
Abstract
RNA velocities and generalizations emerge as powerful approaches for extracting time-resolved information from high-throughput snapshot single-cell data. Yet, several inherent limitations restrict applying the approaches to genes not suitable for RNA velocity inference due to complex transcriptional dynamics, low expression, or lacking splicing dynamics, or data of non-transcriptomic modality. Here, we present GraphVelo, a graph-based machine learning procedure that uses as input the RNA velocities inferred from existing methods and infers velocity vectors lying in the tangent space of the low-dimensional manifold formed by the single cell data. GraphVelo preserves vector magnitude and direction information during transformations across different data representations. Tests on synthetic and experimental single-cell data, including viral-host interactome, multi-omics, and spatial genomics datasets demonstrate that GraphVelo, together with downstream generalized dynamo analyses, extends RNA velocities to multi-modal data and reveals quantitative nonlinear regulation relations between genes, virus, and host cells, and different layers of gene regulation.
Similar content being viewed by others


Introduction
Cells need to constantly detect and adapt to changes in extracellular and intracellular environments. Regulation of their gene transcription is a common mechanism of response. Multiple factors affect the transcriptional activity of eukaryotic genes, including cis and trans regulatory elements and chromatin structure. High throughput single-cell sequencing data provide the landscape of cell genotype. These data lack, however, information on how the cell state changes over time. Continuous efforts have been made to extract information about gene regulation and develop methods for connecting the cell states to temporal sequences of events captured by single-cell snapshot data. One group of methods that has received extensive attention is based on RNA velocity1 for predicting the changes in RNA expression states in the cell. The original RNA velocity method leverages the ratio between nascent and mature transcripts to estimate the rate of change in gene expression. This seminal study has inspired numerous methods for improved RNA velocity estimation based on information from splicing2,3,4,5,6, metabolic labeling7,8, lineage tracing9, and transcriptional factor binding10, etc.
The RNA velocity framework has, however, its inherent limitations. First, none of the RNA velocity estimation methods could be applied to any single cell transcriptomic data without restrictions. For example, the splicing-based method is not applicable to prokaryotes or viruses, or organisms without introns. Erroneous inferences of RNA velocities have also been noticed for genes having complex splicing dynamics11. Furthermore, it is difficult to estimate the RNA velocities of genes with low expression, which excludes most transcription factors. Second, multi-omics sequencing technologies provide multifaceted information on cellular states alongside the transcriptome modality, and there exist limited systematic methods to extend such velocity estimations to other modalities12,13.
The single-cell transcriptome state is usually defined by the instantaneous distribution of RNA levels, represented by a multidimensional vector of RNA levels for all (measurable) genes. The usual practice is to map such single-cell data onto a reduced space, e.g., transform from a principal component (PC) space to a UMAP representation to facilitate the visualization of the time-evolution of cell state, with each cell state being represented by a point in that space. Numerous dimension reduction and manifold learning algorithms have been developed for representation transformation. In comparison, transforming a velocity vector between representations is a nontrivial task not rigorously addressed in the single-cell field. Even worse, a visually correct vector field does not necessarily imply accurate high-dimensional velocity estimation14. La Manno et al. proposed a cosine kernel method to address this challenge, which has been adopted since then in most subsequent studies1. Li et al. mathematically proved that the cosine kernel asymptotically gives the correct direction of a velocity vector in the large sampling limit, but the magnitude information is completely lost due to a normalization procedure15. This loss of information casts concerns when such quantitative information is needed.
In this study, we tackle the above challenges through a graph-theoretical representation of RNA velocities, called GraphVelo, with dynamical systems underpinnings. GraphVelo takes an ansatz that the measured single-cell expression profiles and inferred RNA velocities collectively reflect a dynamical process and are connected through a set of dynamical equations. It exploits such additional constraints that couple a high dimensional velocity field and a corresponding single-cell state manifold, and enables the generalization of the approach in the context of multi-modal single-cell data. While the combined expression and velocity information has been widely used to infer cell state transition trajectories2,16, GraphVelo presents the advantage of enabling downstream analyses such as that performed by dynamo7 to extract quantitative information on causal gene-gene relations that dictate the cell state transitions. Benchmarking of the proposed graph framework against simulated and experimental single-cell data lends support to its broad utility.
Results
GraphVelo infers manifold-consistent single-cell velocity vectors through tangent space projection and transforms between representations through local linear embedding
Consider that the internal state of a cell can be specified by an N-dimensional state vector x, with N ≫ 1 generally. Assume that the temporal evolution of the cell state follows a continuous and smooth curve x(t) (see Methods for more general mathematical formulation). The instant velocity vector v(x, t) = dx(t)/dt is always tangent to the curve of x(t) (as a function of t) at x. One can generalize to the situation that the trajectories of a swarm of cells form a M-dimensional manifold \({{{\mathcal{M}}}}({{{\bf{x}}}})\) embedded in the N-dimensional state space with M ≪ N typically, as revealed by high-throughput single cell omics data. Then under the ansatz that a velocity vector v(x, t) dictates the evolution of a state vector x(t), v must lie in the tangent space of \({{{\mathcal{M}}}}({{{\bf{x}}}})\), denoted as \({T}_{p}{{{\mathcal{M}}}}\). In practice, the RNA velocity vectors inferred from existing methods do not automatically satisfy this tangent space requirement (but see4).
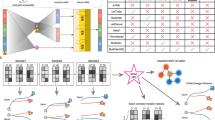
Taking various inferred single-cell RNA velocity vectors, e.g., splicing-based, metabolic labeling-based, or lineage tracing-based, as input, GraphVelo takes advantage of the nature of the low-dimensional cell state manifold to: (1) refine the estimated RNA velocity to satisfy the tangent space requirement; (2) infer the velocities of non-transcriptomic modalities using RNA velocities. GraphVelo thus serves as a plugin that can be seamlessly integrated into existing RNA velocity analysis pipelines, and help process single-cell data for downstream cellular dynamics analyses using methods such as dynamo (Fig. 1a).

a Workflow of RNA velocity-based analyses incoporating GraphVelo. Note GraphVelo takes any form of RNA velocity (i.e., not just splicing-based velocity) as input, and the kNN neighborhood is defined in the full state space (e.g., by both scRNAseq and scATACseq in multi-omics data). b Schematic of tangent space projection and velocity transformation between homeomophic manifolds. Left: RNA velocity vectors are projected onto the tangent space defined by the discretized local manifold of neighborhood cell samples. Right: GraphVelo allows for transformation of velocity vectors from a manifold embeded in a higher dimensional space \(({{{\mathcal{M}}}})\) to that in a lower-dimensional space \((\aleph )\), and vice versa. c The process of minimizing the loss function of tangent space projection. Noisy velocity vectors (left) generated by adding random components orthogonal to those sampled from an analytical 2D manifold were projected back onto the 2D manifold, resulting in smooth velocity vectors that lie in the tangent space (right). d GraphVelo allows whole genome velocity inference based on the robustly estimated MacK genes (see also Fig. 3). Velocities of genes undergoing variable kinetic rates, such as rapid degradation or transcription burst, are difficult to be correctly inferred by other methods, but can be inferred robustly with GraphVelo. e Virus infection dynamics and underlying host-virus interaction mechanisms uncovered by GraphVelo (see also Fig. 4). Upper: pathways involved in host-virus interactions were identified using GraphVelo. Lower: GraphVelo predicted reversed trajectory of viral infection in response to in silico perturbations of viral factors. f GraphVelo provides a consistent view of epigenetic and transcription dynamics (see also Fig. 5). Upper: GraphVelo analyses on multi-omics data revealed that most cell-cycle dependent genes showed decoupling between transcription dynamics and chromatin accessibility change dynamics. Lower: Effective dose-response curves reconstructed from multi-omics data revealed pioneer transcription factors increased chromatin accessibility then transcription of targe genes.
Practically, GraphVelo approximates the tangent space at a cell state x by a k-nearest neighbor (kNN) graph following the local linear embedding algorithm17, and uses the more reliable data manifold \({{{\mathcal{M}}}}\) to refine the velocity vectors by imposing the constraint that the N-dimensional velocity vector \({{{\bf{v}}}}\) should lie in the tangent space (Fig. 1b, c). Consider a given point \({{{{\bf{x}}}}}_{i}\) on a manifold corresponding to the expression state i of the single cell. Its infinitesimal neighborhood forms an Euclidean space that approximates the tangent space \({T}_{p}{{{\mathcal{M}}}}\). With a sufficient sampling of the neighboring cell states j in the state space, the incremental displacement vectors between cell state i and its neighboring cell states, \({{{{\boldsymbol{\delta }}}}}_{{ij}}={{{{\bf{x}}}}}_{j}-{{{{\bf{x}}}}}_{i}\), form a set of complete albeit possibly redundant and nonorthogonal/non-normalized basis vectors of the Euclidean space in the local region. Then the projection of the measured velocity vector onto \({T}_{p}{{{\mathcal{M}}}}\) can be expressed as a linear combination (see also Supplementary Notes 1.1),
where \({{{{\mathcal{N}}}}}_{i}\) is the neighborhood of cell state i, defined by its k nearest neighbors in the feature space determined by sequencing profiles. Direct application of Eq. 1 to determine the coefficients \({\phi }_{{ij}}\) is numerically unstable in real data (see Supplementary Notes 1.2 for detailed discussion). Instead, we performed the projection by optimizing the following tangent space projection (TSP) loss function (Fig. 1c),
where \(\Vert \cdot \Vert\) refers to vector modulus. \({{{{\mathbf{\phi }}}}}_{i}^{{{{\rm{corr}}}}}\) is a heuristic “cosine kernel” widely used in the RNA velocity analyses for projecting velocity vectors onto a reduced space, the elements of which are \({\phi }_{{ij}}\left({withj}\in {{{{\mathcal{N}}}}}_{i}\right)\) (see also Supplementary Notes 1.3); the second term \(\cos \left(\cdot,\cdot \right)\) denotes the cosine similarity. The first term in the loss function learns the correct velocity magnitudes, and the second term retains the reliable direction information based on previous mathematical analyses showing that \({{{{\mathbf{\phi }}}}}_{i}^{{{{\rm{corr}}}}}\) asymptotically gives correct direction of the velocity vector15. The L2 regularization is used to bound parameters \({{{{\mathbf{\phi }}}}}_{i}\). Hyperparameters a, \(b\) and \(\lambda\) are for retaining the projection strength, direction, and for regularization, respectively.
With local linear embedding, it is straightforward to transform velocity between different representations. Assuming a mapping function f exists connecting manifold \({{{\mathcal{M}}}}\) and \(\aleph\) such that for cell i with state vector \({{{{\bf{x}}}}}_{i}\) in \({{{\mathcal{M}}}}\), the coordinate of the same cell in \(\aleph\) is given as \({{{{\bf{y}}}}}_{i}=f\left({{{{\bf{x}}}}}_{i}\right)\). Since a given local patch of a continuous manifold is approximated by an Euclidean space, a locally linear transformation connects the patch in the two representations. Consequently, for a vector described by Eq. 1 in \({{{\mathcal{M}}}}\), the velocity vector in \(\aleph\) is,
where \({{{{\boldsymbol{\delta }}}}}_{{ij}}^{{\prime} }={{{{\bf{y}}}}}_{j}-{{{{\bf{y}}}}}_{i}\). That is, one only needs to change the basis vectors.
Therefore, Eqs. 1–3 form the mathematical and computational foundation of GraphVelo. With Eq. 3 one can extend velocity inference to datasets that velocity inference is not traditionally applicable such as host-virus interactome and multi-omics datasets based on the Whitney embedding theorem18. Details of the mathematical foundation were given in Methods. With velocity vectors refined with GraphVelo, one can readily perform downstream analyses, as exemplified in Fig.1d–f, which will be further elaborated below in the context of specific applications.
Benchmark studies demonstrate the effectiveness of GraphVelo across simulation datasets with diverse topology
To demonstrate the effectiveness of the geometry-constrained projection, we first benchmarked our method on a 3D bifurcation system constrained on a 2D manifold (Methods). We added random components vertical to the tangent plane to mimic the noise. The resulting velocity vectors inferred by GraphVelo through minimizing the TSP loss were consistent with the ground truth vectors (Fig. 2a). Both GraphVelo and cosine kernel successfully removed the normal components (Fig. 2b i) and maintained the directional information (Fig. 2b ii), but only GraphVelo kept the velocity magnitude information (Fig. 2b iii).

a Velocity vectors of an analytical three variables bifurcating vector field constrained to a spherical surface. The data points were colored by simulation time. b Violinplots of: (i) normal component of velocity vectors, (ii) cosine similarity and (iii) root mean square error (RMSE) between ground truth and velocity vectors projected by GraphVelo and cosine kernel, respectively. The number of simulated cells is 2000 for statistical test. c–e Simulation of scRNA-seq data using dyngen under linear, cycling, and bifurcating differentiation models (left), and velocity fields projected on multidimensional scaling (MDS) coodinates (right) using GraphVelo-corrected velocities, respectively. Each simulation consists of 1000 cell states and 100 genes. The cells in different states were colored by their simulation time along trajectory. f–h Comparisons of cosine similarity, and accuracy between the ground truth velocity vectors and dyngen simulated velocities after projection using GraphVelo TSP loss without cosine regularization, GraphVelo TSP loss with cosine regularization, cosine kernel, and random predictor, respectively. The number of dyngen-simulated cells is 1000 for statistical test. In b and f–h, *** indicates Welch’s independent two-sided t-test at p < 0.05. Violinplot in panel b shows the distribution of data points after grouping by projection methods. Boxplots in f–h indicate median (middle line), first and third quartiles (box), and the upper whisker extends from the edges to the largest value no further than 1.5 × IQR (interquartile range) from the quartiles, and the lower whisker extends from the edge to the smallest value at most 1.5 × IQR of the edge. Source data are provided as a Source Data file.
Next, we performed multifaceted evaluations of the ability of GraphVelo to robustly recover the transcriptional dynamics across a range of simulated datasets with different underlining phenotypic structures. We used dyngen19, a multi-modal scRNA-seq simulation engine, to generate gene-wise dynamics defined by gold-standard transcriptional regulatory networks (Methods). We generated simulated scRNA-seq data for networks with a variety of underlying linear, cyclic, and bifurcating topological structures, and recovered the corresponding vector field using GraphVelo-corrected velocity vectors (Fig. 2c–e). To comprehensively assess the outcome, we used three diverse metrics, cosine similarity, root-mean-square error (RMSE), and accuracy, which evaluate the correctness of velocity direction, magnitude, and sign, respectively. We presented (Fig. 2c–e) the comparative results obtained with the cosine kernel and with GraphVelo (TSP with (i.e., Eq. 2 with b ≠ 0) and without (Eq. 2 with b = 0) the cosine regularization term). By minimizing TSP loss, GraphVelo preserved both the direction and magnitude of the vector field (Fig. 2f–h). With an increase of noise level by adding Gaussian noise to the ground truth vectors, GraphVelo refined the distorted velocity and outperformed the cosine kernel projection consistently (Supplementary Fig. 1a-c).
Then, we tested whether manifold constraints could preserve the speed of the cell progression across different representations. GraphVelo was able to scale velocity vectors between the original space and the PCA space, showing a high correlation with the ground truth, even as noise levels increased, whereas the cosine kernel failed (Supplementary Fig. 1d). The results on UMAP showed less agreement, which is not surprising. UMAP is a convenient representation for visualizing single cell data but is not designed for representing quantitative cell state transition dynamics. That is, UMAP is not a continuous transformation from the original gene space and cannot preserve local distances after projection.
To further explore whether GraphVelo could correct the RNA velocity estimated by the splicing kinetics, we took the velocity inferred using different packages (scVelo2, dynamo7 and VeloVI5) as input. The output from GraphVelo agreed significantly better with the ground truth compared to the raw input (Supplementary Fig. 1e), highlighting the significant improvement achieved by GraphVelo in evaluating both the direction and magnitude of the velocity vector fields across all datasets.
GraphVelo achieved consistent improvements across multiple real-world datasets
Although GraphVelo is designed as a velocity-correction and prediction model complementing existing velocity estimation tools, we benchmarked the performance of scVelo-based GraphVelo outputs on five independent datasets (FUCCI20, pancreatic endocrinogenesis21, dentate gyrus1, intestinal organoid22 and hematopoiesis7) against five RNA velocity estimation methods (scVelo2, VeloVI5, UniTVelo23, DeepVelo24, and CellDancer3) (Supplementary Fig. 2–4). GraphVelo achieved noticeably improved cross-boundary correctness (CBC) score25 against input velocity and other advanced methods (see Methods for calculation details). We evaluated velocity consistency2 across two datasets whose trajectories were estimated using different tools (Supplementary Fig. 4). While GraphVelo shows slightly lower overall vector smoothness scores compared to others, we observed that these models tend to produce overly smooth and homogenized velocity fields, which may obscure biologically meaningful heterogeneity. In contrast, GraphVelo preserves fine-grained local transitions and reveals subtle divergence in the vector field, particularly around fate bifurcations in endocrinogenesis data.
We examined the cell cycle datasets annotated by the dynamo package, which features a relatively simple geometry, to quantitatively evaluate our method. First, we focused on the CBC score between cell cycle states. The velocity vectors processed by TSP showed greater consistency with the ground truth compared to the unprocessed inputs (Supplementary Fig. 5a). To demonstrate the effectiveness of GraphVelo in scaling velocities based on the data manifold, we used the L2 norm of velocity vectors to quantify the cell cycle speed. This analysis revealed a peak in velocity within the M and G1 phases, which was also reflected in the distribution of total UMI counts (Supplementary Fig. 5b). We further validated these findings using the cycling A549 cell line sequenced by sci-fate26 (Supplementary Fig. 5c) and through the temporal variation of stratified cell cycle speed based on velocities inferred from metabolic-labeling data (Supplementary Fig. 5d). Leveraging the quantitative velocity vectors generated by GraphVelo, we classified genes by both the phase and peak magnitude of their velocities (Supplementary Fig. 5e). Analysis of phase-magnitude relationships uncovered the sequential activation cascade of marker genes throughout the cell cycle.
GraphVelo infers quantitative genome-wide RNA velocity from a subset of genes with manifold-consistent RNA turnover kinetics
Most RNA velocity methods are based on biophysical models of mRNA turnover dynamics with specific assumptions that may break down in certain cases11. These methods typically provide velocities of only a subset of (~500 or less) genes, termed velocity genes in the subsequent discussions, out of a larger list of (~2-4 k) highly variable genes in a dataset, and some of the velocities are questionable. For example, the splicing-based RNA velocity may have an erroneous sign for processes under active regulation on mRNA degradation or promotors switching between states with different transcription efficiency (Fig. 3a, Supplementary Fig. 6).

a Schematitc of transcritional events mislead RNA velocity estimation in the phase portrait by standard approaches. Left: for genes exhibiting rapid degradation, the cells appear above the steady state line on the phase portrait, whereas the true velocity is negative. Right: For genes exhibiting transcription burst, the transcription rate abruptly increases at intermediate states, leading to a steady state line whose slope is overestimated. b Schematic of manifold-consistent score calculation for robustly estimated velocity genes. c The projected velocity field from GraphVelo are consistent with the erythroid differentiation by using all highly variable genes. d The correlation between GraphVelo vector field-based pseudotime and embryo time for erythroid lineage cells. Spearman correlation coefficients are shown. e GO enrichment analyses of top ranked MacK genes. f The phase portrait of two transcription burst genes (Smim1, Hba-x). g Scatter plots of: i) velocities estimated by scVelo, ii) refined velocities by GraphVelo, and iii) mature mRNA expression of transcription burst genes (Smim1, Hba-x). Cells were colored by corresponding velocity, and mature mRNA abundance, respectively, and visualized on the UMAP representation. h Gene regulatory cascade unraveled by GraphVelo-based vector field analyses that drives cell lineage commitment. Gene set enrichment was performed using one-sided Fisher’s exact test, Benjamini–Hochberg correction. Adjusted p-values represent FDR-corrected significance of gene set enrichment. i Activation of Gata1 inhibitor TF Spi1 lead to reversed velocity flows in gastrulation erythroid maturation investigated through in silico perturbation analyses on GraphVelo-based vector field. j Velocities derived from GraphVelo for the branching lineage in the hematopoiesis development and projected onto a pre-defined TSNE embedding. Directions of the projected cell velocities on TSNE are in agreement with the reported differentiation directions. k Terminal states identified by CellRank based on Markov chain formulation derived from GraphVelo velocities. l Phase portrait, velocity estimated by scVelo, refined velocity by GraphVelo, and gene expression of mature mRNA of identified rapid degradation genes (NPR3, ANGPT1). The cells were colored by the palantir pseudotime31 in the phase portrait. The box plots showed cell-specific \({{{\boldsymbol{\gamma }}}}\) for cells divided into bins according to pseudotime ordering in the phase protrait. The number of cells within each time bins from early to late is 653, 650, 654, 657, 657, respectively. Boxplots indicate median (middle line), first and third quartiles (box), and the upper whisker extends from the edges to the largest value no further than 1.5 × IQR (interquartile range) from the quartiles and the lower whisker extends from the edge to the smallest value at most 1.5 × IQR of the edge, while data beyond the end of the whiskers are outlying points that are plotted individually. Source data are provided as a Source Data file.
GraphVelo uses the velocities of high-confidence genes obtained from any method as input to infer velocities of other genes. One can use several existing approaches to evaluate the confidence scores of inferred RNA velocity values of genes7. Alternatively, we identified a subset of Manifold-consistent Kinetics (MacK) Genes based on their agreement with prior knowledge or additional information acquired from other methods such as lineage tracing (Fig. 3b), We first applied GraphVelo to a mouse erythroid maturation dataset27. This study provided a transcriptional landscape of the erythroid lineage with well-documented differentiation trajectory during mouse gastrulation. Previous analyses have shown that the dataset contains genes with multiple rate kinetics, leading to erroneous prediction of the cell state transition direction27,28. We selected the top 200 out of 450 velocity genes as MacK genes, representing those with robustly estimated velocities (see Methods for details). The projected vector field in UMAP showed consistency with prior knowledge in developmental biology (Fig. 3c). We then used the corrected RNA velocities for dynamo velocity field analyses. The vector field-based pseudotime accurately predicted the lineage with scRNA-seq data of temporal mouse embryos (Fig. 3d).
Previous studies identified multiple rate kinetics (MURK) genes showing transcription bursts in the middle of erythroid differentiation28. For example, two MURK genes (Smim1 and Hba-x) showed complex patterns of phase portrait (Fig. 3f). Consequently, the RNA velocity of Simi1 inferred with scVelo was negative along a major part of the developmental axis (Fig. 3g i), contradicting the trend of increasing Simi1 mRNA levels (Fig. 3g iii). For Hba-x, scVelo even failed to infer its RNA velocity. On the other hand, GraphVelo inferred velocities and predicted correct kinetic patterns of these genes (Fig. 3g ii). Similar performances have been observed in other MURK genes (Supplementary Fig. 7a). To examine the overall prediction of cell state transitions from transcription burst genes, we projected the MURK genes velocity inferred from GraphVelo and scVelo to the predefined UMAP. The velocities from GraphVelo but not scVelo correctly captured the directional flow of differentiation using only MURK genes (Supplementary Fig. 7b). We further recapitulated gene succession and oscillation magnitudes along the erythroid trajectory and evaluated the phase-magnitude relationships of all highly variable genes (Supplementary Fig. 8a). Compared to non-MURK genes, MURK genes exhibited larger average velocity magnitudes and were predominantly enriched in the late stages of lineage progression. Analysis of genes with larger peak velocity amplitudes identified Fth1, Car2, and Hbb-bs as candidates with altered kinetic parameters. These dynamics patterns were evident in their phase portraits and gene expression trends (Supplementary Fig. 8b, c), consistent with previous reports that Car2 transcription in erythroid cells is regulated by both the promoter activity and long-range enhancer interactions29—such complex regulations lead to its transcription dynamics not well-described by the simple transcription model used in the original splicing -based RNA velocity inference.
Next, we evaluated the quantitative performance of GraphVelo in estimating cell-wise transition speed. Specifically, we estimated the speed of cell state transition using the norm of velocity vector in high-dimensional space and identified the transcriptional surge stage (Supplementary Fig. 7c). We hypothesized that the MURK genes, which exhibited a sudden increase in transcription rate during this stage, were responsible for the sharp acceleration in cell state transition speed. Using dynamo, we estimated the acceleration derived from the GraphVelo vector field and found that the acceleration value, as the derivative of the velocity vector, demonstrated its potential as a predictor for transcription burst genes (Supplementary Fig. 7d).
With the velocity estimation extended to the whole gene space, we were able to perform comprehensive mechanistic analyses on the entire genome spectrum. First, we calculated the MacK score for each gene using the corrected RNA velocities. We hypothesized that a gene with a higher MacK score indicated a better agreement between its RNA velocity vector and the developmental axis, suggesting that the gene served as a potential lineage-driver gene. We ranked genes based on their scores and performed GO biological process enrichment analyses for the top genes. Indeed, the enriched processes were associated with erythropoiesis, including the heme biosynthetic process and interleukin-12-mediated signaling pathway30 (Fig. 3e).
Next, we applied dynamo to perform differential geometry analyses of the vector field and mechanistically dissected the activation cascade of erythroid marker gene Klf1 (Supplementary Fig. 9a, b). Jacobian analyses based on GraphVelo vector field revealed sequential activation of driver transcription factors (TFs) Gata2, Gata1, and Klf1 during erythroid lineage differentiation, with Gata1 subsequently repressing the expression of Gata2 (Fig. 3h, Supplementary Fig. 9c)7. To further demonstrate the crucial role of transcriptional factor Gata1 during erythropoiesis, we performed in silico genetic perturbation across all cells. Results showed that both inhibiting Gata1 and upregulating the Gata1 repressor Spi1 lead to a reversal of normal developmental flow (Fig. 3i, Supplementary Fig. 9d). The above analyses collectively suggest that activation of Gata1 in the blood progenitors biased its differentiation to erythropoiesis, agreeing with experimental reports28.
To further evaluate GraphVelo, we tested the method on another dataset of human bone marrow development31. This developmental process has complex progressions from hematopoietic stem cells (HSCs) to three distinct branches: erythroid, monocyte, and common lymphoid progenitor (CLP). Again, we used the top 100 out of 454 velocity genes as MacK genes to predict the RNA velocities of 2000 highly variable genes. The GraphVelo velocity field accurately recovered the fate of cells on the sophisticated transcriptional landscape in contrast to scVelo (Fig. 3j, k, Supplementary Fig. 10a). By combining the likelihood estimated by scVelo with the MacK score, we identified rapid degradation and transcription burst genes whose dynamics deviated from the RNA velocity assumptions (Supplementary Fig. 10b, c). ANGPT1 and RBPMS are two examples which were overall highly expressed in the progenitors and decreased quickly along the trajectories (Fig. 3l), reminiscent of what was shown in Fig. 3a. These genes misled RNA velocity inference with scVelo assuming a constant degradation rate constant. GraphVelo revealed a cell context-specific transcription rate \(\alpha=u+\frac{{du}}{{dt}}\) and degradation constant \(\gamma=(u-\frac{{ds}}{{dt}})/s\), thus a degradation wave along the differentiation path (Fig. 3l, Supplementary Fig. 10d), consistent with simulation result and reports on regulation of ANGPT1 mRNA by microRNAs such as miRNA-153-3p32 (Supplementary Fig. 6d).
It is straightforward to apply Graphvelo to spatial transcriptomics datasets that permit RNA velocity inference. Note that the transcriptional dynamics of a gene can be affected by both the intracellular expression state and extracellular environmental factors. A data manifold containing additional spatial information allows distinction of cell states with similar expression profiles but distinct extracellular environments. Such a refined manifold leads to more accurate inference of the RNA velocities, which are typically performed over averaging the neighborhood of a cell state1 (see Methods) and is also used in GraphVelo for tangent space projection. We applied Graphvelo to the mouse coronal hemibrain dataset33 processed with a bin size 60, which includes spliced and unspliced transcript information at spatial context (Supplementary Fig. 11a). GraphVelo inferred coherent velocity fields across brain regions, with streamlines on UMAP reflecting anatomically structured transitions that align with the spatial annotation (Supplementary Fig. 11b). Compared to the uncorrected RNA velocities using the dynamo build-in module, GraphVelo captured sharper transcription speed patterns, particularly in the dentate gyrus (DG), a neurogenic region where cell proliferation and neuronal differentiation persist into adulthood34 (Supplementary Fig. 11c, d). Spatial mapping of representative genes revealed that GraphVelo velocities were more spatially confined and aligned with known expression domains, whereas the uncorrected velocities were noisier and less localized (Supplementary Fig. 11e). These results highlight the ability of GraphVelo to generate interpretable, spatially structured transcription dynamics in spatial transcriptomic data when splicing information is available.
GraphVelo reconstructs host–pathogen transcriptome dynamics from infection trajectory
The continuous battle between human immune surveillance and viral immune evasion takes place in the host cell system after viral entry. scRNA-seq data provide a massive and parallel way of assessing the time evolution of both host and viral transcripts, unraveling the delicate inherent dynamics of a virus-host system35,36. For splicing-based RNA velocity methods, genes lacking sufficient unspliced transcript counts are typically filtered out—which is inherently the case for viral genes due to their absence of introns. However, several studies37,38,39 have applied tools such as scVelo to host-virus systems by focusing exclusively on host velocity genes that accurately reflect the lineage dynamics, rather than attempting to derive velocities directly from viral transcripts. GraphVelo enables inference of the velocities of virus RNA abundance based on the kinetics of host transcripts velocities, as illustrated next.
We analyzed a human cytomegalovirus (HCMV) viral infection dataset to learn viral transcriptomic kinetics in monocyte-derived dendritic cells (moDCs)39. The result from GraphVelo unraveled how viral infection progressed along the transcriptional space (Fig. 4a). The velocity vectors pointed to directions consistent with an increasing trend of the percentage of viral RNAs in individual cells, which inherently served as an indicator of the infection time course40. Compared to the trend obtained with the raw RNA velocities from scVelo, the vector field-based pseudotime calculated using GraphVelo-corrected RNA velocities consistently showed higher correlation with the (pseudo)temporal progression of viral infection as reflected by viral RNA percentage (Fig. 4b).

a Viral infection captured by the GraphVelo velocity field. Cells were colored by the percentage of viral RNA within a single cell. b Correlation between viral RNA percentage and pseudotime inferred by scVelo or GraphVelo. Correlation was calculated using a two-sided Spearman rank correlation test. c Viral RNA velocities infered by GraphVelo along the viral RNA percentage axis. The black dot line highlights the zero velocity. The solid line denotes the mean trend, dashed lines denote 1 s.d. d Boxplot summarizing the MacK scores of all viral genes calculated by GraphVelo, CellRank pseudotime kernel and random predictor. The number of viral genes is 67 for the statistical test. *** indicates Welch’s independent two-sided t-test at p < 0.05. Boxplots indicate median (middle line), first and third quartiles (box), and the upper whisker extends from the edges to the largest value no further than 1.5 × IQR (interquartile range) from the quartiles and the lower whisker extends from the edge to the smallest value at most 1.5 × IQR of the edge, while data beyond the end of the whiskers are outlying points that are plotted individually. e Correlation between viral infection speed and RNA abundance. Genes were ranked by Spearman correlatioin coefficients. Host and viral genes that contribute to viral DNA synthesis were marked in the left side and those contribute to viral defense response were marked in the right side. Viral genes were highlighted in red. f UMAP representation of host and viral genes with distances defined by their dynamic expression patterns along the viral RNA percentage axis. g Example dynamic expression patterns within specific clusters (Leiden4, 5, 3, 6 from top to bottom) along the viral RNA percentage axis. Zero velocity was highlighted by black dot line. The solid line denotes mean trend, Shaded region represents 1 s.d. h GO enrichment of each cluster in (g). Gene set enrichment was performed using one-sided Fisher’s exact test, Benjamini–Hochberg correction. Adjusted p-values represent FDR-corrected significance of gene set enrichment. i Top host genes inhibited by each viral factor based on dynamo Jacobian analyses. Host effectors were organized by their involved pathways. j Dynamo prediction of total viral RNA change in response to in silico viral factor knockout. Viral factors were ranked by the mean of total viral RNA changes. The number of samples is 1454 for virtual perturbation screen. Boxplots indicate median (middle line), first and third quartiles (box), and the upper whisker extends from the edges to the largest value no further than 1.5 × IQR (interquartile range) from the quartiles and the lower whisker extends from the edge to the smallest value at most 1.5 × IQR of the edge. k Vector field change resultant from infinitesmal inhibition of UL123 during the viral infection process. Source data are provided as a Source Data file.
Furthermore, the examination of individual genes revealed that the RNA velocities consistently predicted the trend of the mRNA expression level change with increasing virus load (Fig. 4c, Supplementary Fig. 12a, b). Most viral genes started with a fast-increase phase, and the expressions of some genes (e.g., UL22A) gradually saturated at high virus load, together with the corresponding RNA velocities approaching zero. One exception is UL122, whose expression profile increased first then decreased to a steady state level lower than the peak value. This overshooting is characteristic of a negative feedback network structure41. Indeed, a recent study reported that UL122 negatively regulates its own promotor42. Furthermore, comparison of MacK scores across GraphVelo, the CellRank pseudotime kernel, and randomized prediction showed that GraphVelo-computed viral RNA velocities aligned best with the transcriptome gradient of viral load (Fig. 4d). Note that the MacK score also served as a reliable predictor for dynamics-driving factors, specifically viral genes in this case (Supplementary Fig. 12d).
We further quantified the velocity norm of all viral factors as infection speed, and observed that the transcription of viral factors was significantly restricted initially, then gradually increased along the trajectory (Supplementary Fig. 12e). Interestingly, most of the genes that exhibited positive correlation with the infection speed were related to viral DNA synthesis, while those negatively correlated to the infection speed were engaged in host viral defense response43(Fig. 4e, Supplementary Fig. 12f).
GraphVelo identifies host genomic response modules and predicts host-virus gene interactions
With GraphVelo-inferred RNA velocities, we probed the time evolution of lytic infection and the complex interplay between host and viral functional genomes. By fitting the GraphVelo velocity trends along the viral load axis, we identified genes with similar kinetic patterns (Fig. 4c). Using the smoothened velocity trends to calculate the distances, we clustered the genes into seven major modules and visualized them on the UMAP space (Fig. 4f). Not surprisingly, viral genes were concentrated in several enclosed regions, indicating that they formed distinct functional genomic modules during the lytic cycle40. The genes showed two major dynamical features: acceleration and deceleration along the viral load axis (Fig. 4g).
To systematically investigate whether the dichotomy between host kinetics genes and viral genes share similar dynamics, we performed gene functional enrichment analyses of host genes residing close to the viral gene clusters. Genes located in the deceleration part were associated with repressing viral genome replication, which includes negative regulation of viral life cycle and known restriction factors in antiviral responses activated in DCs such as the induction of cytokine and chemokine responses as well as interactions with neutrophils44,45. In parallel, the toll-like receptor signaling pathway, required for antiviral defense of the host, was arrested46. Neutrophil related processes, which typically cooperate closely with DCs to modulate adaptive immune responses47, were suppressed. Therefore, the deceleration part also showed how critical set of host factors were silenced by viral entry to achieve immune evasion.
The acceleration groups, on the other hand, demonstrated how viruses hijacked the host cell endogenous cellular programs for virus replication. Notably, the pathways related to viral genome replication were triggered, promoting DNA replication and transcription, such as negative regulation of G1/S transition of mitotic cell cycle48, cellular response to DNA damage stimulus49 and regulation of transcription from RNA polymerase II promoter50. Cells showed a shift towards a transcriptional signature resembling the G1 phase (Supplementary Fig. 12g), agreeing with previous report on HCMV infection51. Along the infection process, antiviral interferon (IFN)-γ response of moDC cells was first activated then suppressed. These results highlighted the organized and antagonistic strategies adopted by both host cells and viruses during their tug-of-war for survival and proliferation.
To investigate the crosstalk between host and viral factors systematically in depth, we performed dynamo Jacobian analyses. We scanned the entire spectrum of viral genomes and delineated how the HCMV factors silence IFN and NFκB signaling (Fig. 4i). A large proportion of the identified viral factors functioned in evading host cell immune responses, a finding supported by several recent studies52,53,54. In silico virus-directed knock out experiments revealed altered accumulation patterns of viral transcripts (Fig. 4j). Notably, inhibition of UL123, which ranked first with the total viral RNA inhibition in our analyses, led to a qualitatively distinct trajectory. These results highlight the multifunctional UL123 locus in the viral genome as a potential target for antiviral intervention40,55. The analyses demonstrated potential usage of GraphVelo-inferred velocities for understanding the interactions between viral and host factors, assessing the effects of perturbations on infection, and designing potential antiviral interventions40.
GraphVelo reaveals an abortively infected cell population in SARS-CoV-2 infection
Although HCMV is renowned for its elaborate transcriptional landscape—characterized by extensive alternative splicing, overlapping transcripts, and diverse isoform expression—the RNA virus SARS-CoV-2 transcriptome reveals an even greater level of complexity within its relatively compact ~30 kb genome56. To characterize the molecular mechanisms of host response which protect cells from productive trajectory, we applied GraphVelo to a SARS-CoV-2 infected Calu-3 cells dataset57. To focus on the host-virus interactions and their corresponding fate outcomes, we subset the infected cell population for downstream analyses. The infected cluster M, characterized by interferon production genes, was hypothesized to represent a subpopulation of abortively infected cells57, similar to those described in herpesviruses HSV-158 and HCMV40. We subsampled the infected cell population for downstream analyses and visualization, where cluster M exhibited distinct connectivity properties compared to the main infected groups (Supplementary Fig. 13a). To confirm that M is one of the terminal states of infection outcomes with such low cell abundance (Supplementary Fig. 13b), we applied GraphVelo to gain the quantitative velocity with dynamo outcomes as input (Supplementary Fig. 13c). Using vector field topology analysis, we classified an initial state with relatively low viral load, two terminal states associated with high apoptosis activity, and a saddle point characterized by high viral load (Supplementary Fig. 13d). Notably, the region corresponding to cluster M was identified as an attractor, confirming it represents an abortively infected cell state with a high death rate57.
We further validated the complex lineage commitments of SARS-CoV-2–infected cells using the CellRank framework16, which successfully identified three potential terminal states as outcomes of host-virus competition (Supplementary Fig. 13e). Interestingly, we characterized the saddle stage in dynamo as a productive terminal state governed by viral genes (Supplementary Fig. 13f), exhibiting high viral transcription speed (Supplementary Fig. 13g). The pathogen-triggered cell death can be driven for protecting the host or for pathogen dissemination purpose59. To investigate the underlying causes for host cell death, we performed gene functional enrichment analyses on the top correlated genes along distinct lineages (Supplementary Fig. 13h). Active programs in the abortive infection lineage were highly enriched for host defense mechanisms against viral invasion, consistent with previous studies40,58. In contrast, the drivers of the apoptosis-associated lineage revealed a distinct profile. Notably, we observed enrichment for cellular responses to unfolded proteins, which have been implicated in facilitating pathogen-mediated dissemination of infected cells60,61. Additionally, ERBB2 inhibition has been shown to suppress SARS-CoV-2 replication62. These findings support the hypothesis that similar cell death outcomes may arise from fundamentally different host responses. Abortively infected cells appear to promote efficient pathogen clearance, likely through cytokine-mediated immune activation that eliminates both the infected cell and the virus. Conversely, cells undergoing virus-induced apoptosis fail to clear the virus, with cell death instead serving as a mechanism for viral escape, immune evasion, and potential dissemination to deeper tissue layers or the bloodstream.
GraphVelo permits multi-omics velocity inference and chromatin dynamics analyses
The molecular anatomy during cell development entails multiple layers, and how different layers coordinate to regulate gene expression is a fundamental problem. For example, the anagen hair follicle features distinct lineages branching from a central population of progenitor cells. Ma et al.63. used SHARE-seq to capture both the transcriptome and the epigenome data simultaneously for the lineage commitment process from transit-amplifying cells (TACs) to the inner root sheath (IRS), cuticle layer, and medulla. Upon robust selection of estimated genes following dynamo criteria (Methods), we further refined the RNA velocities of these genes through tangent space projection and obtained the chromatin open/close dynamics from the corresponding scATAC data using GraphVelo. The resultant vector field in the combined transcriptome-epigenome space proved to reconstruct the correct multilineages differentiation paths during the anagen phase (Fig. 5a).

a GraphVelo velocity fields of mouse hair follicle development. Cells were colored by cell macrostates. b Number of terminal states predicted by CellRank using velocities inferred with different methods. c Driver genes along multiple lineages identified through CellRank. d Topological analyses of GraphVelo vector field identified novel root cells and attractors residing in three terminal states(IRS, hair shaft-cuticle cortex, and medulla). e Expression levels of marker genes in novel root cells and expected root cells. Markers identified by Ma et al.63 were highlighted with stars, and newly identified markers were highlighted in bold. f Regression results of MSD values along the transition path from the expected root or novel root to IRS. Two genes Runx1 and Shh genes with large MSD originating from the novel root were highlighted. The solid line denotes mean of regression result, Shaded region represents 1 s.d. g DTW distance between RNA velocity and chromatin velocity of individual genes. CCD genes were colored in red. The dotted line indicates the elbow point separating the decoupled genes from the rest. h GO enrichment of decoupled genes in (g). i Line plot of nomarlized RNA and chromatin velocity along pseudotime for genes predicted by GraphVelo to have notable decoupling patterns. Chromatin velocity trends were colored as brown and RNA velocity trends were colored as green. j Heatmaps of Jacobian element distribution along the axis of regulator RNA abundance of four regulator effector circuits: i) Lef1 versus Wnt3 chromatin accessibilities. ii) Hoxc13 versus Wnt3 chromatin accessibilities. iii) Lef1 versus Wnt3 transcription. iv) Hoxc13 versus Wnt3 transcription. k Effective dose-response curves obtained from integrating the averaged Jacobian elements over the corresponding normalized regulator mature mRNA regulator level in (j). Source data are provided as a Source Data file.
To test the consistency of dynamics across different modalities, we performed CellRank terminate stage analyses16 from the refined velocity vectors. Using GraphVelo velocities of either the RNA modality or the ATAC modality, we accurately estimated three diverse terminal stages (Fig. 5b). For comparison, we also performed similar analyses using MultiVelo, scVelo with all velocity genes or robustly estimated genes in above GraphVelo studies and pseudotime-based vector field inferred by CellRank. The 2D projection of these vector field functions also exhibited seemingly correct velocity flow direction (Supplementary Fig. 14a). However, none of them captured the cell fate commitment based on coarse-grained transition matrix (Fig. 5b, Supplementary Fig. 14b). Notably, the results from the RNA modality and the ATAC modality of MultiVelo gave inconsistent results. GraphVelo-corrected velocities, on the other hand, helped identify the top-correlating genes towards individual terminal populations which showed agreement with previous study64 (Fig. 5c, Supplementary Fig. 15).
Next, we conducted differential geometry analyses based on the composite GraphVelo vector field. We identified novel root cells, which were also characterized by chromatin potential (Fig. 5d)63. These novel root cells expressed distinct marker genes compared to the expected root cells using the Wilcoxon test (Fig. 5e). Moreover, we unraveled differentially expressed markers identified by the original study63, as well as new differentiation-potent genes and validated their initiation properties in another transcriptome dataset (Supplementary Fig. 16a)64. To further investigate how these two distinct groups of root cells convert to other cell types, we performed a least action path (LAP) analysis between different cell phenotypes. The expected and novel root cells converted to the IRS terminal state following two distinct least action paths in the vector field (Supplementary Fig. 16b, c). The two paths revealed different temporal change patterns of transcription factor expression profiles (Supplementary Fig. 16d). We calculated the mean-squared displacement (MSD) for every transcription factor to explore the dynamics of TFs along the path from novel root to IRS. The result demonstrated that the fate conversion by novel root was mediated by the Shh-Runx1 signaling axis (Fig. 5f, Supplementary Fig. 16d), which has been demonstrated in human embryonic stem cells65 and is crucial for hair development66. In summary, GraphVelo unraveled the multiple molecular mechanisms that orchestrated hair follicle morphogenesis.
With available chromatin velocity and RNA velocity, we set to quantify the coupling/decoupling relationships between chromatin structure and gene expression for each gene (see Methods). Here chromatin structure refers to the extent of exposure/accessibility of the gene locus to the environment as indicated by scATACseq data, shortly called open or closed state; and chromatin velocities refer to changes between these states as inferred from scATACseq counts at the examined gene locus. We used dynamic time warping (DTW) distances between the velocities from different omics layers to quantify the similarity between temporal patterns of these two modalities for each gene. A higher DTW value indicates higher similarity. Using the elbow of the ranked distance curve as a cutoff we identified genes that showed decoupled transcription and chromatin structure dynamics. These decoupled genes had an accumulation of cell cycle-dependent (CCD) genes found in previous study20 (Fig. 5g). This group of genes showed strong involvement in cell cycle-related processes, as indicated by GO enrichment analyses (Fig. 5h). Close examinations indicated that the transcription of cell cycle related genes decreased along the differentiation path, while the chromatin structure at the corresponding loci remained open (Fig. 5i, Supplementary Fig. 17). To validate this hypothesis, we further applied GraphVelo to a recently published 10x Multiome dataset from developing human cortex67 (Supplementary Fig. 18a). Following the same analyses, we identified decoupled genes and found out that most of these genes were related to cell cycle (Supplementary Fig. 18b–f). which has also been reported in a previous MultiVelo study13.
We further performed dynamo differential geometry analyses on the composite transcriptome-chromatin vector field. One intriguing phenomenon observed in lineage dynamics is that Lef1 and Hoxc13 are the driver TFs correlated with domains of regulatory chromatin (DORCs) of Wnt363. Differential geometry analyses on the composite vector field can go beyond correlation analyses and provide an underlying casual mechanism. As a prerequisite for such analyses, GraphVelo-inferred RNA and chromatin velocities of the three genes correctly predicted the trend of change of mRNA and ATAC-seq counts (Supplementary Fig. 19a, b), in contrast to the performance of MultiVelo (Supplementary Fig. 19c). Then, Jacobian analyses on the GraphVelo vector field confirmed that priming activation of Lef1 subsequently activated the Hoxc13 TF68 (Supplementary Fig. 19d, e). Both Lef1 and Hoxc13 were found to activate the Wnt3 target gene, initiating lineage commitment (Supplementary Fig. 19f, g). To quantitatively understand how the two TFs affect Wnt3 chromatin structure and transcription, we plotted the response heatmap to reflect the distributions of Jacobian elements versus the abundance of mature mRNA for each TF (Fig. 5j). The two terms \(\partial {f}_{{Wnt}3-{{{\rm{chrom}}}}}/\partial {x}_{{Lef}1}\) and \(\partial {f}_{{Wnt}3-{{{\rm{chrom}}}}}/\partial {x}_{{Hoxc}13}\) started with positive values at low concentrations of TF mRNA copy numbers then decreased to zero, indicating that increasing the level of either TF lead to further opening of the Wnt3 chromatin region, and the effect saturated at high TF expression. The other two term \(\partial {f}_{{Wnt}3}/\partial {x}_{{Lef}1}\) and \(\partial {f}_{{Wnt}3}/\partial {x}_{{Hoxc}13}\) increased with the TF levels, indicating that these two TFs also activated Wnt3 transcription. Upon integration of the Jacobian elements over regulator expression changes, we obtained the effective dose-response curves obtained (see Methods), which revealed more transparently the TF dose lag between the opening of the target chromatin region and the initialization of transcription (Fig. 5k).
Our results therefore illustrated the sequential events that these two driver TFs, Lef1 and Hoxc13, drove as pioneer transcription factors (PTFs) to initiate local chromatin opening and then activated the transcription of Wnt3. Notably, computational methods and experimental research confirm that Lef1 acts as a nucleosome binder and exhibits diverse binding patterns across various cell lines69. The Hox family of TFs has also been shown to have the capacity to bind their targets in an inaccessible chromatin context and trigger the switch to an accessible state70, consistent with our analyses that Hoxc13 revealing that it shared a regulation mechanism similar to that of PTF Lef1.
Discussion
In this work, we provided a general framework that extends the framework developed for RNA velocity and related approaches to various data modalities such as proteomics, spatial genomics, 3 d genome organization, and imaging data, which were originally beyond the reach of this framework. We validated GraphVelo using various in vivo cellular kinetics models, confirmed its efficacy and robustness in handling complex and noisy multimodal data. Upon application to various datasets, we unraveled gene regulation relations of an extended list of genes, host-virus gene regulations, and coupling between transcription and local chromatin structures. GraphVelo can be seamlessly integrated with broad downstream analyses, such as dynamo continuous vector field analyses, as well as Markovian analyses using graph dynamo or CellRank.
Methods
Dynamical systems theory formulation of cellular state transitions
Assume that a cell state can be represented by the cell volume V (or cellular compartment size) and the copy number of L ≫ 1 pairs of gene products (nm, np), where m and p designate mRNA and protein, and the bold fonts indicate vectors. For simplicity here we only consider m and p. It is straightforward to generalize to finer cell state specifications, for example, with distinction of nuclear and cytosol localizations, posttranslational states of proteins, other species such as microRNAs, epigenetic states, etc.
The temporal evolution of the cell state is described by a set of chemical master equations. When the copy numbers of molecular species are not too small, and the chemical reactions are not strongly coupled, Gillespie showed that the chemical master equations can be approximated by a set of chemical Langevin equations71. With extrinsic noises also included, we assume the ansatz that the dynamics of cell state is described by a set of generic stochastic differential equations,
where the L-dimensional vectors \({{{{\bf{x}}}}}_{m}\) and \({{{{\bf{x}}}}}_{p}\) are cellular concentrations of m and p, and the \(\zeta\) and \(\eta\) are taken as white noises with zero mean.
The low-dimensional manifold assumption is central to machine learning approaches on data analyses. From a dynamical systems theory perspective, after a transient time, a multi-dimensional dynamical system often converges to a low-dimensional slow manifold. In practice, such property has been exploited with techniques such as quasi-steady-state approximation, quasi-equilibrium approximation. For a rigorous formulation, assume that one can identify a set of variables \(\left({{{\bf{z}}}},{{{\bf{Z}}}}\right)\) with (2L-M) dimensional fast variables \({{{\bf{z}}}}={{{\bf{z}}}}({{{{\bf{x}}}}}_{m},{{{{\bf{x}}}}}_{p})\) and M-dimensional slower variables \({{{\bf{Z}}}}={{{\bf{Z}}}}({{{{\bf{x}}}}}_{m},{{{{\bf{x}}}}}_{p})\). Xing and Kim extended the celebrated Zwanzig-Mori projection72,73 to a general dynamical system described by Eq. 474. The projection procedure results in a set of stochastic integral-differential equations of \({{{\bf{Z}}}}\) with colored noises, which are formally equivalent to Eq. 4. Then if one assumes clear time scale separation between \({{{\bf{z}}}}\) and \({{{\bf{Z}}}}\), the equations reduce to a set of Langevin equations with white noises,\(\frac{d{{{\bf{Z}}}}}{{dt}}={{{\bf{A}}}}({{{\bf{Z}}}}({{{{\bf{x}}}}}_{m},{{{{\bf{x}}}}}_{p}))+\eta \left({{{\bf{Z}}}},t\right)\), where \(\left({{{\bf{Z}}}},t\right)\) are white noises with zero mean. Through ensemble averaging over the vicinity of a given point Z, one has
The equations define a M-dimensional manifold embedded in the \(({{{{\bf{x}}}}}_{m},{{{{\bf{x}}}}}_{p})\) space. A scRNA-seq data then measures the corresponding manifold projected to the transcriptomic subspace.
One should note that in practice the reported RNA velocity vector of a cell state i is typically obtained through averaging the raw velocity vectors of cell states within its neighborhood \({{{{\mathcal{N}}}}}_{i}\) on the manifold as a numerical approximation of the ensemble average, \({{{{\bf{v}}}}}_{{{{\bf{x}}}}i}= < \frac{d{{{{\bf{x}}}}}_{{{{\bf{m}}}}}}{{dt}} > \approx {\sum }_{j\in {{{{\mathcal{N}}}}}_{i}}{{{{\bf{v}}}}}_{{{{\bf{x}}}}j}\). In this work, we used the k-nearest neighbor (kNN) algorithm to define the neighborhood in single-modality datasets, including spatial-transcriptomics datasets. For multi-omics data, the neighborhood of a cell state was defined using weighted nearest neighbors (WNN)75 in the composite cell state space. The procedure of applying GraphVelo is the same for different data types, except using the neighborhoods defined on their corresponding data (cell state) manifold.
Mathematical foundation for applying GraphVelo to multi-omics datasets
Equation 3 in the main text applies to the transformation between a manifold embedded in a state space and in a subspace. According to the Whitney Embedding theorem18, any smooth real M-dimensional manifold can be embedded in a 2M-dimensional real space provided that M > 0. Consider a full set of genes versus a subset in a scRNAseq dataset, or a combined scRNAseq/scATACseq multi-omics dataset versus the scRNAseq subset. Assume that the full cell state space has a dimensionality N, while a single cell data manifold is typically low-dimensional with M ≪ N. Then the Whitney Embedding theorem18 suggests that with proper choice of the subset the manifolds in the full space and the subspace are homeomorphic or at least piece-wise homeomorphic (Fig. 1b, Supplementary Notes 1.4), i.e., a one-to-one mapping exists between the two. Then applying Eq. 3 allows one to infer the velocity vectors for the full-space representation from those of the subspace.
Denoise velocity vectors in the space of principal components (PCs)
Learning coefficients \({{{{\mathbf{\phi }}}}}_{i}\) in the gene space directly often fails due to thousands of gene profiles. To avoid the curse of high dimensionality and learn parameters in a compact manifold, we designed a procedure to denoise the velocities in a reduced PCA space. Specifically, we extrapolated the cell state i in the original space using the infinitesimal propagation operator to extrapolate the future state:
Moreover, we estimated an optimal step size dt based on the local density to guarantee the cell states are bound to the manifold:
After utilizing the cell-dependent \({t}_{i}\) to forcing the predictions inhabiting regions of the phenotypical manifold, we applied dimension reduction to project both current and future status from the gene space to the PCA space through linear transformation. Then we obtained the projected velocity vectors as:
where Q is the PC loading matrix estimated using x that serves as the coordinate transformation matrix.
Manifold-consistent kinetic genes
The kinetic assumptions between nascent and mature RNA fail when the underlying parameters shift along the developmental trajectory11, which leads to transcription burst and rapid degradation in the phase portraits (Fig. 3a). An internal clock exists during cell proliferation and differentiation. Current methods rely on different criteria to select confident estimated velocity genes (see Supplementary Notes 1.5 for detailed discussion). Here, we presume that the velocity of robustly estimated genes should be consistent with the (pseudo)time derivative estimated under the manifold assumption. We can utilize any available ts inferred from data manifold by either pseudotime, velocity latent time, or lineage tracing to approximate the temporal information and use k nearest neighbors to define the locally linear plane. After ordering cells within the local Euclidean space, we calculate the MacK score for any gene g as an indicator of whether the sign of estimated velocity agrees with the dynamic cascades within manifold,
Where \({{{{\mathcal{N}}}}}_{i}\) indicates the neighbor points of cell i, \({\mathbb{l}}\) represents the indicator function and sgn returns the sign of the values. \(\Delta {x}_{{ij}}\left(g\right),{v}_{i}(g)\) are the difference in abundance of gene g between cell i and j, and the velocity of gene g in cell i, respectively. We parallelize the calculation to scale efficiently with the number of genes, which is important due to the number of highly variable genes. We want to point out that one can use methods other than the MacK score to identify genes with reliable velocity estimations.
Dynamo criteria
Dynamo7 offers a correction strategy by removing genes with low gene-wise confidence in the phase plane. This allows us to identify genes that appear in incorrect phase portrait positions and contribute to erroneous flow directions (illustrated in Fig3. a). To filter out genes with misleading dynamic patterns, one can supply the established lineage hierarchy information to the dyn.tl.confident_cell_velocities function in dynamo. This function scores each gene based on the agreement of its behavior in the splicing phase diagram with the input lineage hierarchy priors.
Post-GraphVelo analyses
Reconstruction of extended dynamo vector field from multi-omics data
Dynamo is a general framework of reconstructing dynamical models from scRNAseq data, and it is straightforward to generalize to multi-modal data. The framework is based on specific realizations of Eq. 5, \({{{\bf{v}}}}\equiv < \frac{d{{{\bf{x}}}}}{{dt}}{ > }_{{{{\bf{x}}}}}={{{\bf{A}}}}\left({{{\boldsymbol{x}}}}\right)\), with state vector x being the transcript concentrations for scRNAseq data, and combined transcript concentrations and continuous quantification of locus-specific chromatin open-close state for multi-omics scRNAseq/scATACseq data. The variables x can be defined in various representations, e.g., the original gene space, principal component subspace, latent space defined by a variational autoencoder, etc. With GraphVelo it is straightforward to transform \({{{{\bf{v}}}}}_{{{{\bf{x}}}}}\) between different representations.
The continuous vector field functions \({{{\bf{A}}}}\left({{{\bf{x}}}}\right)\) contain quantitative regulation relations between genes that are learned from single cell data points of (x, vx). Various algorithms can be used to learn the analytical forms of \({{{\bf{A}}}}\left({{{\bf{x}}}}\right)\). The original dynamo paper illustrated a Reproducing Kernel Hilbert Space (RKHS) representation method. The method expresses \({{{\bf{A}}}}\left({{{\bf{x}}}}\right)\) as a linear combination of pre-selected basis functions, \({{{\bf{v}}}}={{{\bf{A}}}}\left({{{\bf{x}}}}\right)={\sum }_{\alpha }{{{{\bf{c}}}}}_{{{{\boldsymbol{\alpha }}}}}{{{{\mathbf{\Gamma }}}}}_{{{{\rm{\alpha }}}}}({{{\bf{x}}}})\), similar to the more familiar Taylor expansion that uses a linear combination of polynomial functions to represent a continuous analytical function. It should be noted that the basis functions and so \({{{\bf{A}}}}\left({{{\bf{x}}}}\right)\) are generally nonlinear functions of x. Following Qiu et al.7., we chose Gaussian functions centered at selected reference points \({\widetilde{{{{\bf{x}}}}}}_{{{{\rm{\alpha }}}}},{{{{{\mathbf{\Gamma }}}}}_{a}\left({{{\bf{x}}}}{,}{\widetilde{{{{\bf{x}}}}}}_{{{{\rm{\alpha }}}}}\right)={{{\rm{e}}}}}^{-2w{\|{{{\bf{x}}}}{-}{\widetilde{{{{\bf{x}}}}}}_{\alpha }\|}^{2}}\), with default parameter value of w in the package dynamo. Then we determined the coefficient vectors cα through minimizing the loss function \(\Phi ({{{{\bf{c}}}}}_{1},{{{{\bf{c}}}}}_{2},\ldots,{{{{\bf{c}}}}}_{{{{\rm{m}}}}})={\sum }_{i=1}^{n}{\|{{{{\bf{v}}}}}_{i}-{\sum }_{\alpha }\Gamma ({{{\bf{x}}}},{\widetilde{{{{\bf{x}}}}}}_{\alpha }{)}{{{{\bf{c}}}}}_{\alpha }\|}^{2}+\frac{\lambda }{2}{\sum }_{\alpha=1}^{m}{\sum }_{\beta=1}^{m}{{{{\bf{c}}}}}_{\alpha }^{\top }\varGamma ({\widetilde{{{{\bf{x}}}}}}_{\alpha },{\widetilde{{{{\bf{x}}}}}}_{\beta }{)}{{{{\bf{c}}}}}_{\beta },\) where the first sum was over all the data points, and the second term was Tikhonov regularization weighted by λ. The superscript T means matrix transpose. One can also use neural networks, e.g., variational autoencoders, to learn an optimal set of basis functions, and other algorithms such as neural ODE to learn \({{{\bf{B}}}}\left({{{\bf{x}}}}\right)\). The difference is merely algorithmic under the same framework of dynamical systems theories.
Jacobian analyses and reconstruction of effective dose-response curves of gene regulation
The extended dynamo vector field generally contains a nonlinear relationship about regulation between genes, and between genes and other modalities (e.g., chromatin open/close conformations). Several posterior interpretation methods exist to analyze the vector field. Below, we will describe two of them.
With the analytical form of F(x), one can calculate efficiently the Jacobian field J at any cell state x. Each element of J, \(({J}_{{ij}}=\partial {F}_{i}/\partial {x}_{j})\), can be understood as an in-silico perturbation experiment on how upregulating gene j affects the transcription rate of gene i, with all other gene expression levels kept constant at state x. For example, a positive value of \({J}_{{ij}}\) indicates that at x further increasing the expression of gene j causes increase of the transcription rate of gene i. Note that the sign and value of a Jacobian element alone does not unambiguously reflect the nature of the regulation. A close-to-zero Jacobian element can be associated with either no regulation or the regulator is at a saturating concentration of regulation. The regulation relation can be direct, or indirect through intermediate molecular species not implicitly treated as variables of the vector field function.
Complementary to the local Jacobian analysis is to reconstruct effective dose-response (D-R) curves of a regulator-target gene pair. The curve reveals the rate of change of one quantity, e.g., the transcription rate or the chromatin open/close dynamics of the target gene, as a function of the value of a regulator, e.g., the mRNA level of a regulator or the chromatin open/close status of a specific genomic region. Note that the D-R curve is generally a multi-variate function, we designed a procedure to reconstruct an effective one-variate function7. One can genetically write the regulation on quantity \({x}_{i}\) as two terms with and without dependence on variable \({x}_{j}\),
Notice that \({F}_{i}^{2}\) is not a function of gene j. First, we calculated the Jacobian element \(\frac{\partial {F}_{i}}{\partial {x}_{j}}\) for each measured cell state. Note \(\frac{\partial {F}_{i}}{\partial {x}_{j}}=\frac{\partial {F}_{i}^{1}}{\partial {x}_{j}}\), so the background variation from \({F}_{i}^{2}\) due to effects of other genes has been numerically removed. Then from the histogram of \(\frac{\partial {F}_{i}}{\partial {x}_{j}}\) versus \({x}_{j}\), we binned \(\frac{\partial {F}_{i}}{\partial {x}_{j}}\) over \({x}_{j}\) and calculated \( < \frac{\partial {F}_{i}}{\partial {x}_{j}}{ > }_{\alpha }\), which was averaged over all data points of \(\frac{\partial {F}_{i}}{\partial {x}_{j}}\) within bin \(\alpha\). Next, we performed numerical integration to obtain \(\bar{{F}_{i}^{1}}({x}_{j})={F}_{i0}^{1}+{\int }_{0}^{{x}_{j}} < \frac{\partial {F}_{i}}{\partial {x}_{j}}{ > }_{\alpha }d{x}_{j}\approx {F}_{i0}^{1}+{\sum }_{\alpha } < \frac{\partial {F}_{i}}{\partial {x}_{j}}{ > }_{\alpha }\Delta {x}_{j\alpha }\). In practice, if \(\frac{\partial {F}_{i}}{\partial {x}_{j}}\) shows large variance within each bin of \({x}_{j}\), it may imply that other factors affect the D-R curve. For example, the regulation of \({x}_{j}\) on \({x}_{i}\) may even be opposite at the presence or absence of a specific cofactor. In this case, one should first cluster cells, e.g., grouping cells based on whether an identified cofactor reaches a threshold value, then perform the D-R curve reconstruction on individual clusters.
Markovian analyses
Marius et al.16 have developed a framework named CellRank to study cellular dynamics based on a Markov chain formulation. We use CellRank to identify cell state transitions using a velocity kernel and identify terminal states within datasets by the GPCCA function module. In addition, CellRank pseudotime kernel8 is used for methods comparison in real datasets.
Analytical function form of a vector field and differential geometry analyses
Dynamo learns a nonlinear function form of RNA velocity vector field, providing a physics-informed framework that integrates mechanism modeling and single-cell data analyses. We use dynamo to learn continuous vector field functions and perform differential geometry analyses such as gene acceleration, vector field-based pseudotime, least action path (LAP), Jacobian analyses and in silico perturbation.
Pseudotemporal orderings
GraphVelo itself does not compute an ordering index of cells as we are seeking for a more quantitative method to infer RNA velocity. With an accurate RNA velocity as input, we can approximate the vector field precisely. Thus, we use the scalar potential estimated from the functional form vector field with Hodge decomposition as a proxy of time, which is implemented by dynamo package.
Cross boundary correctness scoring
While the GraphVelo framework is designed to quantify velocity vectors across different representations, known transitions between coarse cell states—such as cell types or cell cycle phases—can be used to evaluate the correctness of velocity directions. Suppose there are two cell populations A and B, with A a progenitor state of B. One can define the set of boundary cells between A and B as
where \({{{{\mathcal{C}}}}}_{{{{\rm{A}}}}}\) or \({{{{\mathcal{C}}}}}_{{{{\rm{B}}}}}\) denote the sets of cells in state A or state B, \({{{{\mathcal{N}}}}}_{c}\) indicates the kNN of cell c. The CBC score is then defined as
where \({\#c}^{\prime} \in {{{{\mathcal{C}}}}}_{{{{\rm{B}}}}}\cap {{{{\mathcal{N}}}}}_{c}\) is the number of cells in state B which is also the kNN of cell c. While the \(({{{{\boldsymbol{x}}}}}_{c},{{{{\boldsymbol{v}}}}}_{c})\) can be represented in different basis (raw count, PCA, or UMAP), we computed the CBC score in the original count space to ensure that all genes contribute to the velocity estimation. We deliberately avoided using 2D embeddings like UMAP for this purpose, as such visualizations may distort the true geometric relationships in the high-dimensional space and could lead to misleading interpretations.
Velocity consistency scoring
For most of the cases, we expect the inferred RNA velocity vectors to be coherent in a uni-directional vector field. To quantify the local consistency of the velocity flow for each cell, we calculate the velocity consistency score2 for each cell i as the mean correlation of its velocity \({v}_{i}\) with velocities from neighboring cells,
where cell j is the neighbor of cell i and cos indicates the cosine similarity. One thing should be clarified is that overly smooth and homogenized velocity fields may obscure biologically meaningful heterogeneity.
Approximate smooth velocity trends with a generalized addictive model
The variation of transcription rates contains the high-order dynamic information of the cell system. To model the dynamic patterns of RNA velocity along the transition path from noisy data, we refine the velocity vectors by local geometry via TSP and further fit GAM to velocity value of each gene that has been refined by GraphVelo. For any gene g, we model the velocity trend for the temporal variable t via
Where \({v}_{{gi}}\) indicates the velocity of gene g in cell i, f is built using penalized B-splines which allow us to automatically model non-linear mapping while maintaining additivity76. To visualize the velocity trends, we select 100 equally spaced testing points along the transition path and predict gene expression at each of them using the fitted model. The estimated velocity trends can be treated as smoothed time series for further analyses.
Clustering velocity trends along infection trajectory
With manifold-constrained velocity estimated by GraphVelo, we are able to cluster genes into different functional modules that are involved in the same regulatory circuit. We recover transcription variation of both host and virus factors along the infection trajectory by fitting GAMs in the temporal indicator, the percentage of viral RNA. Next, we select 100 equally spaced time points and generate the GAM-smoothed velocity trends. We compute a kNN graph and cluster the kNN graph using the Leiden algorithm. We used k = 15 for the velocity-trend kNN graph and the Leiden algorithm with a resolution parameter set to 0.3 to avoid over-clustering the trends. For each recovered cluster, we compute its mean and standard deviation (pointwise, for all generated points that were used for smoothing) and visualize the smoothed trends per cluster.
Characterizing decoupling genes based on the dynamic time warping distance between multi-modality velocities
We perform the DTW distance calculation by dtaidistance package. To eliminate the influence of scale in different modality, we maximum-normalize the chromatin/RNA velocity to the same range of [0, 1]. Then we fit the velocity trends of both modalities along vector field-based pseudotime to yield the smoothed velocity trends. We calculate the DTW distance between velocity trends per gene. To distinguish the decoupling genes based on their multi-modality velocities, we rank the genes based on the DTW distance and identify the elbow point as a cutoff. Genes with a distance metric larger than the cutoff are characterized as decoupling genes and used for visualization and functional analysis.
Synthetic datasets
Generating two genes bifurcation process and mapping it to 3d
The bifurcation data (n = 2000 cells) for the toggle-switch system is simulated using the Gillespie algorithm. We use activation and inhibition Hill functions to model the induction and suppression effects between the two genes:
We use the simulation backend implemented by dynamo with default parameters except the timescale (reset τ = 1) to generate the bifurcating process. We then map the synthetic dataset onto a sphere (radius r = 70) and yield the variable z as:
Then we are able to calculate the correctly-scaled 3d vectors by infinitesimal propagation operator with sufficient small step size (dt = 1 in our case):
Generating scRNA-seq synthetic data with dyngen.\
To generate high-dimensional single-cell transcriptomic data in silico, we use a multi-modal simulation engine, dyngen, to account for different developmental topologies. We constructe module networks to represent regulatory cascades and feedback loops driving progressive changes in gene expression and influencing cell fate decisions. We generate three datasets with 1000 cells and 100 genes using the linear, cyclic, and bifurcating loop backbones provided by dyngen, with all other parameters set to default values. These datasets include simulated nascent and mature mRNA counts along with ground-truth RNA velocities and known manifold structure.
Simulation of rapid degradation and transcription burst events on phase portrait
As for genes with variable degradation rate, we present a minimal regulatory network with linear model in which an external signal both inhibits transcription and promotes microRNA (miRNA). The miRNA exerts a linear influence on the degradation rate of mRNA. We have miRNA’s velocity as
The nascent gene transcription rate and mature mRNA’s degradation rate would change to
where \({{{{\rm{\alpha }}}}}_{0}\) and \({{{{\rm{\gamma }}}}}_{0}\) represent the constant transcription rate and degradation rate without the effect of miRNA, \({k}_{{{{\rm{\alpha }}}}}\) and \({k}_{{{{\rm{\gamma }}}}}\) represent the magnitude of influence from miRNA, t indicates the simulation time to mimic the cell-context change along trajectory.
We set the initial condition to a steady state with \({u}_{0}={\alpha }_{0}/\beta\) and \({s}_{0}={\alpha }_{0}/{\gamma }_{0}\), while the miRNA abundance \({m}_{0}=0\). We simulate \({u}_{t}\) and \({s}_{t}\) as the microRNA signal \({m}_{t}\) gradually increases. The aim is to evaluate whether the estimated RNA velocity consistently aligns in sign with the ground truth RNA velocity.
To generate genes with transcription burst phase portrait, we set the initial condition to \({u}_{0}=0,{s}_{0}=0\), together with \({{{\rm{\gamma }}}}\) as constant and \(\alpha\) promotes to \({\alpha }^{{\prime} }=3\alpha\) when the simulation reaches specific time.
Processing of sequencing data
All sequencing data in this study are downloaded publicly (see details in the ‘Data availability’ section). Though the number of confident-estimated gene sets differed case by case, GraphVelo predicted the velocity of all highly variable genes and used them for downstream calculations such as CBC score, CellRank velocity kernel, and dynamo vector field learning. We parallelized the TSP optimization to scale efficiently and set the hyperparameters in loss with a = 1, b = 10 and λ = 1 as default in all studies (Supplementary Fig. 20).
Analyses of scRNA-seq datasets
For the erythroid lineage of the mouse gastrulation, we follow the standard data pre-processing procedures implemented by scVelo and select 9815 cells and 2000 highly variable genes to construct the kNN graph using 30 nearest neighbors for downstream calculation.
For the human bone marrow dataset, we follow the standard data pre-processing procedures implemented by scVelo and selected 5780 cells and 2000 highly variable genes to construct the k-nearest neighbor graph using 30 nearest neighbors for downstream calculation. To estimate the variation of degradation rate \(\gamma\) along the differentiation lineages, we divide the cells into five discrete time bins based on precomputed Palantir pseudotime. We then estimate cell-specific degradation rates and visualized their distribution shifts along the hematopoiesis trajectory.
Analyses of spatial transcriptomics dataset
For the mouse coronal hemibrain spatial dataset, we follow the monocle preprocessing pipeline implemented in dynamo and select 7,765 cells and 2000 highly variable genes. To include spatial information during manifold, we build a spatial kNN graph with k = 8 and then take the union with the kNN graph based on transcriptomic data. The combined graph is used for downstream analyses.
Analyses of HCMV dataset
We sample the cells from donor 1 to eliminate sample-specific variation and further filter out cells lacking immediate early UL123 gene expression to focus on the viral infection trajectory. We adapt the monocle preprocessing recipe implemented by dynamo and yield 1454 cells and 2000 highly variable genes for further analyses. The number of nearest neighbors were set to 30 as default. Then 1022 velocity genes are used for downstream analyses.
We collect the pathway-related genes from MSigDB and perform the Jacobian analyses implemented by dynamo, using viral genes as regulators and host genes as effectors. We rank the regulation relationships based on the collections of Jacobian elements. We pick the top 50 inhibited effectors of viral genome and select the common set between pathway genes and all the effectors for visualization.
In silico knock-out experiments are performed via Dynamo. We suppressed every single virus factors per a time using dynamo.pd.perturbation function and calculated the change of total viral gene expression after perturbation.
Analyses of SARS-CoV−2 dataset
We subsample the cells based on their infection status. We adapt the monocle preprocessing pipeline implemented in dynamo and exclude the UMI reads from lenti-virus. We obtain 5001 infected cells and 1000 highly variable genes for downstream analyses. We use both highly variable genes in host and virus genes to perform PCA and calculate the nearest neighbors with k = 30. Then 269 velocity genes are used for downstream analyses. We use the apoptosis-related genes collected by KEGG to calculate the apoptosis activity score of each cell using dynamo.tl.score_cells function.
Analyses of multi-modality datasets
RNA velocity estimation for multi-modality datasets
In traditional scRNA-seq datasets, RNA velocity methods use smoothed spliced and unspliced RNA counts through nearest-neighbor pooling, based on the PCA space computed from transcripts alone. However, this approach is not suitable for multimodal scenarios, as it overlooks hidden variables by relying on a single modality. To construct a consistent manifold combining information from multi-modal genomics data, we utilized WNN as implemented in MultiVelo13. The WNN algorithm combines low-dimensional representations from RNA and ATAC omics data. Specifically, we use PCA results from scRNA-seq data and latent semantic indexing (LSI) from scATAC-seq as inputs. The nearest neighbors identified by WNN were then used to calculate the first moment, reducing noise in separate modalities and approximating a unified manifold for GraphVelo.
Mouse skin dataset preprocessing
The preprocessed SHARE-seq mouse skin dataset63 is adopted directly from MultiVelo data resources. All the procedures are consistent with MultiVelo, except we get the LSI representation processed by SCARlink77. We construct the WNN graph using 50 nearest neighbors for downstream calculation. We run scVelo with ‘stochastic’ mode to estimate the RNA velocity based on the WNN graph as we discussed above.
Human cortex dataset preprocessing
The preprocessed human cerebral cortex data is adopted directly from the MultiVelo data resources. All the procedures are consistent with MultiVelo, except we get the LSI representation processed by SCARlink. We construct the WNN graph using 50 nearest neighbors for downstream calculation. We run scVelo with ‘stochastic’ mode to estimate the RNA velocity based on the WNN graph.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the sequencing raw data are publicly accessible. The A549 dataset can be accessed via https://figshare.com/ndownloader/files/53666738. The FUCCI cell cycle data can be downloaded from https://figshare.com/ndownloader/files/53705057. The processed data used for benchmark are available at https://figshare.com/ndownloader/files/53667548. The mouse gastrulation subset to erythroid lineage can be extracted using scVelo’s CLI: scvelo.datasets.gastrulation_erythroid() or from the original work under accession number E-MTAB-6967 of ArrayExpress. The human bone marrow can be extracted using scVelo’s CLI: scvelo.datasets.bonemarrow() or through the Human Cell Atlas data portal at https://data.humancellatlas.org/explore/projects/091cf39b-01bc-42e5-9437-f419a66c8a45. The mouse coronal hemibrain spatial transcriptomic data can be downloaded from (https://www.dropbox.com/s/c5tu4drxda01m0u/mousebrain_bin60.h5ad?dl=0). The original HCMV infected moDC data can be accessed via Zenodo (https://zenodo.org/records/10404879) and the processed data can be downloaded from https://figshare.com/ndownloader/files/53666756. The SARS-CoV-2 data can be accessed via https://figshare.com/ndownloader/files/53666588. The preprocessed mouse skin development dataset can be accessed via https://figshare.com/articles/dataset/Mouse_Hair_Follicle_RNA_Data/22575307 and https://figshare.com/articles/dataset/Mouse_hair_follicle_ATAC_data/22575313. The preprocessed human cortex dataset can be downloaded from https://figshare.com/articles/dataset/Developing_Human_Cortex_RNA_Data/22575376 and https://figshare.com/articles/dataset/Developing_Human_Cortex_ATAC_Data/22575370. Source data are provided with this paper.
Code availability
The source code of python package GraphVelo78 can be downloaded from https://github.com/xing-lab-pitt/GraphVelo and is released under the BSD 3-Clause License. Reproducibility and tutorials can be found in https://github.com/xing-lab-pitt/GraphVelo/tree/main/notebook and https://graphvelo.readthedocs.io/en/latest/. The specific version of the code associated with this publication is archived in Zendo and is accessible via https://doi.org/10.5281/zenodo.15852884.
References
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Li, S. et al. A relay velocity model infers cell-dependent RNA velocity. Nat. Biotechnol. 42, 99–108 (2024).
Lederer, A. R. et al. Statistical inference with a manifold-constrained RNA velocity model uncovers cell cycle speed modulations. Nat. Methods 21, 2271–2286 (2024).
Gayoso, A. et al. Deep generative modeling of transcriptional dynamics for RNA velocity analysis in single cells. Nat. Methods 21, 50–59 (2024).
Gao, C. F., Vaikuntanathan, S. & Riesenfeld, S. J. Dissection and integration of bursty transcriptional dynamics for complex systems. Proc. Natl Acad. Sci. USA 121, e2306901121 (2024).
Qiu, X. et al. Mapping transcriptomic vector fields of single cells. Cell 185, 690–711 e45 (2022).
Weiler, P., Lange, M., Klein, M., Pe’er, D. & Theis, F. CellRank 2: unified fate mapping in multiview single-cell data. Nat. Methods 21, 1196–1205 (2024).
Wang, K. et al. PhyloVelo enhances transcriptomic velocity field mapping using monotonically expressed genes. Nat. Biotechnol. 42, 778–789 (2024).
Li, J., Pan, X., Yuan, Y. & Shen, H. B. TFvelo: gene regulation inspired RNA velocity estimation. Nat. Commun. 15, 1387 (2024).
Bergen, V., Soldatov, R. A., Kharchenko, P. V. & Theis, F. J. RNA velocity-current challenges and future perspectives. Mol. Syst. Biol. 17, e10282 (2021).
Gorin, G., Svensson, V. & Pachter, L. Protein velocity and acceleration from single-cell multiomics experiments. Genome Biol. 21, 39 (2020).
Li, C., Virgilio, M. C., Collins, K. L. & Welch, J. D. Multi-omic single-cell velocity models epigenome-transcriptome interactions and improves cell fate prediction. Nat. Biotechnol. 41, 387–398 (2023).
Zheng, S. C., Stein-O’Brien, G., Boukas, L., Goff, L. A. & Hansen, K. D. Pumping the brakes on RNA velocity by understanding and interpreting RNA velocity estimates. Genome Biol. 24, 246 (2023).
Tiejun Li, T. L., Jifan Shi, J. S., Yichong Wu, Y. W. & Peijie Zhou, P. Z. On the Mathematics of RNA Velocity I: Theoretical. Anal. CSIAM Trans. Appl. Math. 2, 1–55 (2021).
Lange, M. et al. CellRank for directed single-cell fate mapping. Nat. Methods 19, 159–170 (2022).
Roweis, S. T. & Saul, L. K. Nonlinear dimensionality reduction by locally linear embedding. Science 290, 2323–2326 (2000).
Whitney, H. The self-intersections of a smooth n-Manifold in 2n-Space. Ann. Math. 45, 220–246 (1944).
Cannoodt, R., Saelens, W., Deconinck, L. & Saeys, Y. Spearheading future omics analyses using dyngen, a multi-modal simulator of single cells. Nat. Commun. 12, 3942 (2021).
Mahdessian, D. et al. Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590, 649–654 (2021).
Bastidas-Ponce, A. et al. Comprehensive single cell mRNA profiling reveals a detailed roadmap for pancreatic endocrinogenesis. Development 146, dev173849 (2019).
Battich, N. et al. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science 367, 1151–1156 (2020).
Gao, M., Qiao, C. & Huang, Y. UniTVelo: temporally unified RNA velocity reinforces single-cell trajectory inference. Nat. Commun. 13, 6586 (2022).
Cui, H. et al. DeepVelo: deep learning extends RNA velocity to multi-lineage systems with cell-specific kinetics. Genome Biol. 25, 27 (2024).
Qiao, C. & Huang, Y. Representation learning of RNA velocity reveals robust cell transitions. Proc. Natl Acad. Sci. USA 118, e2105859118 (2021).
Cao, J., Zhou, W., Steemers, F., Trapnell, C. & Shendure, J. Sci-fate characterizes the dynamics of gene expression in single cells. Nat. Biotechnol. 38, 980–988 (2020).
Pijuan-Sala, B. et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019).
Barile, M. et al. Coordinated changes in gene expression kinetics underlie both mouse and human erythroid maturation. Genome Biol. 22, 197 (2021).
Lee, J., Krivega, I., Dale, R. K. & Dean, A. The LDB1 Complex Co-opts CTCF for Erythroid lineage-specific long-range enhancer interactions. Cell Rep. 19, 2490–2502 (2017).
Dybedal, I., Larsen, S. & Jacobsen, S. E. IL-12 directly enhances in vitro murine erythropoiesis in combination with IL-4 and stem cell factor. J. Immunol. 154, 4950–4955 (1995).
Setty, M. et al. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 37, 451–460 (2019).
Ning, W. et al. Blocking exosomal miRNA-153-3p derived from bone marrow mesenchymal stem cells ameliorates hypoxia-induced myocardial and microvascular damage by targeting the ANGPT1-mediated VEGF/PI3k/Akt/eNOS pathway. Cell Signal 77, 109812 (2021).
Qiu, X. et al. Spatiotemporal modeling of molecular holograms. Cell 187, 7351–7373.e61 (2024).
Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 624, 317–332 (2023).
Bost, P. et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 181, 1475–1488.e12 (2020).
Ratnasiri, K., Wilk, A. J., Lee, M. J., Khatri, P. & Blish, C. A. Single-cell RNA-seq methods to interrogate virus-host interactions. Semin Immunopathol. 45, 71–89 (2023).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).
Edahiro, R. et al. Single-cell analyses and host genetics highlight the role of innate immune cells in COVID-19 severity. Nat. Genet. 55, 753–767 (2023).
Costa, B. et al. Human cytomegalovirus exploits STING signaling and counteracts IFN/ISG induction to facilitate infection of dendritic cells. Nat. Commun. 15, 1745 (2024).
Hein, M. Y. & Weissman, J. S. Functional single-cell genomics of human cytomegalovirus infection. Nat. Biotechnol. 40, 391–401 (2022).
Alon, U. An introduction to systems biology: design principles of biological circuits, (Chapman & Hall/CRC, 2007).
Ball, C. B. et al. Human Cytomegalovirus IE2 both activates and represses initiation and modulates elongation in a context-dependent manner. mBio 13, e00337–22 (2022).
Manska, S. & Rossetto, C. C. Identification of cellular proteins associated with human cytomegalovirus (HCMV) DNA replication suggests novel cellular and viral interactions. Virology 566, 26–41 (2022).
Gredmark-Russ, S. & Soderberg-Naucler, C. Dendritic cell biology in human cytomegalovirus infection and the clinical consequences for host immunity and pathology. Virulence 3, 621–634 (2012).
Paulus, C., Krauss, S. & Nevels, M. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl Acad. Sci. USA 103, 3840–3845 (2006).
Park, A. et al. HCMV-encoded US7 and US8 act as antagonists of innate immunity by distinctively targeting TLR-signaling pathways. Nat. Commun. 10, 4670 (2019).
Yang, D., de la Rosa, G., Tewary, P. & Oppenheim, J. J. Alarmins link neutrophils and dendritic cells. Trends Immunol. 30, 531–537 (2009).
Bogdanow, B. et al. Human cytomegalovirus tegument protein pp150 acts as a cyclin A2-CDK-dependent sensor of the host cell cycle and differentiation state. Proc. Natl Acad. Sci. USA 110, 17510–17515 (2013).
Luftig, M. A. Viruses and the DNA damage response: activation and antagonism. Annu Rev. Virol. 1, 605–625 (2014).
Ball, C. B. et al. Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III. Viruses 14, 779 (2022).
Bogdanow, B., Phan, Q. V. & Wiebusch, L. Emerging Mechanisms of G(1)/S Cell Cycle Control by Human and Mouse Cytomegaloviruses. mBio 12, e0293421 (2021).
Goodwin, C. M., Ciesla, J. H. & Munger, J. Who’s driving? Human Cytomegalovirus, Interferon, and NFkappaB signaling. Viruses 10, 447 (2018).
Manandhar, T., Ho, G. T., Pump, W. C., Blasczyk, R. & Bade-Doeding, C. Battle between Host Immune Cellular Responses and HCMV Immune Evasion. Int. J. Mol. Sci. 20, 3626 (2019).
Zeng, J. et al. Insights into the Transcriptome of Human Cytomegalovirus: A comprehensive review. Viruses 15, 1703 (2023).
Adamson, C. S. & Nevels, M. M. Bright and early: inhibiting human cytomegalovirus by targeting major immediate-early gene expression or protein function. Viruses 12, 110 (2020).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e10 (2020).
Sunshine, S. et al. Systematic functional interrogation of SARS-CoV-2 host factors using Perturb-seq. Nat. Commun. 14, 6245 (2023).
Drayman, N., Patel, P., Vistain, L. & Tay, S. HSV-1 single-cell analysis reveals the activation of anti-viral and developmental programs in distinct sub-populations. Elife 8, e46339 (2019).
Labbe, K. & Saleh, M. Cell death in the host response to infection. Cell Death Differ. 15, 1339–1349 (2008).
Augusto, L. et al. Toxoplasma gondii Co-opts the Unfolded Protein Response To Enhance Migration and Dissemination of Infected Host Cells. mBio 11, e00915-20 (2020).
Prasad, V. & Greber, U. F. The endoplasmic reticulum unfolded protein response–homeostasis, cell death and evolution in virus infections. FEMS Microbiol. Rev. 45, fuab016 (2021).
Saul, S. et al. Anticancer pan-ErbB inhibitors reduce inflammation and tissue injury and exert broad-spectrum antiviral effects. J. Clin. Invest. 133, e169510 (2023).
Ma, S. et al. Chromatin potential identified by shared single-cell profiling of RNA and Chromatin. Cell 183, 1103–1116.e20 (2020).
Joost, S. et al. The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell 26, 441–457.e7 (2020).
Pethe, P., Noel, V. S. & Kale, V. Deterministic role of sonic hedgehog signalling pathway in specification of hemogenic versus endocardiogenic endothelium from differentiated human embryonic stem cells. Cells Dev. 166, 203685 (2021).
St-Jacques, B. et al. Sonic hedgehog signaling is essential for hair development. Curr. Biol. 8, 1058–1068 (1998).
Trevino, A. E. et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069.e23 (2021).
Wang, S. et al. The inconsistent regulation of HOXC13 on different keratins and the regulation mechanism on HOXC13 in cashmere goat (Capra hircus). BMC Genomics 19, 630 (2018).
Peng, Y. et al. Detection of new pioneer transcription factors as cell-type-specific nucleosome binders. Elife 12, RP88936 (2024).
Desanlis, I. et al. HOX13-dependent chromatin accessibility underlies the transition towards the digit development program. Nat. Commun. 11, 2491 (2020).
Gillespie, D. T. The chemical Langevin Equation. J. Chem. Phys. 113, 297–306 (2000).
Zwanzig, R. Ensemble method in the theory of irreversibility. J. Chem. Phys. 33, 1338–1341 (1960).
Mori, H. Transport, collective motion, and Brownian motion. Prog. Theor., Phys. 33, 423–455 (1965).
Xing, J. & Kim, K. S. Application of the projection operator formalism to non-Hamiltonian dynamics. J. Chem. Phys. 134, 044132 (2011).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Hastie, T. & Tibshirani, R. Generalized additive models, (Chapman and Hall, London; New York, 1990).
Mitra, S. et al. Single-cell multi-ome regression models identify functional and disease-associated enhancers and enable chromatin potential analysis. Nat. Genet 56, 627–636 (2024).
Chen, Y. et al. GraphVelo allows for accurate inference of multimodal velocities and molecular mechanisms for single cells. Zendo https://doi.org/10.5281/zenodo.15852884 (2025).
Acknowledgements
This work was partially supported by the National Institute of General Medical Sciences (1R01GM148525 to J.X. and 1R01GM139297 to I.B.), and the National Science Foundation (2325149 to J.X.). This work was supported by the National Key Research and Development Program of China (2023YFE0112300 to M.C.); National Natural Sciences Foundation of China (32261133526; 32270709; 32070677 to M.C.); the 151 talent project of Zhejiang Province (first level to M.C.), the Science and Technology Innovation Leading Scientist (2022R52035 to M.C.).
Author information
Authors and Affiliations
Contributions
J.X. conceived and formulated the theoretical framework, Y.C. and J.G. performed tests on simulated data, Y.C. and J.X. analyzed scRNA-seq datasets, Y.C., Y.Z., J.G., and K.N. developed the package graphvelo, Y.C. and J.X. wrote the draft, M.C. and I.B. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zheng Hu, Shihua Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Y., Zhang, Y., Gan, J. et al. GraphVelo allows for accurate inference of multimodal velocities and molecular mechanisms for single cells. Nat Commun 16, 7831 (2025). https://doi.org/10.1038/s41467-025-62784-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62784-w
This article is cited by
-
Single cell snapshot analyses under proper representation reveal that epithelial-mesenchymal transition couples at G1 and G2/M
Communications Biology (2026)
-
Geometry-preserving vector field reconstruction of high-dimensional cell-state dynamics using ddHodge
Nature Communications (2025)
