
Abstract
The clinical efficacy of the antidiabetic drug acarbose is hampered by degradation by the acarbose-preferred glucosidase (Apg) from K. grimontii TD1. Understanding the catalytic mechanism of Apg can aid the design of next-generation hypoglycemic pharmaceuticals acarbose analogs. Here, we determine several crystal structures of Apg to identify the catalytic residues and the ligand-binding pocket of Apg. Structural analyses and computational modeling reveal D448 as the active nucleophile, contrasting with prior studies that assumed D336 to be the nucleophile. In addition to E373 proposed as the proton donor in previous reports, we find that R334 might be an alternative proton donor. Our experimental and computational analyses indicate the two-ring product acarviosine is the two-step hydrolyzed product, where the second hydrolysis is the rate-limiting step. Additionally, further investigation of the acarbose analogs acarstatins A and B that are resistant to Apg is conducted by computational analysis. Overall, our studies provide perspectives into the intricacies of Apg’s catalytic mechanism, contributing to the design of next-generation hypoglycemic pharmaceuticals.
Similar content being viewed by others

Introduction
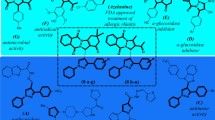
Type 2 Diabetes mellitus (T2DM) is rapidly emerging as one of the greatest global challenges1,2. Acarbose, a pseudotetrasaccharide3, is widely prescribed as the first-line treatment of T2DM in clinical practice4. Acarbose has minimal absorption into the bloodstream, with few systemic side effects. However, a large individual variation in the effectiveness and dosage of acarbose is observed in clinical practice, which is severely resistant in some patients after prolonged treatment5. In recent years, research into the resistance mechanisms to this host-targeted (i.e., non-antibiotic) drug has attracted significant attention6. Recently, Jiang et al.7 reported acarbose-preferred glucosidase (Apg) in K. grimontii TD1, which significantly degrades acarbose to acarviosine-glucose (M1) and acarviosine (M2), thereby disrupting the efficiency of acarbose (Fig. 1).

Acarbose-degrading pathways in K. grimontii TD1 mediated by Apg.
In the CAZy database, Apg is a member of the glycoside hydrolase family 13_21 (GH13_21), which hydrolyze the α−1,4-O-glycosidic bond. Typically, members of this family follow a ‘Koshland’ mechanism8,9 to hydrolyze glycosidic bonds through substitution reactions10,11,12,13. However, acarbose is difficult to be hydrolyzed by standard glycosidases14. Understanding the distinct catalytic mechanism of Apg will benefit the design of antidiabetic drugs. Previous molecular docking simulations7 suggested that acarbose was hydrolyzed by orienting the nucleophile D336 towards at the linkage between the third and the fourth rings, yielding the three-ring product M1, or at the linkage between the second and the third rings, yielding the two-ring product M2. The latter pathway is predominantly observed in the hydrolysis of glycosidic bonds catalyzed by α-amylase15, β-amylase16,17, sucrase-isomaltase18,19, and so on. The question then arises as to whether M2 can be formed via stepwise hydrolysis from M1 or a one-step hydrolysis from acarbose. Certainly, the best insight into this mechanistic hydrolysis comes from X-ray crystallographic analysis to identify the catalytic residues and the ligand-binding pocket of Apg.
Here, we report several X-ray crystal structures of Apg, including WT-Apg, the D448A mutant, the D448N mutant, the D448A mutant complex with acarbose, the D448N mutant complex with acarbose, the D336A mutant complex with M1, the D336A mutant with M2, the ring deletion mutant ∆loop A with glucose, ∆loop B and ∆loop A/B. Our studies show that D448 acts as a nucleophile, and E373 or R334 can act as a proton donor during hydrolysis. Furthermore, acarbose undergoes hydrolysis in two stages to produce M2, involving the conversion of acarbose to M1, followed by a subsequent reaction that transforms M1 into M2. Moreover, comprehensive computational evaluations of the acarbose analogs, acarstatins A and B, demonstrating resistance to Apg, are conducted leveraging our crystallographic structures. Our results not only elucidate the catalytic mechanism of Apg in acarbose degradation but also provide structural insights for the design and development of next-generation diabetes drugs20.
Results and discussion
Overall structure of Apg
According to size exclusion chromatography (SEC) experiments, Apg exists in solution as a dimer with a molecular weight of 138 kDa (theoretical molecular mass of Apg is 69 kDa (Supplementary Fig. 1). The WT-Apg (PDB 9IXW; Supplementary Table 1 and Supplementary Fig. 1) was crystallized in space group P65 2 2. The crystal structure was solved at 2.32 Å resolution by the molecular replacement method using the putative glucosidase structure (maltodextrin glucosidase MaIZ, 79% sequence homology to Apg, PDB 7VT9) as the model21. In the unit cell of Apg crystal, the asymmetric unit contains one Apg chain, and the biological assembly in the form of homodimer is assembled with a symmetric molecule (Fig. 2A).

A The biological assembly in the form of homodimer (molecule A in pink and molecule B in blue). B The catalytic structural domain (residues 114–530) is labeled in pink. The N-terminal (residues 1–113) and C-terminal (residues 531– 604) domains constituting a fully β-folded structure, labeled in slate and wheat, respectively. The cavity volume of the active site is estimated by CayityPlus.
The overall structure of the Apg monomer consists of three major structural domains, namely, the N-structural domain (residues 1–113), the C-structural domain (residues 531–604), and the catalytic structural domain (residues 114–530). The overall structure of Apg adopts the typical TIM barrel fold observed in the glycoside hydrolase family 13_21 (GH13_21) (Fig. 2B and Supplementary Fig. 2A), including maltodextrin glucosidase MaIZ (PDB 7VT9, root-mean-square deviation, r.m.s.d for short, 0.556 Å)21 and TVAI (PDB 1IZJ, r.m.s.d 0.960 Å)22. The active site is a surface-protruding groove at the top of the TIM barrel with an absolutely conserved catalytic triad (Asp-Glu-Asp)7,23 at the bottom of the groove (Fig. 2B and Supplementary Fig. 2B). The total volume of active sites is estimated to be 1392 Å3, which is sufficient to accommodate acarbose (702 Å3, the total molecular volume of acarbose) (Fig. 2B). However, the full alignment with the amylases such as MaIZ (from the GH13_21 subfamily, PDB 7VT9; r.m.s.d = 0.557 Å)21 and MaIS (from the GH13_19 subfamily, PDB 8IM8; r.m.s.d = 4.058 Å)24 indicate that the cleft width of Apg (7.9 Å) is relatively narrow, which is not conducive to the accommodation of bulky substrates such as cyclic oligosaccharides(Supplementary Fig. 3 and Supplementary Fig. 4A). In contrast, the cleft widths of MaIZ (9.1 Å) and MaIS (12.1 Å) are significantly wider, allowing them to accommodate bulky substrates such as cyclodextrins (Supplementary Fig. 4B, C). Binding free energy calculations further showed that Apg had a higher binding affinity for acarbose than MaIZ and MaIS (Supplementary Table 2). In addition, residues 138–143 is missing in the Apg final model due to a lack of density, which suggests high flexibility and/or potential disorder in this region. In the homodimer, the N-domain of Apg is near the catalytic domain of the adjacent molecule with a head-to-tail arrangement, with a dimeric interfacial surface area of ~1748.9 Å2 for each monomer, as calculated by PDBePISA25. This intimate homodimeric architecture resembles that of other glycoside hydrolases21,22,26,27.
Structure of the Apg (D448A) Complex with Acarbose
To understand how Apg recognizes acarbose, we try to obtain a complex structure by co-crystallizing WT-Apg with acarbose or soaking WT-Apg crystal with acarbose. Unfortunately, it is difficult to obtain a complex structure because WT-Apg hydrolyzes acarbose in a very short time. Since the conserved catalytic triad (Asp-Glu-Asp) is essential for catalytic activity7,28,29, the D336A, E373A, and D448A mutants are prepared to capture the complex crystal structure of Apg with acarbose. Ultimately, the complex structure of the D448A mutant with acarbose (PDB 9IVZ) is determined to 2.29 Å resolution (Fig. 3A). The electron density reveals that acarbose is well and fully occupied in the active pocket (Fig. 3A, Supplementary Fig. 5A).

Simulated annealing omit maps are shown in gray mesh and contoured at 1.0 σ. Hydrogen bonding interactions are indicated with gray dashed lines. A Clip diagram of the substrate acarbose in the lumen of the active site of Apg. The hydrophobic surface in blue and the hydrophilic surface in white. Acarbose is indicated by cyan color of stick. The 2Fo-Fc electron density maps of acarbose are shown as gray grids with contours at the 1.0 σ level. B Crystal structure of Apg(D448A)-acarbose (PDB 9IVZ). The substrate acarbose is shown in cyan, residues, in pink, hydrogen bonding interactions in gray dashed lines, and water molecules in red spheres. Validation of the electron density of acarbose-bound and active-ligand water is shown in Supplementary Fig. 7. C Apg(D336A) crystal structure of M1 (PDB 9IZE) (ligand, palegreen; residues, pink). D Crystal structure of Apg(D336A)-M2 (PDB 9IZO). Product M2 is shown in slate, residues, in pink. Stereo views of B-D see Supplementary Fig. 8. E Apg(D448A)-acarbose complex (PDB 9IVZ) (colored as in A) superimposed on mutant Apg(D448A) (PDB 9IXH) (residues, white). Significant conformational changes are observed for W292, Y295. The direct loop domains of Y295 and W292 are shifted to a certain extent. Among them, Y295 has a shift distance of 2.6 Å (atom OH) and W292 has a shift distance of 5.1 Å (atom NH). Maximum displacement of capping loop B to the sugar portion is 4.1 Å (defined by the NH2 atom of D291). F Mutagenesis experiment of residues in the active site of Apg. Asterisks indicate mutants with no measurable activity. Data are presented as mean values ± SD, n = 3. Error bars represent the standard deviation from three repeats.
Overall, the interactions surrounding the active sites are mainly dominated by several hydrogen bonds (Fig. 3B and Supplementary Fig. 6A). The -3 and -2 subsites of acarbose maintain the conformation through a network of hydrogen bonds mediated by a bonded water molecule with R496, D492 and G170. The -1 subsite adopts a semi-chair-like conformation, forming hydrogen bonds with H251 and D336, or through a bonded water molecule with H447, R334, and D336. The +1 subsite forms multiple hydrogen bonds with E373, F375 via a bonded water molecules. In addition, a water molecule locks the -1 and +1 subsites configurations via hydrogen bonds formed with C3-OH (2.50 Å) at the +1 subsite and C6-OH (3.0 Å) at the -1 subsite. Subsequently, a structural similarity search was performed in the PDB database using the DALI server30. By performing multiple sequence comparisons with 13 glycoside hydrolases of the GH13 family, we found that these residues were nearly conserved (Supplementary Fig. 9).
Structure of the Apg(D336A) Complex with M1
To explore the molecular basis of the Apg-catalyzed hydrolysis of acarbose, the structure of the Apg(D336A)-M1 complex subsequently (PDB 9IZE) is obtained with 1.87 Å resolution by co-crystallizing the mutant D336A with acarbose. In this binary complex model, the aminocyclic alcohol (-2 subsite), 4- amino-4,6-dideoxyglucose(−1 subsite), and glucose ( + 1 subsite) units of M1 are clearly observed and well localized in the active site (Fig. 3C and Supplementary Fig. 5B). Similar to the interactions surrounding the active sites in structure of the Apg complex with acarbose, several hydrogen bonding interactions are observed in the active site pocket of the Apg-M1 complex structure (Fig. 3C, Supplementary Fig. 6B and Supplementary Fig. 10A). In the −2 subsite, C3-OH forms a hydrogen bond with a water molecule and C7-OH forms a second hydrogen bond with the carbonyl oxygen of D492. The binding structure of −1 subsite relies on the formation of hydrogen bonding interactions with D492 and R496. The +1 subsite forms a hydrogen bonding network mainly through H251, R334, E373, H447, and D448 (Fig. 3C). These hydrogen bonds may play a pivotal role in precisely determining the orientation and placement of the product.
Structure of the Apg(D336A) Complex with M2
In order to shed light on possible pathway to form M2, a well-defined 1.93 Å structure of the Apg complex with M2 (PDB 9IZO) is revealed by incubated the D336A mutant with M1 (Fig. 3D and Supplementary Fig. 5C). Similar to the bonding model observed on the binary complex structure of Apg-M1, C2-OH and C3-OH of the −1 subsite is stabilized by hydrogen bonds with residues D492 and R496 in the model of the Apg(D336A)-M2 binary complex (Fig. 3D, Supplementary Fig. 6C and Supplementary Fig. 10B). The hydroxyl group located precisely at the C4 position actively participates in hydrogen bonding, forming a robust linkage with two water molecules. Similarly, the 4-amino-4,6-dideoxyglucose moiety is stabilized through hydrogen bonds with R334, E373, H447 and D448, respectively.
The role of flexible capping loops in the mechanism of Apg hydrolysis
The overall structure of Apg(D448A)-acarbose is very similar to that of the ligand-free Apg(D448A) (PDB 9IXH), with a r.m.s.d of all Cα atoms of ~0.211 Å. However, substrate binding triggers a localized structural change that is primarily focused on a pair of flexible capping loops (residues 138–152 are referred to as loop A, residues 257–300 are referred to as loop B) (Fig. 3E and Supplementary Fig. 11A). When Apg binds acarbose, loop A becomes more organized, thereby anchoring the substrate to the enzyme. The segment of loop B, which includes residues W292 and Y295 located at the top of the groove, undergoes significant rearrangement, creating space for substrate binding and thereby optimizing the enzyme-substrate interactions. Similar loops are present in other acarbose hydrolases, such as the α -glucosidase WcAG31 and thermophilic maltose amylase ThMA32 (Supplementary Fig. 12A). In contrast to the significant conformational change in the loop A/B observed when Apg binds acarbose, WcAG did not undergo a significant conformational change in the structural of its corresponding homologous mobile loop when bound to acarbose (Supplementary Fig. 12B). In contrast, ThMA exhibited Apg-like conformational changes in its corresponding loop region upon binding acarbose (Supplementary Fig. 12C). However, it is noteworthy that the binding affinity of ThMA for acarbose was significantly lower than that of Apg (Supplementary Table 2). Further root-mean-square-fall (RMSF) analysis showed that the structural flexibility of this ring region in ThMA was higher, which was consistent with its lower substrate affinity (Supplementary Fig. 13).
To analyze the effect of flexible capping loops on enzyme activity, the mutants of ∆loop A, ∆loop B, and ∆loop A/B were generated. Comparing to WT, the activities of the ∆loop A, ∆loop B, and ∆loop A/B mutants significantly are lower (46%, 22%, and 0.66%, respectively) (Supplementary Fig. 11B). SEC analysis of the above three mutants revealed that the ring deletion mutant did not affect the dimerization state (Supplementary Fig. 14). Further crystal structure data showed that the protein’s structure remains largely unchanged regardless of the tracking of cappling loops, indicating that the capping loops might benefit for substrate binding into Apg’s active site (Supplementary Table 1 and Supplementary Fig. 15). Absence of these capping loops makes it challenging for acarbose to form a stable complex with Apg. 200 ns MD simulations of acarbose and the mutant ∆loop A, ∆loop B and ∆loop A/B complexs reveal that acarbose leaves the active sites at 28 ns, 10 ns and 3 ns, respectively (Supplementary Fig. 16). Furthermore, comparing with WT Apg, the calculated binding energy of the mutant ∆loop A, ∆loop B and ∆loop A/B to acarbose decreases to −10.68, −8.28 and −1.49 kcal/mol, respectively (Supplementary Table 2).
Mutagenesis and computational studies confirm the roles of the key residues
To investigate functions of the key residues, hydrolytic activities of mutants toward acarbose are measured (Fig. 3F). Remarkably, the introduction of the W292A mutant located at the capping loop B region has resulted in a marked enhancement of reactivity by approximately 29%. This improvement may stem from an enlargement of the active pocket’s capacity. Conversely, the Y295A mutant reduces catalytic activity by 74%. However, substituting Y295 with F enhances the activity by 24%, indicating that large size amino acid is crucial for maintaining activity. Presumably, Y295 might act as a substrate-locking gateway. In addition, the mutant G170A, H251A, H447A, D492A, and R496A result in approximately 9%, 66%, 78%, 45%, and 71% loss of activity, respectively, which might cause by the elimination of hydrogen bonding between these residues and the glycosyl unit of acarbose.
In previous studies7, D336, E373, and D448 were considered to be the catalytic nucleophilic reagent, the proton donor and transition state stabilizer, respectively. Mutation of E373 to A or Q with weaker hydrogen bond donating capacity, caused 41% and 60% decrease in activity, respectively. The retention activity of E373A and E373Q suggests the presence of alternative proton donors in proximity to these residues, which was also proposed in previous research33. In order to speculate this unclear mechanism, double mutant from E373A with H447, Y211, and R334 are generated. A further double mutant, E373A/R334A was found to loss of activity (Supplementary Fig. 17). This result uncover that proton donors are not only E337 alone but also R334. The QM/MM results can also support R334 as a proton donor with slightly higher reactive barrier compared with E373 (Supplementary Fig. 18).
Moreover, the D336A mutant exhibited a retention of 48% of its original activity, suggesting that D336 is not the paramount residue. Interestingly, the D448A mutant completely eliminated its catalytic activity towards acarbose. Additionally, the mutant D448N was also found to totally loss of activity(Fig. 3F). To verify the binding ability of the inactivated mutant D448N to acarbose, we performed isothermal titration calorimetry (ITC) experiments (Supplementary Fig. 19). The results showed that D448N had a higher binding affinity for acarbose compared to WT-Apg. Further, we successfully resolved the crystal structure of the Apg(D448N)-acarbose complex (PDB 9UNS; Supplementary Table 1, Supplementary Fig. 20, Supplementary Fig. 21), revealing its binding mode in detail. The structure shows that the D448N residue forms hydrogen bonding interactions with the 2-OH and C1-O atoms of the acarbose −1 subsite. Notably, the distance between the carboxy oxygen atom (OD2) of D448N and the acarbose C1 atom (4.2 Å) is significantly shorter than the distance between the carboxy oxygen atom of D336 and C1 (5.0 Å) (Supplementary Fig. 22). This structural difference suggests that D448, rather than D336, is more likely to act as a nucleophilic reagent. Furthermore, when the D448E mutation, which also has a carboxylic acid side chain, was introduced, the catalytic activity was restored by 15% (Fig. 3F). Taken together, we propose that the carboxylic acid side chain of D448 may function directly as a nucleophilic catalyst in the transition state of the reaction, rather than only as a stabilizing agent. This hypothesis is further supported by computational studies. In multiple 200 ns MD simulations based on the crystal structure of acarbose and WT Apg, the average distance between C1 atom in the −1 subsite of acarbose and OD2 atom in the carboxylic acid group of D448 is much closer than that between C1 atom in the −1 subsite of acarbose and C atom in the carboxylic acid group of D336 (4.90 Å V.S. 6.22 Å) (Supplementary Fig. 23A). Additionally, a greater negative charge is found on OD2 of D448 compared with that of D336 (−0.49 v.s. −0.12) based on density functional theory (DFT) calculations, indicating that D448 is a more favorable nucleophile (Supplementary Fig. 23B). The roles of D336 and D448 during catalysis are further investigated using quantum mechanics/molecular mechanics (QM/MM). The QM/MM calculations also indicated that the energy barrier of D336 as a nucleophilic reagent (26.7 kcal/mol, Supplementary Fig. 24) is much higher than that of D448 (17.1 kcal/mol, Fig. 4).

A In the QM/MM calculations of acarbose hydrolyzed into M1, representative snapshots of the reactant (Michaelis complex, acarbose with Apg, ES), glycosyl-enzyme intermediate state (IC1) and product (M1 with Apg, EP1) states of the catalytic itinerary. B In the QM/MM calculations of M1 hydrolyzed into M2, representative snapshots of the reactant (Michaelis complex, M1 with Apg, EP1ˈ), glycosyl-enzyme intermediate state (IC2) and product (M2 with Apg, EP2) states of the catalytic itinerary. C The catalytic mechanism of acarbose with Apg by schematic drawings. The unit of energy is kcal/mol. The unit of distance involved in the 3D structure diagram is Å.
M2 is the two-step hydrolyzed product with the second hydrolysis as the rate-limiting step
Besides the three-ring product M1, the two-ring product M2 is also formed during hydrolysis of acarbose catalyzed by Apg. The previous molecular docking simulation7 suggested that M2 was yielded by orienting the nucleophile D336 towards the C1 of the second ring of the non-reducing end of acarbose. However, side product maltose was not detected when incubating acarbose with Apg (Supplementary Fig. 25). Notably, QM/MM simulations reveal that the energy barrier of directly hydrolyzing acarbose to M2 and maltose in one step is quite high (36.9 kcal/mol, Supplementary Fig. 26). Moreover, M2 is also observed when incubating Apg with M1, which indicates that M2 might be formed by two steps hydrolysis from acarbose.
To elucidate the detailed catalytic mechanism of the two hydrolysis steps, we perform MD and QM/MM calculations. MD simulations suggest that the Apg binds more tightly with acarbose (−20.5 kcal/mol, Table S2) than with M1 (−13.8 kcal/mol, Supplementary Table 2). Similar to catalytic cycle of producing M1, the second hydrolysis step is also proceeded through a retaining mechanism mediated by D448 and E373 (Fig. 4). However, the energy barrier of constructing the covalent ester linked intermediate from M1 (EP1’ → TS3, 19.7 kcal/mol, Fig. 4) is slightly higher than that calculated from acarbose (ES → TS1, 17.1 kcal/mol, Fig. 4). The results of enzyme kinetic experiments further elucidated the difference in catalytic properties of Apg towards acarbose and M1. When M1 was used as the substrate, the hydrolytic activity of Apg was significantly reduced by 30% compared to the acarbose system (Supplementary Fig. 27A). Further analysis of the kinetic parameters showed that in terms of substrate affinity, the Km value of acarbose (2.600 ± 0.652 µM) was significantly higher than that of M1 (6.642 ± 0.371 µM), suggesting that Apg has a stronger substrate binding ability to acarbose. While in terms of catalytic efficiency, the kcat value of acarbose (5.360 ± 0.320 s−1) was significantly higher than that of M1 (4.895 ± 0.512 s−1), indicating that acarbose has a higher rate of substrate conversion per unit time (Supplementary Fig. 27B). ITC thermodynamic analysis further revealed that the binding affinity of Apg-acarbose was significantly higher than that of Apg-M1 (Supplementary Fig. 19). Therefore, the second hydrolysis step is the slower step in the two-steps hydrolysis reactions. Furthermore, the possibility of cleaving carbon-nitrogen bond, a metabolization of acarbose found in recent study was also invested14. In our QM/MM results, the energy hurdle for the subsequent step is exceedingly high (43.7 kcal/mol, Supplementary Fig. 28), elucidating that M2 is the final product.
Further investigation of acarbose analogs
Meanwhile, the crystal structure we obtained in this paper is also beneficial to the design process of acarbose analogs. Recently, acarbose analogs acarstatins A and acarstatins B are reported that exhibited greater inhibitory activity towards human salivary α-amylase than acarbose and exhibited partial resistance to Apg-mediated hydrolysis20. To explore the structural insights of the resistance to Apg, the MD simulations are conducted based on our crystal structures. Although acarstatins A/B are also located within the Apg active cavity (Fig. 5A, C), the distances between the C1 of −1 subsite in acarstain A/B and OD2 atom of D448 are farther than that in acarbose (Fig. 5B), leading to the sugar ring carbon atoms can’t be attacked by D448. Further QM/MM analysis reveals that the activation energy barriers of the hydrolysis acarstain A/B reactions catalyzed by Apg (Supplementary Fig. 29 and Supplementary Fig. 30) are both higher than the activation energy barriers of the hydrolysis of acarbose (Fig. 4). It is expected that the insights gleaned from our studies will not only significantly contribute to expand the diversity of glycoside hydrolysis, but also to the design of next-generation clinically effective antidiabetic drugs.

A The MD snapshots of WT-Apg with acarstain A at 70 ns. B This figure shows the average distances (solid lines) between C1 atom (−1 subsite of the three sugar molecules for the yellow: acarbose, purple: acarstatins A and red: acarstatins B) and OD2 atom (D448, nucleophilic reagent) as a function of time, with standard errors of the average distances in 3 times MD simulations (shaded region). C The MD snapshots of WT-Apg with acarstain B at 70 ns. The distances are given in Å.
In summary, we present the X-ray crystallographic structures of WT-Apg, the D448A mutant, the D448N mutant, the D448A mutant complex with acarbose, the D448N mutant complex with acarbose, the D336A mutant complex with M1, the D336A mutant with M2, the ring deletion mutant ∆loop A with glucose, ∆loop B and ∆loop A/B. Structural analyses, mutagenesis, and computational studies reveal that D448 act as nucleophilic reagent, D336 act as stabilizer, and E373/R334 act as proton donors, offering insights into glycoside hydrolysis diversity. Ultimately, our exhaustive investigation, incorporating structural analysis, biochemical assays, and computational modeling, has illuminated that the hydrolysis mechanism of acarbose by Apg proceeds through a sequential glucose cleavage process, as opposed to the previously documented direct one-step conversion of acarbose into M2 and maltose (Fig. 6).

Acarbose is first hydrolyzed to +1 subsite (17.1 kcal/mol), then removed to −1 subsite (19.7 kcal/mol), and the second hydrolysis reaction is a rate-determining step. Three key crystal structures were obtained. The energy barrier of acarbose direct hydrolysis to M2 ( + 1/−1 subsites) is too high.
Our results will provide structural insights for the design and development of diabetes drugs, including lengthening the length of sugar chain that enhance the resistance to Apg hydrolysis or selective modification of acarbose moieties to weaken the binding affinity. Further development of acarbose analogs with improved pharmacological activity based on these structural analyses are ongoing.
Methods
Materials
Acarbose was purchased from Sigma Chemical Co. acarviosine-glucose was purchased from biddepharm (Shanghai, China). Isopropyl-beta-D-thiogalactopyranoside (IPTG) were purchased from Macklin (Shanghai, China). All other chemicals and reagents were purchased from Aladdin reagent (Shanghai, China). 2×Phanta Max Master mix (P511-02) was purchased from Vazyme Biotech Co. Ltd. (Nanjing, China).
Site-directed mutagenesis of Apg
Genes encoding Apg sequences (UniProt: WP_049088784.1, GenBank: NZ_CABGKM010000006.1) were synthesized by General Bio (Chuzhou, China) and codon optimized for expression in E. coli cells. The Apg gene was cloned into the pET28a(+) vector, which adds an N-terminal 6×His tag. To express the plasmid, it was transformed into E. coli strain BL21 (DE3). For site-directed mutagenesis (SDM), Phanta Max (Vazyme, Nanjing, China) was used for pcr-based site-directed mutagenesis using plasmid pET28a(+)-Apg as a template. For mutants D336A and D448A, which were cloned into ppSUMO vectors by homologous recombination, respectively, SUMO proteins were fused to the N-terminal end after 6×His tagging. A list of primers used for the construction of plasmids for site-directed mutagenesis studies can be found in Table S3. To remove the template from the PCR product, DpnI (Takara, Beijing, China) was used, and the product was transformed into E. coli DH5α (laboratory product) for sequencing. DNA sequencing was used to confirm identity, including the location of the mutation in the expression plasmid. Next, the plasmid was transformed into E. coli BL21 (DE3) for protein synthesis.
Apg protein expression and purification
All required methods for expression and purification of purified proteins and mutants were performed under the following operating conditions. Specifically, E. coli BL21 (DE3) containing plasmid pET28a (+)-Apg was cultured in LB medium containing 50 mg/L kanamycin at 37 °C with 220 rpm shaking. When the OD600 of cell culture reached 0.8, the protein expression was induced by adding IPTG to a final concentration of 0.2 mM and the cells were cooled to 16 °C. After an 18-h growth period at 16 °C, 220 rpm, cells were harvested by centrifugation at 4000 g, 4 °C for 15 min. All of the following steps were performed at 4 °C or on ice. Cell lysates were resuspended in lysis buffer containing 100 mM Tris-HCl, pH 8.0, 200 mM NaCl, 5% glycerol, and lysed by high-pressure homogenization (Shanghai United Biotechnology, China). The samples were sonicated and centrifuged at 12,000 g for 30 min at 4 °C to remove cellular debris. The resulting supernatant was loaded onto a Ni-NTA agarose column (GE Healthcare) pre-equilibrated with lysis buffer. The column was then washed with buffer (100 mM Tris-HCl, pH 8.0, 200 mM NaCl, 5% glycerol and 50 mM imidazole) to remove nonspecifically bound proteins. His-tagged proteins were eluted with a buffer containing 100 mM Tris (pH 8.0), 200 mM NaCl, 300 mM imidazole, 5% glycerol and further concentrated using an Amicon ultrafiltration unit (Millipore) with a molecular weight cut-off 50 kDa. Subsequently, elution fractions were run on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophores (SDS-PAGE) and used for enzymatic assays directly. For mutants ppSUMO-Apg-D336A, ppSUMO-Apg-D448A, prior to crystallization, in order to remove the His tag, the eluate was incubated with ULP1 protease and dialyzed overnight in 5 L of buffer (25 mM Tris-HCl, 150 mM NaCl, pH 8.0) in buffer. After dialysis, the mixture was loaded onto the Ni-NTA column, and the column was then eluted with equilibration buffer. The enzyme fractions were further purified via gel filtration chromatography using a Superdex 200 10/300 GL column (Shanghai Yonglian Biotechnology, China) and buffer (50 mM TrisHCl, pH 8.0, 100 mM NaCl, 1 mM DTT). The fractions were checked by SDS-PAGE, and the target fractions were concentrated using an ultrafiltration tube (Amicon Ultra-4, molecular weight cut-off 50KD, UFC801024). Finally, protein concentrations were determined using a NanoDrop™ One/OneC Micro UV-vis spectrophotometer (Thermo Fisher, Waltham, USA).
Crystallization of wild-type and mutant Apg
All Apg complexes were prepared by sitting-drop vapor diffusion at 16 °C. Aqueous acarbose solution (3.46 mM) was added to purified Apg and mutant (12 mg/mL, 0.173 mM) at a substrate:protein molar concentration ratio of 20:1. The samples were incubated on ice for 30-60 min and then centrifuged. Apg crystals were incubated in a 1 μL droplet, mixed in a 1:1 ratio with the reservoir solution in a volume of 0.5 μL, and equilibrated with 50 μL of reservoir solution (0.02 M magnesium chloride hexahydrate, 0.1 M HEPES (pH 7.0), and 22% w/v poly(acrylic acid sodium salt) 5100) were equilibrated. Prismatic crystals appeared after 2 days at 16 °C. Mutant D336A was co-crystallized under 0.1 M sodium chloride, 0.1 M HEPES (pH 7.0), and 1.6 M ammonium sulfate to obtain crystals of the ligand-binding complex. Crystals of mutant D448A and the ligand-binding complex were obtained by growth in a buffer containing 2.1 M DL-malic acid (pH 7.0). Crystals of mutant D448N were grown in a buffer containing 0.1 M BICINE, 1.6 M ammonium sulfate. The D448N mutant was co-crystallized at 0.02 M Magnesium chloride hexahydrate, 0.1 M HEPES pH 7.5 and 22% w/v Poly(acrylic acid sodium salt) 5100 to obtain crystals of the ligand binding complex. Crystals of the glucose complex were obtained by co-crystallization of the ∆loop A mutant in a buffer containing 0.1 M HEPES PH = 7.5, 20% w/v Poly(acrylic acid sodium salt)5100. The mutant ∆loop B and ∆loop A/B were crystallized at 0.1 M Citric acid pH 5.0, 5% w/v Polyethylene glycol 6000. Prior to data collection, crystals were cryoprotected in ethylene glycol crystallization solution with the addition of 15% to 20% (v/v), followed by rapid freezing in liquid nitrogen with a nylon ring.
Data collection and structure determination
All X-ray diffraction data were collected at Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China) beamline BL19U1 (λ = 0.979 Å). 360 images of 1° oscillation per frame were collected at a crystal-to-detector distance of 250 mm and an exposure time of 0.2 ~ 0.5 s per image. Initial data were indexing and integration using the XDS software package. Next, merged and scaled using AIMLESS in the CCP4 program suite by using the PHASER program in the CCP4 suite. The crystal structure of Apg was solved by molecular replacement using the maltodextrin glucosidase MaIZ (PDB ID 7VT9) as a search model34. Further model building and iterative refinement was performed using COOT and PHENIX, respectively, to yield the final structure of Apg. In addition, the structures of other Apg mutant complexes were determined by molecular replacement using the atomic coordinates of wild-type Apg as a search model. Atomic models with different complexes were manually adjusted and reconstructed in Coot and improved by iterating with PHENIX. For each structure, the overall quality of the structural model was assessed using MolProbity35 and Procheck36 software. The data collection and refinement statistics for each final model was shown in Supplementary Tables 1. The atomic coordinates and structure factors of all refined models have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB)37. Finally, structures were visualization and graphics were done using the PyMol software program (http://www.pymol.org/)38.
Enzyme activity assays
The activity of the acarbose metabolizing enzyme Apg and its mutants was determined according to the literature7 with minor modifications. Briefly, the general reaction system consisted of a solution containing 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5% (v/v) glycerol, and 3 mM DTT, 1 mM of acarbose, and 1 µM of Apg and its mutants, for a total reaction volume of 200 µL. Incubate at 37 °C for 10 min with shaking every 5 min. Next, the reaction was terminated by heating at 100 °C for 5 min. The reaction mixture was centrifuged at 14,000 g for 12 min at 4°. The supernatant was used to determine the concentration of acarbose by high performance liquid chromatography-Mass Spectrometry (HPLC-MS). Each reaction was repeated three times.
To determine the Km and kcat values of Apg for acarbose and M1, the enzyme was tested at a concentration of 0.00090 μM ([E]) and at different concentrations ([S]) of substrates (acarbose and M1) in the reaction system. The reaction system was consistent with the above. Containing 0.00090 μM purified Apg and different concentrations of acarbose (0.30, 0.44, 0.50, 1.00, 2.00, 3.00, 4.00, 5.00, 6.00, 7.00 µM), different concentrations of M1 (0.50, 0.90, 1.00, 2.00, 3.00, 4.00, 5.00, 6.00, 7.00, 8.00 µM) and incubated at 37 °C for 5 min. The reaction was terminated by heating at 100 °C for 5 min. The reaction mixture was then centrifuged at 14,000 g for 10 min at 4 °C. The supernatant was used to determine acarbose concentration by LC-MS. The initial rate of substrate consumption (V) was obtained by linear regression of the data points (substrate consumption versus time) over the initial reaction time (0–5 min). The observed rate constants (kobs) were calculated according to the equation (kobs = V/[E ]) and then fitted to kobs = kcat × [S]/(Km + [S]) in Graphpad Prism to obtain Km and kcat values7.
To analyze the enzyme activity of Apg and the different mutants, HPLC-MS samples were injected into an Agilent 6470 HPLC-MS system (Agilent Technologies, Palo Alto, USA). The separation was carried out using two-component mobile phases A and B, where the solvent a consisted of water containing 0.1% (v/v) formic acid and the eluent B consisted of acetonitrile containing 0.1% (v/v) formic acid. The flow rate was 0.3 mL/min (T = 0 min, 50% B; T = 3 min, 95% B; T = 12 min, 95% B; T = 12.1 min, 50% B; and T = 14 min, 50% B). For the detection of maltose, an isocratic elution of 70% B was used for 14 min. The column temperature was maintained at 30 °C and the autosampler temperature was set at 4 °C. The mass spectrometer was operated in positive mode using electrospray ionization (ESI), while data acquisition was performed using the the multiple reaction monitoring (MRM) mode. MassHunter workstation software version 10.1 (Agilent) was used for data acquisition and post-run processing.
Isothermal Titration Calorimetry (ITC) assays
Isothermal titration calorimetry experiments were performed at 25 °C using a MicroCal VP-ITC apparatus (Malvern Panalytical). Protein and ligand solutions were prepared in dialysis buffer containing 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl. For binding experiments, 20 μM Apg and Apg(D448N) proteins were titrated to saturation with 200 μM of different ligand solutions. For the titration, the sample cells were stirred at 750 rpm and the syringe was filled with ligand injected 19 times every 2.0 μL (without pre-injection of 0.4 μL) at 150 s intervals, with the first injection discarded when the data were analyzed. A control experiment was performed along with each titration to measure the heat of dilution by titrating different ligands into dialysis buffer. The heat of dilution of the ligand after the control titration was subtracted from the experimental titration data. Data were processed using MicroCal VP-ITC analysis software (Malvern Panalytical). Nonlinear regression analysis was performed using a single-point fit model (MicroCal Origin). Results are mean ± standard deviation of repeated experiments.
Molecular dynamics simulation
The initial structures for the simulations were taken from crystallographic structures of the complexes of our experiment. The crystal structure of the nucleophile mutant Apg_D448A in complex with acarbose (PDB: 9IVZ) was used to build the Michaelis complex. To revert the D448A mutation, the side chain atom coordinates of the D448 residue were taken from the resolved apo-Apg crystal structure (PDB: 9IXW). Similarly, the crystal structure of the nucleophile mutant Apg_D336A in complex with M1 (PDB: 9IZE) was used to build another Michaelis complex. To revert the D336A mutation, the side chain atom coordinates of the D336 were taken from the apo-Apg structure (PDB: 9IXW). For computational modeling where the crystal structure of the complex is unavailable—such as Apg (PDB: 9IXW) with acarstatin A/B, or the Apg complexes involving ∆loopA (PDB: 9VD8), ∆loopB (PDB: 9VDG), or ∆loopAB(PDB: 9VDW) with acarbose—structural alignment was employed to constrain molecular conformations by superimposing the corresponding glycosyl units (PDB: 9IVZ). Subsequently, molecular docking was performed using AutoDock Vina39, based on the Apg_D448A–acarbose complex structure (PDB: 9IVZ).
In the MD simulations, hydrogen atoms, solvation box, and counter ions necessary to neutralize the system’s charge were added with the AmberTools2240 utility tLeaP. The following force fields (FF) were used: ff14SB41 (enzyme), and TIP3P42 (water solvent). GLYCAM06j-143 and GAFF244 atom types and parameters were employed for the sugar moieties (−1/ + 1 glycosyl unit) and the inositol group (−3/−2 glycosyl unit). This combination is particularly advantageous for hybrid systems such as acarbose, which contains both saccharide and non-saccharide components. Additionally, the AmberTools40 supports flexible parameter fitting, and RESP charge derivation45 with VMD plugin Force Field Toolkit46 and ORCA 5.0.447, for system preparation and analysis.
Molecular dynamics (MD) simulations were performed with the Gromacs 2023.348. To perform molecular dynamics simulations with energy minimization, NVT and NPT equilibration, and production runs were performed. The energy minimization was carried out using the steepest descent algorithm until the maximum force dropped below 100 kJ/mol/nm, with a step size of 0.01 nm. Periodic boundary conditions (PBC) were applied in all directions. Each simulation sets a different random number seed. NVT equilibration for 100 ps with position restraints on heavy atoms. Temperature was maintained at 300 K using the V-rescale thermostat49 with a time constant of 0.1 ps. NPT equilibration for 100 ps with pressure controlled at 1 bar using the Parrinello-Rahman barostat50 with a coupling constant of 2.0 ps and isotropic pressure coupling. The production MD simulation was run for 200 ns with a time step of 2 fs using the Verlet integrator51. All bond lengths involving hydrogen atoms were constrained using the LINCS algorithm52, allowing the use of a 2 fs timestep. Electrostatics were handled using the Particle Mesh Ewald (PME) method53 with a real-space cutoff of 1.0 nm. Van der Waals interactions were truncated at 1.0 nm, and dispersion corrections were applied to the energy and pressure to account for the cutoff. A neighbor list with a cutoff of 1.0 nm was updated every 10 steps. The all production simulations were further continued for a total of 200 ns with 3 times. The 200 ns duration was selected based on refs. 23,28,54 of similar systems, where key conformational states and energetic stabilization are typically observed within this timescale. The production MD trajectories were analyzed with in-house VMD 1.9.455 and Python scripts. The CAVITY Plus server was used to identify the pockets56.
DFT method
Density functional theory (DFT) calculations were performed at the B3LYP-D3BJ level using the Orca 5.0.447. Geometries were fully optimized in conjunction with the CPCM continuum solvation model at the B3LYP/def2-SVP level57 of theory. To get Gibbs free energies, harmonic frequency calculations were performed to get the thermal and entropic corrections at 300 K. Intermediates and transition states were confirmed by frequency calculations to have zero and only one imaginary vibration, respectively. The electronic energies were further corrected with the def2-TZVP basis set. The Gibbs free energy correction at B3LYP/def2-SVP level and D3 dispersion correction was included in single-point energy calculations.
QM/MM method
QM/MM simulations were used to describe the acarbose hydrolysis reaction in the active site of the Apg. The simulations were started with a suitable snapshot from the classical MD simulations. The QM region was described using the B3LYP-D3BJ functional58 with the def2-TZVP57 basis set. Inclusion of dispersion corrections improves the calculation of reaction barriers in enzymes at a negligible computational cost. QM/MM calculations were also carried out with NAMD2.1459 for MM in combination with ORCA 5.0.447 for QM calculations. All QM/MM simulations employed the charge-shift method to handle link atoms. Additionally, a shift function was applied to the surrounding classical partial charges within the electrostatic embedding scheme.
To investigate the free energy profile (also as potential of mean force, PMF) for the hydrolysis reaction, we defined collective variables, such as interatomic distances and distances between the centers of mass of atom groups. These collective variables (Colvars)60 were utilized to monitor changes in the system throughout the transformation and served as a basis for defining the reaction coordinate. For example, in acarbose hydrolysis QM/MM simulation, the QM region includes the acarbose, the catalytic triad D336/E373/D448, and the catalytic water molecule. Fours collective variables (CVs) were defined for driving the system from the initial state (reactants, ES) to the reaction intermediate (the glycosyl-enzyme covalent intermediate, IC1). The first CV, C1Glc2-O4Glc1 (CV1) for the cleavage of the O-glycosidic bond. This CV1 accounts for the breaking of the glycosidic bond. The CV2, distance between C1Glc2 and OD2D448, for the nucleophilic attack of D448 on the anomeric carbon of acarbose. The CV3, was selected as the distance O4Glc1-HE2E373, thus describing the proton transfer from the general acid E373 to the glycosidic oxygen O4. And the CV4 is HE2E373-OE2E373 for O–H bond cleavage (Supplementary Fig. 31A). Next, the deglycosylation reaction, the second step of the hydrolysis mechanism of Apg with acarbose, was modeled starting from a trajectory snapshot of the QM/MM MD simulation of glycosylation reaction. The string method also used 4 CVs to drive the deglycosylation reaction (Supplementary Fig. 31B). For comparative QM/MM calculations in other system (one-step hydrolysis, hydrolysis reaction of Apg with M2, hydrolysis reaction of Apg with acarstain A/B, hydrolysis reaction which D336 as nucleophilic reagent), only the energy barrier of the activation step (glycosylation) is computed to compare the relative difficulty of reactions. The details can be found in Supporting Information.
To compute the PMF, we employed the string method and the extended-system adaptive biasing force (eABF) method61, which are implemented within the colvars module of NAMD 2.14. The string method is an iterative approach that optimizes the reaction coordinate to identify the least-resistance pathway from the initial to the final state of the system. Once optimized, the string provides images that delineate a continuous path for the transformation, and eABF is utilized to calculate free energy changes. Initial configurations for each eABF window were generated using the qmCSMD module in NAMD 2.14. The PMF and the associated gradient, derived using the corrected z-averaged restraint (CZAR) estimator62, were output by the colvars module from the eABF data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The structure factors and coordinates of WT-Apg, the D448A mutant, the D448N mutant, the D448A mutant complex with acarbose, the D448N mutant complex with acarbose, the D336A mutant complex with M1, the D336A mutant with M2, the ring deletion mutant ∆loop A with glucose, ∆loop B and ∆loop A/B have been deposited in the Protein Data Bank under the accession numbers 9IXW, 9IXH, 9V0I, 9IVZ, 9UNS, 9IZE, 9IZO, 9VD8, 9VDG and 9VDW, respectively. The coordinate and system setup files of MD simulations and QM/MM simulations generated in this study are deposited in Zenodo [https://doi.org/10.5281/zenodo.15922738]. Source data are provided with this paper.
References
Chiasson, J.-L. et al. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet 359, 2072–2077 (2002).
Kumar, R., Kerins, D. M. & Walther, T. Cardiovascular safety of anti-diabetic drugs. Eur. Heart J. Cardiovasc. Pharmacother. 2, 32–43 (2016).
Sharma, P., Grewal, A. S., Sharma, N. & Singh, S. Role of glucokinase enzyme in the treatment of type 2 diabetes. ECS Trans. 107, 9065–9072 (2022).
Hollander, P. Safety profile of acarbose, an α-glucosidase inhibitor. Drugs 44, 47–53 (1992).
Breuer, H.-W. M. Review of acarbose therapeutic strategies in the long-term treatment and in the prevention of type 2 diabetes. Int. J. Clin. Pharmacol. Ther. 41, 421–440 (2003).
Wang, K. et al. Microbial-host-isozyme analyses reveal microbial DPP4 as a potential antidiabetic target. Science 381, eadd5787 (2023).
Tian, J. et al. Inactivation of the antidiabetic drug acarbose by human intestinal microbial-mediated degradation. Nat. Metab. 5, 896–909 (2023).
Ben Bdira, F. et al. Dynamics of ligand binding to a rigid glycosidase. Angew. Chem. Int. Ed. 59, 20508–20514 (2020).
Thompson, A. J. et al. Evidence for a boat conformation at the transition state of GH76 α-1,6-mannanases—key enzymes in bacterial and fungal mannoprotein metabolism. Angew. Chem. Int. Ed. 54, 5378–5382 (2015).
Xie, J. et al. Structural analysis of the catalytic mechanism and substrate specificity of anabaena alkaline invertase InvA reveals a novel glucosidase *. J. Biol. Chem. 291, 25667–25677 (2016).
Morais, M. A. B., Nin-Hill, A. & Rovira, C. Glycosidase mechanisms: sugar conformations and reactivity in endo- and exo-acting enzymes. Curr. Opin. Chem. Biol. 74, 102282 (2023).
Gao, D. et al. Increased enzyme binding to substrate is not necessary for more efficient cellulose hydrolysis. Proc. Natl. Acad. Sci. USA 110, 10922–10927 (2013).
Zechel, D. L. & Withers, S. G. Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res. 33, 11–18 (2000).
Nasseri, S. A. et al. An alternative broad-specificity pathway for glycan breakdown in bacteria. Nature 631, 199–206 (2024).
Fujimoto, Z. et al. Crystal structure of a catalytic-site mutant α-amylase from Bacillus subtilis complexed with maltopentaose 1. J. Mol. Biol. 277, 393–407 (1998).
Nag, M., Lahiri, D., Garai, S., Mukherjee, D. & Ray, R. R. Regulation of β-amylase synthesis: a brief overview. Mol. Biol. Rep. 48, 6503–6511 (2021).
van der Maarel, M. J. E. C., van der Veen, B., Uitdehaag, J. C. M., Leemhuis, H. & Dijkhuizen, L. Properties and applications of starch-converting enzymes of the α-amylase family. J. Biotechnol. 94, 137–155 (2002).
Danialifar, T. F., Chumpitazi, B. P., Mehta, D. I. & Di Lorenzo, C. Genetic and acquired sucrase-isomaltase deficiency: a clinical review. J. Pediatr. Gastroenterol. Nutr. 78, 774–782 (2024).
Barber, E., Houghton, M. J., Visvanathan, R. & Williamson, G. Measuring key human carbohydrate digestive enzyme activities using high-performance anion-exchange chromatography with pulsed amperometric detection. Nat. Protoc. 17, 2882–2919 (2022).
Zhang, X., Huang, Q., Guo, Z., Cai, F. & Kang, Q. Acarbose glycosylation by AcbE for the production of acarstatins with enhanced α-amylase inhibitory activity. Synth. Syst. Biotechnol. 9, 359–368 (2024).
Ahn, W.-C. et al. Dimeric architecture of maltodextrin glucosidase (MalZ) provides insights into the substrate recognition and hydrolysis mechanism. Biochem. Biophys. Res. Commun. 586, 49–54 (2022).
Ohtaki, A. et al. Mutual conversion of substrate specificities of Thermoactinomyces vulgaris R-47 α-amylases TVAI and TVAII by site-directed mutagenesis. Carbohydr. Res. 338, 1553–1558 (2003).
Neves, R. P. P., Fernandes, P. A. & Ramos, M. J. Role of enzyme and active site conformational dynamics in the catalysis by α-amylase explored with QM/MM molecular dynamics. J. Chem. Inf. Model. 62, 3638–3650 (2022).
An, Y. et al. The distinctive permutated domain structure of periplasmic α-amylase (MalS) from glycoside hydrolase family 13 subfamily 19. Molecules 28, 3972 (2023).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Kim, J.-S. et al. Crystal structure of a maltogenic amylase provides insights into a catalytic versatility. J. Biol. Chem. 274, 26279–26286 (1999).
Kamitori, S. et al. Crystal structures and structural comparison of Thermoactinomyces vulgaris R-47 α-Amylase 1 (TVAI) at 1.6 Å Resolution and α-Amylase 2 (TVAII) at 2.3 Å resolution. J. Mol. Biol. 318, 443–453 (2002).
Pengthaisong, S., Piniello, B., Davies, G. J., Rovira, C. & Ketudat Cairns, J. R. Reaction mechanism of glycoside hydrolase family 116 utilizes perpendicular protonation. ACS Catal. 13, 5850–5863 (2023).
Wallace, M. D. et al. Constrained catalytic itinerary of a retaining 3,6-anhydro-D-galactosidase, a key enzyme in red algal cell wall degradation. Angew. Chem. Int. Ed. 63, e202411171 (2024).
Holm, L., Laiho, A., Törönen, P. & Salgado, M. DALI shines a light on remote homologs: one hundred discoveries. Protein Sci. 32, e4519 (2023).
Wangpaiboon, K. et al. A GH13 α-glucosidase from Weissella cibaria uncommonly acts on short-chain maltooligosaccharides. Acta Cryst. D. 77, 1064–1076 (2021).
Kim, T.-J. et al. Modes of action of acarbose hydrolysis and transglycosylation catalyzed by a thermostable maltogenic amylase, the gene for which was cloned from a ThermusStrain. Appl. Environ. Microbiol. 65, 1644–1651 (1999).
Yoshioka, Y., Hasegawa, K., Matsuura, Y., Katsube, Y. & Kubota, M. Crystal structures of a mutant maltotetraose-forming exo-amylase cocrystallized with maltopentaose1. J. Mol. Biol. 271, 619–628 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Williams, C. J. et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993).
Michalska, K. & Joachimiak, A. Structural genomics and the protein data bank. J. Biol. Chem. 296, 100747 (2021).
Yuan, S., Chan, H. C. S. & Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 7, e1298 (2017).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Case, D. A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183–6191 (2023).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Kiss, P. T. & Baranyai, A. Sources of the deficiencies in the popular SPC/E and TIP3P models of water. J. Chem. Phys. 134, 054106 (2011).
Kirschner, K. N. et al. GLYCAM06: a generalizable biomolecular force field. Carbohydr. J. Comput. Chem. 29, 622–655 (2008).
He, X., Man, V. H., Yang, W., Lee, T.-S. & Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 114502 (2020).
Cornell, W. D., Cieplak, P., Bayly, C. I. & Kollman, P. A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 115, 9620–9631 (1993).
Zeng, Y. et al. Broadening access to small-molecule parameterization with the force field toolkit. J. Chem. Phys. 160, 242501 (2024).
Neese, F. Software update: the ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Páll, S. et al. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 153, 134110 (2020).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Verlet, L. Computer ‘Experiments’ on Classical Fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 159, 98–103 (1967).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Morais, M. A. B. et al. Two distinct catalytic pathways for GH43 xylanolytic enzymes unveiled by X-ray and QM/MM simulations. Nat. Commun. 12, 367 (2021).
Spivak, M. et al. VMD as a platform for interactive small molecule preparation and visualization in quantum and classical simulations. J. Chem. Inf. Model. 63, 4664–4678 (2023).
Xu, Y. et al. CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 46, W374–W379 (2018).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Hénin, J., Lopes, L. J. S. & Fiorin, G. Human learning for molecular simulations: the collective variables dashboard in VMD. J. Chem. Theory Comput. 18, 1945–1956 (2022).
Melo, M. C. R. et al. NAMD goes quantum: an integrative suite for hybrid simulations. Nat. Methods 15, 351–354 (2018).
Lesage, A., Lelièvre, T., Stoltz, G. & Hénin, J. Smoothed biasing forces yield unbiased free energies with the extended-system adaptive biasing force method. J. Phys. Chem. B 121, 3676–3685 (2017).
Acknowledgements
This work was supported by grants from Guangdong S&T Program (2024B1111160007 to J.-H.Z.), National Natural Science Foundation of China (22201295 to Y.G., 22207119 to L.W.), Shenzhen Science and Technology Program (JCYJ20220818100804010 to Y.G.). We thank the staff of the beamline BL19U1 of Shanghai Synchrotron Radiation Facility for help in X-ray data collection.
Author information
Authors and Affiliations
Contributions
J.H., Y.G., J.-H.Z. and J.-W.Z. conceived and designed the project. J.H. designed the experiments, conducted the experiments, and wrote the manuscript. Z.S. performed the computational analysis of the protein crystal data and contributed to the writing of the paper. J.H. and X.X. contributed to plasmid construction, protein expression, and purification. J.H. performed the crystallization experiments, and J.H. and L.W. collected crystallographic data and solved the structures. J.H. designed and performed all the mutation and enzymatic assays. All the authors analyzed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, J., Shen, Z., Xiao, X. et al. Molecular insights of acarbose metabolization catalyzed by acarbose-preferred glucosidase. Nat Commun 16, 7839 (2025). https://doi.org/10.1038/s41467-025-62855-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62855-y
