
Abstract
The innate immune system can develop a form of memory called priming, where prior exposure to a stimulus enhances subsequent responses. While well-characterized in peripheral immunity, its function in brain-resident cells such as astrocytes under non-disease conditions remains unclear. Here we show that human astrocytes derived from the induced pluripotent stem cells of healthy female donors, but not microglia, acquire a primed state following transient immune stimulations. Upon subsequent exposure to amyloid-β (Aβ), these astrocytes secrete elevated levels of cytokines and promote microglial Aβ uptake. In contrast, astrocytes carrying the Alzheimer’s disease (AD) risk allele APOE4 exhibit reduced priming and fail to support microglial phagocytosis. These findings are validated in astrocyte-microglial co-cultures, cerebral organoids, and male mice, where astrocyte priming enhances Aβ clearance in an APOE4-sensitive manner. Our findings identify astrocytic immune memory as a modulator of microglial function and Aβ pathology, providing insights into how early protective responses in AD may be disrupted by genetic risk factors.
Similar content being viewed by others


Introduction
In response to diverse immunogenic stimuli, such as bacterial and viral infections, tissue injury, and social stress, the body continuously adapts through dynamic remodeling of the immune system. One of its key features is the capacity to develop “immune priming,” a phenomenon in which innate immune cells exhibit enhanced or altered responses upon secondary stimulation. This form of innate immune memory, also known as “trained immunity,” was first identified in plants and invertebrates and is now increasingly recognized as a regulatory mechanism in peripheral myeloid cells and epithelial barriers in vertebrate systems, including mice and humans1.
Immune primed cells are phenotypically similar to naïve ones but display a lower threshold for subsequent immune activation, resulting in exaggerated responses and elevated cytokine production, as observed in various cell types, including monocytes, natural killer cells, and epithelial cells2,3. In murine models, priming with microbial ligands such as β-glucan, muramyl dipeptide, or CpG oligonucleotides provides protection against lethal infections even in the absence of adaptive immune components. Similarly, studies in both animals and humans have demonstrated that live attenuated vaccines, such as Bacillus Calmette-Guérin (BCG) or measles, can induce long-term reprogramming of innate immune cells, enhancing their responsiveness and reducing overall infectious mortality. Recent epidemiological studies also report an association between vaccination and a reduced prevalence of Alzheimer’s disease (AD), raising important mechanistic questions4,5,6,7.
The inflammatory response of brain glial cells, such as microglia and astrocytes, plays critical roles in the central nervous system during environmental insults, including tissue injury and microbial infections, through the production of pro-inflammatory cytokines. While immune priming may enhance glial surveillance and facilitate the clearance of pathological proteins, excessive or dysregulated activation of the immune system has been implicated in the progression of various neurological disorders. For instance, systemic inflammation has been shown to induce microglial priming in the brain, leading to exacerbated neurodegenerative features in prion disease models and enhanced amyloid-β (Aβ) accumulation in AD mouse models7,8. These dual aspects suggest a potential convergence in AD pathogenesis: immune memory mechanisms that are beneficial when appropriately regulated may become harmful in a chronically inflamed neurodegenerative environment.
Astrocyte reactivity in pathological conditions is of great interest because abnormal astrocytic inflammation and synapse engulfment are strongly associated with the pathogenesis of various neurodegenerative diseases, including AD. However, it remains unclear whether astrocytes can be primed specifically under non-disease conditions, and if so, whether and how this priming contributes to the onset or progression of neurodegeneration. Most previous studies, especially those on microglial priming, have employed disease-associated or pathogen-driven models7 or have used protocols that repeatedly stimulate cells without sufficient recovery time, a key factor for the establishment of a primed state9. To more accurately model immune challenges occurring before disease onset, we utilized astrocytes derived from induced pluripotent stem cells (iPSCs) generated from healthy individuals. We applied transient immune stimulation followed by a full recovery period, confirmed by the return of cytokine and chemokine levels to baseline, and evaluated priming activity. We found that astrocyte priming was evident under these conditions, whereas no such effect was observed in iPSC-derived microglia (iMGL). Notably, we discovered that primed astrocytes exerted a protective effect in AD-related models by enhancing microglial phagocytosis of Aβ42 in both co-culture and 3D cerebral organoid systems. Moreover, we demonstrated that human astrocytes carrying the APOE4 allele, the strongest genetic risk factor for late-onset AD, exhibited impaired priming capacity, which in turn hindered microglial clearance of Aβ. Finally, we validated these findings in vivo using humanized APOE3/E4 knock-in (KI) mice.
Results
Formation of immune priming in hiPSC-derived astrocytes
To determine whether astrocytes can achieve an immune priming state, we first generated astrocytes from human induced pluripotent stem cells (hiPSCs) derived from a 75-year-old healthy female donor carrying the APOE3 genotype, using previously reported methods10. We then induced an astrocyte-specific inflammatory response by treating the cells with polyinosinic:polycytidylic acid (Poly I:C), an agonist of Toll-like receptor 3 (TLR3), followed by RNA-seq analysis (Fig. 1A). As expected, many genes involved in the inflammatory response were upregulated. Notably, these changes were mostly reversed after a 72-h recovery period following the Poly I:C washout (3,516 out of 3,800 genes), with the top gene ontology (GO) terms associated with these genes including “regulation of programmed cell death” and “response to cytokines” (Fig. 1B). Quantification of changes in cytokine levels in media secreted by astrocytes revealed a consistent increase in the levels of numerous cytokines following Poly I:C treatment. However, after 48 h of washout, secretion levels decreased although transcript levels for interleukin (IL)−6, IL-1β, and tumor necrosis factor α (TNFα) remained elevated (Fig. 1C, Supplementary Fig. 1A). This discrepancy may be due to a transient inhibition of secretion or translation, resulting in an “undershoot” in cytokine release that ultimately returns to baseline levels by 72 h (Fig. 1C). To confirm immune priming, characterized by an amplified inflammatory response to subsequent stimuli, we re-treated the 72-h recovered astrocytes with Poly I:C. The transcript levels of IL-6, IL-1β, and TNFα were significantly increased compared to the first Poly I:C stimulation (Fig. 1D). RNA-seq analysis further confirmed that many genes upregulated by the secondary Poly I:C treatment were associated with the inflammatory response, indicating that astrocytes can achieve an immune priming state (Fig. 1E). Microglia are the primary cell population responsible for the inflammatory response in the brain. Therefore, we investigated microglial priming using the same system. In contrast to hiPSC-derived astrocytes, iMGL did not exhibit a priming effect (Fig. 1F, G).

A Schematic overview of the experimental process for inducing and verifying immune priming in hiPSC-derived astrocytes. B Number and ontology of genes that are upregulated by Poly I:C treatment and subsequently downregulated during recovery in human astrocytes. C Patterns of cytokine secretion change by Poly I:C treatment and the subsequent recovery period. D Changes in IL-6, IL-1β, and TNFα transcript levels by Poly I:C treatment, recovery, and second Poly I:C treatment. N = 24, from four independent batches. E Genes with increased reactivity upon second Poly I:C treatment induced by immune priming and their gene ontology. Differentially expressed genes (DEGs) were identified using Cuffdiff. Statistical significance was determined by adjusted q-values (<0.05). F Schematic overview of the experimental process for inducing and verifying immune priming in iMGL. G Changes in IL-6, IL-1β, and TNFα transcript levels by Poly I:C treatment, recovery, and second Poly I:C treatment in iMGL. N = 4, from two independent batches. All schematics were created in BioRender. Seo, J. (2025) https://BioRender.com/09a0byv. *p < 0.05, ****p < 0.0001; ns, not significant. (One-way ANOVA test followed by Tukey’s post hoc analysis). Exact P values are provided in the Source Data. Error bar ± S.E.M.
To ensure that the observed phenomenon is not unique to a specific hiPSC line, we used another hiPSC line derived from an 87-year-old female donor carrying the APOE4 allele. We previously generated isogenic APOE3 hiPSCs from this line to serve as a control for studying the effects of APOE4 on AD pathology10. Astrocytes generated from these control iPSCs were assessed for priming potential. A similar increase in the inflammatory response to the second challenge confirmed the presence of astrocyte priming in this line as well (Supplementary Fig. 1B). We further investigated whether repeated stimulation would continuously amplify the inflammatory response or if the priming-induced immune activation would reach a saturation point. After a second challenge and a subsequent 3-day recovery period, a third Poly I:C stimulation was administered. A significant increase in IL-1β, but not in IL-6 or TNFα was observed, suggesting that the priming effect is not perpetually enhanced (Supplementary Fig. 1C). While Poly I:C treatment and subsequent TLR3 activation model inflammatory reactions induced by various stimuli, including viral infections in humans, we sought to determine whether immune memory formation in astrocytes is not restricted to specific antigens. Therefore, we explored whether astrocyte priming could be induced by stimuli other than Poly I:C. Astrocytes were pre-treated with a mixture of TNFα, IL-1α, and complement component 1q (C1q), cytokines known to be secreted by microglia and capable of reactivating astrocytes11. These pre-treated astrocytes were subsequently challenged with Poly I:C. The results showed a significant increase in IL-6 and TNFα expression with IL-1β also trending upward (Supplementary Fig. 1D), indicating that reactive stimuli other than Poly I:C can also induce astrocyte priming. These findings demonstrate that astrocyte priming is a generalizable phenomenon, not limited to astrocytes derived from specific hiPSC lines, and can be triggered by a variety of inflammatory stimuli.
In response to Aβ42, primed human astrocytes enhance inflammatory signaling and promote microglial phagocytosis
To investigate how astrocyte priming under non-disease conditions may influence the pathogenesis of neurological diseases, particularly AD, a representative neurodegenerative disorder characterized by the accumulation of toxic Aβ aggregates, we primed astrocytes with Poly I:C and subsequently exposed them to Aβ42, assessing changes in their uptake activity (Fig. 2A). We observed that primed astrocytes exhibited a significantly reduced Aβ42 uptake compared to non-primed cells (Fig. 2B). Notably, the secretion levels of various cytokines were altered, with a higher proportion of inflammatory cytokines (Fig. 2C). To confirm whether Aβ42 increases cytokine synthesis in primed astrocytes, we examined the transcript levels of IL-6, IL-1β, and TNFα, all of which showed increased secretion in the media. In non-primed astrocytes treated with Aβ42, we did not observe any increase in IL-6, IL-1β, or TNFα transcript levels. Generally, under astrocyte culture conditions that mimic physiological situations (by depriving fetal bovine serum), Aβ does not induce a significant inflammatory response. This contrasts with most previous experiments, which often used FBS-supplemented media or treated astrocytes with Aβ following immune stimulation or under pathological conditions, leading to an inflammatory response. In contrast, IL-6 and IL-1β transcripts levels were significantly increased in primed astrocytes compared to those in non-treated astrocytes (Fig. 2D). Consistently, RNA-seq analysis of transcriptome changes in non-primed and primed astrocytes treated with Aβ42 revealed that mRNA levels of genes associated with lysosomes, lipid metabolism, and immune response specifically increased in primed astrocytes (Fig. 2E). Conversely, genes related to the cytoskeleton, cell junctions, and extracellular matrix organization were downregulated (Fig. 2F).

A Schematic overview of the experimental process exploring the effects of priming on human astrocyte reactivity to Aβ42. B Decreased Aβ42 uptake capability in primed astrocytes. N = 11, from three independent batches. C Changes in cytokine secretion patterns in primed astrocytes compared to non-primed astrocytes upon Aβ42 treatment. Cytokines that were significantly increased or decreased are indicated in red and blue, respectively. In each experiment, three samples are pooled, and the entire experiment is repeated three times using cells derived from independent batches. D Increased levels of IL-6 and IL-1β transcripts in primed astrocytes in response to Aβ42. N = 12, from three independent batches. E Ontology analysis of genes uniquely upregulated by Aβ42 in primed astrocytes compared to non-primed astrocytes. F Ontology analysis of genes uniquely downregulated by Aβ42 in primed astrocytes compared to non-primed astrocytes. Differentially expressed genes (DEGs) were identified using Cuffdiff. Statistical significance was determined by adjusted q-values (<0.05). G Schematic overview of the experimental process analyzing the impact of cytokines secreted by primed astrocytes in response to Aβ42 on microglial Aβ42 uptake. H Increased microglial Aβ42 uptake in response to astrocyte-conditioned media (ACM) from primed astrocytes exposed to Aβ42. N = 20, from two independent batches. Scale bar = 5 μm. All schematics were created in BioRender. Seo, J. (2025) https://BioRender.com/09a0byv. *p < 0.05, **p < 0.01, ****p < 0.0001; ns not significant. (One-way ANOVA test followed by Tukey’s post hoc analysis or two-tailed Student’s t-test). Exact P values are provided in the Source Data. Error bar ± S.E.M.
These results suggest that priming reduces Aβ uptake capacity while inducing an inflammatory response in astrocytes, which may contribute to AD pathogenesis. However, intriguingly, the elevated cytokines included Fractalkine (CX3CL1), monocyte chemoattractant protein-1 (MCP-1[CCL2]), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-3, all of which significantly influence microglial activity12,13. To explore the roles of these factors on microglia surrounding astrocytes, we collected the conditioned media from the non-primed and primed astrocytes exposed to Aβ42, followed by a washout. Human microglia were then exposed to this collected media (Fig. 2G). Notably, microglia cultured with conditioned media from Aβ42-treated primed astrocytes exhibited a threefold increase in Aβ42 phagocytosis compared to those treated with conditioned media from Aβ-treated non-primed astrocytes (Fig. 2H).
We then sought to determine whether the transcriptomic changes in Aβ42-treated primed astrocytes would resemble or diverge from those observed in astrocytes from AD brains. To address this, we compared RNA-seq data from patients with AD and unaffected individuals14 with the transcriptome profiles of Aβ42-treated primed astrocytes. We observed a strong inverse correlation between the two conditions, particularly pronounced in early-stage AD (Supplementary Fig. 2A). These results suggest that immune priming, when induced under non-disease conditions, may exert a protective effect against the onset of AD, in contrast to previous studies where priming was primarily associated with disease contexts or pathological models. Additionally, we found that many of the differentially expressed genes (DEGs) negatively correlated between Aβ42-treated primed astrocytes and AD brains were associated with lipid biosynthesis (Supplementary Fig. 2B).
APOE4 impairs astrocyte priming and reduces cytokine secretion in response to Aβ42, leading to decreased microglial phagocytosis
Given that APOE4, the most significant genetic risk factor for late-onset AD, is strongly expressed in astrocytes and has been shown to dysregulate lipid metabolism, we next aimed to explore how the APOE4 isoform influences astrocyte priming formation and its subsequent effect on microglial Aβ uptake10,15,16,17,18. We used previously established isogenic APOE4 iPSCs derived from control iPSCs with APOE3 alleles to generate astrocytes and applied the same priming protocol followed by RNA-seq analysis10. The results showed that, similar to control astrocytes, many genes related to immune responses were upregulated by Poly I:C treatment in APOE4 astrocytes and returned to baseline after recovery (Fig. 3A). Quantitative polymerase chain reaction (qPCR) experiments confirmed that the transcript levels of IL-6, IL-1β, and TNFα were significantly increased upon the second challenge, indicating a priming effect in APOE4 astrocytes, similar to that in APOE3 (control) astrocytes (Fig. 3B). Additionally, RNA-seq analysis revealed that many of the genes further upregulated by the second challenge in APOE4 astrocytes were related to defense and cytokine responses (Fig. 3C). However, the priming potential, defined as the increased inflammatory reactivity to a second stimulus compared to the initial response, was significantly lower in APOE4 astrocytes than in APOE3 astrocytes (Fig. 3D).

A Number and ontology of genes upregulated by Poly I:C treatment and subsequently downregulated during recovery in APOE4 hiPSC-derived astrocytes. B Changes in IL-6, IL-1β, and TNFα transcript levels by Poly I:C treatment, recovery, and second Poly I:C treatment. N = 24, from four independent batches. C Genes with increased transcripts upon second Poly I:C treatment induced by immune priming and their gene ontology. Differentially expressed genes (DEGs) were identified using Cuffdiff. Statistical significance was determined by adjusted q-values (<0.05). D Reduced increase in IL-6, IL-1β, and TNFα transcript levels in primed APOE4 astrocytes compared to APOE3 astrocytes by the second Poly I:C challenge. N = 24 (IL-6 and TNFα) and N = 22 (IL-1β), derived from four independent batches. E Changes in cytokine secretion patterns in primed APOE4 astrocytes compared to non-primed astrocytes. Cytokines that were significantly increased or decreased are indicated in red and blue, respectively. In each experiment, three samples are pooled, and the entire experiment is repeated three times using cells derived from independent batches. F Increased IL-1β transcript levels in primed APOE4 astrocytes in response to Aβ42. N = 6, from three independent batches. G Ontology analysis of genes uniquely upregulated by Aβ42 in primed APOE4 astrocytes compared to non-primed astrocytes. H Ontology analysis of genes uniquely downregulated by Aβ42 in primed APOE4 astrocytes compared to non-primed astrocytes. DEGs were identified using Cuffdiff. Statistical significance was determined by adjusted q-values (<0.05). I Increased Aβ42 uptake capability in primed APOE4 astrocytes compared to non-primed astrocytes. N = 11, from three independent batches. J Decreased microglial Aβ42 uptake in response to astrocyte-conditioned media (ACM) from primed APOE4 astrocytes exposed to Aβ42. N = 20, from two independent batches. Scale bar=5 μm. **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant. (One-way ANOVA test followed by Tukey’s post hoc analysis or two-tailed Student’s t-test). Exact P values are provided in the Source Data. Error bar ± S.E.M.
To determine the impact of this weakened priming on Aβ-induced responsiveness, we treated non-primed and primed APOE4 astrocytes with Aβ42 and measured cytokine secretion. Notably, unlike APOE3 astrocytes, primed APOE4 astrocytes treated with Aβ42 showed a decrease in the secretion of many cytokines, including IL-6, IL-1β, and TNFα, with only IL-1β showing an increase at the transcript level by qPCR (Fig. 3E, F). RNA-seq analysis of Aβ42-treated primed APOE4 astrocytes compared to non-primed ones revealed that upregulated genes were more related to neuronal development and cellular morphogenesis rather than immune response, while downregulated genes were associated with cytoskeletal organization and cell adhesion (Fig. 3G, H). To assess the functional changes induced by these transcriptomic alterations, we examined the effect of priming on Aβ42 uptake in APOE4 astrocytes. Unlike APOE3 astrocytes, primed APOE4 astrocytes showed a significant increase in Aβ42 uptake activity (Fig. 3I). Moreover, consistent with altered cytokine secretion, conditioned media from Aβ42-treated primed APOE4 astrocytes significantly reduced phagocytosis of Aβ42 by human microglia by approximately 50% compared to those treated with conditioned media from Aβ42-treated non-primed APOE4 astrocytes (Fig. 3J).
We next investigated whether these effects were consistent in iMGL. To this end, we differentiated iMGL from the same hiPSCs as previously reported19 and treated them with conditioned media from non-primed or primed APOE3 astrocytes exposed to Aβ42. iMGL cultured with media from Aβ42-treated primed APOE3 astrocytes exhibited higher Aβ42 uptake (Supplementary Fig. 3A). Conversely, conditioned media from Aβ42-treated primed APOE4 astrocytes reduced Aβ42 uptake by iMGL, consistent with results in human microglia cell lines (Supplementary Fig. 3A). To investigate whether these changes were influenced by the APOE isoform in microglia, we differentiated iMGL from isogenic APOE4 hiPSCs and repeated the experiments. The results showed the same pattern regardless of the APOE isoform in iMGL (Supplementary Fig. 3B). Next, to confirm that these effects were not specific to a particular hiPSC line, we used another isogenic pair (as shown in Supplementary Fig. 1B) and repeated the experiments with human microglia treated with conditioned media. The outcomes were consistent with earlier findings, showing differential effects of factors secreted by primed APOE3 versus APOE4 astrocytes exposed to Aβ42 on microglia (Supplementary Fig. 3C). Additionally, we generated an APOE4 isogenic line in a human microglia cell line using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome editing. Aβ42 treatment of primed APOE3 and APOE4 astrocytes resulted in distinct effects in APOE4 human microglia mirroring observations in APOE4 iMGL (Supplementary Fig. 3D). Finally, we examined whether priming astrocytes with TNFα, IL-1α, and C1q, instead of Poly I:C, would similarly enhance the inflammatory response to Aβ and influence microglial Aβ42 uptake. We found that astrocytes primed with these cytokines showed increased inflammatory responses upon Aβ42 treatment (Supplementary Fig. 3E), which in turn the increased Aβ42 uptake by iMGL (Supplementary Fig. 3F). However, similar to those primed with Poly I:C, APOE4 astrocytes primed with these cytokines secreted factors that reduced Aβ42 uptake by microglia (Supplementary Fig. 3F). These results confirm that astrocyte priming and its APOE isoform-dependent differential effects on microglial Aβ uptake are consistent across microglial APOE genotypes, hiPSC lines used for astrocyte differentiation, and different priming stimuli.
Taken together, we found that when astrocytes were primed, they did not primarily internalize Aβ42 themselves, but instead increased cytokine synthesis and secretion, thereby promoting Aβ42 uptake by surrounding microglia. In contrast, in the presence of APOE4, astrocytes tended to internalize Aβ directly, which appeared to reduce the Aβ42 uptake by nearby microglia.
Co-culture and cerebral organoids models provide evidence for primed astrocyte-mediated enhancement of microglial Aβ phagocytosis
In previous experiments, we applied the astrocyte priming protocol to iMGL and found that priming did not occur in these cells. Furthermore, using fluorescently tagged Aβ treatment and imaging, we confirmed that this treatment did not alter the Aβ uptake activity of iMGLs (Supplementary Fig. 4A). Similarly, no changes in priming or Aβ uptake activity were observed in APOE4 iMGL (Supplementary Fig. 4B, C).
In the brain, astrocytes and microglia interact to perform and coordinate their functions. To mimic this interaction, we co-cultured hiPSC-derived astrocytes and iMGL, induced priming, and treated them with Aβ42 to assess cell-specific uptake activity (Fig. 4A). When priming was induced, we observed an increase in Aβ42 uptake by microglia, while the Aβ42 signal in astrocytes remained relatively low. Moreover, microglial activation was confirmed by an increase in Iba1 expression (Fig. 4B). However, in co-culture of APOE4 astrocytes and APOE3 iMGL, microglial Aβ uptake was significantly decreased compared to non-primed conditions, and no change in Iba1 expression was observed (Fig. 4C).

A Schematic overview of the experimental process for the astrocyte-microglia co-culture system. B, C After applying the priming protocol, increased Aβ42 clearance and enhanced Iba1 signal are observed in APOE3 microglia when co-cultured with APOE3 astrocytes, but not with APOE4 astrocytes. Scale bar=5 μm. D Schematic overview of the experimental process for APPswe organoids embedded with APOE3 astrocytes. E Organoids embedded with APOE3 astrocytes contain only GFAP-positive cells, with no Iba1 signal. Similar glial cell embedding was confirmed across all organoids used for the analysis. Scale bar=50 μm. F Priming does not reduce accumulated Aβ42 in organoids embedded solely with astrocytes. N = 12, from four different batches. Scale bar=50 μm. G Schematic overview of the experimental process for APPswe organoids embedded with APOE3 microglia. H Organoids embedded with APOE3 microglia contain only Iba1-positive cells, with no GFAP signal. Similar glial cell embedding was confirmed across all organoids used for the analysis. Scale bar=50 μm. I Priming does not reduce accumulated Aβ42 in organoids embedded solely with microglia. N = 12, from four different batches. Scale bar = 50 μm. J Schematic overview of the experimental process for APPswe organoids embedded with both APOE3 astrocytes and microglia. K Organoids embedded with both cell types contain GFAP-positive and Iba1-positive cells. Similar glial cell embedding was confirmed across all organoids used for the analysis. Scale bar=50 μm. L Priming reduces accumulated Aβ42 in organoids embedded with both astrocytes and microglia. N = 12, from four different batches. Scale bar=50 μm. All schematics were created in BioRender. Seo, J. (2025) https://BioRender.com/09a0byv. *p < 0.05, ***p < 0.001; ns not significant. (Two-tailed Student’s t-test). Exact P values are provided in the Source Data. Error bar ± S.E.M. test.
We then employed a cerebral organoid model carrying the APPswe mutation, which is known to elevate Aβ levels and promote Aβ aggregation20. Consistently, we observed an increase in Aβ accumulation as culture progressed (Supplementary Fig. 5). In this model, astrocytes become detectable after more than 3 months of culture, whereas microglia are absent because they originate from a lineage distinct from that of neural cells10. Therefore, after 40 days of culture in these AD cerebral organoid models, we embedded astrocytes and microglia either individually or together to assess the effect of glial priming on Aβ accumulation. Although changes in Aβ aggregates were not observed when cerebral organoids were embedded with astrocytes or microglia alone with priming induced (Fig. 4D–I), after embedding both astrocytes and microglia and inducing priming, we observed a significant decrease in Aβ accumulation (Fig. 4J–L). These findings suggest that astrocytes can develop an immune memory through cycles of immune stimulation and recovery and that signals from these primed astrocytes can prompt microglia to respond more effectively, ultimately exerting a beneficial effect against Aβ accumulation.
Astrocyte priming enhances Aβ42 clearance in an in vivo mouse model, while it is inhibited by APOE4
To determine the presence of astrocyte priming and its impact on Aβ clearance, as well as how the APOE4 genotype modifies this process in vivo, we first conducted experiments using wild-type 6-month-old mice (Fig. 5A). We administered Poly I:C via intraperitoneal injection21, followed by a 3-day recovery period, and confirmed that this recovery was sufficient to return the elevated inflammatory response in the hippocampus to pre-treatment levels (Supplementary Fig. 6A). To assess the exacerbated inflammatory effect caused by priming, we re-injected Poly I:C into primed mice and compared the resulting hippocampal inflammatory responses with those observed after the initial challenge. As observed in hiPSC-derived astrocytes, the inflammatory response to the second immune challenge showed a significant increase in IL-6 and TNFα compared to the first challenge (Fig. 5B). IL-1β also showed an increasing trend, although it was not statistically significant (Fig. 5B).

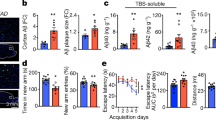
A Schematic overview of the experimental process for inducing and verifying astrocyte priming in an in vivo mouse model. B Confirmation of increased IL-6, IL-1β, and TNFα transcript levels upon second Poly I:C treatment induced by in vivo astrocyte priming. N = 8 (IL-6 and TNFα) and N = 7 (IL-1β) animals. C Schematic overview of the experimental process using a humanized APOE knock-in (KI) mouse model to verify the changes in priming effects due to APOE4. D Increased Aβ42 clearance by in vivo priming in APOE3 KI mice. N = 26 (non-primed group) and N = 32 (primed group), from four animals. Scale bar=200 μm. E Reduced Aβ42 clearance by in vivo priming in APOE4 KI mice. N = 40 (non-primed group) and N = 32 (primed group), from four animals. Scale bar=200 μm. F–H Microglial activation is assessed using Iba1 and CD11b staining. G Microglia in primed APOE3 KI mice exhibit a more activated morphology, characterized by a reduced number of intersections when Aβ42 is injected, whereas microglia in primed APOE4 KI mice show no such changes with Aβ42 injection. N = 11, from four animals. Scale bar=20 μm. H Increased CD11b intensity in primed APOE3 KI mice when Aβ42 is injected, but microglia in APOE4 KI mice show decreased CD11b intensity with Aβ42 injection. N = 20, from four animals. Scale bar=20 μm. I Increased GFAP intensity in primed APOE3 KI mice when Aβ42 is injected, but microglia in APOE4 KI mice show decreased GFAP intensity with Aβ42 injection. N = 23, from four animals. Scale bar=20 μm. All schematics were created in BioRender. Seo, J. (2025) https://BioRender.com/09a0byv. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant. (Two-tailed Student’s t-test). Exact P values are provided in the Source Data. Error bar ± S.E.M.
To investigate whether astrocyte priming exerts a beneficial or detrimental effect on Aβ clearance in the brain, where both astrocytes and microglia are present and interact, we primed mice with Poly I:C and then administered fluorescently tagged oligomeric Aβ42 via intracerebroventricular injection, as previously described to model early-stage Aβ accumulation in AD22,23. Aβ42 clearance was assessed at 1, 12, and 24 h post-injection (Supplementary Fig. 6B). Our results showed that by 24 h, most of the Aβ42 signal had disappeared, with substantial clearance already evident at 12 h. At the 1-h mark, however, a strong Aβ42 signal persisted. Based on these observations, we determined that the 6-h time point would be optimal for detecting differences in clearance under various conditions. Accordingly, 6 h after Aβ42 injection, we euthanized the mice and measured Aβ42 accumulation in the hippocampus. To examine the influence of the APOE4 isoform, we used humanized APOE3 KI mice as controls (Fig. 5C). Primed APOE3 KI mice showed significantly reduced residual Aβ42 compared to non-primed APOE3 KI mice (Fig. 5D). Furthermore, microglia in primed APOE3 KI mice exhibited a more reactive morphology following Aβ42 injection, as indicated by decreased processes determined by Sholl analysis (Fig. 5F, G). Notably, this microglial response was not observed in APOE4 KI mice (Fig. 5E–G). Similarly, while CD11b intensity was increased in primed APOE3 KI mice compared to that in non-primed APOE3 KI mice following Aβ42 injection, it was decreased in APOE4 KI mice (Fig. 5H). Furthermore, increased expression of GFAP was observed in Aβ42-injected primed APOE3 KI mice, an effect that was absent in Aβ42-injected primed APOE4 KI mice (Fig. 5I).
Discussion
Inflammatory memory, also referred to as immune priming, is well established in the immune cells. However, recent reports indicate that this phenomenon is not limited to immune cells, because various other cell types, including epithelial cells, also possess this capability2,3,24. This raises the possibility that glial cells in the brain, which are key mediators of inflammatory responses to diverse immune stimuli, such as microbial invasion or toxic protein accumulation, may similarly acquire immune memory. Given the aberrant inflammatory responses observed in glial cells during neurodegenerative diseases such as AD, understanding the relationship between immune priming and the onset or progression of neurological disorders is of critical importance.
Previous studies have shown that microglia, a major cell type in the brain that responds to external or internal immune stimuli, can be primed upon systemic inflammation in models of neurodegenerative disease conditions. In these models, systemic stimuli triggered accelerated immune responses and accelerated neurodegenerative pathology, suggesting that inflammation could be a causative factor in AD7,8,25,26,27. However, it remains unclear whether similar transformations in glial cells occur under non-disease conditions. While the formation of immune priming in glia under non-disease conditions may share characteristics with the increased inflammatory responses found in neurodegenerative diseases, their role in pathogenesis, especially against toxic proteins, could differ fundamentally.
In this study, we investigated the functional changes in astrocytes undergoing immune priming. Unlike astrocytes, we did not observe excessive inflammatory activity in microglia following acute immune challenge. The discrepancy from previous studies reporting microglia priming upon systemic inflammation in models of neurodegeneration is possibly owing to using unaffected (non-disease) conditions or a different protocol for immune stimulation. We rather observed that microglia displayed enhanced phagocytotic activity against Aβ by conditioned media from Aβ-treated, primed astrocytes, while these astrocytes had reduced Aβ42 uptake activity. This change seems to be more effective in clearing Aβ from the brain. Co-culture of hiPSC-derived astrocytes and microglia, embedding these cells in AD cerebral organoids, and in vivo experiments with mouse models all confirmed the enhanced Aβ clearance effect of astrocyte priming. Although we controlled Aβ input by injecting equal volumes into the mouse hippocampus, supporting the interpretation that reduced Aβ levels following priming are likely due to glial-dependent uptake, the decreased Aβ observed in APPswe cerebral organoids embedded with astrocytes and iMGL upon priming could also be partially attributed to reduced neuronal APP expression or altered Aβ processing. This possibility represents an interesting avenue for future investigation. In APOE4 astrocytes, priming resulted in increased Aβ42 uptake by the astrocytes while simultaneously reducing cytokine secretion. This reduction in cytokine signaling may contribute to the decreased Aβ42 uptake by microglia. In summary, inflammation-induced priming appears to enhance the ability of astrocytes to promote microglia-mediated clearance of Aβ in the brain. The APOE4 variant, however, impairs this beneficial response, contributing to the development of AD.
We aimed to propose a potential mechanism by which primed astrocytes enhance microglial Aβ phagocytosis. For the donor cells (astrocytes), given that the priming status results in the normalization of most transcriptome profile changes following stimulation, leading to fewer DEGs compared to controls, it is likely that the activity of primed astrocytes is regulated epigenetically2. Therefore, epigenetic profiling will be essential to investigate this mechanism. Thus, we performed an assay for transposase-accessible chromatin with sequencing (ATAC-seq) to examine the differences in chromatin accessibility before and after priming and subsequent Aβ42 treatment. Interestingly, after Aβ42 treatment, primed astrocytes exhibited 80 regions with increased accessibility compared to non-primed astrocytes, with no regions showing decreased accessibility (Supplementary Fig. 7A). Examining the genes near these regions revealed a significant enrichment of genes involved in cholesterol/lipid regulation. When comparing these genes with those upregulated by Aβ42 treatment in primed astrocytes, the overlapping genes identified by both ATAC-seq and RNA-seq were also related to lipid functions as well as phagocytosis (Supplementary Fig. 7A). For the recipient cells (microglia), we compared the DEGs in microglia treated with Aβ42-exposed astrocyte conditioned media (ACM) derived from APOE3 versus APOE4 astrocytes. Our RNA-seq analysis indicates that microglia treated with conditioned media from Aβ42-treated primed APOE3 astrocytes exhibited only six DEGs compared to those treated with media from Aβ42-treated non-primed APOE3 astrocytes (four upregulated, two downregulated) (Supplementary Fig. 7B). In contrast, microglia treated with conditioned media from Aβ42-treated primed APOE4 astrocytes showed 130 DEGs compared to those treated with media from Aβ42-treated non-primed APOE4 astrocytes (34 upregulated, 96 downregulated) (Supplementary Fig. 7C). Although no particular ontological terms were noticeable, examining the functions of these individual genes and conducting future perturbation studies will be necessary to elucidate the key mediators and mechanisms involved. Overall, these findings underscore a potential mechanism by which astrocytic priming modulates microglial Aβ clearance through lipid signaling, including cholesterol pathways. This is in agreement with the GO terms observed in the transcriptomic comparison of Aβ42-treated primed astrocytes with human astrocytes derived from AD brains. In addition, the cytokines that were increased in Aβ-treated primed APOE3 astrocytes (which were not seen in primed APOE4 astrocytes; Figs. 2C vs 3E) may also contribute to these changes.
While our analysis from in vitro culture systems provides valuable insights, they are limited by the lack of a comprehensive in vivo microenvironment, including vascular structures. Nevertheless, by validating in more complex 3D cerebral organoids and in vivo mouse models that astrocyte priming enhances protective glial function under non-disease conditions, our work supports the hypothesis that controlled, context-specific immune activation could modulate disease progression. Therefore, our study provides a foundational experimental framework to investigate how immune experience influences central nervous system resilience through glial responses.
Methods
Human-Induced Pluripotent Stem Cells (hiPSCs)
The Institutional Review Board (IRB) of Daegu Gyeongbuk Institute of Science and Technology (DGIST) approved the use of hiPSCs (Permit Number: DGIST-190829-BR-071-01). Unaffected hiPSCs carrying the APOE3 genotype were obtained from a 75-year-old female donor (Coriell #AG09173). Isogenic APOE4 hiPSC lines were generated from these APOE3 hiPSCs using CRISPR/Cas9 technology as previously reported28. Additionally, hiPSCs carrying the APOE4 genotype from an 87-year-old female donor (Coriell #AG10788) were utilized, and isogenic APOE3 hiPSC lines were generated from these APOE4 hiPSCs using the same CRISPR/Cas9 methodology as previously reported10. The APPswe isogenic hiPSC line was generated using CRISPR/Cas9 genome editing as previously reported29. All hiPSC lines were obtained from the Coriell Institute, and their generation and use for research were approved with written informed consent from the donors.
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Korea Institute of Oriental Medicine (Protocol Numbers: 22-091, 22-112, and 23-057). All experiments were performed using male mice (Mus musculus). C57BL/6 J inbred mice (8 weeks old) were obtained from Doo Yeol Biotech. Humanized APOE3 (B6.129P2-Apoetm2(APOE*3)MaeN8) and APOE4 (B6.129P2-Apoetm3(APOE*4)MaeN8) knock-in (KI) mice on a C57BL/6 genetic background were purchased from Taconic Biosciences and used at 6 months of age. All animals were housed and handled in a specific pathogen-free (SPF) facility maintained on a 12-h light/dark cycle, at an ambient temperature of 23 ± 2 °C and a relative humidity of 50 ± 10%. For all surgical procedures, mice were anesthetized with isoflurane. Anesthesia was induced at 3.0% and was maintained at 2.0% after confirming a deep anesthetic plane. In accordance with IACUC guidelines, humane endpoints were determined using a scoring system based on changes in body weight, behavior, and the condition of the fur, eyes, and nose. Mice that reached a score of 8 or higher were euthanized using carbon dioxide (CO2) gas.
hiPSCs culture
The hiPSCs were cultured on hESC-qualified Matrigel (Corning)-coated 6-well plates using mTeSR1 media (STEMCELL Technologies). After reaching 70% to 80% confluency, cells were dissociated with ReLeSR (STEMCELL Technologies) and passaged onto new Matrigel-coated 6-well plates with mTeSR1 media supplemented with 10 μM ROCK inhibitor Y-27632 (Tocris) at a 1:20 split ratio.
Neural Progenitor Cells (NPCs) differentiation
Upon reaching full confluency, the hiPSCs were switched to neuronal induction media (NM), composed of DMEM/F-12 GlutaMAX (Gibco), Neurobasal (Gibco), 0.5x N-2 (Gibco), 0.5x B27 (Gibco), 0.5x GlutaMAX (Life Technologies), 5 μg/ml insulin (Gibco), 0.5x NEAA (Sigma-Aldrich), 100 μM 2-mercaptoethanol (Sigma-Aldrich), and 1x Penicillin/Streptomycin (Gibco). To induce NPC differentiation, 1 μM dorsomorphin (Tocris) and 10 μM SB431542 (Tocris) were added. After 11 days, cells were passaged onto a Matrigel (Corning)-coated 6-well plates and cultured in NM. They were then transferred and cultured in NM containing 20 ng/ml FGF2 (PeproTech) for 4 days to promote NPC proliferation.
Astrocytes Differentiation
NPCs were seeded at a density of 3 × 106 cells/well in a Matrigel-coated 6-well plate. The following day, the media was replaced with NM containing 20 ng/ml FGF2 and 10 ng/ml BMP4 (PeproTech). The media was replenished every 1-2 days. After 28 days, astrocytes were sorted using an anti-GLAST-PE antibody (Miltenyi Biotec) and a cell sorter (Sony SH800). The sorted astrocytes were cultured in astrocyte media (AM, ScienCell).
Differentiation of hiPSCs to Hematopoietic Progenitor Cells (HPCs)
hiPSC-derived HPCs were generated as previously described19 using the STEMdiff Hematopoietic Kit (STEMCELL Technologies). Briefly, one day before differentiation, hiPSCs were washed with 1x DPBS (without Ca2+ and Mg2+) and detached using ReLeSR. Approximately 160 colonies were seeded onto Matrigel-coated 6-well plates with mTeSR1 media containing 10 μM Y-27632. On Day 0, mTeSR1 was replaced with 2 ml of basal media containing supplement A (basal sup A) at a 1:200 dilution. Half of the media was replaced with fresh basal sup A after two days. On Day 3, basal sup A was removed and replaced with 2 ml of basal media containing supplement B (basal sup B) at a 1:200 dilution. An additional 1 ml of basal sup B was added every two days. On Day 11, floating HPCs were collected by centrifugation at 300 x g for 5 min and subsequently used for differentiation into microglia-like cells.
Differentiation of HPCs to Microglia-like Cells (iMGL)
HPCs were suspended in basal media and seeded at 500,000 cells per well in a Matrigel-coated 6-well plates. The basal media consisted of DMEM/F12, insulin-transferrin-selenite (Gibco), B27 (Gibco), N2 (Gibco), GlutaMAX (Gibco), NEAA (Sigma-Aldrich), 400 μM monothioglycerol (Sigma-Aldrich), and insulin (Sigma-Aldrich) 5 μg/ml. On Day 0, HPCs were seeded in 2 ml basal media supplemented with three cytokines: IL-34 (100 ng/ml, PeproTech), TGF-β1 (50 ng/ml, Miltenyi Biotec), and M-CSF (25 ng/ml, Miltenyi Biotec). Every two days, 1 ml of fresh basal media containing the three cytokines was added. On Day 12, the media was partially changed by centrifugation and re-suspension in fresh media, maintaining the cells in the plate. The same cytokine-containing media (1 ml) was added every two days until Day 24. On Day 25, the media was partially changed again, and two additional cytokines, CD200 (100 ng/ml, Novo protein) and CX3CL1 (100 ng/ml, PeproTech), were added. From this point, basal media containing all five cytokines were added every two days. On Day 28, mature microglia-like cells were used for experiments.
Astrocytes and microglia-like cells co-culture
hiPSC-derived astrocytes and mature iMGL were seeded onto coverslips in 24-well plates at a ratio of 2:1 (astrocytes to iMGLs). Cells were cultured in astrocyte media (AM, ScienCell) without FBS, supplemented with M-CSF and CD200 to support iMGL survival. Although five cytokines (M-CSF, IL-34, TGF-β1, CD200, and CX3CL1) are typically used for iMGL culture, only M-CSF and CD200 were used in co-culture to avoid potential immunosuppressive effects. After 2 days of co-culture, the cells were used for experiments.
Immortalized human microglia
Immortalized human microglia, derived from a healthy female donor who provided informed consent for research use, were purchased from Applied Biological Materials Inc. (ABM; Cat. No. T0251). The use of immortalized human microglia was approved by the IRB of DGIST (Permit Number: DGIST-190829-BR-071-01). Sanger sequencing confirmed that these cells carried the APOE3 genotype. Isogenic APOE4 microglia were generated using CRISPR/Cas9 genome editing with the same sgRNA and ssODN as previously reported10. Cells were cultured in Prigrow III media (ABM) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin in 6-well plates. Upon reaching 70–80% confluency, cells were dissociated using 0.05% trypsin (Sigma-Aldrich) and replated at a 1:4 ratio in fresh media.
Cerebral organoid generation from hiPSCs
hiPSCs cultured on Geltrex-coated plates were dissociated using Accutase (STEMCELL Technologies) and seeded at a density of 12,000 cells/well into V-shaped 96-well plates (Thermo Fisher Scientific) pre-coated the previous day with 1% Pluronic (Sigma-Aldrich). Cells were maintained in embryoid body (EB) formation media consisting of Glasgow’s MEM (Gibco), 20% KnockOut Serum Replacement (Gibco), 1x sodium pyruvate, 1x NEAA, 100 μM 2-mercaptoethanol, 3 µM endo-IWR-1 (Tocris), and 5 µM SB431542, supplemented with 20 µM ROCK inhibitor, for 18–20 days. Additionally, 2 µM dorsomorphin was added to the EB formation media for the first 3 days. The media was changed every other day. On Days 18–20, individual EBs were transferred to 24-well plates using cut pipette tips, and cultured in neural induction media composed of DMEM/F-12 GlutaMAX, 1x N-2, and 1x Chemically Defined Lipid Concentrate (Gibco). Cells were maintained in an incubator with 40% oxygen. On Day 35, the media was supplemented with 10% FBS (Gibco), 5 µg/ml heparin, and 1% growh factor (GF)-reduced Matrigel (Corning). Cerebral organoids were used for experiments after 1 month of differentiation.
Glial cell embedding in AD Organoids
To model elevated Aβ levels in organoids, we used hiPSCs harboring the APPswe mutation, which increases Aβ production in neurons. On Day 40, glial cells were embedded into the organoids using 96-well U-bottom ultra-low binding plates (Corning). Organoids were then shaken horizontally at 60 rpm overnight in the existing organoid media without Matrigel. The following day, the media was completely replaced, and daily media changes were performed for 5 days to facilitate glial embedding. On Day 46, the organoids were primed with 100 µg/ml Poly I:C for 6 h. After a recovery period, the organoids were prepared for image analysis on Day 50.
Chemical treatment
To induce an immune response in astrocytes, cells were stimulated with Poly I:C (10 μM; Sigma-Aldrich) or a cytokine mixture: IL-1α (3 ng/ml; R&D Systems), TNFα (30 ng/ml; PeproTech), C1q (400 ng/ml; ProSpec). To evaluate the effect of priming on astrocytic response to Aβ42, primed astrocytes were treated with 10 μM Aβ42 oligomers for 6 h. Aβ42 oligomers were prepared by dissolving the peptide (AnaSpec) in 1% NH4OH, followed by dilution in PBS, and incubation at 37 °C for 24 h.
RNA extraction, cDNA synthesis and quantitative polymerase chain reaction (qPCR)
Cells were lysed using Ambion Trizol Reagent (Invitrogen) under RNase-free conditions for RNA extraction. The lysate was separated into an aqueous RNA phase using chloroform (Daejung) and centrifugation. The RNA pellet was washed with RNase-free 75% ethanol, air-dried, and resuspended in RNase-free water. RNA was reverse transcribed into cDNA using PrimeScriptTM Reverse Transcriptase (Takara Bio) and Oligo(dT)15 primer (Takara Bio). cDNA synthesis was performed using a Thermal Cycler (Bio-Rad) with the following conditions: 30 °C for 10 min, 50 °C for 45 min, and 70 °C for 15 min. Synthesized cDNA was subjected to qPCR using iTaqTM Universal SYBR (Bio-Rad) and the primer sequences described in Supplementary Data 1. qPCR was conducted on a CFX96TM System (Bio-Rad) with a cycling program of 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 59 °C for 30 s, and 72 °C for 30 s, ending with a melting curve analysis. Relative mRNA expression levels were normalized to GAPDH.
Cytokine array
To analyze 80 secreted cytokines from astrocytes, we used the Human Cytokine Antibody Array (Abcam) according to the manufacturer’s instructions. In brief, equal amounts of astrocyte-conditioned media were collected from identically seeded astrocyte cultures and incubated overnight at 4 °C with the antibody-printed membrane. Membranes were washed twice with washing buffer, then incubated with biotin-conjugated anti-cytokines at 4 °C for 2 h. Afterward, membranes were incubated overnight with HRP-conjugated streptavidin at 4 °C. The next day, the biotin-streptavidin-HRP-labeled membrane was detected using a detection buffer and visualized with a LuminoGraph II system (ATTO). Image was analyzed using ImageJ software (NIH).
RNA-seq analysis
Extracted total RNA was subject to RNA-seq library preparation. Libraries were pooled for sequencing using the Illumina NGS platform. The raw fastq data were aligned to the human hg38 assembly using STAR (version 2.4.0) RNA-seq aligner30. Gene raw counts were generated from the mapped data using the featureCounts tool31. The mapped reads were also processed by Cufflinks (version 2.2)32 to estimate transcript abundances. Gene differential expression test between sample groups was performed using the Cuffdiff module with adjusted q-value < 0.05 for statistical significance. The geometric method was chosen as the library normalization method for Cuffdiff. To explore concordant gene expression changes in the human AD brain14, DEG lists from the original AD brain single-cell RNA-seq paper for comparing (1) no pathology vs. pathology; (2) no pathology vs. early pathology; (3) early pathology vs. late pathology were used. Genes with significant changes in both iPSC-derived cell lines (adj p-value < 0.05) and the human brain (IndModel adj p-value < 0.25) were selected for making scatterplots and for overlap statistical analysis.
ATAC-seq analysis
ATAC-seq was performed following the protocol previously published33. In brief, 50,000 cells were collected by centrifugation at 300x g for 3 min at 4 °C. The cells were lysed in 50 μl of cold lysis buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween-20, 0.01% Digitonin [Promega]) and centrifuged at 500x g for 10 min. The pellet was gently resuspended in the transposition reaction buffer (25 μl 2X Tagment DNA buffer [Illumina], 2.5 μl Tagment DNA Enzyme 1 [Illumina Tagment DNA TDE1 Enzyme and Buffer Kit], 0.1% Tween-20, 0.01% Digitonin, 5 μl nuclease-free water). The transposition reaction was carried out by incubating the samples at 37 °C for 30 min. Following incubation, DNA was isolated using the Qiagen MinElute Reaction Cleanup Kit. The purified DNA was then amplified using NEBNext 2X PCR Master Mix and Illumina/Nextera i5 common adapters and i7 index adapters (sequences are provided in Supplementary Data 1). DNA libraries were amplified to 1/3 of the maximum qPCR RFU and purified using Agencourt AMPure XP magnetic beads (Beckman Coulter). The DNA libraries were sequenced on an Illumina HiSeqX sequencer. A snakemake batch processing script was generated to pre-process bulk ATAC-seq paired-end sequencing data as part of our standard pipeline (a similar script has been published previously http://github.com/gaofan83/snakemake_atac_alignment). In brief, 40-bp end sequences were trimmed for bowtie2 (version 2.3.4) alignment against the hg38 human genome with specific parameters (--end-to-end --very-sensitive --no-mixed --no-discordant -q --phred33 -I 10 -X 700). Aligned bam files were sorted and indexed using samtools (version 1.6). After the alignment step, fragment size histograms were plotted using R as quality control. Genrich peak caller34 was used for ATAC-seq peak calling. ATAC-seq signals across the union peak regions across all the samples were correlated to generate a heatmap using the multiBamSummary function of deeptools. Differential peak calling was done using DiffBind (version 2.14)35. Differential regions were selected with an adjusted p-value < 0.05.
Astrocytic Aβ42 uptake
Astrocytes were seeded in 12-well plates at a density of 3 × 105 cells/well for Aβ42 uptake analysis. For priming, astrocytes were treated with 10 μM Poly I:C. After a 72 h of recovery period, 250 ng/ml of oligomeric Aβ42 was added to the astrocytes. The total levels of Aβ42 were measured 48 h after treatment using a human Aβ42 ELISA kit (Invitrogen), following the manufacturer’s instructions. The amount of Aβ42 taken up by astrocytes was calculated by subtracting the residual Aβ42 in the media from the total amount added. The uptake index was determined by normalizing the amount of Aβ42 internalized to the number of cells.
Microglial Aβ42 uptake
To visualize Aβ42 clearance by human microglia and hiPSC-derived microglia-like cells (iMGL), Aβ42 labeled with HiLyte Fluor-555 peptide (AnaSpec) was used. Human microglia and iMGL were seeded at a density of 4 × 104 cells/well in 24-well plates and cultured for 2 days prior to experimentation. Cells were treated with 250 ng/ml Aβ42 for 30 min, fixed with 4% paraformaldehyde (Biosesang), and mounted with ProLong Diamond (Thermo Fisher Scientific) for imaging. Confocal microscopy (Zeiss) was used for image acquisition.
Systemic Poly I:C challenges
Six-week-old male C57BL/6 J mice were subjected to systemic Poly I:C challenges. After one week of acclimatization, the mice were divided into groups and challenged with either sterile water or Poly I:C. The animal experiments were conducted following the guidelines of the KIOM Animal Care and Use Committee (Approval Numbers: 22-091 and 22-112). Male APOE3 and APOE4 knock-in mice were used at an average age of 8 weeks. Poly I:C was dissolved in sterile saline, heated to 50 °C at a concentration of 2.4 mg/ml to ensure complete solubilization, then cooled naturally to room temperature to allow annealing of double-stranded RNA. Aliquots were stored at −20 °C until use. Mice received an intraperitoneal (i.p.) injection of Poly I:C (12 mg/kg) or sterile saline to assess the CNS inflammatory response21.
Oligomerization of Fluor 555-labeled Aβ42
The procedure was performed as previously reported36,37. Briefly, HiLyte Fluor 555-labeled Aβ42 was dissolved in DMSO and diluted to 100 µM with cold PBS. The solution was vortexed thoroughly and incubated at 4 °C for 24 h. Oligomeric Aβ42 was stored at −70 °C and used within one week of preparation.
Stereotaxic surgery for oligomeric Aβ42 injection
Stereotaxic injection was performed as described previously22,23. Oligomeric Aβ42 was bilaterally administered into the brain using a small animal stereotactic apparatus (Kopf Instruments). During the surgical procedure, anesthesia was induced and maintained with 3% isoflurane. A 33-gauge internal cannula (Plastics One) was positioned at the following coordinates: anterior-posterior (AP): −2.1 mm, medial-lateral (ML): ±1.4 mm, dorsal-ventral (DV): 2.1 mm. Aβ42 was injected at a rate of 0.2 µg (0.2 µl/min) per side. The incision site was sutured with Silkam Black Silk, and mice were placed on a 37 °C heating pad for recovery.
Immunohistochemistry of mouse brain tissue
Mice were sacrificed 6 h after the oligomeric Aβ42 injection. Brains were excised and fixed in 4% paraformaldehyde overnight. Following fixation, brains were washed with PBS and dehydrated in 30% sucrose at 4 °C for 48 h. Each brain was sectioned into 30 µm-thick slices using a cryostat (CM1800, Leica). Sections were then blocked in PBT (0.03% Tween 20 in 1x PBS) containing 0.3% Triton X-100 (Sigma-Aldrich) and 5% donkey serum (Genetex) for 1 h at room temperature. Immunofluorescence staining was performed using the following primary antibodies: anti-Iba1 antibody (Abcam, ab5076, 1:200), anti-GFAP antibody (Abcam, ab53554, 1:200), and anti-β-Amyloid (D54D2) XP® Rabbit monoclonal antibody (Cell Signaling Technology, #8243, 1:200). Brain sections were incubated with the primary antibodies overnight at 4 °C and then washed three times with PBT. Sections were subsequently incubated for 1 h with Alexa Fluor 488-conjugated donkey anti-goat secondary antibody (1:500, Invitrogen). After further washing three times with PBT, sections were mounted with Mounting Media (Vectashield) and visualized using an EVOS M7000 microscope. Fluorescence intensity in the images was quantified using ImageJ software (NIH).
Sholl analysis of microglia morphology
Morphological analysis of microglia located within 100 μm of Aβ42 was performed using Sholl analysis to quantify the complexity of cellular processes. Using Fiji (ImageJ) with the Sholl Analysis plugin, concentric circles were drawn around each cell’s soma at 2 μm intervals, from 0 to 20 μm. The number of intersections between these circles and the cell processes was counted, providing a measure of process complexity at various distances from the cell body. For each distance interval (0-20 μm at 2 μm steps), the average number of intersections was calculated across all cells, yielding a quantitative representation of the overall morphological complexity of the cell population.
RNA extraction, cDNA synthesis, and qPCR from mouse brain tissue
Following euthanasia, hippocampal tissue was lysed using QIAzol lysis reagent (Qiagen) in 2 mL soft tissue homogenizing tubes (CK14, Bertin Instruments). Samples were incubated on ice for 15 min and homogenized with a Precellys 24 tissue homogenizer (Bertin Instruments) for 20 s at 5000 rpm. Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA was reverse transcribed using the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). qPCR was performed using Power SYBR™ Green PCR Master Mix (Thermo Fisher Scientific) on a CFX-96 system (Bio-Rad), with gene-specific primers. Relative mRNA expression levels were normalized to 36B4. Primer sequences used are described in Supplementary Data 1.
Statistical analysis
All statistical analyses were performed using GraphPad Prism (version 9) or R software (edgeR version 4.4.2). Data in graphs are presented as mean ± S.E.M. The sample size for each experiment is indicated in the respective figure legend and represents the number of biological replicates (e.g., independent batches or individual animals). Statistical significance between two groups was determined using a two-tailed Student’s t-test. For comparisons among three or more groups, a one-way ANOVA followed by Tukey’s post-hoc test was employed. Significance levels are denoted in the figures as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Exact pvalues for each comparison are provided in the Source Data files. For transcriptome and chromatin accessibility analyses, statistical significance was determined using adjusted q-values or p-values < 0.05, as detailed in the RNA-seq and ATAC-seq analysis sections. All key experiments were successfully repeated with independent biological samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA seq and ATAC-seq data that support the findings of this study are deposited in the Gene Expression Omnibus. RNA-seq from astrocytes. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE274868. RNA-seq from microglia. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE274869. ATAC-seq data. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE274870 Source data are provided with this paper.
References
Netea, M. G. et al. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098 (2016).
de Laval, B. et al. C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell 26, 657–674.e8 (2020).
Larsen, S. B. et al. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell 28, 1758–1774.e8 (2021).
Bukhbinder, A. S. et al. Risk of Alzheimer’s disease following influenza vaccination: a claims-based cohort study using propensity score matching. J. Alzheimer’s. Dis. 88, 1061–1074 (2022).
Pomirchy, M. et al. Herpes Zoster vaccination and dementia occurrence. JAMA 333, (2025).
Eyting, M. et al. A natural experiment on the effect of herpes zoster vaccination on dementia. Nature 641, 438–446 (2025).
Perry, V. H. & Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224 (2014).
Sheng, J. G. et al. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145 (2003).
Neher, J. J. & Cunningham, C. Priming microglia for innate immune memory in the brain. Trends Immunol. 40, 358–374 (2019).
Lin, Y.-T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7 (2018).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Kwon, H. S. & Koh, S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9, 42 (2020).
Bright, J. J., Natarajan, C., Sriram, S. & Muthian, G. Signaling through JAK2-STAT5 pathway is essential for IL-3-induced activation of microglia. Glia 45, 188–196 (2004).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Sienski, G. et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 13, (2021).
TCW, J. et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185, 2213–2233.e25 (2022).
Kim, J., Basak, J. M. & Holtzman, D. M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303 (2009).
Lee, H. et al. ApoE4-dependent lysosomal cholesterol accumulation impairs mitochondrial homeostasis and oxidative phosphorylation in human astrocytes. Cell Rep. 42, 113183 (2023).
McQuade, A. et al. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13, 67 (2018).
Raja, W. K. et al. Self-Organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS ONE 11, e0161969 (2016).
Field, R., Campion, S., Warren, C., Murray, C. & Cunningham, C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNα/β and IL-1β responses in the diseased brain and exacerbates chronic neurodegeneration. Brain, Behav., Immun. 24, 996–1007 (2010).
Zhang, Y. et al. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 548, 52–57 (2017).
Oh, S.-Y. et al. Central administration of afzelin extracted from Ribes fasciculatum improves cognitive and memory function in a mouse model of dementia. Sci. Rep. 11, 9182 (2021).
Bekkering, S., Domínguez-Andrés, J., Joosten, L. A. B., Riksen, N. P. & Netea, M. G. Trained immunity: reprogramming innate immunity in health and disease. Annu. Rev. Immunol. 39, 1–27 (2021).
Krstic, D. et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm.9, 151 (2012).
Tejera, D. et al. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 18, 391–16 (2019).
Kitazawa, M., Oddo, S., Yamasaki, T. R., Green, K. N. & LaFerla, F. M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 25, 8843–8853 (2005).
Meyer, K. et al. REST and neural gene network dysregulation in iPSC models of Alzheimer’s disease. Cell Rep. 26, 1112–1127.e9 (2019).
Park, J.-H. et al. Lomerizine inhibits LPS-mediated neuroinflammation and tau hyperphosphorylation by modulating NLRP3, DYRK1A, and GSK3α/β. Front. Immunol. 14, 1150940 (2023).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2012).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Cooper, C. Living through extremes in process drama. Theatron 16, 157–164 (2022).
Christian, M. Transcriptional fingerprinting of “browning” white fat identifies NRG4 as a novel adipokine. Adipocyte 4, 50–54 (2014).
Choi, J.-W. et al. PyrPeg, a blood–brain-barrier-penetrating two-photon imaging probe, selectively detects neuritic plaques, not Tau aggregates. ACS Chem. Neurosci. 11, 1801–1810 (2020).
Stine, W. B., Jungbauer, L., Yu, C. & LaDu, M. J. Alzheimer’s disease and frontotemporal dementia, methods and protocols. Methods Mol. Biol. 670, 13–32 (2010).
Acknowledgements
We thank Dr. Tsai at MIT and Dr. Yankner at Harvard Medical School for sharing their iPSCs lines. This work was supported by the National Research Foundation (NRF) grant funded by the Ministry of Science and ICT (MSIT), South Korea (grant number: 2020M3A9D8039920) to M.S.K., the grant of the Korea Health Technology R&D project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (MOHW), South Korea (grant number: HI23C1368), the Korea Institute of Oriental Medicine (KIOM) research program funded by the MSIT (grant number: KSN2411012) to Y.G., the grant from the Korea Dementia Research Project through the Korea Dementia Research Center (KDRC), funded by the MOHW and MSIT (grant number: RS-2024-00343370), the Korea Brain Research Institute (KBRI) basic research program funded by the MSIT (24-BR-03-02), and the NRF grant funded by MSIT (2022R1A2C4001611) to J.S.
Author information
Authors and Affiliations
Contributions
S.I.L., J.Y., H.L., Y.G. and J.S. designed the project. S.I.L., J.Y., H.L., Y.G., and J.S. wrote the manuscript. S.I.L., J.Y., H.L., B.K., M.J., H.J., N.Y.K. M.E.P., J.K.K., and S.C. performed experiments. S.I.L., J.Y., H.L., B.K., M.J., H.J., H.H.W., M.S.K., F.G. and J.S. analyzed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, SI., Yu, J., Lee, H. et al. Astrocyte priming enhances microglial Aβ clearance and is compromised by APOE4. Nat Commun 16, 7551 (2025). https://doi.org/10.1038/s41467-025-62995-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62995-1
This article is cited by
-
NRC31 limits the imprinting of astrocyte epigenetic inflammatory memory early in life
Nature Communications (2025)
