
Abstract
Mitochondrial function supports energy and anabolic metabolism. Pathogenic mitochondrial DNA (mtDNA) mutations impair these processes, causing mitochondrial diseases. Their role in human cancers is less clear; while some cancers harbor high mtDNA mutation burden, others do not. Here we show that a proofreading mutant of DNA polymerase gamma (PolGD256A) increases the mtDNA mutation burden in non-small-cell lung cancer (NSCLC). This mutation promotes the accumulation of defective mitochondria, reduces tumor cell proliferation and viability, and improves cancer survival. In NSCLC, pathogenic mtDNA mutations enhance glycolysis and create a glucose dependency to support mitochondrial energy, but at the expense of a lower NAD+/NADH ratio that hinders de novo serine synthesis. Thus, mitochondrial function in NSCLC is essential for maintaining adequate serine synthesis, which in turn supports the anabolic metabolism and redox homeostasis required for tumor growth, explaining why these cancers preserve functional mtDNA.
Similar content being viewed by others


Introduction
The balance between energetic and anabolic metabolism is a sine quo-non condition for tumor cell fitness1. While various cancer cells employ diverse strategies to achieve this balance, mitochondrial function plays a critical role in this equilibrium2,3,4. Inducing metabolic stress by limiting the availability or blocking the synthesis of specific nutrients in tumor cells can be an effective strategy for improving cancer therapies5,6,7,8,9. Therefore, understanding the metabolic mechanisms that support mitochondrial function and nutrient utilization for maintaining energy and anabolic balance is crucial.
Mitochondria serve as a prime example of this interplay between energetic and anabolic metabolism as they provide both energy and anabolic metabolites to sustain tumor cell growth and survival10. Nevertheless, only a limited number of cancer treatments target mitochondrial function, possibly due to concerns about potential adverse effects11. Noteworthy examples of therapeutic strategies include studies involving the Complex I inhibitor Metformin12,13 folate inhibitors14, nucleosides/nucleotides analogs15, and serine/glycine starvation16. These strategies target key metabolic pathways: mitochondrial respiration, one-carbon (1 C)-metabolism, nucleotide synthesis, and the serine synthesis pathway (SSP)17,18,19.
One unique characteristic of mitochondria as organelles is that they harbor mitochondrial DNA (mtDNA). As mtDNA encodes essential proteins for the function of the electron transport chain (ETC), pathogenic mtDNA mutations can directly impact that important mitochondrial function20,21,22.
mtDNA mutations accumulate over time and are linked to mitochondrial dysfunction, which may play a role in mammalian aging23,24, and age represents the most significant risk factor for most cancers25,26. However, high levels of pathogenic mtDNA mutations are observed in only a few types of cancer, specifically in cancers of the colon27, thyroid4, and kidney28. In contrast, most other cancers such as lung cancer tend to selectively avoid truncating mtDNA mutations. The goal is to determine why this is the case. Despite the age-related accumulation of mtDNA mutations in the host, most cancers—particularly when compared to chromophobe renal cell carcinoma —tend to exhibit relatively low to moderate levels of pathogenic mtDNA mutations28 presumably because these mutations may be deleterious to metabolic homeostasis. The response of cancers, including lung cancer, to mtDNA mutations and the resulting mitochondrial dysfunction is unclear.
Lung cancer has a high mortality rate, affecting around 2.2 million people, leading to 1.8 million deaths worldwide annually29. Non-small-cell lung cancer (NSCLC) is the most common type of lung cancer30 and has been studied in the genetically engineered mouse model (GEMM) KP, (Trp53flox/flox; KRasLSL-G12D/+)31. NSCLC initiation in this model is induced by inhaling adenovirus encoding Cre-recombinase32,33. KP mimics the NSCLC naturally found in humans, including relatively low levels of mtDNA mutation burden28,34. Thus, to evaluate the impact of high mtDNA mutation burden in NSCLC, we generated the PGKP (PolGD256A/D256A; KRasLSL-G12D/+;Trp53flox/flox) by breeding KP with the PolG mutator mouse (PolGD256A/D256A) models35.
The PGKP animals, like KP, can conditionally generate NSCLC but have a high mtDNA mutation burden due to mutated PolG. PolG, a nuclear-encoded polymerase, replicates only mtDNA. The PolGD256A mutation eliminates its proofreading exonuclease activity, increasing mtDNA mutations. PolG mutator mice have whole-body PolGD256A mutation, causing mtDNA mutations across all tissues, leading to aging-related diseases like severe erythroid dysplasia and megaloblastic anemia, resulting in a compromised body condition, which reduces their lifespan to less than 18 months24,36,37,38.
In this work, which addresses the role of mtDNA mutations and mitochondrial function in NSCLC, we compare PGKP and KP mouse models. Introducing the PolGD256A mutator allele into the KP mouse model of lung cancer causes the accumulation of pathogenic mtDNA mutations, dysfunctional mitochondria, reduced tumor burden, and increased lifespan. This effect is more pronounced in older mice with PGKP tumors, despite their poor body condition caused by premature aging. Moreover, mitochondrial dysfunction induced by PolGD256A leads to defective respiration, making tumor cells heavily reliant on glucose to fuel metabolism. Respiratory defects cause NADH accumulation and a decreased NAD+/NADH ratio, suppressing the SSP and the synthesis of glutathione (GSH) and nucleotides. Dietary serine/glycine depletion further represses the growth of respiration-defective tumors, while increased dietary carbohydrates improve their growth. In vitro, GSH or nucleoside supplementation during serine/glycine starvation improves the growth of respiration-defective tumor cells, highlighting the critical role of the SSP in tumor growth. Our findings demonstrate the importance of mitochondrial function in supporting serine synthesis for nucleotide production and redox homeostasis in NSCLC.
Results
High mtDNA mutation burden increased the survival of mice with NSCLC
The KP and PolG mouse models were crossed to generate PGKP (Fig. 1a-upper panel. For breeding strategy, refer to Suppl Fig. S1). Cohorts of young (2 months of age) and old (10 months of age) KP and PGKP animals were generated, NSCLC was induced, and the extent of mtDNA mutation burden in tumor and normal lung were examined. mtDNA sequencing of normal lungs from PGKP and KP pups (one day old) was performed to ascertain the initial mtDNA mutation burden (Fig. 1a-lower panel). Normal lung and tumors were harvested from all mice 15 weeks post-NSCLC initiation (Fig. 1a and Suppl Fig. S2a). Over time, PGKP animals accumulated more single nucleotide variants (SNVs) and insertions/deletions (indels) in mtDNA in normal lungs and tumors than observed in KP animals (Fig. 1b). This accumulation was consistently higher in tumors than in normal lungs of PGKP mice and was much higher than in KP tumors and normal lungs at all three-time points. The high accumulation of mtDNA mutations results from homozygous PolGD256A/D256A, which continuously fails to correct random mutations in mtDNA. Moreover, cytosine and guanosine misincorporation into mtDNA increased over time in PGKP tissues (Suppl Fig. S2b), suggesting exposure to ROS or imbalanced nucleotide pools39. Most mtDNA genes in PGKP tumors accumulated mutations (Suppl Fig. S2c). These mutations might directly impact the activity of the ETC. To investigate this possibility, we used the sorting-intolerant from tolerant mutations (SIFT) tool, which predicts whether an amino acid substitution affects the protein function40. In young and old animals, the predicted tolerated and deleterious mtDNA mutations were increased in PGKP compared with KP tumors (Suppl Fig. S2d), suggesting a possible alteration of mitochondrial function.

a At the top is a diagram illustrating the generation of the NSCLC conditional model with a high mtDNA mutation burden, PGKP. For breeding strategy, refer to Suppl Fig. S1. Below is a representation of mtDNA sequencing for KP and PGKP animals at various ages (n indicated per animal genotyping). b Accumulation of mtDNA mutations, depicted by single nucleotide variants (SNVs) and Indels, in tissues and conditions presented in panel A. p-values comparing young (p = 0.0001) and old (p = 0.0007) KP and PGKP tumors. Two pup lungs were used as a baseline for both KP and PGKP groups. Three animals were included in the tumor-free groups, and five animals were used for the NSCLC groups. c Survival curve of KP and PGKP, young and old animals. The median survival is illustrated to the right of the survival curve. d In young animals, tumor burden and number, 15 weeks after cancer initiation. H&E shows lung lobes affected by NSCLC. e Wet lung weight after 15 weeks of NSCLC initiation, with accompanying images illustrating normal lungs and NSCLC conditions. Only the relevant statistics are displayed in the figure. f–g Tumor area, number, and wet lung weight after six weeks of cancer initiation. h–j IHC for ki67, cleaved-caspase 3, and TUNEL assay were conducted on PGKP and KP tumors 15 weeks post NSCLC initiation (see Suppl Fig. S3a for representative images). For each group, we used 6 animals, capturing at least 12 images of distinct tumors from each animal. Data is shown as ± SEM. In H-J, individual sample distributions are represented by dots, while the gray bar indicates the median ± SEM. Two-tailed t-tests determined P values for B, D, F, H-J, while one-way ANOVA with Tukey correction was used for Figs. C, E, and G. Log-rank (Mantel-Cox) calculated P values for the survival curve. Significance levels are denoted as *≤0.05, **≤0.01, ***≤0.001, and ****≤0.0001. For detailed individual P values, please refer to the Source Data table. The Figures a and c were created in Biorender. Cararo Lopes, E and White, E (2025).
To evaluate the survival of KP and PGKP mice under cancer conditions, we initiated NSCLC in young animals. PGKP mice demonstrated a longer survival than KP mice (Fig. 1c). This increased survival was attributed to reduced tumor burden, as evidenced by histology, hence reducing wet lung weight 15 weeks post-NSCLC induction (Fig. 1d, e)41.
PolGD256A mutation in PGKP mice leads to the accumulation of mtDNA mutations over time (Fig. 1b)35. Therefore, we explored whether inducing NSCLC in old animals would further impact their survival. The survival in old PGKP with NSCLC was further increased (15.5%) compared to young PGKP (Fig. 1c). The increased survival of old PGKP mice with NSCLC was due to remarkably reduced tumor burden 15 weeks post-NSCLC induction (Fig. 1e).
We also tested mice carrying a single, homoplastic pathogenic mtDNA mutation associated with optic atrophy and Leigh Syndrome in humans. This MT-ND6 gene mutation (MT-ND6P25L) reduces complex I activity42 and was introduced into KP to form ND6KP mice. Consistent with results from PGKP animals, the ND6KP mice also showed increased survival (17.5%) compared to KP after NSCLC induction (Suppl Fig. S2e). Altogether, the results suggest that pathogenic mtDNA mutations are deleterious to NSCLC.
mtDNA mutations impair NSCLC progression but not initiation
We hypothesized that the tumor burden in PGKP mice was lower than in KP mice because tumor initiation could be compromised. To test this hypothesis, NSCLC was induced in both models, and the lungs of the animals were collected at six rather than 15 weeks post-tumor induction to assess the frequency of tumor initiation. At six weeks, there was no statistically significant difference in tumor burden by histology and wet lung weight in both groups, suggesting that the initiation in the PGKP model is unaffected (Fig. 1f, g).
Since we did not observe a significant difference in tumor initiation, we investigated whether tumor progression was affected by performing immunohistochemistry (IHC) for the proliferation marker Ki67, which was reduced in PGKP compared to KP tumors (Fig. 1h, Suppl Fig. S3a). Additionally, we evaluated tumor cell viability using IHC for cleaved caspase-3 and TUNEL assays. Both methods revealed elevated labeling in PGKP tumors, indicating lower viability than KP tumors (Fig. 1i, j, Suppl Fig. S3a). These results showed that the accumulation of mtDNA mutations reduced tumorigenesis by decreasing tumor cell proliferation and viability, likely contributing to the increased survival of PGKP mice with NSCLC.
PolG D256A elevates mtDNA mutations without affecting nuclear DNA
We investigated whether an increased mtDNA mutation burden can indirectly promote mutations in nuclear DNA (nDNA), which has been proposed as a means of promoting cancer and aging23,43. Since the tumors of PGKP animals presented elevated levels of mtDNA mutations (Fig. 1b), we generated lung tumor-derived cell lines (TDCLs) from young and old KP and PGKP mice, and then we used these TDCLs to perform mtDNA genome and whole exome sequencing (Fig. 2a). The complete loss of sequencing read coverage in the floxed exons of Trp53 confirms the purity of NSCLC KP and PGKP clones, showing the absence of non-tumor cells (Suppl Fig. S4a). Additionally, the clonality of these TDCLs can be inferred from the somatic mutation allelic frequencies (AF), which display distinct peaks near 0.5 (indicating heterozygous mutations) and 1.0 (indicating homozygous mutations due to loss of heterozygosity [LOH]) (Suppl Fig. S4b). Moreover, the pattern of AF distribution is consistent with one dominant clone in each TDCL. The levels of SNVs and indels in the mtDNA of PGKP TDCLs were higher than in KP TDCLs, independent of whether the tumor cells were generated from young or old animals (Fig. 2b, c). Despite the increased mtDNA mutation burden found in PGKP TDCLs, the levels of nDNA mutations are similar between PGKP and KP TDCLs (Fig. 2d, e). These findings suggest that impaired tumorigenesis of PGKP animals results from alterations in mitochondrial function caused by pathogenic mutations in mtDNA and not in nDNA.

a Schematic illustrating the independently generated TDCLs from NSCLC tumors in KP, PGKP, young, and old animals. b, c The mtDNA mutation burden is represented by SNVs and Indels in PGKP and KP TDCLs. The n for each group is informed in A d,e The nDNA mutation burden is represented by SNVs and Indels. Only the relevant statistics are displayed in the figure. f Growth curve of KP and PGKP TDCLs (Incucyte) derived from young animals. This curve is a combination of four experiments. Supplementary Fig. S11 shows individual growth curves. g The confluence of TDCLs at the end of 80 h. This experiment represents the last time point of the previous growth curves. h The viability of the TDCLs was measured during 13 passages twice. Each point represents one viability test per clone using four KP and eight PGKP clones. i The allograft tumor growth assay was performed using three KP and five PGKP clones in C57/B6 mice. The lines represent the mean values ± SEM. j–l Flow cytometry assessing mitochondrial mass and mitochondrial membrane potential in TDCLs. The graph depicted the data of three independent experiments using four KP and eight PGKP clones m, n OCR and ECAR were measured to evaluate mitochondrial respiration and glycolysis in TDCLs. The graph represents the average of two independent experiments using four KP and eight PGKP clones. Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) and 2-deoxy-glucose (2-DG) were employed in the study. In H, individual sample distributions are represented by dots, while the gray bar indicates the median ± SEM. Boxplot (Min-to-Max) displaying the interquartile range (box), median (line inside the box), and whiskers extending from the minimum to the maximum observed values. Two-tailed t-tests determined P values for G, H, J, K, L, M, and N, while one-way ANOVA with Tukey correction was used for Fig. s B - E. Significance levels are denoted as *≤0.05, **≤0.01, ***≤0.001, and ****≤0.0001. For detailed individual P values, please refer to the Source Data table. The Figures a and i were created in Biorender. Cararo Lopes, E and White, E (2025).
High mtDNA mutation burden leads to the accumulation of dysfunctional mitochondria
To address whether the defective tumor growth seen in PGKP mice was tumor cell-intrinsic rather than a host effect, we evaluated the growth and viability of PGKP and KP TDCLs in vitro. PGKP TDCLs exhibited reduced proliferation and viability compared to KP (Fig. 2f–h). Furthermore, PGKP demonstrated slower growth than KP TDCLs when implanted into syngenetic C57B/6 mice that lack the PolGD256A allele (Fig. 2i). This indicates that PGKP tumor cells inherently possess defects in cell proliferation and viability, replicating the same phenotype observed in induced PGKP tumors (Fig. 1h-j, Suppl Fig. S3a). It is noteworthy that the PGKP phenotype persisted irrespective of the experimental strategy employed, whether in vitro or in vivo, and whether conditional or allograft models were utilized.
We used flow cytometry to assess the mitochondrial mass and membrane potential of NSCLC cells. PGKP TDCLs exhibited increased mitochondrial mass compared to KP TDCLs. This observation aligns with findings from mouse tumors, where higher levels of TOM20, a mitochondrial marker, indicated greater mitochondria accumulation in PGKP tumors. (Fig. 2j; Suppl Fig. S3b). However, their mitochondrial membrane potential was similar (Fig. 2k), suggesting that PGKP tumor cells accumulate dysfunctional mitochondria (Fig. 2l).
To directly evaluate mitochondrial function, we measured the oxygen consumption rate (OCR) in TDCLs (Fig. 2m). As suggested by cytometry data, mitochondrial function in PGKP tumor cells was compromised, presenting with a lower basal and maximum respiration expected to correspond to lower production of ATP by oxidative phosphorylation (OXPHOS).
Tumor cells with lower mitochondrial respiration rely on glycolysis to produce ATP and sustain their growth44,45. We, therefore, measured the extracellular acidification rate (ECAR) of TDCLs (Fig. 2n). As expected, the glycolytic activity was increased in PGKP compared to KP TDCLs. Interestingly, PGKP TDCLs had a much lower level of glycolytic reserve than KP TDCLs, indicating maximizing glycolysis in PGKP cells for their metabolic needs. These results demonstrate that the elevated mtDNA mutation burden in NSCLC cells results in the accumulation of dysfunctional mitochondria with respiration defects and enhanced glycolysis.
PolG mice deplete circulating glucose, which contributes to defective tumor growth
Despite the tumor cell-intrinsic defects from the PolG mutation, mitochondrial dysfunction in PGKP mice may lead to a systemic metabolic deficit that impairs tumor growth. To test this hypothesis, we assessed metabolic parameters in blood plasma and solid tissues (Fig. 3A), comparing PGKP with KP in young and old mice. As controls for PGKP and KP, we utilized cancer-free animals, specifically PolG (PolGD256A/D256A; Trp53fl/fl) and PolG wild type (Trp53fl/fl), respectively.

A Experimental design schematic: Old and young KP, WT, PGKP, and PolG animals were infected with adenovirus-Cre. After 15 weeks, euthanasia was performed, and plasma, NSCLC tumors (only KP and PGKP develop NSCLC), and normal lungs (from WT and PolG) were collected. The gathered material underwent pool-size metabolomics and glucose meter measurements. B Blood glucose levels. C Survival curve and median survival of KP and PGKP animals under a high carbohydrate diet (70% sucrose). D, E Representative volcano plot and pathway impact analysis for pool-size metabolomics comparing old KP and PGKP tumors. f, g Serine and GSH levels are shown for normal lungs (WT and PolG) and NSCLC tumors (PGKP and KP) at different ages. n indicates the number of animals for solid tissue for both, serine and GSH. H, I Serine and GSH plasma levels. n indicates the number of animals for plasma for both, serine and GSH. j Survival curve and median survival of KP and PGKP animals under amino acid control (AA-crtl) and serine/glycine-free diet. In B and F-I, individual sample distributions are represented by dots, while the gray bar indicates the median ± SEM. Two-tailed t-tests determined P values for C (median survival), while one-way ANOVA with Tukey correction was used in B, C, and F-J. Log-rank (Mantel-Cox) calculated P values for survival curves C and J. Significance levels are denoted as *≤0.05, **≤0.01, ***≤0.001, and ****≤0.0001. Only the relevant statistics are displayed in the figures. For detailed individual P values, please refer to the Source Data table. The Figures A, C and J were created in Biorender. Cararo Lopes, E and White, E (2025).
Lower blood glucose levels were observed in KP animals with NSCLC after 15 weeks of initiation compared with control wild-type mice (Fig. 3B). In PGKP and PolG animals, alterations in blood glucose levels were more pronounced, suggesting a potential age-related trend, though this effect did not reach statistical significance. Moreover, it was reported that the PolG mutator mouse exhibits hypoglycemia in a starved condition46. However, we observed that the blood glucose levels of PolG and PGKP, mainly in old animals, were within the range of hypoglycemia even in a fed state (Fig. 3B)47.
We hypothesized that the metabolic changes imposed by the accumulation of mtDNA mutations and defective mitochondria in normal tissues of PGKP animals could decrease glucose availability to tumors, thereby contributing to a reduction in tumor growth. Therefore, we tested if dietary manipulation of glucose would rescue tumor growth and survival defects caused by PolG by using a high carbohydrate (HC) diet composed of 70% sucrose (Fig. 3c). We found that the HC diet eliminated the survival difference observed between PGKP and KP animals with NSCLC (Fig. 1c), suggesting that systemic glucose limitation in highly glycolytic tumors and tissues may contribute to defective NSCLC growth.
Defective mitochondria sensitized NSCLC to dietary serine/glycine starvation
We performed untargeted metabolomic profiling on tumors from PGKP and KP animals and their controls (Fig. 3A). Remarkably, when compared to PGKP tumors, KP tumors exhibited a significant increase in 3-phosphoserine, an SSP precursor that uses glycolytic intermediates (Fig. 3D). Furthermore, we observed reduced levels of aspartate in PGKP tumors, which serves as a biomarker of electron transport chain inhibition. This can be caused by the low ratio of NAD + /NADH, which promotes oxaloacetate reduction to malate, rendering oxaloacetate unavailable as a substrate for aspartate synthesis48. Consequently, aspartate deficiency may pose a challenge for nucleotide synthesis. Indeed, the pathway impact analysis showed that the alterations in PGKP compared with KP tumors were concentrated in energetic and anabolic pathways (Fig. 3E).
Considering the altered anabolic metabolism and reduced 3-phosphoserine in PGKP tumors, we examined the underlying metabolic mechanism and its impact on tumor growth. In KP mice, NSCLC tumors had higher serine levels compared to normal lung tissue from wild-type mice at any age, but the serine levels in PGKP tumors and normal PolG lung tissue were not significantly different (Fig. 3F). This suggests that an elevated level of serine in tumors is a distinctive feature of KP NSCLC that was absent from PGKP tumors. The GSH levels in PGKP tumors from old mice were notably lower compared to normal lungs from old PolG and old KP animals (Fig. 3G). These variations were not evident in the plasma, suggesting that alterations in serine and GSH are characteristics of tumors (Fig. 3H, I). These results suggested that limited substrate levels (e.g., glucose) in PGKP tumors may impair the synthesis of essential downstream metabolites, making them more sensitive to external nutrient reduction, such as serine. To test this hypothesis, we fed KP and PGKP animals with a control amino acid diet (AA-crt) and a serine/glycine-free diet and monitored the survival of these mice post-NSCLC initiation. The KP animals fed with these different diets had similar median survival (Fig. 3J). However, PGKP animals with NSCLC showed 16% improved survival under serine/glycine starvation compared to PGKP animals on the AA-crtl diet and 25% compared to KPs on both diets (Fig. 3J). This suggests that mitochondrial dysfunction in PGKP tumors hinders serine synthesis from glucose, increasing dependency on dietary serine for tumor growth.
PGKP tumors synthesize less serine from glucose
Serine is a non-essential amino acid that can be synthesized from glucose and obtained from food sources (Fig. 4a)49. We showed that plasma glucose and tumor serine levels were lower in PGKP than in KP animals. We then investigated if KP and PGKP tumors differed in using glucose to make serine. All experimental groups were pre-fed an AA-crt or ser/gly-free diet for one week, then NSCLC was induced, and the same diets continued for 12 weeks. Then, we performed in vivo isotope tracing with [U-13C]-Glucose and measured tissue metabolic changes (Fig. 4b)8,9,50,51.

a Schematic depicting glycolysis→TCA with 13C distribution on pathway intermediates using [U-13C]-glucose. Intermediates include Fructose 6-phosphate (F6P), glyceraldehyde 3-phosphate (GAP), 3-phosphoglycerate (3PG). b Schematic experimental design for in vivo isotope tracing with [U-13C]-glucose infusion. KP, PGKP, WT, and PolG animals were infected with adenovirus-Cre and maintained on AA-crt or serine/glycine-free diets for 12 weeks. After fasting for six hours, animals underwent 2h30 of [U-13C]-glucose infusion and were then euthanized. Muscle, liver, plasma, and tumors or lungs were snap frozen. The collected samples were subjected to pool-size metabolomics, and tissue metabolite enrichment or isotopologue were normalized by the plasma glucose enrichment of the corresponding animal. c, d Dilution of 13C across the first cycle of glycolysis→TCA intermediates in normal (cancer-free) lungs and NSCLC tissues of mice infused with [U-13C]-glucose. e Statistical analysis of the main altered metabolites enrichment presented in D. f Serine M + 3 fractions (first turn of glycolysis→SSP). g Total pool size levels of serine in lung, tumor, and their matched plasma. The experiment was conducted with the following number of animals per group: WT = 5, WT-SG = 7, PolG = 6, PolG-SG = 6, KP = 5, KP-SG = 7, PGKP = 7, and PGKP-SG = 8. For animals with tumors, metabolites were extracted from two independent tumors per animal. For animals without tumors, metabolites were extracted from two independent lung lobes per animal. Data is shown as ± SEM. For all graphs, the P values were determined using one-way ANOVA with Tukey correction. P values are indicated as ≤0.05*, ≤0.01**, ≤0.001***, and ≤0.0001****. For detailed individual P values, please refer to the Source Data table. The Figure b was created in Biorender. Cararo Lopes, E and White, E (2025).
The 13C enrichment downstream of glucose in the lungs of wild-type animals was progressively diluted (Fig. 4c). Curiously, wild-type lungs had less 13C labeling in glycolytic and TCA cycle intermediate metabolites than PolG lungs, suggesting that PolG animals have altered glucose metabolism and/or increased uptake of these isotopologues from the bloodstream in normal lungs. Indeed, the levels of glucose M + 6 in PGKP tumors are higher than in KPs and increase even further during serine/glycine starvation (Suppl Fig. S5a). We also observed this behavior in vitro, where the consumption of glucose is also elevated in PGKP tumor cells in comparison with KP cells (Suppl Fig. S5b).
The metabolic profile in tumors markedly contrasted with that of normal lungs. The 13C labeling fraction of glycolytic intermediates showed notable similarity between PGKP animals on both diets and KP animals with the AA-ctr diet (Fig. 4d). However, labeled intermediates like pyruvate, lactate, and alanine were lower in KP tumors under ser/gly starvation (Fig. 4e), indicating that KP tumors induce adaption to glucose metabolism, while PGKP tumors lost this flexibility.
Alterations in glucose metabolism can directly affect the synthesis of serine, which requires glycolytic intermediates and NAD+ to be synthesized. Serine M + 3 is the product of the first turn of the SSP from glycolysis (Fig. 4a). The labeling fraction of serine M + 3 in all tissues of cancer-free PolG animals subjected to ser/gly starvation tends to be higher than that from the AA-crt diet; whereas in wild-type mice, except for lung tissues, the level of serine M + 3 is lower in ser/gly-starvation compared to AA-crt diet (Fig. 4f). Nevertheless, this serine M + 3 pattern was altered in NSCLC. Serine M + 3 levels in KP tumors were higher than in any other tissue or condition. However, PGKP tumors showed significantly reduced 13C labeling fraction from glucose to serine M + 3 compared to KP tumors, indicating less glucose use for serine biosynthesis. Bloodstream serine levels, however, were unaffected by serine/glycine starvation in NSCLC animals (Fig. 4g). Therefore, the decreased serine levels in PGKP tumors (Figs. 3F, 4g) could be attributed to the reduced de novo serine synthesis from glucose. These findings suggest that pathogenic mtDNA mutations and ETC defects increase glucose dependency for glycolysis while suppressing serine synthesis, increasing the need for exogenous serine and glycine to support anabolic metabolism and tumor growth.
Low NAD + /NADH levels may fail to adequately sustain both SSP and glycolysis in tumors with respiration defects
To determine if glucose use for serine production differed between KP and PGKP TDCLs, we performed untargeted metabolomics (Suppl Fig. S5c). PGKP TDCLs showed a distinct profile compared to KP TDCLs, with significant changes in glycolysis and SSP intermediates. Pathway impact analysis indicated that differences were focused on glycolysis, serine/glycine, and purine/pyrimidine metabolism pathways (Suppl Fig. S5d), suggesting mitochondrial dysfunction in TDCLs altered glycolysis flow to the SSP.
These results align with in vivo observations, demonstrating that PGKP tumor cells depend more heavily on external glucose to sustain glycolysis and its downstream pathways as SSP. This is underscored by the lactate levels in the PGKP medium, which are almost three times higher than those in the KP medium after 72 h of incubation (Suppl Fig. S5b, e). However, despite this elevated glucose usage, glucose was not depleted in either KP or PGKP cultures after the same period.
To better evaluate nutrient sources, we conducted a kinetic experiment in vitro, extracting metabolites from TDCLs at three-time points without replacing the culture medium (Fig. 5a). Most glycolytic intermediates in the preparatory phase (Glucose-6P→Glyceraldehyde-3P) were higher in PGKP than in KP TDCLs after 24 h, but their levels decreased over time. In contrast, these intermediates increased over time in KP TDCLs (Fig. 5b). Interestingly, intermediates in the pay-off phase (3-Phosphoglycerate→Pyruvate) showed a different pattern, starting from the GAP → 1,3BPG step, which is also a hub for the SSP. At this point, most of these intermediates were in lower levels in PGKP than in KP TDCLs. The oxidation of GAP is catalyzed by the enzyme GAPDH, which requires NAD + 52. Notably, GAPDH is subject to allosteric inhibition by NADH53,54. In PGKP tumor cells, NADH levels are consistently elevated when compared to KP, leading to a low NAD + /NADH ratio (Fig. 5c). This disparity likely accounts for the observed glycolytic pattern.

a The experimental design of pool-size metabolomic kinetics. The culture medium was not changed during the 72 h. b Kinetic of key metabolic intermediates of glycolysis and SSP. Dihydroxyacetone phosphate (DHAP), 1,3-Biphosphosglycerate (1,3BPG), 3-Phosphohydroxypyruvate (3PHP), and phosphoenolpyruvate (PEP). c Kinetics of NAD + , NADH, and the resulting NAD + /NADH ratio over time. d adenylated energy charge variation over time. e, f KP and PGKP TDCLs growth curve (Incucyte) under glucose and serine/glycine starvation. The control graph depicted in panel F is a projection of Fig. 2f. This curve is a combination of two and three experiments respectively. Refer to Suppl Fig. S11 for individual growth curves. g, h Kinetics of metabolic intermediates in Purine and Pyrimidine metabolisms. Green-shaded intermediates exemplify metabolites involved in nucleotide synthesis, while those in gray represent metabolic intermediates associated with nucleotide degradation. i Metabolites of the pentose phosphate pathway and sugar nucleotides. UDP-glucose (UDP-Glc). Uridine diphosphate N-acetylgalactosamine (UDP-GalNac). j GSH, GSSG levels, and their ratio variation over time. All experiments used four KP and eight PGKP-independent clones. Data is shown as ± SEM. P was calculated using one-way ANOVA with Tukey correction and indicated as ≤0.05*, ≤0.01**, ≤0.001***, and ≤0.0001****. For detailed individual P values, please refer to the Source Data table. The Figure a was created in Biorender. Cararo Lopes, E and White, E (2025).
Over time, acetyl-CoA gradually accumulated in PGKP TDCLs. The reduced citrate levels suggested limited condensation of oxaloacetate with acetyl-CoA, hindering the TCA cycle flow (Suppl Fig. S6a). Additionally, nearly all TCA cycle intermediates were significantly lower in PGKP compared to KP TDCLs, confirming TCA cycle dysfunction in PGKP tumor cells, as supported by the reduced OCR (Fig. 2m). Moreover, the low NAD + /NADH ratio may further impede the proper functionality of the TCA cycle55, potentially affecting the fitness of PGKP tumor cells. As a result, PGKP tumor cells have defective energetics over time without replenishment of external nutrients, which ultimately was reflected in decreased energy charge (Fig. 5d).
The energetic and glycolysis alterations suggested that PGKP TDCLs might depend more on glucose. To test this, we removed glucose from the culture media (Fig. 5e, Suppl Fig. S6b). While KP TDCLs showed slightly reduced proliferation, most clones reached at least 70% confluence by 80 h. In contrast, PGKP TDCLs showed complete growth impairment, confirming that NSCLC cells with dysfunctional mitochondria are highly dependent on glucose. Remarkably, as in vivo (Fig. 3D), PGKP TDCLs exhibited significantly low levels of 3-phosphoserine (Suppl Fig. S5c). This finding underscores the heightened dependence of PGKP TDCLs on external glucose, serine, and glycine, potentially affecting downstream metabolic pathways like the SSP. Notably, the initial committed step in the SSP, which necessitates NAD + , is catalyzed by the enzyme phosphoglycerate dehydrogenase (PHGDH)56,57, thereby implicating a potential link between low NAD + /NADH ratio and reduced serine synthesis. To test this hypothesis, we starved TDCLs of serine and glycine (Fig. 5f, Suppl Fig. S6b). Proliferation in KP TDCLs was slightly repressed (about 15%) in serine/glycine-free medium, but PGKP TDCLs decreased by nearly 60%. Thus, respiration defects in NSCLC increased dependency on extracellular glucose, serine, and glycine, further exacerbated by low NAD + /NADH suppressing the SSP.
Defects in respiration increase dependency on external sources of glucose, serine, and glycine to maintain GSH and nucleotide pools
Serine produced from glucose through glycolysis is a main source of newly synthesized glycine, extensively used in anabolic processes like 1 C-metabolism58,59. Alterations in one-carbon pools can affect nucleotide availability due to missing intermediates in the methionine cycle, folate cycle, and purine/pyrimidine synthesis18,60.
The intermediate levels in purine/pyrimidine synthesis differed significantly between PGKP and KP TDCLs (Fig. 5g, h). In PGKP TDCLs, biosynthetic intermediates were very low or decreased over time, while intermediates representing degradation increased. Similar trends were observed in other anabolic metabolites, such as pentose phosphate pathway intermediates and nucleotide sugars (Fig. 5i). In contrast, in KP TDCLs, newly synthesized metabolites increased while metabolites representing degradation remained stable or decreased. This suggests NSCLC cells boost de novo synthesis of anabolic metabolites as external nutrients deplete. However, NSCLC cells with mitochondrial dysfunction decreased de novo synthesis and failed to maintain essential anabolic metabolite levels, such as nucleotides.
Another important downstream product of the SSP and synthesis of glycine is GSH. GSH helps maintain redox homeostasis and is also synthesized in response to ROS generation61,62,63. Unlike nucleotides, GSH was higher in PGKP TDCLs (Fig. 5j), likely due to ROS accumulation (Sup Fig. S6c). Interestingly, in PGKP cells, glutathione disulfide (GSSG) levels drop between 24 h and 48 h, in contrast to the modest increase observed in KP cells. These results suggest that the turnover between GSH and GSSG may be faster and/or that GSH synthesis could be elevated in PGKP cells, likely due to their unique physiological conditions. However, mitochondrial respiration defects and SSP suppression are expected to hinder GSH synthesis by limiting substrate availability and ATP production, both essential for GSH formation. To explore this hypothesis, we performed in vitro isotope tracing by feeding TDCLs with [U-13C]Glucose in both complete and serine/glycine-free media to evaluate the fate of glucose-derived carbons in these tumor cells. (Fig. 6a and Sup Fig. S7a).

a Schematic of carbons flux from glucose to serine, glycine, and GSH using the glycolysis→SSP → 1C-metabolism axes. M + 0 to M + 7 represents the number of 13C present in the isotopologue. b Pool size levels of each isotopologue of serine, glycine, and GSH (labeled fractions + M + 0) in TDCLs cultivated in both control and serine/glycine-starved medium. The statistical analysis presenting the fractions of the isotopologues is outlined in Supp. Fig. S7. c This graph displays the ratio between the isotopologues GSH M + 2 and Serine M + 3. d NAD+ and NADH levels and resulting NAD + /NADH ratio of KP and PGKP TDCLs in complete and serine/glycine-free medium. The graphs (b-d) display data from the median of three technical replicates of three KP and five PGKP clones, as shown in Supp. Fig. S7a. e Growth of KP and PGKP TDCLs cultivated for 72 h in both control and serine/glycine-free medium, with supplementation (or without) of GSH (5 mM) and nucleosides. The graph depicts the fold change of the control group compared to the supplemented group. The absolute quantifications are presented in Suppl Fig. S11. In B data is shown as ± SEM. In C-E boxplot (Min-to-Max) displays the interquartile range (box), median (line inside the box), and whiskers extending from the minimum to the maximum observed values. For Fig. C, the P values were determined using one-way ANOVA with Dunnett’s multiple comparisons test using KP as control. For Fig. D, the P values were determined using one-way ANOVA with Tukey correction. Two-tailed t-tests determined P values for E. P values are indicated as ≤0.05*, ≤0.01**, ≤0.001***, and ≤0.0001****. For detailed individual P values, please refer to the Source Data table.
In regular media, KP cells used more glucose than PGKP TDCLs to synthesize serine and glycine (Fig. 6b, Sup Fig. S7b, c). Additionally, PGKP TDCLs showed impaired synthesis of GSH from glucose (Fig. 6b, Sup Figure S7d), indicating that mitochondrial dysfunction affects glucose utilization for serine, glycine, and GSH production. Under serine/glycine starvation, glucose usage increased in KP TDCLs, partially restoring internal serine and glycine levels and fully restoring GSH levels (Fig. 6b, Sup Fig. S7d enrichment).
The reliance of NSCLC cells on glucose to synthesize GSH during serine/glycine starvation is evidenced by the increased levels of GSH M + 2 (Gly or Glu) and M + 4 (Gly+Glu) isotopologues in KP cells (Fig. 6b and Sup Fig. S7d). Additionally, GSH synthesis from serine can be inferred from the GSH M + 2/Ser M + 3 ratio (Fig. 6c)50, which highlights PGKP’s dependence on serine (and consequently glucose) for GSH synthesis. However, during serine/glycine starvation, this optimal ratio is not achieved in PGKP, preventing full recovery of GSH levels. Moreover, while KP cells consistently show a higher contribution of glucose to pyrimidine synthesis regardless of the medium used, they significantly increase their reliance on glucose for purine synthesis during serine/glycine starvation. In contrast, PGKP cells fail to exhibit this adaptive response (Suppl. Fig. S8a).
Although KP cells consume serine more rapidly and in greater quantities than PGKP cells, the serine levels in the medium are drastically decreased in either case over a 72 h period, which might impact cell proliferation (Suppl Fig. S8b, c). Furthermore, KP cells release significantly higher levels of newly synthesized serine (serine M + 3) derived from glucose into the medium than PGKP cells. Under serine/glycine starvation conditions, NSCLC cells further increase their release into the medium (Sup. Fig. S8d).
While both KP and PGKP cells demonstrate an adaptive increase in SSP activity under serine/glycine starvation, KP cells release serine at levels three times higher than PGKP cells under the same conditions. This finding suggests that NSCLC cells with mitochondrial dysfunction are unable to produce sufficient serine to sustain robust proliferation. This underscores NSCLC’s strong dependence on serine and its ability to replenish serine pools from glucose during serine/glycine starvation, a process that suggests being tightly linked to mitochondrial function. Once again, the NAD + /NADH ratio emerges as the critical factor in this process (Fig. 6d). NAD+ levels were lower in PGKP than in KP TDCLs when deprived of serine/glycine. This supports the hypothesis that NSCLC cells with respiratory defects have an NAD + /NADH imbalance exacerbating redox activity impairment, particularly during serine/glycine starvation.
Under serine starvation, p53−/− tumor cells rapidly increase glycolytic flux into the TCA cycle as an adaptive response64. Similarly, KP TDCLs show elevated levels of most TCA cycle intermediates, though notably without an increase in glucose carbon consumption. This observation is evidenced by the rise in total TCA cycle intermediates levels (ion counts) with constant not labeled fractions (M + 0) (see Suppl Fig. S9). Collectively, these findings suggest that, during serine/glycine starvation, only cells with adequate mitochondrial function can elevate TCA cycle intermediates levels to maintain energetic balance6. In summary, NSCLC cells under nutrient-deprived conditions preserve their energetic and anabolic balance by maintaining adequate NAD + /NADH levels. Tumor cells with impaired mitochondrial respiration or those subjected to serine/glycine starvation struggle to achieve this balance, as a proper NAD + /NADH ratio is crucial for supporting de novo serine synthesis56,65,66,67,68.
In an attempt to rescue PGKP TDCL growth under serine/glycine starvation, we supplemented the TDCLs with GSH and nucleosides. This significantly improved growth compared to KP TDCLs, indicating these metabolites are key fates for external serine/glycine (Fig. 6e). Curiously, GSH supplementation also improved KP TDCL growth under serine/glycine starvation, but to a lesser extent. Additionally, the GSH levels in serine/glycine-starved KP TDCLs tend to rise, surpassing even those in KP cells cultured in a complete medium (Fig. 6b; Supp Fig. S7d). Moreover, their reliance on glucose for GSH synthesis notably escalates compared to any other group (Fig. 6b higher enriched GSH, Suppl Fig. S7d lower M + 0 fraction). This data suggests that NSCLC cells rely on external serine and glycine to maintain optimal GSH levels, a need exacerbated by mitochondrial dysfunction. To connect external serine/glycine availability, ROS accumulation, and GSH levels, we measured ROS under control and serine/glycine-starved conditions (Suppl Fig. S10). Under serine/glycine starvation, KP tumor cells showed increased ROS levels after 72 h, unlike PGKP cells. This is likely due to the already elevated basal ROS levels in PGKP cells or because cells with higher ROS levels in the PGKP group did not survive, allowing us to measure ROS only in the surviving population. These results suggest that KP NSCLC controls ROS using external serine/glycine to synthesize GSH, switching to SSP under starvation as a compensatory mechanism. Moreover, serine/glycine starvation, most likely due to an accelerated TCA cycle (as showed elsewhere17,64,69 and here in KP TDCLs) and/or defects in mitochondrial respiration, generates even more ROS to compensate for the lack of glucose that might be deviated to SSP and, thereby, nucleotides and GSH synthesis17.
Discussion
Mitochondria play a major role in many cellular functions such as energy generation, biosynthesis (e.g., amino acids, iron-sulfur clusters, heme), redox control, differentiation, and signaling22,69,70, but the role of mitochondria in cancer is not well understood11,63. A subset of tumors, mostly arising in the kidney, are characterized by pathogenic mtDNA mutations71,72,73. In these tumors, pathogenic mtDNA mutations are often homoplastic and appear to be founder events, suggesting tumorigenesis can be initiated by mitochondrial function loss. Other notable examples include thyroid and colon cancers, which also show a high burden of mtDNA mutations and an accumulation of dysfunctional mitochondria. In colon cancer, for instance, the accumulation of mtDNA mutations drives metabolic adaptations in tumor cells, enhancing their fitness and significantly promoting tumorigenesis while reducing the lifespan of affected mice4,28,74. However, in most of these cases, the specific mechanistic role of mtDNA mutations remains poorly understood. Now that we know that lung cancers rely on mitochondrial function, we have identified a metabolic vulnerability that was not previously known and can be therapeutically exploited.
Most cancers, however, appear to preserve mitochondrial function by selecting against the accumulation of pathogenetic mtDNA mutations28,75. There are also examples where tumor cells engineered to have mtDNA eliminated reacquire mtDNA from their host76. Indeed, knocking out the essential mitochondrial transcription factor Tfam results in impaired tumor growth63. Given mitochondria’s role in energy generation, biosynthesis, redox control, and signaling, most tumors likely benefit from maintaining mitochondrial function. This suggests distinct mitochondrial roles in various cancers: most rely on them, while some rare cases may initiate tumorigenesis with mitochondrial function loss. We sought to identify which tumors benefit from mitochondrial function and what aspects of their function are important in cancer.
Tumor growth has been found to require the ETC to oxidize ubiquinol, which is essential to drive the oxidative TCA cycle, and dihydroorotate dehydrogenase (DHODH) activity essential for de novo pyrimidine synthesis77,78. Restoring NAD+ levels in respiration-defective tumor cells via Lactobacillus brevis NADH oxidase (LbNOX), which mimics complex III activity, fails to rescue tumor cell growth68. An important prediction from our work is that rescuing the NAD + /NADH, rather than just elevated NAD+ levels, balance by expressing the NADH oxidase LbNOX will restore metabolic function, which is the subject of ongoing work.
We found that engineering NSCLCs to accumulate pathogenic mtDNA mutations via a PolGD256A allele significantly impairs tumor growth and extends mouse survival, indicating reliance on mitochondrial function. These PolGD256A/D256A mutator tumors and TDCLs show decreased proliferation and viability. To determine the cause of these defects, we examined metabolic alterations in the tumors and TDCLs. The mtDNA mutations caused expected glucose dependency and glycolytic metabolism with impaired respiration but also decreased SSP metabolites necessary for glutathione and nucleotide synthesis. Indeed, PolGD256A/D256A mutator tumor cells were more dependent on exogenous serine/glycine for survival. Supplementation with nucleosides and glutathione partially rescued their growth and survival. Finally, a serine/glycine-free diet did not affect mice with NSCLC but further impaired tumorigenesis and extended survival in those with pathogenic mtDNA mutations. This shows that mitochondrial dysfunction creates a metabolic liability, increasing glucose reliance due to respiratory impairment. This respiratory impairment causes the accumulation of NADH and an imbalanced NAD + /NADH ratio, which is crucial for maintaining the redox balance necessary for both glycolysis and the SSP, affecting the availability of 1 C units crucial for biosynthetic reactions such as purine synthesis68,79,80. Serine is the primary source of 1 C units, and NSCLC cells with respiratory deficiencies develop a critical dependence on external serine for growth. Our findings reveal that the combination of mitochondrial dysfunction and serine/glycine starvation induces a unique metabolic stress, which we can define as “mitochondria-driven serine deprivation stress.” Moreover, SSP impairment limits nucleoside and GSH production, causing redox stress and reduced tumor growth (Fig. 7). A high-carbohydrate diet, compensating for limited glucose to fuel glycolysis and the SSP, reversed the survival advantage of PolGD256A/D256A mutator NSCLC. Thus, NSCLC preserves mtDNA to avoid metabolic liabilities from respiration defects, including increased glucose dependency limiting serine uses for biosynthesis. How respiration defects lead to suppression of the SSP is likely related to elevation of NADH and lowering of the NAD + /NADH ratio. Elevated NADH levels can impair glycolysis, as Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) is allosterically inhibited by NADH53,54. This inhibition affects glycolysis and the availability of crucial substrates like 3-phosphoglycerate (3PG)81. Since a significant portion of 3PG is rerouted into the serine/glycine biosynthetic pathway82, an imbalanced NAD + /NADH ratio can directly influence the serine synthesis flux.

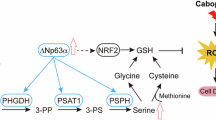
Graphical abstract illustrating the mechanisms discussed in this study. From top to bottom: NSCLC cells with functional mitochondria and no limitation of external substrates; the same cells under serine/glycine starvation; and NSCLC cells with impaired mitochondrial function combined with serine/glycine starvation. The size of the metabolite labels represents the relative increase or decrease of the indicated substrate. Green triangles denote increased levels of the substrate or phenotype, while red triangles indicate decreased levels. Red Xs represent the absence of the specified metabolite. This Figure was created in Biorender. Cararo Lopes, E and White, E (2025).
These findings raise a few important points. First, as metabolism is distinct across different cancers, do other cancers similarly depend on preserving mtDNA and mitochondrial function to fuel the SSP? Does the upregulation of the SSP alone sustain NSCLC growth and survival, or do these tumor cells essentially depend on a balanced NAD + /NADH ratio to sustain optimal growth? Upregulation of the SSP through amplification or upregulation of PHGDH expression is seen in some cancers, which illustrates the functional importance of this pathway7,83,84, but whether this is in addition to the preservation of mitochondrial function or an adaptation to mitochondrial deficiency is poorly understood. Second, our findings suggest that a deeper understanding of cancer metabolism can reveal novel metabolic dependencies for targeted chemotherapy. Third, we show that a deep understanding of cancer metabolism can also reveal metabolic vulnerabilities that can be targeted by dietary manipulation. While diet as therapy is not new, identifying specific diets for cancer prevention and treatment is worth further exploration. New approaches could include combining the mitochondrial Complex I inhibitor Metformin with a serine/glycine-free diet in NSCLC or treating rare tumors with pathogenic mtDNA mutations (as evidenced by ND6KP model) using dietary serine/glycine deprivation or SSP inhibitors. Although we have not yet characterized the metabolism of MT-ND6 tumors, this analysis will be crucial—not only for advancing cancer research but also for gaining deeper insights into the severe toxicities associated with mitochondrial diseases.
Methods
Genetically engineered mice models
All animal care procedures, experiments, and maintenance complied with the Rutgers University Institutional Animal Care and Use Committee (IACUC). The animals were kept in the 12 h light/night cycles at 22-24 °C and 40-60% of humidity. The KP mouse model (The Jackson Laboratory RRID: IMSR_JAX:032435)31 was cross-bred with the PolG mouse model (The Jackson Laboratory RRID:IMSR_JAX:017341) to generate the PGKP mouse model. For colony maintenance, we pair heterozygous (PolGD256A/+) males with wild-type females (PolG+/+).
To establish experimental groups, we breed homozygous males (PolGD256A/D256A) with heterozygous females (PolGD256A/+). The homozygous males are used for breeding only within the first three months, as their breeding performance declines progressively, and they become infertile by approximately four months of age. Notably, homozygous females are never used for breeding, as they are infertile in our cohorts.
The ND6 mouse model (generously provided by Dr. Douglas C. Wallace)42 was cross bred with the KP mouse model to generate the ND6KP mouse model. NSCLC is initiated through intranasal infection with adenovirus-Cre 4 × 107 pfu per mouse at 8–10 weeks (young) and 10 months old (old). We proportionally infected male and female mice, as there were no significant differences between the sexes. All animals in our colony have wild-type nicotinamide nucleotide transhydrogenase (NNT).
For survival analysis, the animals were infected and monitored weekly until the 10th week. After this period, they were monitored daily until they reached the humane endpoint (body condition score 2). The animals’ weight and food intake were measured weekly (when indicated) until the end of the experiment. The controls cancer-free animals PolG (PolGD256A/D256A; Trp53fl/fl) and PolG wild type (Trp53fl/fl), respectively were infected with adeno-Cre, but they did not develop NSCLC due to the lack of KRasLSL-G12D/+.
Purified diets
The animals were weighed one week before tumor induction. In the same period, special food was provided and kept until the end of the experiment. The food intake and animal weight were taken weekly. The special diets were obtained from Envigo according to the product code: Serine/Glycine-free diet (TD.180296), Amino acid defined (AA-crt) diet (TD.01084), and 70% High Carbohydrate diet (TD.220722).
Blood glucose measurement
Fresh blood was collected from the animals’ tails and immediately submitted to measurement using FreeStyle Optimum Neo glucometer (Abbott Laboratories) to determine blood glucose levels. The blood was always collected between 1–3 p.m. in ad libitum conditions.
Tumor burden quantification
For tumor area measurements, H&E-stained lung samples were imaged at Rutgers Cancer Institute of New Jersey Biomedical Informatics shared resource using an Olympus VS120 whole-slide scanner at 20X magnification. The image analysis protocol was custom-developed on the Visiopharm image analysis platform to identify tissue area and number and compute tumor burden based on semiautomatically detected tumors as described previously45. For tumor number, the prepared H&E-stained lung sections were analyzed using a Nikon Eclipse 80i microscope.
Cell lines generation
All KP and PGKP tumor-derived cell lines (TDCLs) were generated in-house directly from their corresponding mouse tumors. Mice were infected at 8–10 weeks of age (young group) or at 10 months of age (old group) and maintained for 15 weeks to allow lung tumor growth. After this period, tumors were harvested and dissociated, with cells initially cultured in dishes for five days before being re-seeded into flasks. Each clone was derived from a distinct lung tumor located in a separate lung lobe, ensuring the generation of independent clones. The TDCLs were cultivated in RPMI with 10% fetal bovine serum and 1% PenStrep solution. Additionally, serine/glycine-free RPMI (Teknova) was used as indicated.
Cell proliferation and viability assays
TDCLs were plated 5 ×104 cells per well in 6-well plates in RPMI conditions for 24 h. The next day, the medium was aspirated, washed with PBS, and changed for complete RPMI or serine/glycine-free RPMI plus the indicated treatments: 5 mM of reduced glutathione (Sigma) or nucleoside mix (EmbryoMax®Nucleosides 100x, Millipore). The cells were kept without medium change for 72 h, harvested, and then counted in triplicate using a Vi-CELL™XR cell counter and viability (Trypan blue) analyzer system with a cell viability assay using a flow cytometer (Beckman Colter). To generate Incucyte growth curves, TDCLs were plated 5 × 104 cells per well in 12-well plates in complete, glucose-free, or serine/glycine-free RPMI conditions for 80 h. For viability assays using Trypan blue in Zcounter, the KP (n = 4) and PGKP (n = 8) TDCLs clones were tested for 13 passages twice in a regular RPMI medium. All experiments and conditions were repeated at least twice. All experiments used at least three independent TDCLs clones per genotype.
Growth of TDCLs in mice
C57Bl/6 J male mice (The Jackson Laboratory) aging 8 weeks were injected (subcutaneously) with 105 cells per animal flank. The tumors were measured at least twice weekly until the animal reached the humane endpoint as determined by the protocol (by 1000 mm3 determined by π//6 x L x W x H).
Tissue Collection and Dissection
At the end of the experimental period, animals were euthanized, and tissues were carefully dissected. For KP and PGKP mice, tumors were isolated from the lungs to prevent contamination with non-tumor cells. In WT and PolG mice, the entire healthy lungs were collected to serve as controls.
WES and mtDNA genome sequencing
Total DNA from tissues from tumors n = 5, normal lung tissues n = 3, and normal pup lung tissue n = 2 from each genotype and age group were isolated. The healthy pup lung tissues were included solely to establish a baseline for mtDNA mutation burden and are not intended for comparison among the groups. The same procedure was adopted for TDCLs (KP n = 4 and PGKP n = 8 from each age group)
Library preparation was carried out using the Agilent SureSe-lectXT mouse mitochondrial custom enrichment protocol (Agilent Technologies). Each pooled sample was clustered and sequenced on an Illumina MiSeq instrument (Novogene Corp.) using two 150-base-pair (bp) paired-end reads and∼2.6 million reads per sample.
mtDNA genome and whole exome sequencing data analysis
The analysis of mtDNA genome raw fastq files used FastQC v0.11.985 for quality checks and Trimmomatic v0.4086 for read trimming. To align the reads to the reference genome GRCm38.p6, we employed bwa (Burrow-Wheeler Aligner) v0.7.17-r118887. The alignment data were then evaluated using Qualimap v2.388. We used samtools v1.3.1 to index and sort the resulting bam files for further processing. Duplicate marking was carried out using web-based gatk v4.1.8 (https://gatk.broadinstitute.org). To identify somatic SNVs and indels, we utilized gatk mutect2 with mitochondrial mode. To ensure the quality of the variants, we applied specific filters based on allele fractions (AF) > 0.01, approximate read depth (DP) > 2000, and the log 10 likelihood ratio score of variant existence (TLOD) > 10. For variant annotation, we employed VEP (Variant Effect Predictor)89. IGV v2.8.2, R, and Python were used for data visualization.
A similar pre-processing pipeline for whole exome sequencing was employed. However, for somatic single nucleotide variants (SNVs) and indel calling90, the gatk mutect2 tool in tumor-normal mode was applied. Variant annotation was performed using SnpEff v4_3t91. For post-processing, we applied the following filter settings: SNVs and indels were retained if they had a variant allele frequency of ≥10%, a minimum read coverage of 10x in both the tumor and normal samples at that specific position, a minimum of three reads supporting the variant allele in the tumor sample, and no reads supporting the variant allele in the matched normal tissue. To detect copy number variants, we employed CopyrightR v1.392. We also utilized R and Python to identify cancer-drive genes and visualize the data.
Histology, TUNEL and IHC
Lungs with tumors from each experimental group were harvested after 15 weeks of NSCLC initiation (except for the tumor initiation experiment that was harvested after six weeks), fixed in 10% buffer formalin solution overnight, and then transferred to 70% ethanol for paraffin-embedded sections. Tissue sections were deparaffinized, rehydrated, and boiled for 45 min in 10 mM pH 6 citrate buffer. All Paraffin-embedded blocks were submitted to be stained with hematoxylin-eosin (H&E) for quality check. Blank slides were used for IHC preparation with antibodies for TOM20 (Abcam, ab186735 1:200), Ki67 (Abcam, ab15580 1:250), and cleaved caspase 3 (Cell Signaling, 9661S 1:300). Paraffin-embedded slides were also used for the TUNEL assay and processed according to manufacturer instructions (Abcam, TUNEL Assay Kit - HRP-DAB ab206386). The prepared sections were analyzed using a Nikon Eclipse 80i microscope. ImageJ was used to extract optical density (CC3) and positive nuclei count (Ki67 and TUNEL) for quantification. We used six animals per group for immunohistochemistry (IHC) and TUNEL assays. For each animal, we randomly selected and imaged at least 12 distinct tumors, ensuring that each image represented a separate lung tumor. Quantification was then performed following the established protocol.
Assessment of oxygen consumption rate (OCR) and extracellular acidification ratio (ECAR measurement)
OCR in KP and PGKP TDCLs OCR was measured using a Seahorse Biosciences extracellular flux analyzer (XF24) as described previously93. Briefly, cells were seeded at 104 cells per well in the XF24 plates overnight before XF assay. Real-time OCR measurements were performed in RPMI, HBSS, and HBSS with 2 mM glutamine or 1 mM dimethyl-α-KG for 3 h, and measurements were taken every 15 min. Relative OCR (percentage) was normalized to the 0 min time point. ECAR and glycolysis rates were also measured using a Seahorse XF24, as previously described93. Briefly, cells were seeded at 104 cells per well in the XF24 plates overnight and then subjected to XF assays according to the manufacturer’s instructions to determine basal glycolysis, glycolytic levels, glycolytic capacity, and glycolytic reserve.
Flow cytometry
Cells were plated in 6 well plates and proceeded as indicated by each experiment. All samples were run in the Attune NxT Blue-Red-Violet (Thermo Fisher), collecting 3 × 103 events in at least three biological duplicates (independent clones) and in at least two independent experiments. MytoTracker green (Thermo) was used to measure mitochondria mass; MytoTracker red was used to measure mitochondria membrane potential according to manufacture specifications. CellROX™ Green Flow Cytometry Assay Kit (Thermo) was used to quantify the accumulation of total ROS according to the manufacturer’s instructions. The raw data were treated and analyzed by FlowJo® V10.2. to extract the Median Fluorescence Intensity (MFI) per sample.
Extraction of polar metabolites from TDCLs and conditioned medium
TDCLs were plated in 6 multiwell plates (Corning) and incubated (37 °C in 5% CO2) for 24 h h. After this period, the medium was replaced, and cells were further incubated and extracted at the indicated time points. Three wells were used for the metabolite extraction, and the other three were harvested using trypsin 0.25% to measure the wet cell volume using a PCV-packed cell volume tube (TPP). One aliquot of the medium (15 µL) of each well was collected and immediately introduced in extraction buffer 40:40:20 (Methanol: Acetonitrile: Water) with 0.05% formic acid allowed to rest on ice for 5 min. The metabolites extraction was performed by washing the wells twice with cold PBS, and 400 µL of extraction buffer 40:40:20 with 0.05% formic acid was added to each well. The plate was allowed to rest on ice for 5 min, and then the cells and buffer were scraped. The samples, extraction from medium and cells, were placed in a 1.5 mL microtube with 22 µL of 15% NH4HCO3 vortexed and centrifuged for 10 min at 15,000 g at 4 °C. 380 µL of the supernatant were collected and stored in −80 °C freezer until analysis by LC-MS.
For isotope tracing experiments, TDCLs were seeded in 6-well plates (Corning) and cultured in order to reach 60–80% confluence at the time of [U-¹³C]D-glucose labeling. After 24 h, the medium was replaced with a fresh culture medium, and the cells were incubated for an additional 72 h. Following this incubation period, the medium was replaced with fresh RPMI containing 2 g/L of [U-¹³C]D-glucose (Cambridge Isotope Laboratories), and cells were incubated for 12 h prior to metabolite extraction. For each condition, three wells were used for metabolite extraction, while the remaining three wells were harvested using 0.25% trypsin to measure wet cell volume, using PCV (packed cell volume) tubes (TPP). For Kinect release, the conditioned medium was collected at three time points every 4 h after the introduction of [U-13C]D-glucose, followed by metabolite extraction as previously described. To determine the absolute concentrations of glucose and serine in the culture medium, we extracted metabolites from fresh medium (both complete RPMI and serine/glycine-free RPMI). The complete medium contained 2 g/L glucose and 30 mg/L serine, while the serine/glycine-free medium contained only 2 g/L glucose. Metabolite extraction was then performed as outlined previously. The energy charge was calculated through the equation [ATP] + 1/2[ADP] / [ATP] + [ADP] + [AMP] using the ion counts provided by metabolomics.
Extraction of polar metabolites from solid tissues
20 to 30 mg of tissues were weighed and added to a 2 mL round-tipped microtube with a – 80 °C cold Yttria Grinding Ball per tube. The tissues were pulverized in CryoMill (Retsch) following alternating three cycles at 5 Hz for 2 min with three cycles at 25 Hz for 2 min. Buffer was added to each 2 mL microtube (sample weight x 40)/2 volume of the buffer) 40:40:20 buffer with 0.5% formic acid, samples were vigorously vortexed and incubated on ice for 10 min, vortexed, and incubated for an additional 10 min. After the samples were centrifuged for 10 min at 16,000 g at 4 °C, the supernatant A was collected and saved, and the pellets were submitted to re-extraction following the same procedure to generate supernatant B. Supernatant A and B were mixed and transferred to a clean 1.5 mL microtube with the appropriated volume of 15% NH4CO3. The samples were stored in a −80 °C freezer until analysis by LC-MS.
Extraction of polar metabolites from plasma
To extract polar metabolites from mouse plasma 40 µL of cold methanol was added to a 15 µL of mouse plasma in a 1.5 mL microtube. This mixture was vortexed for 10 seconds and incubated for 20 min in a −20 °C freezer. Samples were centrifuged for 10 min at 16,000 g at 4 °C. Next, supernatant A was collected in a new tube, and the pellet was saved for re-extraction. For re-extraction, the pellet was resuspended in 200 µL of 40:40:20 buffer, vortexed, and allowed to sit on ice for 10 min. The samples were centrifuged for 10 min at 16,000 g at 4 °C. Supernatant B was collected and mixed with supernatant A. This mixture was further processed to remove phospholipids using 1 mL Phenomenex (Phenomenex Inc.) tubes according to the manufacturer’s instruction and stored at −80 °C until analysis by LC-MS.
in vivo isotope tracing
KP and PGKP animals were put in an AA-crt or serine/glycine-free diet one week before NSCLC induction. After 11 weeks following NSCLC induction, the animals were subjected to aseptic surgical jugular vein catheterization (Instech, C20PV-MJV1301, 2 French, 10 cm) to implant the catheter to a vascular access button (Instech, VABM1B/25, 25 gauge, one-channel button) implanted under the back skin of the animal. The animals were left to recover for five days, according to the procedure previously described3. A fully recovered mouse was fasted for six hours and placed in a plastic harness (SAI Infusion Technologies), and the catheter was connected to an infusion pump (New Era Pump System) through a mouse tether and swivel system (Instech Laboratories). [U-13C]Glucose isotope tracer (110187-42.3 Cambridge Isotope Laboratories) was dissolved in sterile saline and infused at a rate of 3.5 nmol/g/min (0.1 μl/g/min) for 2.5 h without a priming dose. Mice were euthanized after infusion and serum, tumor, lung, liver, and muscle (gastrocnemius) were collected. For each animal, one aliquot of serum, liver, and muscle was used to extract metabolites. For lungs and tumors were used two aliquots from different lung lobes.
For the normalization of labeled isotopologues metabolites, ion counts per isotopologue were used to determine the percentage of each labeled isotopologue per metabolite (\({\sum }_{i=0}^{n}({{\rm{M}}}+i)=1\)). The obtained results were normalized by plasma glucose enrichment level determined by: \({\sum }_{i=1}^{6}(\left({{\rm{M}}}+i\right)*i)/6\).
LC-MS/MS analyses
Metabolomics Shared Resource performed the LC-MS/MS on a Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer coupled to a Vanquish Horizon UHPLC system (Thermo Fisher) with an XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μm particle size, Waters, Milford, MA). The HILIC separation used a gradient of solvent A (95%:5% H2O: acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, (pH 9.4) and solvent B (20%:80% H2O: acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4). The gradient was 0 min, 100% B; 3 min, 100% B; 3.2 min, 90% B; 6.2 min, 90% B; 6.5 min, 80% B; 10.5 min, 80% B; 10.7 min, 70% B; 13.5 min, 70% B; 13.7 min, 45% B; 16 min, 45% B; 16.5 min, 100% B; and 22 min, 100% B94. The flow rate was 300 μL/min. The column temperature was set to 25 °C. The autosampler temperature was set to 4 °C, and the injection volume was 5 μL. MS scans were obtained in negative and positive ionization modes with a resolution of 70,000 at m/z 200 and an automatic gain control target of 3 × 106, and m/z scan range of 72–1000. The itaconate was monitored by a PRM event (m/z 129.02@HCD53.33). Metabolite data was obtained using the MAVEN software package95 (mass accuracy window: 5 ppm) based on their retention time, polarity, and or ion count abundance to ensure accurate peak identification and quantification.
Pathways impact analysis and metabolites classification
To evaluate the impact of the metabolic alterations found in KP versus PGKP TDCLs, we submitted the raw data obtained from the metabolic extraction on MAVEN (ion counts) to log transformation using the web-based software MetaboAnalyst 5.0.77. The transformed data were used to obtain the class of the metabolites significantly altered and the impact of these alterations on TDCLs and tissues96.
Software, graphs, and Figures
ImageJ 1.53 J was used for image quantification using densitometry and labeled cell count. R 4.2.1 studio was used to generate heat maps. All charts and statistics presented in this article were built using GraphPad Prism 10 (GraphPad Prism, RRID: SCR_002798). The metabolites pick extraction was performed in El-Maven v0.12.097. For tumor burden and quantification, OLYMPUS OlyVIA 2.9. All Illustrations were drawn using Bio Render Premium (all figures are publicly available) and Adobe Illustrator 2023 (Adobe Illustrator, RRID:SCR_010279).
Statistics and reproducibility
All statistical analyses were performed using GraphPad Prism 10.0, Excel Office, and MetaboAnalyst 5.096, with the box representing the interquartile range (25–75th percentile), the line indicating the median, and whiskers extending to the dataset’s minimum and maximum values, or shown as ±SEM. Samples sizes were chosen in advance based on the common practice of the described experiment and indicated by n or the number of points in the graph. Moreover, the initial sample size was determined based on a vast experience with tumor growth, survival, and manipulation of KP animals. The sample sizes for repeated experiments were based on the first one performed. Each experiment with mice was conducted with biological (at least three animals each) and technical (repeated at least twice) replicates. The experiments with TDCLs were also conducted with biological (at least three independent clones per genotype), technical (at least two independent wells seeded per clone), and experimental (repeated at least twice) replicates. Based on Grubb’s test, outlier data were automatically excluded (GraphPad Prism). Outliers were removed from the plasma metabolic panel. Outliers in the metabolomics triplicate data were identified using Dixon’s Q Test and Standard Deviation Thresholds to assess statistical deviations and detect extreme values. For clarity, some graphs display statistics only for the most relevant comparisons due to the large number of groups. For detailed individual p-values, please refer to the Source Data table. All experiments randomly allocated mice into experimental groups. The investigators were not blinded during the experiments and outcome assessments. Blinding was not performed as the genotype and sex of the mice required identification for housing purposes. Sex differences were not identified among the genotypes and experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data used to generate the graphs and their respective individual p-Values are available in the Source Data Table. The DNA sequencing data (mtDNA and nDNA) generated in this study have been deposited as follows: Bioproject: PRJNA1119555. The metabolomic raw data were deposited in the Metabolomic Workbench under the following https://doi.org/10.21228/M8RN89. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Poillet-Perez, L. et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 1, 923–934 (2020).
Poillet-Perez, L. et al. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018).
Cararo-Lopes, E. et al. Integrated metabolic and genetic analysis reveals distinct features of human differentiated thyroid cancer. Clin. Transl. Med. 13, e1298 (2023).
Labuschagne, C. F., van den Broek, N. J. F., Mackay, G. M., Vousden, K. H. & Maddocks, O. D. K. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7, 1248–1258 (2014).
Bartman, C. R. et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 614, 349–357 (2023).
Tajan, M. et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat. Commun.12, 1–16 (2021).
Bhatt, V. et al. Inhibition of autophagy and MEK promotes ferroptosis in Lkb1-deficient Kras-driven lung tumors. Cell Death Dis.14, 1–14 (2023).
Lan, T. et al. Glucose-6-phosphate dehydrogenase maintains redox homeostasis and biosynthesis in LKB1-deficient KRAS-driven lung cancer. Nat. Commun.15, 1–18 (2024).
Liu, Y. & Birsoy, K. ı. Metab. Sens. Control Mitochondria. Mol. Cell 83, 877–889 (2023).
Zong, W., Rabinowitz, J. D. & White, E. Mitochondria and cancer. Mol. Cell 61, 667–676 (2016).
Liu, Q. et al. Metformin inhibits prostate cancer progression by targeting tumor-associated inflammatory infiltration. Clin. Cancer Res. 24, 5622–5634 (2018).
Hirsch, H. A., Iliopoulos, D., Tsichlis, P. N. & Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 69, 7507–7511 (2009).
Zarou, M. M., Vazquez, A. & Vignir Helgason, G. Folate metabolism: a re-emerging therapeutic target in haematological cancers. Leukemia35, 1539–1551 (2021).
Jordheim, L. P., Durantel, D., Zoulim, F. & Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov.12, 447–464 (2013).
Maddocks, O. D. K. et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 (2017).
Yang, M. & Vousden, K. H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 16, 650–662 (2016).
Ducker, G. S. & Rabinowitz, J. D. One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017).
Amelio, I., Cutruzzolá, F., Antonov, A., Agostini, M. & Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 39, 191–198 (2014).
Lee, Y. S., Johnson, K. A., Molineux, I. J. & Yin, Y. W. A single mutation in human mitochondrial DNA polymerase Pol γA affects both polymerization and proofreading activities of only the holoenzyme. J. Biol. Chem. 285, 28105–28116 (2010).
Stumpf, J. D. & Copeland, W. C. Mitochondrial DNA replication and disease: insights from DNA polymerase γ mutations. Cell. Mol. Life Sci. 68, 219–233 (2011).
Weinberg, S. E. & Chandel, N. S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 11, 9 (2015).
Wallace, D. C. A Mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev. Genet 39, 359–407 (2005).
Kujoth, G. C. et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian. Aging 309, 481–485 (2005).
Cancer Statistics Review, 1975-2017 - SEER Statistics.
Prag, H. A. & Murphy, M. P. mtDNA mutations help support cancer cells. Nat. Cancer 1, 941–942 (2020).
Smith, A. L. M. et al. Age-associated mitochondrial DNA mutations cause metabolic remodeling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer1, 976–989 (2020).
Yuan, Y. et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet.52, 342–352 (2020).
International Agency for Research, o. C. in IARC Biennial Report 1–163 (IAR, 2021).
Travis, W. D. et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 10, 1243–1260 (2015).
DuPage, M., Dooley, A. L. & Jacks, T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 4, 1064–1072 (2009).
Jackson, E. L. et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 65, 10280–10288 (2005).
Walters Robert, W., Dongsheng, D., Engelhardt John, F. & Welsh Michael, J. Incorporation of adeno-associated virus in a calcium phosphate coprecipitate improves gene transfer to airway epithelia in vitro and in vivo. J. Virol. 74, 535–540 (2000).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Li-Harms, X. et al. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood 125, 162–174 (2015).
Chen, M. L. et al. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood 114, 4045–4053 (2009).
Vermulst, M. et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 39, 540–543 (2007).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Sim, N. L. et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452 (2012).
Guo, J. Y. et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27, 1447–1461 (2013).
Lin, C. S. et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 109, 20065–20070 (2012).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Guo, J. Y. et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717 (2016).
Karsli-Uzunbas, G. et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 915–927 (2014).
Lim, E. W. et al. Progressive alterations in amino acid and lipid metabolism correlate with peripheral neuropathy in Polg D257A mice. Sci. Adv. 7, 4077–4092 (2021).
Saleem, A. et al. Polymerase gamma mutator mice rely on increased glycolytic flux for energy production. Mitochondrion 21, 19–26 (2015).
Birsoy, K. et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015).
Mattaini, K. R., Sullivan, M. R. & Vander Heiden, M. G. The importance of serine metabolism in cancer. J. Cell Biol. 214, 249–257 (2016).
Faubert, B., Tasdogan, A., Morrison, S. J., Mathews, T. P. & DeBerardinis, R. J. Stable isotope tracing to assess tumor metabolism in vivo. Nat. Protoc. 16, 5123–5145 (2021).
Faubert, B. et al. Lactate metabolism in human lung tumors. Cell 171, 358–371 (2017).
Diehl, F. F., Lewis, C. A., Fiske, B. P. & Vander Heiden, M. G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 1, 861–867 (2019).
Tristan, C., Shahani, N., Sedlak, T. W. & Sawa, A. The diverse functions of GAPDH: views from different subcellular compartments. Cell. Signal. 23, 317–323 (2011).
Mohr, S., Stamler, J. S. & Brüne, B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J. Biol. Chem. 271, 4209–4214 (1996).
Arnold, P. K. et al. A non-canonical tricarboxylic acid cycle underlies cellular identity. Nature 603, 477–481 (2022).
Murphy, J. P. et al. The NAD + salvage pathway supports PHGDH-driven serine biosynthesis. Cell Rep. 24, 2381–2391.e5 (2018).
Pacold, M. E. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 12, 452–458 (2016).
Newman, A. C. & Maddocks, O. D. K. One-carbon metabolism in cancer. Br. J. Cancer2 116, 1499–1504 (2017).
Oslund, R. C. et al. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat. Chem. Biol. 13, 1081–1087 (2017).
Schober, F. A. et al. The one-carbon pool controls mitochondrial energy metabolism via complex I and iron-sulfur clusters. Sci. Adv. 7, eabf0717 (2021).
Ribas, V., García-Ruiz, C. & Fernández-Checa, J. C. Glutathione and mitochondria. Front. Pharmacol. 5 JUL, 151 (2014).
Cararo-Lopes, E. et al. Autophagy buffers Ras-induced genotoxic stress enabling malignant transformation in keratinocytes primed by human papillomavirus. Cell Death Dis. 12, 1–16 (2021).
Weinberg, F. et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 107, 8788–8793 (2010).
Maddocks, O. D. K. et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2012).
Xiao, W., Wang, R. S., Handy, D. E. & Loscalzo, J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal. 28, 251–272 (2018).
Wu, J., Jin, Z., Zheng, H. & Yan, L. J. Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes, Metab. Syndr. Obes. 9, 145–153 (2016).
Yang, Y. & Sauve, A. A. NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochimica et. Biophysica Acta - Proteins Proteom. 1864, 1787–1800 (2016).
Titov, D. V. et al. Complementation of mitochondrial electron transport chain by manipulation of the NAD + /NADH ratio. Science 352, 231–235 (2016).
Han, S. H. et al. Mitochondrial integrated stress response controls lung epithelial cell fate. Nature 620, 890–897 (2023).
Poor, T. A. & Chandel, N. S. SnapShot: mitochondrial signaling. Mol. Cell 83, 1012–1012 (2023).
Gopal, R. K. et al. Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc. Natl. Acad. Sci. USA 115, E6283–E6290 (2018).
Joshi, S. et al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 13, 1895–1908 (2015).
Zecchini, V. et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature 615, 499–506 (2023).
Tallini, G. Oncocytic tumours. Virchows Arch. 433, 5–12 (1998).
Stratton, M. R., Campbell, P. J. & Futreal, P. A. The cancer genome. Nature 458, 719–724 (2009).
Berridge, M. V., Dong, L. & Neuzil, J. Mitochondrial DNA in tumor initiation, progression, and metastasis: role of horizontal mtDNA transfer. Cancer Res. 75, 3203–3208 (2015).
Vasan, K., Werner, M. & Chandel, N. S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 32, 341–352 (2020).
Martínez-Reyes, I. et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020).
Gui, D. Y. et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 24, 716–727 (2016).
Hosios, A. M. & Matthew, G. V. The redox requirements of proliferating mammalian cells. J. Biol. Chem. 293, 7490–7498 (2018).
Zhu, X., Jin, C., Pan, Q. & Hu, X. Determining the quantitative relationship between glycolysis and GAPDH in cancer cells exhibiting the Warburg effect. J. Biol. Chem. 296, 100369–100370 (2021).
Locasale, J. W. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011).
Engel, A. L. et al. Serine-dependent redox homeostasis regulates glioblastoma cell survival. Br. J. Cancer 122, 1391–1398 (2020).
Reid, M. A. et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 9, 1–11 (2018).
Babraham Bioinformatics - FastQC A. Quality control tool for high throughput sequence data.
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. (2013).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
ensemblorg/ensembl-vep - Docker Image | Docker Hub.
Sawant, A. et al. Immune checkpoint blockade delays cancer and extends survival in murine dna polymerase mutator syndromes. Cancer Res. 85, 1130–1144 (2025).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Kuilman, T. et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 16, 1–15 (2015).
Guo, J. Y. et al. Activated RAS requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 (2011).
Su, X. et al. In-Source CID ramping and covariant ion analysis of hydrophilic interaction chromatography metabolomics. Anal. Chem. 92, 4829–4837 (2020).
Melamud, E., Vastag, L. & Rabinowitz, J. D. Metabolomic analysis and visualization engine for LC - MS data. Anal. Chem. 82, 9818–9826 (2010).
Chong, J., Wishart, D. S. & Xia, J. Using metaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinforma. 68, e86 (2019).
Agrawal, S. et al. El-MAVEN: a fast, robust, and user-friendly mass spectrometry data processing engine for metabolomics. Methods Mol. Biol. 1978, 301–321 (2019).
Acknowledgements
This work was supported by R01 CA243547 to EW, ECL (Lattime), and SG and RO1 CA259635 to DCW. Duncan and Nancy MacMillan Center of Excellence in Cancer Immunology and Metabolism to CSH, EW, SG and CSC. The Biospecimen Repository and Histopathology Service Shared Resource from the Cancer Institute of New Jersey provided all the specimens and associated services (P30CA072720-5919); Biometrics Shared Resource (P30CA072720-5918); Metabolomics Shared Resource (P30CA072720-5923). Biomedical Informatics Shared Resources (P30CA072720-5917).
Author information
Authors and Affiliations
Contributions
E.C.L. performed or participated in all experimental work, PGKP and KPND6 GEMM generation and characterization, data analysis, and data interpretation, and wrote the paper. E.W. is the leading investigator who conceived the project, supervised research, and edited the paper. J.R., J.Y.G., S.G., and E.C.L. provided intellectual input for the experiments, data analysis, and data interpretation. A.S., M.I., and M.G.J. assisted with mouse experiments, data analysis, and interpretation. Z.H. performed jugular vein catheterization and assisted with KPND6 generation. Z.H. and P.M. assisted with mouse colony management and mouse experiments. V.B. assisted with OCR and ECAR experiments. F.S. and C.C. processed, interpreted, and analyzed mtDNA and WES sequencing data. C.H. participated in the paper editing process. W.W. assisted with in vivo isotope tracing experiments. X.S. performed metabolomic processing and assisted with data analysis. D.C.W. contributed advice on mouse models of mitochondrial disease.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cararo Lopes, E., Shi, F., Sawant, A. et al. Respiration defects limit serine synthesis required for lung cancer growth and survival. Nat Commun 16, 7621 (2025). https://doi.org/10.1038/s41467-025-62911-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62911-7
