubated for 6 h. Following incubation, the culture medium was replaced, and an exosome biogenesis/release inhibitor, GW4869 (20 μM), was added. The cells were then further incubated for 12, 24, and 36 h. After the specified incubation periods, the DC2.4 cells were fixed using a cell fixation solution and stained with DAPI for 10 min to visualize the cell nuclei. The cells were subsequently examined using the CLSM.
Confirmation of exosomal protein components on EMVs@SS-Toy
OMVs@SS-Toy was stained with 10 μM DiI for 30 min, centrifuged (20,000 g, 30 min), and then washed three times with PBS. CLSM was used to observe the co-localization of DiI with SS-MSNFITC. EMVs@SS-Toy was blocked with 5% BSA for 30 min and then incubated with APC-labeled CD63 antibody (Biolegend, 143905, 1:200) for 30 min at room temperature. CLSM observed the co-localization of CD63 and EMVs@SS-Toy.
DCs, DEX and EMVs@SS-Toy were lysed in RIPA lysate followed by western blot analysis. The primary antibodies used were anti-CD63 antibody (Abclonal, A19023), anti-TSG101 antibody (Abclonal, A2216), and anti-CD9 antibody (Abclonal, A19027). All primary antibodies were diluted to 1:10000. OMVs, OMVs@SS-Toy, DEX, and EMVs@SS-Toy were lysed in RIPA lysate followed by SDS-PAGE electrophoresis.
LC-MS/MS (proteomics analysis)
DEX@SS-Toy, OMVs@SS-Toy, and EMVs@SS-Toy samples were lysed with SDT (4% SDS, 100 mM Tris/HCl, 0.1 M DTT, pH 7.6) for protein extraction, followed by protein quantification using the BCA assay. An appropriate amount of protein from each sample was trypsinized by the filter aided proteome preparation (FASP) method, and then the hydrolyzed peptides were desalted using a C18 Cartridge. The peptides were lyophilized and redissolved in 40 μL dissolution buffer and quantified (OD280). Samples were separated by chromatography and analyzed using LC-MS/MS (Orbitrap Astral mass spectrometer coupled to a Vanquish Neo UHPLC system). The MS1 scan range was set to 380–980 m/z (resolution 240,000 at 200 m/z), with data-dependent MS2 scans acquired in 299 variable isolation windows. Raw data were analyzed using DIA-NN 1.8.1 software, applying a 99% confidence threshold and ≤1% false discovery rate (FDR) for protein and peptide identification.
Transcriptomic analysis
Total RNA was extracted from tissues using TRIzol and quantified for concentration and purity with a NanoDrop 2000. RNA integrity was assessed with an Agilent 2100 Bioanalyzer. Total RNA ( ≥ 1 μg) was used for library preparation with the NEBNext Ultra II RNA Library Prep Kit for Illumina or NEBNext Ultra Directional RNA Library Prep Kit (strand-specific). Sequencing was performed on the Illumina platform, generating raw FASTQ data. Raw data were processed with fastp to remove adapter sequences and low-quality reads (Q20). The filtered data (Clean data) were used for further analysis. The reference genome index was built with HISAT2. Gene expression was quantified using HTSeq to count reads per gene, and expression levels were normalized using FPKM. Differential expression was analyzed with DESeq. GO enrichment analysis was performed with topGO (P value < 0.05), and KEGG pathway analysis was done with clusterProfiler (P value < 0.05). GSEA was applied to identify enriched gene sets without predefined differential expression thresholds.
ICP-MS assessment for Si content
The lymph node samples were placed in 5 mL EP tubes, and 100 μL perchloric acid was added for digestion. After complete digestion, the samples were diluted to 5 mL with 1% nitric acid, filtered through a membrane, and set aside for further analysis. An appropriate volume of silicon standard solution was accurately transferred into EP tubes and diluted with nitric acid to prepare silicon standard solutions of suitable concentrations as stock solutions. Using completely digested blank lymph node sample solutions as the matrix, the stock solutions were successively diluted with nitric acid to obtain a series of lymph node sample standard solutions with gradient concentrations, ready for sample injection.
Under Kinetic Energy Discrimination (KED) mode, the silicon content in the series of lymph node sample solutions was determined by iCAP™ RQ Inductively Coupled Plasma Mass Spectrometer (ICP-MS, Thermo Scientific Co. Ltd., USA), measuring the response values (Y). Raw files generated by the ICP-MS were processed using Qtegra Intelligent Scientific Data Solution v.2.6, involving baseline correction, internal standard normalization with Sc (1 ppm) to correct signal drift, and blank subtraction using reagent blanks. A calibration curve was established by plotting the standard concentrations (C) on the x-axis and the corresponding response values (Y) on the y-axis.Data organization was performed using Microsoft Excel 365, two-tailed Student’s t-tests were used for statistical analyses and visualization were conducted with GraphPad Prism 8.0.
Uptake and exocytosis of EMVs@SS-Toy
To test cell uptake, FITC-labeled Si-Toy, SS-Toy, OMVs@Si-Toy, OMVs@SS-Toy or EMVs@SS-Toy were incubated with DCs (4 × 105) at specified time points. At the end of incubation, the intensity of FITC was analyzed by flow cytometry. Additionally, 1 mL of 4% paraformaldehyde was added to fix the cells for 10 min, Nuclei were stained with Hoechst 33342 (10 μg/mL, 10 min), and CLSM observed the cell uptake at 6 h.
To quantify the exocytosis efficiency, SS-Toy and OMVs@SS-Toy were incubated with DCs for 4 h, after which the cell medium was replaced with a serum-free medium and cultured further. The supernatant was collected to extract exosomes at 12, 24 and 36 h. Cells were collected for BCA protein quantification, and ICP-MS was used to detect silicon content.
Ex vivo drug release and stability
To assess the release kinetics of Toy, 150 μL of Si-Toy, SS-Toy, OMVs@SS-Toy and EMVs@SS-Toy solutions were analyzed for Toy absorbance at 279 nm using UV-Vis spectroscopy, respectively. PBS buffer (pH 7.4) was prepared with 10 mM H2O2 and 10 mM DTT, respectively. A 1 mL aliquot of each solution was sealed in a dialysis bag (MWCO = 3500 Da) and incubated in a 50 mL centrifuge tube containing 9 mL of the respective buffer at 37 °C. At intervals (1, 2, 4, 6, 8, 10, 24 h), 1 mL of buffer was replaced with fresh buffer, and the Toy concentration was measured via UV-Vis spectroscopy to calculate cumulative release.
Blood from BALB/c mice was centrifuged at 1000 g for 10 min to collect serum. OMVs@SS-Toy and EMVs@SS-Toy solutions were then incubated in PBS containing 10% serum (pH 7.4) at 37 °C. Solution samples (1 mL) were collected at 0, 1, 2, 4, 6, 8, and 12 h to determine particle size.
Generation of TADCs model
We used a co-culture model involving BMDCs and 4T1 cells to simulate TADCs conditions, specifically under high oxidative stress and endoplasmic reticulum (ER) stress. This model was established in a Transwell system to mimic the microenvironment that TADCs experience within a tumor31. BMDCs (4 × 105 cells) were seeded into a 12-well plate with 2.5 mL of RPMI 1640 complete medium. Separately, 200 μL of 4T1 cells, adjusted to a 1:1 ratio with BMDCs, were placed into a 0.4 μm Transwell insert and co-cultured. ROS and lipid content in the TADCs were measured at 0, 6, 12, and 24 h using specific dyes: DCFH-DA (10 μM, 30 min, 37 °C) for ROS detection, and BODIPY 493/503 (5 μM, 20 min, 37 °C) for lipid detection. Hoechst 33342 was used for Nuclei staining. CLSM was employed to observe lipid and ROS intensity.
Examination of ROS consumption capability
The Transwell chamber was removed to terminate the co-culture of the TADCs after 24 h. The culture medium of the TADCs was replaced with medium containing either PBS, MSN or SS-MSN and incubated for 6 h. DCFH-DA levels were analyzed using flow cytometry and observed by CLSM.
Lipid production blocking evaluation
The Transwell chamber was removed to terminate the co-culture of the TADCs after 24 h. The culture medium of the TADCs was replaced with medium containing either PBS, Toy and EMVs@SS-Toy and incubated for 6 h. BODIPY 493/503 levels were analyzed quantitatively using flow cytometry and observed by CLSM.
Evaluation of immune-stimulating activity of EMVs@SS-Toy ex vivo
The Transwell chamber was removed after 24 h, terminating the co-culture of TADCs. Cells were then incubated with PBS, SS-Toy, OMVs@SS-Toy, EMVs@SS, EMVs@Si-Toy, EMVs@SS-Toy, and OVA peptide (1 mg/mL) for another 24 h. Post-incubation, TADCs cells were centrifuged and washed twice with PBS. Fluorescence labeling was performed using anti-CD11c-APC, anti-CD80-FITC, and anti-SIINFEKL-H-2Kb-PE, followed by flow cytometry analysis.
CD3+ T cells isolated from the murine spleen were co-incubated with TADCs for 24 h. Cells were labeled with anti-CD3-FITC, anti-CD4-PE, and anti-CD8-APC for flow cytometry analysis. Additionally, supernatants from the cultures were collected to measure cytokine levels (IFN-γ, IL-6, IL-12, TNF-α, and IL-1β) using ELISA kits.
UHPLC-MS/MS (pharmacokinetic analysis)
Female BALB/c mice weighing 20–22 g were fasted for 12 h (with free access to water) and randomly divided into three groups, then intravenously administered Toy solution, SS-Toy, and EMVs@SS-Toy at a dose of 4 mg/kg via the tail vein. Blood samples were collected at predefined by centrifuging at 500 g for 10 min at 4 °C to isolate plasma. The residue was dissolved in 100 μL methanol, followed by mixing for 1 min, sonication for 5 min, centrifugation for 10 min at a temperature of 4 °C and a speed of 8000 g in a centrifuge, and the upper liquid layer was injected into the UHPLC-MS/MS for analysis.
Liquid chromatographic separation was carried out on an XR LC-30AD Prominence UHPLC system (Shimadzu, Japan), which was equipped with a binary, a degasser, pump, an autosampler, and a column oven. In a steadystate environment at 40◦C, analytes were separated on a Waters CORTECS T3 column (2.1 mm × 100 mm, 3 μm). The mobile phase system consisted of 0.1% formic acid and acetonitrile solution was used to gradient elution at a flow rate of 0.4 mL/min with a sample injection volume of 5 μL. The gradient program was 5–5% B (acetonitrile) for 0.01–1 min, 5–90% B for 1–3 min, 90–95% B for 3–5 min, 95% B for 5–6 min and 5% B for 6.1–8.5 min. MS quantitative analysis was operated on a SCIEX QTRAP 6500+ system (Sciex, USA), and then the MultiQuant 3.0.2 (Sciex, USA) was applied to acquire and process data. In the MRM mode, ESI + was adopted to detect four analytes with nitrogen taken as the circulating gas. Specific MS parameters were set as follows: 50 psi for gas 1, 50 psi for gas 2, 20 psi for air curtain gas, 5500 V for ion spray voltage, 500◦C for ion source temperature. The MRM measurements of the analytes were performed using eclustering potential (DP), entrance potential (EP), collision energy (CE), and cell exit potential (CXP) values optimized for analytes.
The pharmacokinetic parameters of the three sets of analytes were treated by using DAS 2.1 pharmacokinetic software. Next, the parameters of each group were compared by SPSS 20.0 v, and P < 0.05 was considered statistically significant.
In vivo biodistribution
Orthotopic breast tumor models were established by implanting 4T1 cells (1 × 106) into the left mammary fat pads of mice. When tumors reached approximately 100 mm3, mice were i.v. administered FITC-labeld OMVs@SS-Toy or EMVs@SS-Toy at a dose of 10 mg/kg SS-MSNFITC. Tissues, including tumor, heart, liver, spleen, lung, kidney, and inguinal LNs were collected at 6, 12, and 24 h post-administration. LNs were subject to H&E staining for pathological examination and analyzed for SS-MSNFITC fluorescence using the IVIS Spectrum imaging system (IVIS Lumina Series III). ICP-MS quantified the silicon enrichment.
Orthotopic breast tumor models were established by implanting 4T1 cells (1 × 106) into the left mammary fat pads of mice. When tumors reached approximately 100 mm3, mice were i.v. administered Toy, SS-Toy or EMVs@SS-Toy at a dose of 2 mg/kg Toy. Inguinal LNs were collected at 6, 12, and 24 h post-administration. The concentrations of Toy were measured by UHPLC-MS/MS.
Detection of lipid content and ROS levels in DCs within LNs
Orthotopic breast tumor models were established by implanting 4T1 cells (1 × 10⁶) into the left mammary fat pads of mice. When tumors reached approximately 100 mm³, mice were i.v. administered FITC-labeld OMVs@SS-Toy or EMVs@SS-Toy at a dose of 10 mg/kg SS-MSNFITC. After 24 h, mice were euthanized, and LNs were collected for further analysis. For lipid content detection, frozen LN sections were incubated overnight at 4 °C with a primary anti-CD11c antibody. After washing with PBS, sections were incubated with a secondary antibody for 30 min at room temperature, followed by staining with BODIPY 493/503 and DAPI. Lipid content and DCs localization were visualized using CLSM. For ROS detection, excised LNs were digested with 2 mL of collagenase IV for 1 h at 37 °C, filtered through a 70 µm strainer, and centrifuged at 300 × g for 10 min. The resulting cell suspension was stained with anti-CD11c antibody and the DCFH-DA probe for 30 min at 4 °C in the dark. After washing, samples were analyzed by flow cytometry. Additionally, frozen LN sections were incubated with DCFH-DA and imaged using CLSM to assess ROS levels.
Antitumor activity in orthotopic 4T1 breast cancer model
BALB/c mice were inoculated with 1 × 106 4T1 cells into the breast fat pad. When tumor volumes reached approximately 100 mm3, mice were randomized into six groups and treated with PBS, DOX, DOX + SS-Toy, DOX+OMVs@SS-Toy, DOX+EMVs@SS-Toy, and DOX+aPD1+EMVs@SS-Toy. Following an initial DOX dose (2.5 mg/kg, intratumoral) on day 1, subsequent treatments with PBS, SS-Toy, OMVs@SS-Toy, EMVs@SS-Toy, and EMVs@SS-Toy+aPD1 (aPD1: 50 μg/mouse, i.v.; Toy: 1 mg/kg, i.v.) were administered every two days, totaling three administrations. Tumor volume and body weight were monitored throughout the treatment period. On day 19 post-tumor inoculation, mice were euthanized, and tumor tissues and inguinal LNs were harvested for weighing and photography. Tumor necrosis was evaluated using H&E staining, while cell proliferation was assessed using Ki67 staining.
In vivo antitumor immune responses
To prepare single cell suspensions, tumor and LNs’ tissues were immersed in 2 mL collagenase IV solution (1.5 mg/mL), minced, shaken for 30 min, and then digested at 37 °C for 1 h. The digestion was halted by adding an equal volume of complete culture medium and straining through a 70 μm filter. The resulting suspensions were centrifuged (300 g, 10 min), and the pellet was resuspended in 200 μL Cell Staining Buffer for immunophenotyping. Cells were stained with fluorescently labeled antibodies against various immune cell markers and resuspended in 200 μL Cell Staining Buffer, passed through a sieve into a flow tube, and analyzed by flow cytometry. DCs were identified with anti-CD11c-APC, anti-CD86-PE, and anti-CD80-FITC; CD8+ T cells with anti-CD3-FITC, anti-CD4-Percp, and anti-CD8a-APC; Tregs with anti-CD4-Percp and anti-Foxp3-PE; and memory T cells with anti-CD3-Percp, anti-CD44-PE, anti-CD8-FITC, and anti-CD62L-APC/Cy7.
For histological analysis, LNs’ sections underwent antigen retrieval in repair solution at 85 °C for 6 min, blocked with 5% goat serum at 37 °C for 1 h, and incubated with anti-XBP1s antibody and anti-CD8 antibody at 4 °C for 12 h. After PBS washes, sections were treated with a secondary antibody for 90 min, stained with DAPI for 15 min, and examined with CLSM.
In vivo toxicity of EMVs@SS-Toy
All mice were euthanized after treatment, and the major organs (heart, liver, spleen, lungs, and kidneys) were harvested and subjected to the H&E staining. Additionally, serum was collected to analyze levels of hepatotoxic or nephrotoxic parameters including AST, ALT, and Crea.
In vivo antitumor effects against lung metastasis
To establish a lung metastasis model of breast cancer, 100 μL of 4T1-Luc cell suspension was intravenously injected into each mouse. Three days post-inoculation, mice were allocated into five groups and treated with PBS, DOX, DOX+aPD1, DOX+EMVs@SS-Toy, and DOX+aPD1+EMVs@SS-Toy. Treatments were administered one day after the first DOX dose (2.5 mg/kg, i.v.), including PBS, DOX, DOX+aPD1, DOX+EMVs@SS-Toy, and DOX+aPD1+EMVs@SS-Toy (aPD1: 50 μg/mouse, i.v.; Toy: 1 mg/kg, i.v.), every other day for a total of three administrations. On day 11 post-tumor inoculation, mice were euthanized, and lung tissues were excised for fluorescence imaging and metastatic nodule counting. H&E staining was performed on lung and hilar LNs’ tissues to assess pathology. Survival assessment was conducted after different treatment regimens.
Lung sections were stained overnight at 4 °C with antibodies against CD8, CD11c, MHC I, and Foxp3, followed by incubation with respective secondary antibodies at room temperature for 30 min. DAPI staining was conducted in dark and analyzed with CLSM.
The orbital blood of mice was collected prior to euthanasia, with serum isolated via centrifugation (1500 g, 10 min). Memory T cells were identified using anti-CD3-Percp, anti-CD44-PE, anti-CD8-FITC, and anti-CD62L-APC/Cy7 at 4 °C. Serum cytokine levels (IL-6, IL-1β, IFN-γ, IL-12, TNF-α and IL-10) were quantified using ELISA kits.
Statistics & Reproducibility
Statistical analysis was analyzed using Excel 2021, SPSS version 26.0, R software version 4.2.1, and GraphPad Prism 8.0. Data are presented as means ± SD. Kaplan-Meier curves and log-rank tests were used to compare OS among the LNs’ status. The student’s T-test (two-tailed) and one-way analysis of variance (ANOVA) were used to analyze the comparison between groups. All experiments were repeated at least 3 times, and representative results shown in the figures are consistent across replicates. A P value < 0.05 was considered statistically significant. The levels of significance were set at the probability of *P < 0.05, **P < 0.01, ***P< 0.001 and ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data generated in this study are provided in the Supplementary Information/Source Data file. The source data generated in this study have been deposited in the Figshare database at: https://doi.org/10.6084/m9.figshare.29361992. The single-cell RNA sequencing data utilized by this study were sourced from the NCBI Gene Expression Omnibus (GEO) database under accession number GSE158399, and the clinical data for TNBC patients were obtained from the SEER database (https://seer.cancer.gov). The RNA-seq data generated in this study have been deposited to GEO database under accession number: GSE270025. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD052986 (https://www.iprox.cn/page/PSV023.html;?url=1718701694060hOIH, secure token: RNpQ). All the other data that support the findings of this study are available within the Article, Supplementary Information or source data files. Source data are provided with this paper.
References
Leon-Ferre, R. A. & Goetz, M. P. Advances in systemic therapies for triple negative breast cancer. BMJ 381, e071674 (2023).
Topalian, S. L. et al. Neoadjuvant immune checkpoint blockade: a window of opportunity to advance cancer immunotherapy. Cancer Cell 41, 1551–1566 (2023).
Delclaux, I., Ventre, K. S., Jones, D. & Lund, A. W. The tumor-draining lymph node as a reservoir for systemic immune surveillance. Trends Cancer 10, 28–37 (2024).
Rahim, M. K. et al. Dynamic CD8+ T cell responses to cancer immunotherapy in human regional lymph nodes are disrupted in metastatic lymph nodes. Cell 186, 1127–1143.e1118 (2023).
du Bois, H., Heim, T. A. & Lund, A. W. Tumor-draining lymph nodes: at the crossroads of metastasis and immunity. Sci. Immunol. 6, eabg3551 (2021).
Dammeijer, F. et al. The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell 38, 685–700 e688 (2020).
de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403 (2023).
Morris, G., Gevezova, M., Sarafian, V. & Maes, M. Redox regulation of the immune response. Cell Mol. Immunol. 19, 1079–1101 (2022).
Li, Y. L. et al. Single-cell analysis reveals immune modulation and metabolic switch in tumor-draining lymph nodes. Oncoimmunology 9, 1830513 (2020).
Yang, K. et al. The role of lipid metabolic reprogramming in tumor microenvironment. Theranostics 13, 1774–1808 (2023).
Martin-Perez, M., Urdiroz-Urricelqui, U., Bigas, C. & Benitah, S. A. The role of lipids in cancer progression and metastasis. Cell Metab. 34, 1675–1699 (2022).
Yu, W. et al. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J. Hematol. Oncol. 14, 187 (2021).
Cubillos-Ruiz, J. R. et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161, 1527–1538 (2015).
Lei, X. et al. Mitochondrial fission induces immunoescape in solid tumors through decreasing MHC-I surface expression. Nat. Commun. 13, 3882 (2022).
Wu, H. et al. Polychlorinated biphenyls-153 induces metabolic dysfunction through activation of ROS/NF-kappaB signaling via downregulation of HNF1b. Redox Biol. 12, 300–310 (2017).
Ding, Y., Li, Z., Jaklenec, A. & Hu, Q. Vaccine delivery systems toward lymph nodes. Adv. Drug Deliv. Rev. 179, 113914 (2021).
Cheng, L. & Hill, A. F. Therapeutically harnessing extracellular vesicles. Nat. Rev. Drug Discov. 21, 379–399 (2022).
Ghorbaninezhad, F. et al. Dendritic cell-derived exosomes: a new horizon in personalized cancer immunotherapy?. Cancer Lett. 562, 216168 (2023).
Li, J. et al. Dendritic cell derived exosomes loaded neoantigens for personalized cancer immunotherapies. J. Control Release 353, 423–433 (2023).
Pitt, J. M. et al. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Invest 126, 1224–1232 (2016).
Wang, S. et al. Macrophage-tumor chimeric exosomes accumulate in lymph node and tumor to activate the immune response and the tumor microenvironment. Sci. Transl. Med. 13, eabb6981 (2021).
Ma, J. et al. Tumor microenvironment targeting system for glioma treatment via fusion cell membrane coating nanotechnology. Biomaterials 295, 122026 (2023).
Man, X. et al. Survival analysis and nomogram for early-stage occult breast cancer with positive lymph nodes based on the SEER database. Ann. Transl. Med. 10, 1351 (2022).
Patwardhan, S., Mahadik, P., Shetty, O. & Sen, S. ECM stiffness-tuned exosomes drive breast cancer motility through thrombospondin-1. Biomaterials 279, 121185 (2021).
Sun, B. et al. Disulfide bond-driven oxidation- and reduction-responsive prodrug nanoassemblies for cancer therapy. Nano Lett. 18, 3643–3650 (2018).
Schudel, A. et al. Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol. 15, 491–499 (2020).
Wang, Y. et al. Cooperative self-assembled nanoparticle induces sequential immunogenic cell death and toll-like receptor activation for synergistic chemo-immunotherapy. Nano Lett. 21, 4371–4380 (2021).
Feng, H. Y. et al. Targeted micellar phthalocyanine for lymph node metastasis homing and photothermal therapy in an orthotopic colorectal tumor model. Nanomicro Lett. 13, 145 (2021).
Qing S., et al. Biomineralized bacterial outer membrane vesicles potentiate safe and efficient tumor microenvironment reprogramming for anticancer therapy. Adv. Mater. 32, 2002085 (2020).
Song, T. et al. Engineering the deformability of albumin-stabilized emulsions for lymph-node vaccine delivery. Adv. Mater. 33, e2100106 (2021).
Lu, Y. et al. A therapeutic DC vaccine with maintained immunological activity exhibits robust anti-tumor efficacy. J. Control Release 349, 254–268 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81803442), the Scientific Research Construction Project (2023A01-12), Liaoning Provincial Department of Science and Technology Joint Project No. 2024-MSLH-555, the Shenyang Special Fund for the Cultivation of Young and Middle-aged Scientific and Technological Innovation Talents (RC230527) and the Outstanding Youth lifting Program in the Shenyang Pharmaceutical University (YQ202301).
Author information
Authors and Affiliations
Contributions
J.S., S.C. and Z.H. conceived the project. Q.L. and M.S. designed the experiments. Y.W., Z.C., M.S., H.L., T.H., and X.L. performed all the experiments. Y.W., B.Z., X.L., P.O., L.C., and P.C. analyzed all data. All authors reviewed this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Natalie Trevaskis, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, M., Wu, Y., Chen, Z. et al. Chimeric exosomes-derived immunomodulator restoring lymph nodes microenvironment for sensitizing TNBC immunotherapy. Nat Commun 16, 7116 (2025). https://doi.org/10.1038/s41467-025-62543-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62543-x
