
Abstract
Homeostasis relies on a precise balance of fate choices between renewal and differentiation. Although progress has been done to characterize the dynamics of single-cell fate choices, their underlying mechanistic basis often remains unclear. Concentrating on skin epidermis as a paradigm for multilayered tissues with complex fate choices, we develop a 3D vertex-based model with proliferation in the basal layer, showing that mechanical competition for space naturally gives rise to homeostasis and neutral drift dynamics that are seen experimentally. We then explore the effect of introducing mechanical heterogeneities between cellular subpopulations. We uncover that relatively small tension heterogeneities, reflected by distinct morphological changes in single-cell shapes, can be sufficient to heavily tilt cellular dynamics towards exponential growth. We thus derive a master relationship between cell shape and long-term clonal dynamics, which we validated during basal cell carcinoma initiation in mouse epidermis. Altogether, we propose a theoretical framework to link mechanical forces, quantitative cellular morphologies and cellular fate outcomes in complex tissues.
Similar content being viewed by others

Introduction
Epithelial tissues have a central barrier function in multicellular organisms and undergo extensive turnover during homeostasis. The skin epithelium, the largest organ in our body, renews itself completely within four weeks for example1,2. This crucial process of homeostatic self-renewal relies on the basal proliferative layer of the interfollicular epidermis (IFE). This layer is composed of stem cells (SCs) and progenitors, which divide and give rise to differentiated suprabasal cells, showing balanced cell fate during homeostasis and unbalanced behaviors that can lead to wound healing or cancer initiation3,4,5,6,7,8,9,10,11,12. Although lineage tracing datasets have proved instrumental over the last 15 years in defining the potential, hierarchy and modes of divisions of different cellular populations, key open questions remain as to the mechanisms underlying fate choices. For instance, in the context of the homeostatic skin epithelium which follows long-term neutral drift due to stochastic basal cell renewal, what are the mechanisms which determine whether a given basal cell division will give rise to two basal (or suprabasal) cells, i.e., a symmetric division, or one basal and one suprabasal cell, i.e., an asymmetric division? Furthermore, at homeostasis, these processes have to be balanced at the population level to ensure a conserved cell number, and this balance is lost in a predictable manner in processes such as wound healing9,10,13 or basal cell carcinoma (BCC) initiation11,12. Based on previous works, determinants of cell fate decisions can be grouped into two generic categories—(a) cell-intrinsic, i.e., the underlying gene regulatory networks14 and (b) cell-extrinsic factors, i.e., local niche-specific biochemical cues15,16 or biochemical interactions between different SCs17,18. However, from a theoretical perspective, all of these types of models give rise to very similar long-term behavior. For instance, both stochastic zero-dimensional models (where cellular decisions are cell-intrinsic and do not consider space) or stochastic Voter models on spatial grids (where a cell can differentiate/move suprabasally upon the symmetric division of neighboring cells) give rise to identical clone size distribution and long-term patterns of clonal growth5,8,10,18,19,20.
While the predictive power of such minimal models has been impressive, the next emerging question is thus to understand more mechanistically how the fate decision probabilities that underlie these stochastic models are encoded. In spermatogenesis, it has been recently shown that competition for diffusible fate determinants is a simple homeostatic mechanism able to collectively balance cellular fate decisions, which can explain a number of homeostatic and perturbed conditions21. Furthermore, the impact of cellular forces and mechanics has been increasingly recognized over the last decade. Since the classical observation that substrate rigidity can modulate single cell fate decisions in vitro22, a number of emerging evidence also suggest an impact for cell and substrate mechanics in tissues10,23,24,25,26. However, the impact of mechanical forces on 3D cellular fate choices remain poorly explored from a theoretical perspective27, which we tackle in this work.
Over the past decade, extensive efforts in the field of mechanobiology have been devoted to developing minimal theoretical descriptions of 2D and 3D tissue mechanics. One of the simplest and most successful description of confluent tissues has been the vertex and Voronoi models, where cells are represented by polygons with tensions associated with their shared cell–cell contacts28,29,30,31,32,33,34. This is based on the fact that most cellular forces are concentrated at interfaces, from the cell cortex and adhesion machinery28,35. The equilibrium shape of cells and tissues can then be derived by minimizing the total mechanical energy of the system, which can predict a number of complex collective behavior, such as density-independent unjamming transition, novel mechanisms for cell sorting or shear-thickening33,34,36,37,38,39,40,41,42 many of which have been observed in biological tissues43,44,45,46,47. However, these models have traditionally been confined to describing tissues in the absence of cellular renewal or fate choices, e.g., tissues made of a single cell type, or of two independent populations undergoing sorting. To develop a unified theoretical description taking into account both tissue mechanics and cellular fate choices, we choose to concentrate on the skin IFE, which comprises of several confluent layers of cells in homeostasis. Only cells localized to the bottom, or basal, layer (in contact with the basement membrane (BM)) can divide in this system, while suprabasal cells are lost at a constant rate, creating a steady flow of cells upwards. As a first approximation and based on the “open-niche” nature of the skin epithelium8, we assume that all basal cells are identical.
We first show that a “balanced” mode of proliferation emerges organically from simply allowing the SCs to make mechanically favorable fate choices i.e., in absence of any spacial bias or pre-assignment of fates. We then test how fate choices can be modulated by inhomogeneities in the mechanical properties of some cells, in particular, differences in basal adhesion to the BM. We find that clones with lower basal tension undergo a distinct change in their proliferation mode, due to the fact that the remaining basal after division becomes mechanically favorable. Interestingly, fate outcomes are highly sensitive to mechanical inhomogeneities, with a 10% decrease in tensions being sufficient to transform clones into a nearly exponential growth mode. Furthermore, we show that the amount of tension inhomogeneity can be extracted based on cell shape alone, providing a key relationship between cell shape and cell fate outcomes—two experimentally accessible parameters. We test these in silico predictions in mouse tail IFE, where oncogenic Hedgehog signaling pathway activation has been shown to drive BCC initiation due to a well-defined tilt of fate outcomes towards symmetric renewal11. We find a small but significant change in cellular shape upon oncogenic activation, which can predict the range of clonal fate imbalance observed in the data.
Model
We start by using the vertex model, a classical framework to study two and three-dimensional tissues. The mechanical energy of the tissue is a sum of its contributions from the surface (BM) and the bulk (individual cells). Each cell is associated with an energy, written as34
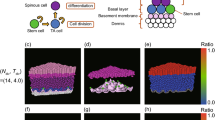
where S0 is the preferred surface area of each cell, V0 its preferred volume, and KS and KV the compressibilities associated to how strong these regulations towards these preferred areas and volumes are. The preferred cell shape index, defined as \({s}_{0}={S}_{0}/{V}_{0}^{2/3}\), is a simplest representation of the different types of forces acting at the interface of different cells, and has been shown to be a simple predictor of the collective physical state of the tissue, with s0 > 5.41 giving rise to fluid tissues (where cell-cell re-arrangements cost vanishing energy) and s0 < 5.41 to solid tissues (where energy barriers associated to cell-cell re-arrangements result in tissues able to sustain mechanical load). Here we use a solid-like value of s0 = 5.30 for all the cells, and KV = 140. Preferred volume is set as V0 = 1, thereby setting the length scale of the simulation. We build an in silico epithelial tissue composed of three layers of cells, as shown in Fig. 1a–c, where each cell is a polyhedral unit created using the Voronoi tessellation of all the cell centers—which are the degrees of freedom of this system41. Random noise is added at each time point to the cell centers, as sketched in Fig. 1a to explore the effect of random motility and/or mechanical fluctuations in the system, which has been shown to be an alternative route to tissue fluidization36,48,49,50 compared to changes in shape index.

a Schematic of our model for multilayered tissues on top of a basement membrane (BM) shown in black. Each cell has a preferred cell volume V0(t) and cell shape s0. The color scheme denotes the distance of the cell from the BM -- the closest to the furthest cells are colored from blue to red. Noise is added to the cell center (blue sphere). Cell-BM contacts have an additional interfacial tension Ti, rendering the interface relatively flat. b Cell division event is depicted first in the the basal cross-sectional view and below in 3D space: the randomly selected cell (in blue) divides into two daughter cells, that gradually grow back to the original volume. Cell extrusion event occur organically, defined as a basal cell (in blue) being pushed out towards the suprabasal layer (in red), i.e., shrinking its basal area to zero. c Tissue is shown in its steady state (after one complete turnover time of τD). The tissue dimensions are 8 × 8 × 3 (cell units) and contains a total of 192 cells that are divided into basal layer (blue) and the supra-basal layers above (red + orange). Boundary conditions are periodic along the horizontal x and y axes.
Next, we sought to model the interface between the epithelium and the basement membrane. To do this in a minimal manner, we assume a tension Tj at the basal-BM interface. As shown in Fig. S1, we use values of interface tension large enough that the BM interface is relatively flat, but small enough to reduce artificial pinning between basal cells and the medium below (which has been observed in Voronoi models), so that results are largely insensitive to the exact choice of interfacial value (see Section: “Methods” Text for more details and sensitivity analysis)38,41.
Finally, in IFE, as in other stratified tissues, the bottom-most cellular layer (composed of basal cells) adheres to a rigid BM, as sketched in Fig. 1a, and can proliferate, whereas layers above (suprabasal cells) cannot divide and are progressively shed as they undergo differentiation. Thus, in simulations, we allow basal cells i.e., any cell sharing a contact with the BM, to grow in size and divide at a random rate kD (see Section: “Methods” text for more details, and Supplementary Video 1). The growth of the mother cell is implemented by gradually increasing the preferred volume V0. After this occurs, we select a random plane of division (perpendicular to the BM, shown in Fig. 1b) and add another cellular center. To conserve the total number of cells, at each division, we also randomly pick a suprabasal cell for deletion. This means that we do not impose any cellular fate balance in basal cells, and each cell delamination, i.e., the transition from the basal to the suprabasal compartment, occurs in a spontaneous manner as a result of force balance and mechanical competition for space, see Fig. 1b and Supplementary Video 2.
In silico tissues follow balanced neutral-drift dynamics
Importantly, we find that simulating this minimal system organically displays a spatial and dynamical organization close to their in vivo counterpart. Firstly, the basal layer displays apico-basal oriented prismatic shapes whereas suprabasal cells are more isotropic (as visible in Fig. 1c and quantified in Fig. S1a, b), which comes from the constraint with the basement membrane (similar to previous observation at the boundary of two different tissues41). However, because of low tension values, there is negligible crowding due to basal tension (details in Fig. S1d). Secondly, the system organizes itself into a state of permanent flux from the basal to the suprabasal layer. This is because cells staying in the basal layer after division would result in crowding, which is energetically unfavorable compared to moving to suprabasal layers. However, basal proliferation still results in an increased basal density, as quantified in Fig. S1d. Thirdly, given that all basal cells are modeled as identical, the ones mechanically poised to extrude towards the suprabasal layers is a random process, resulting in neutral drift dynamics within the epithelium. To visualize this and measure it more quantitatively, we performed computational lineage tracing in our simulations, assigning to each cell a given ID that is retained upon division. Given the difficulty of long-term live imaging in this system, lineage tracing has been a key experimental method to assess cellular fate choices, by irreversibly labeling sparse populations of cells as well as all their future progeny (clones), and measuring clone size at given time points post labeling11.
In accordance with previous analytical results, we found that as cells divided and extruded from the basal layer, the number of surviving clones declined at 1/t as the average surviving clone size grew linearly (Fig. 2a–c and Supplementary Video 3). Reconstructing lineages was consistent with this picture, with around 25% of basal cell divisions resulting in two basal cells, 50% in one basal and one suprabasal cells and 25% in two suprabasal cells. These are the results one would expect if, following a division, the two daughter cells have an uncorrelated 50–50 probability to extrude suprabasally. Interestingly, this is quite close to values observed in short-term live imaging in mouse ear epidermis51.

Clonal growth dynamics of a representative clone is depicted in 3D (a) and in the 2D basal plane (b). Cells that belong to the same clone are tagged in red. The same clone is shown in the 2D basal cross-section (b), where in addition to the initial and final frames, one-forth, middle, and three-fourth frames are also shown. c The average number of basal cells is plotted with respect to time (t) in units of turnover/division timescale τD. When averaged over all the clones, the size remains almost constant at unity-- only to be expected as the total number of cells is conserved. Averaging over the surviving clones, it shows an initial transience, followed by linear growth-- as seen in neutral-drift processes. d The ratio of surviving clones decreases inversely with time, in line with the above quantification. Parameter set: total number of cells = 192, s0 = 5.3, T = 1.0 and zero noise.
Next, we tested if these results were sensitive to the mechanical state of the tissue, by varying the shape index of cells (i.e., simulating fluid vs solid tissues), the basal tension, as well as the amount of noise in the simulations. Interestingly, we found that the results were largely unaffected in terms of cellular fate dynamics and clone size, as shown in Fig. S1d–j, at least in the range of parameter values where we avoid artificial pinning effects to the BM from the Voronoi model. However, the mechanical state of the tissue did have an impact on geometrical features of clones: for both solid and fluid tissues with high noise, the clonal footprints were more elongated and fragmented (see Fig. S1g, h for solid and fluid systems, respectively). Such features could be readily compared to experimental lineage-tracing datasets, which have tended to historically concentrate on overall clonal sizes rather than geometric cellular arrangements. At homeostasis, clones in skin epidermis tend to be highly compact and cohesive11, which would support a solid state for the tissue. Furthermore, we sought to test whether the mechanical state of the tissue could change its response to dynamical perturbations. For this, we abruptly stopped any proliferation in the simulation at a given time point. Interestingly, in the solid regime, this completely stopped any extrusion to the suprabasal layer (Fig. S4e), even though the basal density is higher than what it would have been without any divisions in the first place (see “Methods”). We reasoned that this could be due to the fact that, despite extrusion being energetically favorable, there are energy barriers preventing it in the solid regime. To test this, we performed the same simulations in the fluid regime and confirmed that some extrusions could continue after proliferative arrest (Fig. S4f). This suggests that the mechanical state of stress could be an important variable when considering the coupling of division and suprabasal extrusion, in particular outside of homeostasis23,52.
Driving unbalanced fate choices by differential cellular mechanics
Next, we sought to relax the assumption of identical basal cells to understand processes such as tumor initiation. Lineage tracing combined with targeted oncogenic activation has made it possible to study the long-term clonal dynamics leading to tumor growth in an in vivo context. For instance, Hedgehog signaling pathway activation in single cells results in formation of preneoplastic lesions (dysplasia) or invasive BCC depending on the cell of origin (progenitor or SC)11. In previous works, we had shown that the early stages of tumor formation could be well-recapitulated with a stochastic model of cell fate reminiscent of homeostasis, but with a constant imbalance towards symmetric basal divisions11. Surprisingly, the main driver of tumor formation was therefore not enhanced basal proliferation, but instead by decreased differentiation. Recent studies have also investigated the role of the mechanics of the BM and stroma on later stages of tumor invasion26. However, these previous works leave open the key question of what sets the imbalance of cellular fates in tumors. Driven by previous works showing the importance of mechanical forces for determining the mode of cell fate choices10,23,24,46,53,54, we sought to test whether these imbalances could have a mechanical origin.
Given the simplicity of our model, there are only a few mechanical parameters that can be different in tumor cells. As described above, enhanced proliferation is not correlated experimentally to unbalanced fate11, so we investigated the effect of differences in mechanical tensions. In our model, the choice to differentiate is based on a mechanical competition for finite access to the basal layer, and we therefore reasoned that differences in basal adhesion/tension would have a profound impact on cellular fate choices and clonal dynamics.
We therefore simulated a system where a few mutant (oncogene-expressing) cells and their progeny were endowed with decreased tension to the BM by a ratio R = TB/TT > 1, where TB and TT are the base-line wild-type (WT) and mutant basal tensions respectively as shown in Fig. 3a. Mechanistically, this could be due either to decreased basal cytoskeletal contractility or increased adhesion to the BM in tumor cells, which are equivalent from a modeling perspective, as both result in larger preferred basal areas compared to WT cells55. Strikingly, very small differences in basal tensions were sufficient to drive profound changes in clonal behaviors. Indeed, cellular fate choices became heavily biased towards symmetric divisions, with much larger basal clone sizes (Fig. 3b and Supplementary Videos 4, 5). This can be quantified in terms of overall clonal growth dynamics, which became exponential rather than the linear trend observed for homeostasis, with a mere 10% decrease in basal tension (Fig. 3c) being sufficient to give rise to nearly exclusive symmetric divisions. Moreover, by investigating different values of basal tension differences, we found that the effect was progressive, with a monotonic increase in average basal clone size with tension ratio R (Fig. 3d).

a Schematic for introducing tension disparity is shown, wherein normal cells in blue have basal tension TB, and mutant cells in pink (and their progenies) have a lowered basal tension i.e., TT < TB, making the ratio R = TB/TT ≥ 1. Inter-cellular/bulk tensions are represented as T0, and are left unchanged. b Final 3D morphology is shown for a single clone (red cells) with either normal properties (R = 1, left) and a mutant clone with decreased basal tension (R = 1.15, right). c 2D basal cross-section of the same mutant clone, where initial, middle and final frames are shown, depicting a much faster growth as compared to Fig. 2b. d Number of basal cells, averaged over surviving clones and plotted with respect to rescaled time (t/τD), undergoes a transition from linear for R = 1 to exponential for R = 1.1 (lighter to darker shade of pink). e The final basal size, plotted against tension changes R, shows a monotonically increasing trend. Error bars mean ± SEM (at least 60 simulations per value of R). Parameter set: Total number of cells = 192, s0 = 5.3, base-line/WT tension value TB = 1.0 and medium noise(v0 = 0.1).
Again, we tested how these results were affected by the mechanical state of the tissue (Fig. S2a, b), the degree of noise (Fig. S3) and the baseline WT value of the basal tension, finding that the changes in clonal dynamics with different tension ratio R are robust (Fig. S2c). In particular, the number of basal cells remains largely unchanged by these model variations and is mainly affected by the basal tension ratio R, although, as discussed in the homeostatic case, the spatial shape of clones becomes more dispersed with increasing noise, as shown in Fig. S3. We also observe that the basal growth dynamics depend mainly on the tension ratio R (rather than the absolute difference between both tensions, Fig. S2c). Overall, these simulations suggest a key role for basal tension in favoring the adhesion of basal cells to the BM in tumors, a wetting-like phenomena which favors them to stay basal after divisions and thus providing a competitive advantage over surrounding WT cells.
To describe more quantitatively how individual fate choices are affected by tension differences between tumor and WT cells, we plotted individual clonal lineages to extract the probability of all three fate outcomes (basal-basal, basal-suprabasal, suprabasal), defining population asymmetry-Δ, as the imbalance between symmetric division and symmetric differentiation Fig. 4a–c. We found that the asymmetric fate outcome starts from 50% and slowly declines with increasing R. This population is compensated by an increase in symmetric divisions. While both kinds of symmetric fates are around 25% in the homeostatic case (R = 1), they gradually deviate such that by R = 1.1, the symmetric differentiation plummets to less than 5% and the symmetric divisions increase to over 70% as shown in Fig. 4d increasing the population asymmetry Δ with respect to tension ratio R(Fig. S2d).

a Schematic shows the probability distribution of homeostatic and tumor fate choices, where Basal and supra-basal cells are depicted in magenta and blue respectively. In balanced mode, the distribution between all three fates (basal-basal, basal-suprabasal, suprabasal) is symmetric, with r denoting the basal cell doubling. In tumors, the symmetric fates get imbalanced by Δ. b, c Representative examples of lineage trees are shown for tension ratios--R = 1.0(above) and R = 1.1 (below). Being a basal cell, magenta cells have the power to divide, whereas blue cells typically lose it upon extrusion. All the trees are to scale with time, shown in units of t/τD on the right. Magenta cells at the end of a tree have an unknown fate as they survive in the basal layer till the simulation ends. The division time corresponds to the appearance of daughter cells, and not the mother cell. d Proportion of all three fates-- basal-basal in pink, basal-suprabasal in gray, suprabasal in blue, of all cell divisions that took place within the first turnover i.e., time ≤ 1, is plotted with respect R. The distribution is initially symmetric with values-- 25%, 50%, 25%. Δ becomes the difference between magenta and blue curves, that increases with increasing R because the proportion of suprabasal cells plummet to zero, and is balanced by the ever-increasing ratio of basal-basal fate. The asymmetric fate shows a moderate decline. e Schematic to show the qualitative change in cell shape from prism-like to more pyramidal frustums. Basal area AT is represented in red. f AT is plotted with respect to R. Insets show the representative polyhedral cells that corroborate the increasing trend of AT with their increasingly wider base. Error bars mean ± SEM (at least 60 simulations per value of R). g Connection between fate imbalance and cell-shape changes. Fate choice imbalance Δ is plotted with respect to basal area AT (normalized by the cell volume for the given value of R) in a black dashed curve, and shows a linear relationship between both.
A strong prediction of our model of mechanically-driven fate choice imbalance is that the shape of single tumor cells should be modified for increasing tension ratio R. Indeed, we verified in the simulations that for increasing tension ratios, tumor cells tended to adopt an increasingly pyramidal shape with expanded basal area, as shown in Fig. 4e, f—which intuitively reflected their preference for basal adhesion. Importantly, given that this change in cell shape was gradual with increasing R, we reasoned that it could be used as an intuitive read-out for tension disparities, which can be readily quantified in experiments. An alternative and complementary metric was the contact angle between lateral areas of the WT-mutant clone interface, which became increasingly tilted for increasing R as expected in a simple wetting argument.
Crucially, this means that the model predicts a highly nontrivial, yet robust, correlation between the shape of a tumor cell (a static morphological feature) and the bias in its fate choices between division and differentiation (a complex dynamical property), which is predicted to be roughly linear, as shown in Fig. 4g.
Testing the model in basal cell carcinoma initiation
To test this prediction, we used a genetic mouse model of BCC that consists of the activation of the Hedgehog signaling pathway, using a constitutive form of Smoothened fused to yellow fluorescent protein (SmoM2-YFP) under the control of the Keratin-14 (Krt14-CREER) upon tamoxifen, thereafter referred to as Krt14-CREER/SmoM2. Administration of a low dose of tamoxifen leads to the activation of the oncogene in a few basal cells of the epidermis in Krt14-CREER mice, leading to well-separated clones. We compared the clones generated upon oncogenic activation to those clones generated in homeostatic conditions by inducing the expression of YFP in the basal cells of the epidermis (Krt14-CREER) upon tamoxifen administration, thereafter referred to as Krt14-CREER/YFP. Tail whole mount stainings followed by confocal imaging and quantification, shows that SmoM2-expression in the basal cells of the epidermis leads to an expansion of the basal compartment, as shown by an increase in the number of basal cells/clone, detected as early as 1 week upon tamoxifen administration (1.1 vs 2.1) and at 2 weeks upon tamoxifen (1.3 vs 7.5) administration, in Krt14-CREER/YFP and Krt14-CREER/SmoM2 respectively (Fig. 5a, b). As previously reported, this is largely due to an increase in symmetric renewal/impaired differentiation11, with relatively little cellular loss via apoptosis in Krt14-CREER/SmoM2 clones at 2 weeks following tamoxifen administration and absence of apoptosis (cleaved caspase 3 positive cells) at 1w in Krt14-CREER/SmoM2 clones. Apoptosis was not observed in Krt14-CREER/YFP clones at 1 or 2 weeks following tamoxifen administration (Fig. S2e, f).

a Immunostaining for β4-integrin, YFP and SmoM2 in Krt14-CREER/YFP and Krt14-CREER/SmoM2 epidermis at different time points upon tamoxifen administration. Scale bars: 10 μm. b Quantification of the basal clone size in clones from Krt14-CREER/YFP and Krt14-CREER/SmoM2 at different time points upon tamoxifen administration (n = number of clones quantified from 4 animals). Error bars represent the mean ± SEM. c Schematic depicts the quantity that is used to measure tension homogeneity between adjacent clones-- contact angle (θc). For more details see Section: “Methods”. Using representative z-stack images of mouse skin (IFE), showing Phalloidin (gray), DAPI (blue) for nuclei, immunostaining for β4-integrin (red) for BM and SmoM2 (green), an example for contact angle measurement is shown for a 3-cell clone. More details in Section: “Methods”. Scale bars: 10 μm. Contact angle distributions (d) and averages (e) are shown for week 1 (top in (d), from 3 animals each in (d) and (e)) and week 2 (bottom in (d), from 2 animals each in (d) and (e)). Dotted gray line is drawn at 90∘ for reference, with smaller angles found on average for Krt14-CREER/SmoM2 cells. Error bars represent the mean ± SEM. f Basal contact area of individual cells, normalized by cellular volumes, shown for Krt14-CREER/YFP and Krt14-CREER/SmoM2 clones (n = number of clones quantified from 3 animals). Error bars represent the mean ± SEM. g Representative images of mouse skin IFE are shown for Krt14-CREER/YFP and Krt14-CREER/SmoM2 clones showing the 3D reconstruction of the BM (β4-integrin) in brown and clones in green, for times—1 and 2 weeks after tamoxifen administration. Images are representative of the clones found in the 3 animals analysed per group. On the left panel, yellow highlights the contact area with the BM, i.e., the overlapping region between cells and BM reconstruction. Right panel: green highlights the clone in 3D. Scale bars: 5 μm. All statistical analyses were determined using the two-tailed Mann–Whitney test.
To test whether tumor cells have distinct morphological features and guided by our theoretical results, we performed two types of quantifications. Firstly, taking 2D cross-sections across tumor and WT clones, we calculated the “wetting” angle between lateral membranes at the clonal boundary and the BM as outlined in Fig. 5c. The average was 90° for WT cells, as expected, because lateral membranes need, on average, to be vertical with respect to the BM (Fig. 5d, e). In the tumor, however, we found that the contact angles are significantly lower, with a decrease compared to WT of 4° for week-1 tumors and 7° for week-2 (Fig. 5d, e). Using these contact angles to estimate (see details in Fig. S2g) the tension ratio R between WT and SmoM2-expressing cells predicted R ~ 1.05 and ~1.10 for week 1 and week 2, respectively.
Secondly, to verify these results independently, we used a different quantification strategy based on the basal area Ab of WT and tumor cells. To compute it, we calculated the area of a clone (YFP-positive) that was in contact with the basal lamina (stained using β4-integrin positive) using Imaris software (see “Methods” for more details), and divided it by the number of basal cells in the clone (Fig. 5d and Supplementary Video 6). To correct for the fact that tumor cells have smaller volumes than WT cells, we rescaled this basal area by cellular volume \({A}_{0}/{V}_{0}^{2/3}\), calculated from 3D reconstruction based on YFP labeling of the different clones using Imaris, mirroring the dimensionless ratio calculated in our simulations (Fig. 5). This revealed that individual SmoM2-expressing cells were more basally attached than WT cells, by around 12% at 1w and 8% at 2w, in qualitative agreement with the model. More quantitatively, this translated into a predicted tension ratio (Fig. 4f) of around R = 1.05–1.1, highly consistent with the first quantification method.
Importantly, a tension ratio of around R = 1.05 predicted from our simulations a clonal imbalance of around Δ = 40%. This is comparable in magnitude to clonal imbalances of 25–30% that we had found by fitting long-term clonal dynamics over several months upon Smoothened activation in Involucrin-expressing progenitors or Krt14-expressing SCs. Overall, this validates experimentally some of the key assumptions and predictions of the model, in particular on its ability to relate single-cell morphometrics and fate choice decisions.
Discussion
Using a 3D vertex-based model, with proliferation restricted to the bottom-most layer of a multilayered tissue, we show that mechanics and competition for finite space can organically give rise to homeostasis via balanced stochastic fate choices. Indeed, overcrowding in the basal layer is mechanically unstable, which leads to initially symmetric divisions being resolved into asymmetric fate outcomes at the population level. Thus, without any explicit fate assignment to the progenies, we can recover salient aspects found in lineage tracing experiments and stochastic models in the absence of mechanics, such as effectively stochastic fate choices and neutral drift of clones at the population level. This generically predicts a distribution of 1/4–1/2–1/4 for basal-basal, basal-suprabasal and suprabasal-suprabasal fate outcomes, consistent in observations in several systems, such as mouse tail or ear epidermis11,51. Interestingly, other organs display rarer symmetric divisions (e.g., 10–15% instead of the predicted 25%)4,8, which is a signature of more complex regulation that could be added to our model in the future. One simple assumption that could be easily relaxed is the one that divisions take place in the basal plane: if divisions are also allowed to be perpendicular to the BM, this will clearly bias the fraction of asymmetric divisions via purely geometrical effects. Another, more complex, more of regulation would be to consider more explicitly fate and transcriptional heterogeneity in the basal layer. Indeed, although we have, for the sake of simplicity, considered a homeostatic model with a single basal cell population, extensive heterogeneities have been uncovered, for instance, in their transcriptome by single cell RNA sequencing24,56,57, although debates remain as to the functional role of this cellular and molecular heterogeneity on long-term fate outcomes8. It remains elusive whether the different cell populations and cell states present within the IFE basal layer present differences in geometry (i.e., contact with the BM). This description will be key to understand whether the implemented model, where fate is simply dependent on geometry, could be used or if other parameters would be needed to model such cellular heterogeneity and plasticity reported between the different IFE cell populations and cell states. Another facet of heterogeneity which could be added to the simulations is explicit modeling of signaling interactions between two daughter cells, to coordinate their respective fates58. Furthermore, beyond clone size evolution, our numerical simulations also make a number of predictions on how the mechanical state of the tissue affects not only cellular shape index (as studied over recent years), but also clonal shape and dispersion, generalizing previous findings in 2D monolayers linking tissue fluidity to clonal dispersion59,60. In adult skin, clones are typically highly cohesive11, indicative of a solid tissue rheology, something which would be tested more quantitatively in the future by mechanical measurements.
Surprisingly, upon introduction of mechanical inhomogeneity in the form of lowered basal tension for a sub-population of cells, we discover that the homeostatic balance rapidly becomes tilted towards symmetric divisions. For instance, as little as 10% of disparity in basal tension is enough to induce tumor-like invasiveness within the basal layer, without any change in division rate. Given previous findings which have connected the impaired differentiation, rather than enhanced proliferation, of basal cells in BCC progression, this hints at the idea that differential mechanical changes could drive differential fate and invasion in tumors. One advantage of our mechanical model is that its core constituent is cellular shape, meaning that tension differences between WT and tumor cells manifest themselves in predictable differential shapes between the two populations. In particular, both the contact angle of lateral surfaces with the BM and the fraction of cellular area in contact with the BM depend predictably on the tension differences, facilitating the inference of this critical parameter in the data. We then proceeded to measure detailed cellular morphometrics in both WT and tumor cells at different time points, and found that both metrics were consistent in pointing towards a tension imbalance in tumor cells of 5–10%, which is sufficient in the model to reproduce the magnitude of imbalance towards symmetric renewal observed in long-term lineage tracing experiments11. One important assumption, which could be relaxed in future studies, is that we made these mechanical inhomogeneities constant over time. More realistic implementations could, for instance, take into account the fact that tumor cell properties evolve over time, or conversely that the surrounding WT tissue actively adapts its properties to resist tumor expansion61. We have also considered in our model that all cells have identical volumes, whereas volume differences are seen between cell types at homeostasis, and also in tumors compared to WT cells. This has been suggested to drive clonal dispersion40, for instance, in Drosophila62. Finally, we have not included direct interactions between tumor and WT cells, akin to mechanical cell competition models due to surface tension at the heterotypic boundary61,63,64,65, as this competition in our model is simply due to intrinsic mechanical differences, although capturing both these effects in a single framework would be interesting.
Overall, this model underlines how mechanical forces can be key to tissue dynamics not only as regulators of tissue shape, but also by modulating cellular fate choices in settings where proliferative potential depends on tissue location. Although we have concentrated here on the skin epidermis, our model is highly general and could apply to any stratified tissue. In addition to such mechanical effects, a logical next step would be to consider more direct feedback of forces through fate via mechano-sensing24,25, as well as considering more complex tissues with more heterogeneous fates66. Another interesting aspect of differential mechanics would be to consider the influence of spatial modulation of ECM mechanics, which has recently been shown to critically modulate the competence of tumor initiation in skin12. This could be particularly relevant at later stages of BCC progression that involve invasion of the subjacent tissue (e.g., dyplasia and BCC stage), where extensive BM and ECM remodeling must occur. These late stages are also characterized by the presence of apoptosis, both in the basal and suprabasal compartments, indicating that apoptosis would need to be included. Finally, although we have concentrated on confluent tissues of fixed densities, wound healing from basal cell ablation has been shown to be linked to changes in tissue fluidity67. Overall, this work shows that mechanical models can also be a powerful tool for stem cell biology, by connecting easily accessible morphological signatures such as cell shape to fate and clonal expansion in the initial stages of cancer.
Methods
Mechanical parameters in the simulation
We begin with the simplest homeostatic model where every cell has the same preferred cell shape index s0, irrespective of where it belongs in the three-layered tissue. While we also explore for a fluid-like shape of s0 = 5.5 (Fig panels: S1, S2, and S3) to find both- homeostatic and tumor behavior comparable to the solid shape of s0 = 5.3, we stick to the later as it resembles the physiological tissue better, in particular due to the absence of significant clonal fragmentation in experiments (Fig. S3). These cells are contained between two almost-flat surfaces with a fixed base-line tension value of TB = 1.0 (for s0 = 5.5, the value is much smaller of 0.1). More specifically, the stroma below the BM/epithelial cells is represented in the simulations by “ghost cells”, i.e., cells which play no role in the simulations as we don’t update their position throughout, apart from the first time steps during which the BM flattens from surface tension. Formally, BM tension Tb is thus added at the interface between basal cells and “ghost cells”. These values of tension Tb are chosen based on the region of the shape-tension phase space with no tension-induced pinning41, as Voronoi models have been shown to display pinning artefacts when large values of interfacial tensions are present compared to vertex models68. We augment this further by zooming into the tension range and making sure to have no artificial pinning behavior, as shown in Fig. S1e, f. The cells also have the same noise level throughout the tissue set by self-propulsion speed of v0 = 0.1, which is not a special value, as the dynamical behavior is highly robust with respect to noise (Fig. S1g, h) till a threshold value above which the basement membrane fails to stabilize into a flat surface.
Details about noise implementation
To test the effect of noise, we add cellular activity on top of the static model. As we use Voronoi cells in our system, there is one degree of freedom per cell, which is the Voronoi center. The equation of motion is implemented in a straight-forward way41. Mechanical force on a cell is computed using the derivative of the non-dimensionalised energy functional (e) from Eq. (2) i.e., fi = ∂e/∂ri. Cellular activity, with respect to the cellular surroundings (such as extracellular matrix, for instance), is then added such that it has a polarization vector \({\hat{{{{\bf{n}}}}}}_{i}\) and active force due to self-propulsion of v0/μ where v0 is the propulsion speed, and μ is the cell mobility (it is set to unity).
The polarization vector evolves via white Gaussian noise on a unit sphere with diffusion coefficient Dr as shown below-
where I is a 3 × 3 identity tensor, \({\hat{{{{\bf{n}}}}}}_{i}{\hat{{{{\bf{n}}}}}}_{i}\) is the dyadic product of the polarization vector with itself, and ζi is the white Gaussian noise.
Timescales in the simulations
A natural time unit in the simulations is \(\tilde{t}=1/{K}_{V}{V}_{0}^{4/3}\) which we get by non-dimensionalising the dynamical equations, and thus is set to unity. For implementing noise, we use a self-propulsion speed of v0 = 0.1 and rotational noise Dr = 1, essentially transitioning to a Brownian regime as quickly as \(t > \tilde{t}/{D}_{r}\). The characteristic diffusion timescale in a homogeneous three-dimensional system with s0 = 5.3 is \({\tau }_{0}=1{0}^{5}\tilde{t}\) (for s0 = 5.5 it is \(1{0}^{3}\tilde{t}\)). Note that we do not focus here on the effect of self-propulsive activity and persistence, which have been extensively explored36, given our focus in this system is on the role of cellular division and mechanics-dependent fate choices. We use an integration time-step of \(\Delta=0.01\tilde{t}\).
Cell divisions happen exclusively in the basal layer, that is defined as all the cells which have their centers within a one cell length distance from the BM and have a finite connection to the BM. In this work, the division timescale of a given basal cell is on an average- \({\tau }_{D} \sim 3900\tilde{t}\), making the division rate \({k}_{D}=0.00026{\tilde{t}}^{-1}\). This frequency is chosen in order to have less division-induced-fluidization of the tissue (to resemble a physiological tissue) and at the same time be computationally feasible i.e., a full simulation completes under 24 h. It has been observed that for a 2D confluent monolayer in the solid regime, the chosen division rate diffuses cells only by a couple of cell lengths by the end of our simulation time60.
An individual cell division event occurs in every \(50\tilde{t}\). A randomly chosen mother cell is allowed to grow to a preferred cell volume of 3.5 for \(25\tilde{t}\). Note that this value of preferred volume is larger than the volume actually reached by cells due to the properties of the regular Voronoi tessalation, which is the reason we use a relatively larger value of 3.5. In the future, different tessalation methods such as Radical Voronoi could be used to dampen this effect. The preferred cell surface area is modified accordingly to maintain the preferred shape index. After this, the mother cell is replaced by two daughter cells with centers of mass at the same height as the mother (i.e., in-plane division) and placed 0.25 unit lengths (a unit length refers to the typical cell length l0) from the mother cell’s position in a colinear fashion. This axis of division in the plane of the BM is constructed using a randomly chosen angle with respect to the X axis, and is bisected by the division plane. The daughters inherit all the mechanical attributes and the clonal group number of the mother cell. To balance the total cell number, we also randomly pick a suprabasal cell and delete it (suprabasal cell loss) from the simulations. Note that given that we have minimal movements from the suprabasal to the basal cell layer, the exact loss dynamics of suprabasal cells do not impact on the long-term clonal outcomes of the simulations. The system has evolved for another \(25\tilde{t}\) to adjust to the new configuration. After this cycle of 50 timesteps, another mother cell is chosen at random, and the process is sequentially repeated.
The time it takes for the system to reach steady-state comprises of two stages—in the first stage, the system is evolved for \({T}_{SS1}=1000\tilde{t}\) with no cellular divisions so that the basement membrane stabilizes to a flat surface41. In the second stage, we run simulations with divisions for \({T}_{SS2}=4000\tilde{t}\) to ensure at least all basal cells have divided once, and to let the system come to a steady state with respect to proliferation. By this time, the basal density therefore achieves a steady state as shown in (Fig. S1c, d). Only after this stage, every basal cell is allotted a clonal group number, and for the tumor case, inhomogeneities in basal tensions are implemented for four cells equidistant from each other. The simulation is then run for a total of \(11000\tilde{t}\) that is ~2.8τD. The linear fitting of clonal growth dynamics is analysed for the last ~1τD window, which is when the homeostatic clones achieve a steady growth state, as one can observe in Fig. 2. The transition from linear to exponential is captured really well with Eq. (5), which we use to complement the fate distribution findings. The definition of Δ that we use for fate distributions is slightly different from this, since it is easier to understand in terms of population asymmetry as opposed to a ratio– \(\Delta \longrightarrow 2r\tilde{\Delta }\)69.
From a geometrical perspective, we run simulations with box sizes allowing for three layers of cells. The mouse epidermis contains four different layers (basal, spinous, granular, and cornified layer). The spinous and granular layers can be composed of up to 2 and 3 layers of cells, respectively70. Remarkably, the only proliferative layer in the skin is the basal layer, while the suprabasal layers (spinous, granula, and cornified) are composed of non-proliferative differentiated keratinoctyes. Given that suprabasal cells to not contribute to long-term clonal dynamics, increasing the number of layers would not change the results of the model, and so we simulate only three for computational efficiency. As open-boundaries in Voronoi models are highly nontrivial to implement, we allow three more layers comprising of pseudo cells below the basement membrane, that sandwich the tissue via periodic boundary conditions along z-axis. Since their only job is to sustain the basal surface, these cells are only active during the SS1 stage (with the same diffusion profile as real cells) and are mechanically pinned for the entirety of the simulation. They will have negligible feedback to the real cells across the BM because we use low basal tension values that are much below the tension-induced-pinning regime, which is when the cells across the interface become coupled41.
Analysis protocol for cell-fate distribution in simulations
Lineage trees for every clone are plotted using the mother-daughter cell information. Every cell has a unique ID, and we construct lineage information between mothers and daughters at each division event. Using this information, we construct lineage trees with respect to time as shown in Fig. 4b. At the end of the simulation, for every lineage tree, we have the list of all the clonal members both—past (eliminated through the top layer) and present (basal and non-basal). For the clones that survive till the end, we also have the list of all the final basal members. To identify the set of members that have extruded, we simply remove the set of final basal members from the set of all members. This way, we also identify the daughters that continued to divide or have survived till the end (nodes are shown in pink) and the ones that extruded (nodes shown in blue). The daughters that survive till the final time point of the simulation are not included in fate distribution because they need more time for their fate to become apparent (i.e., either extrude or divide again).
Next, we use this information to find the probabilities of each fate outcome as a function of simulation parameters. For each cell division event, we check how many of the daughter cells have extruded, and we measure the statistics of each of the three possible fate outcomes, i.e., none (basal-basal division), one (basal-suprabasal), or both (suprabasal-suprabasal) of the cells have extruded. We then filter out the divisions that occur after one complete turnover i.e., t ≥ 1τD, because not all the basal cells have gotten the chance to differentiate, thereby artificially biasing the distribution towards “symmetric basal” fate. From the remaining division events, we compute the proportion for each fate, for the given value of tension ratio R, as plotted in Fig. 4.
Quantitative morphometric analysis of clone and cell shapes
We next quantify morphological features of the simulations at both- single-cell and clone level.
As for the quantification of single cell basal area AT, we use the final snapshot i.e., \(11000\tilde{t}\), to quantify it. This includes cells from every kind of clone including the singlets. We isolate all the facets that are in contact with the BM and sum it up.
With an increase in noise, we observe that the solid clonal shapes in solid parameter regime becomes less compact and sprawl more. For fluid systems we see that while the tumor clones are compact in zero noise, they undergo explicit fragmentation for higher noise levels (Fig. S3).
To infer tension inhomogeneity between adjacent clones in a manner that can be mapped to experimental measurements, we use the contact angle (θc) that their lateral contact forms with the BM. In simulations, we typically use a tumor consisting of a single cell, so shorter ~1τD-simulations are also used to identify the lateral edges and their coordinates. As the basement membrane is, on average, parallel to the XY plane, we compute the angles made by the edges with the XY plane. This is plotted for different tumors in Fig. S2e.
To measure the same in experimental images, Figure 5c, d explains the entire protocol with a schematic and example. For a clone with a relatively flat BM, an outer-basal cell is chosen (depicted in pink in Fig. 5c) to measure the angle between its lateral edges and the BM. Figure 5d shows an example for contact angle measurement for a 3-cell clone shown using an orthogonal view on the top-left. Using the apical view on the right (Z-stack corresponding to CS), several cross-sectional cuts are made, two of which are shown for this case—CS1 and CS2. Angles—θ1 and θ2 are measured between the outer-most lateral edges and the BM line (solid gray line) for cross-sections CS1 and CS2 in sub-figures (1) and (2).
Mice and ethics
Krt14-CREER (RRID:IMSR JAX:005107), Rosa-YFP (RRID:IMSR JAX:006148) and Rosa-SmoM2-YFP (RRID:IMSR JAX:005130) mice were obtained from the JAX repository. Mouse colonies were maintained in a certified animal facility in accordance with European guidelines for the laboratory animal use and care based on the 2010/63/EU Directive. Experiments involving mice presented in this work were approved by the Animal Welfare and Ethics Body, Direção-Geral da Alimentação e Veterinária (DGAV, Portuguese Authority) under protocol DGAV protocol number 011681.
In this study, we generated preneoplastic lesions that were microscopic and did not ulcerate. In any of the experiments, we observed ulceration or macroscopic lesions, which are described as the limit that should not be exceeded in DGAV protocol number 011681. Female and male animals were used for all experiments, and equal gender ratios were respected in the majority of the analysis. Analysis of the different mutant mice was not blind, and the sample size was calculated to reach statistical significance. The experiments were not randomized.
Skin tumor induction
For tumor induction, 1.5-month-old mice were used. Krt14-CREER/Rosa-SmoM2-YFP and received an intraperitoneal injection of 0.1 mg (0.5 mg/ml) of tamoxifen (ref. T5648-0005, Sigma). Mice were sacrificed and analysed at different time points following tamoxifen administration. Krt14-CREER/Rosa-SmoM2-YFP mice were heterozygous for the Rosa-SmoM2-YFP mutation in the Rosa Locus. The tail of these animals was used in our analysis.
Lineage tracing experiments during skin homeostasis
For the lineage tracing experiments, 1.5-month-old mice were used. Krt14-CREER/Rosa-YFP mice received an intraperitoneal injection of 0.1 mg (0.5 mg/ml) of tamoxifen (ref. T5648-0005, Sigma). Mice were sacrificed and analysed at different time points following tamoxifen administration. All mice were heterozygous for Rosa-YFP.
Whole-mounts of tail epidermis
Whole mounts of tail epidermis were performed as previously described11. Specifically, pieces of tail were incubated for 1 h at 37 °C in EDTA 20 mM in PBS in a rocking plate, then using forceps, the dermis and epidermis were separated and the epidermis was fixed for 30 min in 4% Formaldehyde methanol-free (ref FB002, ThermoFisher Scientific) in agitation at room temperature and washed 3× with PBS. For the immunostaining, tail skin pieces were blocked with blocking buffer for 3 h (PBS, horse serum 5%, Triton 0.8%) in a rocking plate at room temperature. After, the skin pieces were incubated with primary antibodies diluted in blocking buffer overnight at 4 °C, the next day they were washed with PBS-Tween 0.2% for 3 × 10 min at room temperature, and then incubated with the secondary antibodies diluted in blocking buffer for 3 h at room temperature, washed 2 × 10 min with PBS-Tween 0.2% and washed for 10 min in PBS. Finally, they were incubated in DAPI (1:1000) diluted in PBS for 30 min at room temperature in the rocking plate, washed 3 × 10 min in PBS and mounted in DAKO mounting medium supplemented with 2.5% Dabco (Sigma). Primary antibodies used were the following: Goat anti-GFP (1:800, ref. ab6673, Abcam), Rat anti-β4-integrin (1:500, ref. 553745, BD Pharmingen), rabbit anti-cleaved caspase-3 (1:600, ref. AF835, R&D Systems). Secondary antibodies conjugated to AlexaFluor488 (Goat ref A-11055, Invitrogen), to Rhodamine Red-X (rat, ref. 712-295-153, Jackson ImmunoResearch; rabbit, 711-295-152, Jackson ImmunoResearch), and to Alexa Fluor 647 (rat, ref. 712-605-153, Jackson ImmunoResearch). Samples were also stained with Phalloidin conjugated to AlexaFluor647 (1:400, ref A30107, ThermoFisher Scientific). Images were acquired using Z-stacks with an inverted confocal microscope, LSM 980 (Carl Zeiss).
Analysis of clone size and apoptosis
The quantification of the the basal and total clone size were determined by counting the number of SmoM2-positive cells (in Krt14-CREER/Rosa-SmoM2-YFP mice) and YFP-postive cells (in Krt14-CREER/Rosa-YFP mice) in each clone using orthogonal views of the whole-mount tail epidermis in the interscale region of interfollicular epidermis, as well as the number of cleavedcaspase3-positive cells in each, as described in ref. 11. β4-integrin staining was used to classify the clones according to their location in the basal or suprabasal layers.
Image analysis
The “Surface” module that is part of the Imaris 9.6 software (Andor Inc.) was used to carry out the 3D rendering of the clone structure labeled with anti-GFP and the basal membrane structure labeled with anti-β4-integrin. The 3D-rendering of the objects was performed using smoothing 1 μm, the background subtraction was dependent on the cell size (the value per cell was on average 12 μm) and similar thresholding settings across all experiments for the morphological analysis of the clone volume and the clone area. Structures outside of the rendered objects were excluded from the final analysis. The proximity between the basal cell surface object and the basal membrane object was determined using “Surface Contact Area” Imaris XTension integrated plugin71. The contact area between basal cells from the clone and the basal membrane was calculated using the following formula: \(\frac{SCA}{C{V}^{2/3}}\).
Statistical analysis
All statistical analyses were performed using GraphPad Prims v.8.0.1 software. Results are presented as mean ± SEM. Data normality was assessed using the Shapiro–Wilk test. For datasets that met the assumption of normality, P values were calculated using unpaired t-tests. For non-normally distributed datasets, P values were determined using the two-tailed nonparametric Mann–Whitney test. The number of mice and clones analysed is specified in each figure legend.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data supporting the findings of this study are available within the manuscript. Source data are provided with this paper.
Code availability
Simulation results supporting the findings of this study are available within the manuscript.
References
Mascré, G. et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489, 257–262 (2012).
Hsu, Y. C. & Fuchs, E. Building and maintaining the skin. Cold Spring Harb. Perspect. Biol. 14, a040840 (2022).
Mackenzie, I. C. Relationship between mitosis and the ordered structure of the stratum corneum in mouse epidermis. Nature 226, 653–655 (1970).
Doupé, D. P., Klein, A. M., Simons, B. D. & Jones, P. H. The ordered architecture of murine ear epidermis is maintained by progenitor cells with random fate. Dev. Cell 18, 317–323 (2010).
Klein, A. M. & Simons, B. D. Universal patterns of stem cell fate in cycling adult tissues. Development 138, 3103–3111 (2011).
Blanpain, C. & Simons, B. D. Unravelling stem cell dynamics by lineage tracing. Nat. Rev. Mol. Cell Biol. 14, 489–502 (2013).
Barker, N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 15, 19–33 (2014).
Piedrafita, G. et al. A single-progenitor model as the unifying paradigm of epidermal and esophageal epithelial maintenance in mice. Nat. Commun. 11, 1–15 (2020).
Doupé, D. P. et al. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 337, 1091–1093 (2012).
Roshan, A. et al. Human keratinocytes have two interconvertible modes of proliferation. Nat. Cell Biol. 18, 145–156 (2016).
Sánchez-Danés, A. et al. Defining the clonal dynamics leading to mouse skin tumour initiation. Nature 536, 298–303 (2016).
Bansaccal, N. et al. The extracellular matrix dictates regional competence for tumour initiation. Nature 623, 828–835 (2023).
Aragona, M. et al. Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat. Commun. 8, 14684 (2017).
Mongera, A. et al. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature 561, 401–405 (2018).
Azkanaz, M. et al. Retrograde movements determine effective stem cell numbers in the intestine. Nature 607, 548–554 (2022).
Corominas-Murtra, B. et al. Stem cell lineage survival as a noisy competition for niche access. Proc. Natl. Acad. Sci. USA 117, 16969–16975 (2020).
Lander, A. D., Gokoffski, K. K., Wan, F. Y., Nie, Q. & Calof, A. L. Cell lineages and the logic of proliferative control. PLoS Biol. 7, e1000015 (2009).
Jörg, D. J., Kitadate, Y., Yoshida, S. & Simons, B. D. Stem cell populations as self-renewing many-particle systems. Annu. Rev. Condens. Matter Phys. 12, 135–153 (2021).
Klein, A. M., Doupé, D. P., Jones, P. H. & Simons, B. D. Mechanism of murine epidermal maintenance: cell division and the voter model. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 77, 031907 (2008).
Krapivsky, P. L., Redner, S. & Ben-Naim, E. Spin Dynamics https://www.cambridge.org/core/books/kinetic-view-of-statistical-physics/spin-dynamics/C24348C6CF06480DBB054B6CF6CAAEC1 (Cambridge University Press, 2011).
Kitadate, Y. et al. Competition for mitogens regulates spermatogenic stem cell homeostasis in an open niche. Cell Stem Cell 24, 79–92.e6 (2019).
Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006).
Miroshnikova, Y. A. et al. Adhesion forces and cortical tension couple cell proliferation and differentiation to drive epidermal stratification. Nat. Cell Biol. 20, 69–80 (2018).
Aragona, M. et al. Mechanisms of stretch-mediated skin expansion at single-cell resolution. Nature 584, 268–273 (2020).
Zöllner, A. M., Holland, M. A., Honda, K. S., Gosain, A. K. & Kuhl, E. Growth on demand: reviewing the mechanobiology of stretched skin. J. Mech. Behav. Biomed. Mater. 28, 495–509 (2013).
Fiore, V. F. et al. Mechanics of a multilayer epithelium instruct tumour architecture and function. Nature 585, 433 (2020).
Brückner, D. B. & Hannezo, E. Tissue active matter: integrating mechanics and signaling into dynamical models. Cold Spring Harb. Perspect. Biol. 17, a041653 (2025).
Farhadifar, R., Röper, J. C., Aigouy, B., Eaton, S. & Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Curr. Biol. 17, 2095–2104 (2007).
Staple, D. B. et al. Mechanics and remodelling of cell packings in epithelia. Eur. Phys. J. E Soft Matter 33, 117–127 (2010).
Nagai, T. & Honda, H. A dynamic cell model for the formation of epithelial tissues. Philos. Mag. B 81, 699–719 (2001).
Chiou, K. K., Hufnagel, L. & Shraiman, B. I. Mechanical stress inference for two dimensional cell arrays. PLoS Comput. Biol. 8, e1002512 (2012).
Hannezo, E., Prost, J. & Joanny, J. F. Theory of epithelial sheet morphology in three dimensions. Proc. Natl. Acad. Sci. USA 111, 27–32 (2014).
Bi, D., Lopez, J. H., Schwarz, J. M. & Manning, M. L. A density-independent rigidity transition in biological tissues. Nat. Phys. 11, 1074–1079 (2015).
Merkel, M. & Manning, M. L. A geometrically controlled rigidity transition in a model for confluent 3d tissues. New J. Phys. 20, 022002 (2018).
Otani, T., Ichii, T., Aono, S. & Takeichi, M. Cdc42 gef tuba regulates the junctional configuration of simple epithelial cells. J. Cell Biol. 175, 135–146 (2006).
Bi, D., Yang, X., Marchetti, M. C. & Manning, M. L. Motility-driven glass and jamming transitions in biological tissues. Phys. Rev. X 6, 021011 (2016).
Sahu, P. Fluidization and segregation in confluent models for biological tissues. PhD thesis, Syracuse University (2020).
Sussman, D. M., Schwarz, J. M., Marchetti, M. C. & Manning, M. L. Soft yet sharp interfaces in a vertex model of confluent tissue. Phys. Rev. Lett. 120, 058001 (2018).
Sahu, P., Kang, J., Erdemci-Tandogan, G. & Manning, M. L. Linear and nonlinear mechanical responses can be quite different in models for biological tissues. Soft Matter 16, 1850–1856 (2020).
Sahu, P. et al. Small-scale demixing in confluent biological tissues. Soft Matter 16, 3325–3337 (2020).
Sahu, P., Schwarz, J. M. & Manning, M. L. Geometric signatures of tissue surface tension in a three-dimensional model of confluent tissue. New J. Phys. 23, 093043 (2021).
Malinverno, C. et al. Endocytic reawakening of motility in jammed epithelia. Nat. Mater. 16, 587–596 (2017).
Park, J. A. et al. Unjamming and cell shape in the asthmatic airway epithelium. Nat. Mater. 14, 1040–1048 (2015).
Sanematsu, P. C. et al. 3d viscoelastic drag forces contribute to cell shape changes during organogenesis in the zebrafish embryo. Cells Dev. 168, 203718 (2021).
Erdemci-Tandogan, G., Clark, M. J., Amack, J. D. & Manning, M. L. Tissue flow induces cell shape changes during organogenesis. Biophys. J. 115, 2259–2270 (2018).
Villeneuve, C. et al. Mechanical forces across compartments coordinate cell shape and fate transitions to generate tissue architecture. Nat. Cell Biol. 26, 207–218 (2024).
Yanagida, A. et al. Cell surface fluctuations regulate early embryonic lineage sorting. Cell 185, 777–793.e20 (2022).
Kim, S., Pochitaloff, M., Stooke-Vaughan, G. A. & Campàs, O. Embryonic tissues as active foams. Nat. Phys. 17, 859–866 (2021).
Pinheiro, D., Kardos, R., Hannezo, É. & Heisenberg, C. P. Morphogen gradient orchestrates pattern-preserving tissue morphogenesis via motility-driven unjamming. Nat. Phys. 18, 1482–1493 (2022).
Mitchel, J. A. et al. In primary airway epithelial cells, the unjamming transition is distinct from the epithelial-to-mesenchymal transition. Nat. Commun. 11, 1–14 (2020).
Rompolas, P. et al. Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352, 1471–1474 (2016).
Cockburn, K. et al. Gradual differentiation uncoupled from cell cycle exit generates heterogeneity in the epidermal stem cell layer. Nat. Cell Biol. 24, 1692–1700 (2022).
Ning, W., Muroyama, A., Li, H. & Lechler, T. Differentiated daughter cells regulate stem cell proliferation and fate through intra-tissue tension. Cell Stem Cell 28, 436 (2021).
Niessen, C. M., Manning, M. L. & Wickström, S. A. Mechanochemical principles of epidermal tissue dynamics. Cold Spring Harb. Perspect. Biol. 17, a041518 (2024).
Turlier, H., & Maitre, J. L. Mechanics of tissue compaction. Semin. Cell Dev. Biol. 47-48, 110–117 (2015).
Joost, S. et al. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst. 3, 221–237 (2016).
Negri, V. A. et al. Single-cell RNA sequencing of human epidermis identifies lunatic fringe as a novel regulator of the stem cell compartment. Stem Cell Rep. 18, 2047–2055 (2023).
Mesa, K. R. et al. Homeostatic epidermal stem cell self-renewal is driven by local differentiation. Cell Stem Cell 23, 677–686 (2018).
Bocanegra-Moreno, L., Singh, A., Hannezo, E., Zagorski, M. & Kicheva, A. Cell cycle dynamics control fluidity of the developing mouse neuroepithelium. Nat. Phys. 19, 1050–1058 (2023).
Czajkowski, M., Sussman, D. M., Marchetti, M. C. & Manning, M. L. Glassy dynamics in models of confluent tissue with mitosis and apoptosis. Soft Matter 15, 9133–9149 (2019).
Brás-Pereira, C. & Moreno, E. Mechanical cell competition. Curr. Opin. Cell Biol. 51, 15–21 (2018).
Ramanathan, S. P., Krajnc, M. & Gibson, M. C. Cell size pleomorphism drives aberrant clone dispersal in proliferating epithelia. Dev. Cell 51, 49 (2019).
Bielmeier, C. et al. Interface contractility between differently fated cells drives cell elimination and cyst formation. Curr. Biol. 26, 563–574 (2016).
Valon, L., Matamoro-Vidal, A., Villars, A. & Levayer, R. Interfacial tension and growth both contribute to mechanical cell competition. Preprint at bioRxiv: https://doi.org/10.1101/2025.01.09.632135 (2025).
Schoenit, A. et al. Force transmission is a master regulator of mechanical cell competition. Nat. Mater. 24, 966–976 (2025).
Rübsam, M. et al. Polarity signaling balances epithelial contractility and mechanical resistance. Sci. Rep. 13, 7743 (2023).
Sarate, R. M. et al. Dynamic regulation of tissue fluidity controls skin repair during wound healing. Cell 187, 5298–5315.e19 (2024).
Lawson-Keister, E., Zhang, T., Nazari, F., Fagotto, F. & Manning, M. L. Differences in boundary behavior in the 3d vertex and voronoi models. PLoS Comput. Biol. 20, e1011724 (2024).
Klein, A. M., Brash, D. E., Jones, P. H. & Simons, B. D. Stochastic fate of p53-mutant epidermal progenitor cells is tilted toward proliferation by UV B during preneoplasia. Proc. Natl. Acad. Sci. USA 107, 270–275 (2010).
Rübsam, M. et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat. Commun. 8 https://pubmed.ncbi.nlm.nih.gov/29093447/ (2017).
Gastinger, M. Surface surface contact area—imaris learning centre—oxford instruments. https://imaris.oxinst.com/open/view/surface-surface-contact-area.
Acknowledgements
We thank Alois Schlögl, Paula Sanematsu, Susana Moreno Flores, Bernat Corominas-Murtra, Stefania Tavano, Gayathri Singharaju, and Hannezo group members for helpful discussions, the Bioimaging facility at ISTA, as well as Matthias Merkel and Lisa Manning for sharing the 3D Voronoi code. We also thank the Champalimaud animal facility, Anna Pezzarossa and the Champalimaud ABBE platform for the help with microscopy and image processing. This work was supported by EMBO (ALTF 522-2021), a Fundação para a Ciência e Tecnologia grant to A.S.D. (PTDC/MED-ONC/5553/2020), as well as the European Research Council (grant 851288 to EH). A.S.D., S.C., and R.M.S. are supported by QuantOCancer Project Horizon European Union’s Horizon 2020 program (grant agreement No 810653).
Author information
Authors and Affiliations
Contributions
Conceptualization: P.S., A.S.-D., and E.H. Simulations: P.S., Experiments: S.M.-F, S.C., and R.M.S.; Supervision: A.S.-D., E.H., Manuscript writing: P.S., E.H., and A.S.-D with inputs from all authors
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Klaas Mulder and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sahu, P., Monteiro-Ferreira, S., Canato, S. et al. Mechanical control of cell fate decisions in the skin epidermis. Nat Commun 16, 8440 (2025). https://doi.org/10.1038/s41467-025-62882-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62882-9
