
Abstract
While dysregulation of polyamine metabolism is frequently observed in cancer, it is unknown how polyamines alter the tumor microenvironment (TME) and contribute to therapeutic resistance. Analysis of polyamines in the plasma of pancreatic cancer patients reveals that spermine levels are significantly elevated and correlate with poor prognosis. Using a multi-omics approach, we identify Serpinb9 as a vulnerability in spermine metabolism in pancreatic cancer. Serpinb9, a serine protease inhibitor, directly interacts with spermine synthase (SMS), impeding its lysosome-mediated degradation and thereby augmenting spermine production and secretion. Mechanistically, the accumulation of spermine in the TME alters the metabolic landscape of immune cells, promoting CD8+ T cell dysfunction and pro-tumor polarization of macrophages, thus creating an immunosuppressive microenvironment. Small peptides that disrupt the Serpinb9-SMS interaction significantly enhance the efficacy of immune checkpoint blockade therapy. Together, our findings suggest that targeting spermine metabolism is a promising strategy to improve pancreatic cancer immunotherapy.
Similar content being viewed by others

Introduction
Pancreatic cancer, particularly pancreatic ductal adenocarcinoma (PDAC), is among the deadliest solid organ malignancies, with a five-year survival rate of less than 10%1,2. Recent advancements have identified immune checkpoint blockade (ICB) therapy as a promising treatment for solid tumors3. Despite its potential, only a fraction of patients respond to ICB therapy, and its effectiveness in PDAC has been limited4,5. The tumor microenvironment plays a critical role in shaping the response to ICB therapy, with metabolic factors emerging as key regulators of immune function6,7. The interaction between metabolic states and immune cell functionality defines the TME8,9. Among the metabolic pathways implicated in tumor progression, polyamine metabolism—including putrescine (PUT), spermidine (SPD), and spermine (SPM)—is dysregulated in many cancers10. Ornithine decarboxylase 1 (ODC1) catalyzes the rate-limiting step in polyamine biosynthesis, converting ornithine into PUT. PUT is then sequentially converted into SPD and SPM by spermidine synthase (SRM) and SMS11. Because of their direct association with oncogenes, polyamine metabolism has been targeted for cancer therapeutic development12,13. Research indicates that polyamines have anti-inflammatory and immunosuppressive properties, suggesting that reducing polyamine levels could enhance antitumor immune responses11,14. In pancreatic cancer, polyamine synthesis has been shown to be essential for tumor growth15, and SAT1 (spermidine/spermine N1-acetyltransferase 1)-mediated polyamine metabolism contributes to PDAC aggressiveness16. While the effects of dysregulated polyamine metabolism in tumor cells are well-documented, its molecular mechanisms and impact on tumor immune microenvironment remain less understood.
The progression of pancreatic tumors is intricately governed by molecular changes within the tumor cells and corresponding alterations in the TME17,18. Serpinb9, a member of the serine protease inhibitor family, plays a crucial role in regulating immune responses and cellular homeostasis19,20. In normal cells, Serpinb9 primarily functions to inhibit granzyme B, a serine protease involved in inducing apoptosis in target cells. By modulating granzyme B activity, Serpinb9 prevents excessive immune cell-mediated tissue damage and maintains immune balance21. Cancer cells often exploit Serpinb9 to protect themselves from apoptosis induced by immune cells. This increased expression of Serpinb9 in tumors can hinder the effectiveness of immune-mediated cell killing, allowing cancer cells to survive and proliferate despite the presence of an active immune response22,23,24. Emerging evidence from CRISPR screens has identified Serpinb9 as a key factor in modulating the response to ICB therapy in lung cancer25 and melanoma26, where it is associated with T cell dysfunction and impaired CTL infiltration. Despite these insights, its role in tumor metabolism and TME reprogramming remains largely unexplored.
In this work, we report that plasma levels of spermine are significantly elevated in patients with PDAC compared to healthy individuals, and are associated with poorer clinical outcomes. Mechanistically, we identify Serpinb9 as a key player of spermine biosynthesis in tumor cells. Serpinb9 physically interacts with SMS and thus prevents SMS from lysosome-mediated degradation, leading to increased spermine biosynthesis and secretion. Within the TME, elevated spermine levels suppress mTORC1 signaling, thereby dampening oxidative phosphorylation in mitochondria and impairing CD8+ T cell function. Additionally, spermine also activates TGF-β signaling, promoting M2 polarization of tumor-associated macrophages (TAMs), further reinforcing an immunosuppressive TME. To target the Serpinb9-SMS-spermine metabolic pathway, we develop peptide inhibitors aimed at curtailing spermine production in tumors. This approach is designed to enhance the effectiveness of ICB therapy in treating PDAC.
Results
Serpinb9 is a therapeutic vulnerability associated with spermine metabolism
Metabolism of polyamines, including putrescine, spermidine, and spermine, is often dysregulated in human cancers. We were interested in examining the levels of three polyamines in the plasma of PDAC patients as well as healthy individuals. For the PDAC patients, plasma samples were collected before treatment as part of a clinical study involving Sintilimab (anti-PD-1 monoclonal antibody) plus modified FOLFIRINOX (folinic acid, fluorouracil, irinotecan, and oxaliplatin)27. These samples were then categorized into two groups based on clinical outcomes: Partial Response (PR) and Progressive Disease (PD). Among the three polyamines, spermine levels were significantly increased in the Partial Response group compared to the healthy control group (0.0675 ± 0.078 μM vs. 0.0454 ± 0.0073 μM, p = 0.049). Furthermore, spermine levels were also elevated in the Progressive Disease group compared to the Partial Response group (0.2263 ± 0.0815 μM vs. 0.0675 ± 0.078 μM, p = 0.033), while putrescine and spermidine levels had minimal changes (Fig. 1a). Clinical data analysis also showed that high plasma levels of spermine are significantly correlated with worse overall survival (OS) of PDAC patients (Fig. 1b and Fig. S1a). Elevated spermine concentrations in the tumor interstitial fluid (TIF) of PDAC patient tissues were associated with reduced levels of intratumoral CD8+ T cells. (Fig. 1c). Surprisingly, the mRNA expression levels of key enzymes in the polyamine metabolic pathway, including ODC1, SAT1, spermine Oxidase (SMOX), SMS, SRM, and polyamine oxidase (PAOX), were not associated with the clinical outcome of PDAC patients. No significant differences were observed in their expression levels between OS-high and OS-low patients according to the analysis of The Cancer Genome Atlas (TCGA) datasets (Fig. S1b). Therefore, there must be a factor that determines the spermine metabolism in a post-transcriptional manner.

a Quantification of polyamines in plasma from healthy individuals (n = 10), PR (n = 20) patients, and PD patients (n = 8) with PDAC. b OS of PDAC patients with high or low plasma spermine concentrations (n = 40 patients). c Correlation between spermine levels in TIF and intratumoral CD8+ T cells (n = 40 patients). d Intersection of differentially expressed genes from four pancreatic cancer datasets with T cell dysfunction-related genes. e Extracellular relative spermine levels in BxPC-3 cells after the top seven genes knockdown (n = 3 biologically independent samples). f Positive correlation between Serpinb9 expression and spermine levels in PDAC patients (n = 40 patients). g Representative staining and negative correlation between Serpinb9 expression and CD8+ T cell infiltration in PDAC tissues (n = 40 patients). Scale bars: 50 μm. H-score: histological score. h Uniform manifold approximation and projection (UMAP) of scRNA-seq from PDAC tumors showing 10 clusters. i Dot plot of canonical marker expression in each cluster. j Representative UMAP plot displaying the distribution of Serpinb9 expression in tumor versus normal tissues. k Quantification of Serpinb9 in different cell types, including ductal, T, myeloid, and CAFs. l Representative staining of Serpinb9, pan-CK, CD3, CD68, and ASMA in tumor and paired normal tissues. m Quantification of Serpinb9 in different cell types (n = 7 patients). n, o Association between Serpinb9 expression and tumor grade or TNM stage (n = 148 patients). p OS of all patients with pancreatic cancer, stratified by high versus low Serpinb9 expression (n = 112 patients). q, r OS (q), and disease-specific survival (DSS) (r) of pancreatic cancer patients in TCGA, based on Serpinb9 expression. For a, e, k, m, n, and o, data were presented as mean ± S.D. For b and p–r data were generated using Kaplan-Meier survival curves based on log-rank tests. For c, f, and g Spearman correlations and P values were calculated using a two-sided Spearman’s test. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. Box plots show the median (center line), 25th–75th percentiles (box), and whiskers (1.5×IQR range). Source data are provided as a Source Data file.
To identify pivotal determinants of spermine biosynthesis in pancreatic tumors, we performed a comprehensive bioinformatics analysis leveraging gene expression profiles sourced from four distinct pancreatic cancer datasets (TCGA, GSE15471, GSE28735, and GSE71729). By analyzing differentially expressed genes specific to pancreatic cancer compared to normal controls within each dataset, we subsequently identified a conserved subset of genes common across all datasets. This subset was further cross-referenced with genes implicated in T cell dysfunction26, leading to the identification of 7 candidate genes (Fig. 1d). Next, we knocked down each of the seven genes individually in a pancreatic cancer cell line BxPC-3, and detected intracellular and extracellular polyamine levels. Knockdown (KD) of Serpinb9 significantly reduced intracellular and extracellular spermine levels, while knockdown of the other six genes had no significant effect. Additionally, Serpinb9 knockdown modestly increased intracellular spermidine levels, but no significant changes were observed in extracellular spermidine or putrescine (Fig. 1e and Fig. S1c). The results were also validated in other human PDAC cell lines (PANC-1, SW1990, MIA PaCa-2, and ASPC-1) and mouse KPC (KrasLSL-G12D; Trp53LSL-R172H; Pdx1-Cre) pancreatic tumor cells (Fig. S1d, e). In a cohort of 40 pancreatic cancer patients, Serpinb9 expression correlated positively with spermine levels (Fig. 1f). Next, based on Tumor Immune Dysfunction and Exclusion (TIDE) scores26, which measure T cell dysfunction and exclusion in tumors to predict immune checkpoint blockade (ICB) response, Serpinb9 was ranked as one of the genes most closely related to T-cell dysfunction (Fig. S2a). Additionally, using publicly available CRISPR–Cas9 screens of cancer cell lines, we obtained the CERES scores for these seven genes, which measure the necessity of genes for tumor cell viability28 (Fig. S2b). Serpinb9 is one of the top-ranked essential genes for tumor cell viability. Furthermore, immunohistochemical (IHC) staining of pancreatic tumor tissue samples demonstrated that higher Serpinb9 levels were associated with more reduced CD8+ T cell infiltration (Fig. 1g). These results suggest that Serpinb9 plays a crucial role in regulating spermine levels and CD8+ T cell dysfunction in pancreatic tumors.
Serpinb9 is highly overexpressed in pancreatic tumor tissues compared to paired normal tissues, evidenced by the analysis of the TCGA dataset (Fig. S2c). The result was further substantiated by immunoblotting and IHC staining analyses of in-house paired clinical samples of pancreatic tumors and adjacent normal tissues (Fig. S2d, e), as well as in mouse KPC and KTC (KrasLSL-G12D; TgfbR2fl/fl; Ptf1a-Cre) tumors and adjacent normal tissues (Fig. S2f). To determine the main cell type responsible for the elevated Serpinb9 expression in pancreatic tumor tissues, we analyzed publicly available datasets of single-cell RNA sequencing (scRNA-seq), encompassing 32 samples and over 100,000 cells. All the cells were annotated into 10 major cell populations according to the canonical cell markers (Fig. 1h, i, and Table S1). Serpinb9 expression is most prominently elevated in ductal cells, which represent tumor cells, compared to other cell types (Fig. 1j, k). Multiplex-IHC (mIHC) staining of PDAC tissues and paired normal tissues further confirmed this result (Fig. 1l, m). To explore the clinical significance of Serpinb9 in pancreatic cancer, we assessed its levels in an in-house tumor tissue microarray including 156 PDAC cases. Serpinb9 levels were significantly associated with tumor grade (P = 0.0015) and tumor-node-metastasis (TNM) stage (P = 0.0009) (Fig. 1n, o). Serpinb9 levels were also positively correlated with serum CA19-9 levels (Table S2). Higher Serpinb9 levels correlated with worse OS of patients in the full cohort (Fig. 1p), which is also supported by the analysis of the TCGA dataset (Fig. 1q, r).
Collectively, these results suggest that Serpinb9 is a key factor that determines spermine levels in pancreatic cancer.
Serpinb9 prevents lysosomal degradation of SMS
To elucidate how Serpinb9 mediates the upregulation of SPM levels, proteomics analysis with liquid chromatography-tandem mass spectrometry (LC-MS/MS) was conducted on negative control (NC) or Serpinb9-KD KPC cells. A total of 220 differentially expressed proteins were identified, including 77 down-regulated proteins and 143 up-regulated proteins (fold change > 1.2, p < 0.05) (Fig. S3a). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis revealed an enrichment of proteins associated with three metabolic pathways upon Serpinb9 knockdown (Fig. 2a). Particularly, the arginine and proline metabolism pathway includes spermine biosynthesis. Among the differentially expressed proteins within these pathways, SMS exhibited the most significant change after Serpinb9 knockdown (Fig. 2b). SMS is highly expressed in human pancreatic tumors as well as in KPC and KTC mouse tumors (Fig. S3b, c). A significant correlation between Serpinb9 and SMS levels was observed in the analysis of a pancreatic tumor tissue microarray (n = 148) (Fig. 2c and Fig. S3d, e). Immunofluorescence analysis and fluorescence intensity plots demonstrated significant Serpinb9/SMS colocalization (Serpinb9: green; SMS: red) in PDAC cell lines (KPC, Panc02, BxPC-3) (Fig. S3f–h) and PDAC tumor tissues (human and mouse) (Fig. S3i, j). Co-immunoprecipitation (co-IP) and immunoblotting assay revealed a physical interaction between Serpinb9 (FLAG tag) and SMS (HA tag) in PDAC cell lines (Fig. 2d and Fig. S3k). Direct interaction between these two proteins was also confirmed using purified GST-Serpinb9 and His-tagged SMS proteins in a GST pull-down assay (Fig. 2e). In live cells, there was the real-time interaction between Serpinb9 and SMS (Fig. 2f and Fig. S3l). Next, we used AlphaFold-Multimer29,30 to construct the atomic model of the binding complex of Serpinb9-SMS. F336 at Serpinb9 was predicted to contribute significantly to the intramolecular interaction within the complex based on Free Energy Perturbation (FEP) calculations (Fig. 2g). To validate it, we generated an F336A mutant of Serpinb9, as well as an E306A/R307A/D308A mutant as a distal negative control. Co-IP assay showed that F336A mutation drastically reduced the binding of Serpinb9 to SMS while the E306A/R307A/D308A mutations had minimal effect (Fig. 2h). SMS protein levels changed concomitantly with Serpinb9 knockdown or overexpression in both mouse and human pancreatic cancer cell lines (Fig. S4a, b). The F336A mutation at Serpinb9 led to a reduction in SMS level (Fig. S4c). Additionally, SMS levels were significantly downregulated in Serpinb9-KD KPC tumors compared to control KPC tumors (Fig. S4d). Similarly, immunofluorescence staining results showed that Serpinb9 (green) and SMS (red) levels were reduced in Serpinb9-depleted PDAC cells (Fig. S4e). Importantly, there was literally no change in the transcription of SMS (indicated by mRNA levels) following Serpinb9 KD or overexpression (Fig. S4f), suggesting that Serpinb9 regulates SMS at the posttranslational level.

a KEGG enrichment analysis of differentially expressed proteins in Serpinb9 KD vs. Serpinb9 NC KPC cells (n = 3 biologically independent samples). b Western blotting analysis of proteins within three metabolic pathways in Serpinb9 NC and KD KPC cells. c Correlation analysis of Serpinb9 and SMS expression in PDAC tissue microarrays (n = 148 patients). d Co-IP and western blotting analysis showing the interaction between HA-tagged SMS and Flag-tagged Serpinb9. e GST-pull-down assay of SMS-His and Serpinb9-GST proteins. f Duolink immunofluorescence images of Serpinb9-SMS interaction in KPC and BxPC-3 cells; red dots indicate interaction. Scale bars: 10 μm. g The predicted complex of the Serpinb9 (blue) and SMS (pink), showing the relative binding free energy difference (ΔΔG) from alanine scanning of crucial residues in Serpinb9 (n = 10 biologically independent samples). h Co-IP and quantification of SMS binding to wild-type or mutant Serpinb9 (F336A; E306A/R307A/D308A). i SMS stability in Serpinb9 NC and KD KPC cells after CHX treatment (200 μg/mL). j SMS levels in KPC cells treated with MG132 (5 μM, 12 h) or CQ (50 μM, 12 h). k Representative images and quantification of lysosomes in KPC cells (n = 5 biologically independent samples). Scale bars: 0.5 μm. l SMS distribution in Serpinb9 NC vs. KD cells via confocal microscopy. Scale bar: 25 μm. m SMS expression in CQ-treated NC and KD cells. n Co-IP of HA-tagged SMS in KPC cells. o SMS interaction with Lamp2A in Serpinb9 NC vs. KD cells. p Schematic and binding free energy of Serpinb9 and Lamp2A competitively binding to SMS (n = 50 biologically independent samples). For h, i, j, m, o data were presented as mean ± S.D., n = 3 biologically independent samples. For k data were presented as mean ± S.D., n = 5 biologically independent samples. For g data were presented as mean ± S.D., n = 10 biologically independent samples. For p, data were presented as mean ± SEM, n = 50 independent samples. For c Spearman correlations and P values were calculated using a two-sided Spearman’s test. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. NS, no significance. Source data are provided as a Source Data file.
In the absence of de novo protein synthesis induced by cycloheximide treatment, Serpinb9 knockdown (Serpinb9-KD) in PDAC cells led to a markedly accelerated decline in SMS levels compared to control cells expressing normal levels of Serpinb9 (half-life: 4 hours vs 16 hours) (Fig. 2i and Fig. S4g), implicating a role for Serpinb9 in SMS protein degradation. In mammalian cells, protein degradation pathways primarily involve the ubiquitin-proteasome system and lysosomal proteolysis. Treatment with the lysosome inhibitor chloroquine (CQ) significantly stabilized SMS, whereas treatment with the proteasome inhibitor MG132 had no such effect, indicating that SMS degradation predominantly occurs via lysosomal proteolysis (Fig. 2j and Fig. S4h). Electron microscopy revealed an increase in lysosome numbers in Serpinb9-KD PDAC cells (Fig. 2k and Fig. S4i), alongside elevated levels of Lamp2, a lysosomal marker (Fig. S4j). Moreover, Serpinb9 knockdown resulted in the accumulation of SMS proteins in the lysosomal fraction (Fig. S4k). Immunofluorescence co-staining of lysotracker and SMS in KPC cells further confirmed that SMS localization within lysosomes was enhanced upon Serpinb9 knockdown (Fig. 2l). Consistent with these findings, the reduction of SMS levels due to Serpinb9 depletion was significantly attenuated by chloroquine treatment (Fig. 2m and Fig. S4l). To explore the underlying mechanism of Serpinb9-mediated lysosomal SMS degradation, we investigated SMS interactions with key lysosomal proteins associated with lysosomal function. Notably, SMS was found to specifically interact with the lysosomal membrane protein Lamp2A, but not with other membrane proteins such as LAMP1, Sortilin, and M6PR (Fig. 2n and Fig. S4m). Interestingly, Serpinb9 knockdown enhanced the binding of SMS to Lamp2A (Fig. 2o and Fig. S4n). Predictive modeling using AlphaFold-Multimer indicated that SMS binding energies to both Serpinb9 and Lamp2A were comparable (Fig. 2p), suggesting that Serpinb9 may compete with Lamp2A for SMS binding and stabilize SMS by inhibiting its lysosome-mediated degradation.
Spermine promotes PDAC progression and confers immunotherapy resistance
Because SMS is a rate-limiting enzyme in the SPM biosynthesis13, Serpinb9 may up-regulate SPM levels by inhibiting the lysosomal degradation of SMS (Fig. 3a). To test it, we generated an SMS knockout (KO) KPC cell line using CRISPR/Cas9 technology, and then overexpressed Serpinb9 in both control and SMS-KO KPC cells. Cas9 expression was confirmed in these cells (Fig. S5a). Serpinb9 overexpression increased intracellular and extracellular SPM levels in the control KPC cells, a trend that was abolished by SMS knockout. Additionally, Serpinb9 overexpression reduced intracellular SPD levels, but caused no significant change in extracellular SPD or PUT (Fig. S5b). In an orthotopic KPC mouse model, Serpinb9 overexpression significantly promoted the growth of control KPC tumors, but not SMS-KO KPC tumors (Fig. 3b). IHC staining of tumor-infiltrated lymphocytes revealed a reduction in CD8+ and GZMB (Granzyme B)+ cells following Serpinb9 overexpression. Furthermore, in the SMS-KO tumors, Serpinb9 overexpression did not lead to a reduction in CD8+ and GZMB+ cells (Fig. 3c and Fig. S5c). Similarly, SPM concentrations in the tumor interstitial fluid were elevated in Serpinb9-overexpressing tumors compared to the controls, while SMS knockout eliminated this increase (Fig. 3d). No significant changes in PUT and SPD concentrations in the tumor interstitial fluid were observed between the experimental groups (Fig. S5d). Additionally, we used Serpinb9-KO KPC cells supplemented with spermine in vivo. Spermine supplementation restored tumor growth due to Serpinb9 depletion (Fig. 3e and Fig. S5e). Moreover, the increased infiltration of CD8+ and GZMB+ cells observed in Serpinb9-KO tumors was reversed upon spermine supplementation, further supporting that the immunosuppressive effects of Serpinb9 are mediated through spermine-dependent mechanisms (Fig. S5f, g). To determine whether the effects of Serpinb9/SMS are specifically mediated by CD8+ T cells, we depleted CD8+ T cells in C57BL/6 mice prior to inoculating them with Serpinb9-overexpressing tumor cells. The results showed that the tumor progression induced by Serpinb9 overexpression was completely abrogated following CD8+ T cell depletion (Fig. 3f and Fig. S5h, i). Moreover, the levels of polyamine transporters, including ATP13A3, SLC3A2, SLC7A5, SLC22A16, SLC7A1, and SLC12A8, were not significantly affected in KPC and BxPC-3 cells after Serpinb9 knockdown (Fig. S5j). These findings suggest that Serpinb9 promotes tumor progression through a mechanism dependent on SMS-mediated spermine biosynthesis and CD8+ T cells.

a Diagram of the polyamine metabolic synthesis pathway. b Photographs and weights of tumors from each group (n = 5 mice). c IHC quantification of CD8+ and GZMB+ cells (n = 5 mice). d Spermine concentrations in TIF from tumors (n = 5 mice). e Photographs and tumor weights of orthotopic tumors from mice implanted with Serpinb9-KO KPC cells, with or without spermine (n = 5 mice). f Photographs and tumor weights from mice with or without CD8+ T cell depletion, inoculated with Serpinb9-overexpressing tumor cells (n = 5 mice). g, h Photographs and weights of KPC orthotopic pancreatic tumors from C57BL/6 J and nude mice treated with saline or spermine (10 mg/kg) (n = 7 mice). i, j Survival of KPC-bearing C57BL/6 J and nude mice treated with saline or spermine (n = 10 mice). k Representative images and statistical results of CD8+ T cells, GZMB+CD8+ T cells, IFN-γ+CD8+T cells, Ki67+CD8+T cells, Tregs, and flow cytometric analysis of PD-1+CD8+ T cells, CD80+TAMs, CD206+TAMs, and PD-L1+ TAMs (n = 7 mice). l Tumor photographs and weights from mice treated with SPM, αPD-1 (100μg/mouse), or combination (n = 5 mice). m Survival of mice treated with SPM, αPD-1, or combination (n = 8 mice). n Flow cytometric analysis of CD8 + T cells and GZMB + CD8 + T cells (n = 5 mice). o Schematic of the protocol of SMS knockout combined with αPD-1 therapy in orthotopic KPC SMS NC and SMS KO cells (5 × 105)-bearing mice. p Tumor photographs and weights from each group (n = 5 mice). q Survival of mice in each group (n = 10 mice). For b–f, l, n, and p, data were presented as mean ± S.D., n = 5 biologically independent samples. For g, h, and k data were presented as mean ± S.D., n = 7 biologically independent samples. For i, j, m, and q data were generated using Kaplan-Meier survival curves based on log-rank tests. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. NS, no significance. o was created in part with BioRender. Hanshen, Y. (https://BioRender.com/nfthfna). Source data are provided as a Source Data file.
To explore the role of spermine in pancreatic cancer immune responses, orthotopic KPC tumor-bearing C57BL/6 or nude mice (Nu/J) were administered intraperitoneally with spermine (10 mg/kg, q.d. for 2 weeks). Tumor growth was significantly increased in the immunocompetent C57BL/6 mice, but not in the immunodeficient nude mice (Fig. 3g, h). No notable changes in spleen weight and body weight were observed upon spermine administration (Fig. S6a). Additionally, spermine supplementation markedly reduced the survival of the tumor-bearing C57BL/6 mice, an effect not observed in the nude mice (Fig. 3i, j). The cells isolated from the tumor tissues of C57BL/6 mice treated in vivo with spermine were used for flow cytometry analysis. Flow cytometry revealed that spermine administration significantly decreased CD8+ T cell infiltration, proliferation, and production of cytotoxic effectors, including GZMB and interferon-gamma (IFN-γ), while increased the expression of exhaustion markers, along with increased regulatory T cells (Tregs) (CD25+Foxp3+) and TAMs expressing CD206 and PD-L1 (M2-polarized), decreased TAMs expressing CD80+ (M1-polarized) (Fig. 3k). Minimal effects were observed on other types of immune cells, such as CD4+ T cells, NK cells, myeloid-derived suppressor cells (MDSCs), and dendritic cells (Fig. S6b). These results were confirmed by IHC staining in the TME, with an increase in Ki67+ proliferative tumor cells (Fig. S6c). Additionally, plasma spermine concentrations were significantly elevated in the SPM-treated group compared to controls. Notably, the achieved plasma levels were comparable to those observed in PDAC patients, indicating the physiological relevance of the treatment dose (Fig. S6d). We then assessed whether spermine mitigated the responsiveness of pancreatic tumors to ICB therapy. In the KPC orthotopic tumor models, spermine supplementation significantly reduced the efficacy of anti-PD-1 immunotherapy (Fig. 3l) and shortened the survival of the tumor-bearing mice (Fig. 3m), with no notable changes in body weight (Fig. S6e). It decreased both the number and activity of tumor-infiltrated cytotoxic CD8+ T cells (Fig. 3n and Fig. S6f). C57BL/6 mice bearing orthotopic control or SMS-KO KPC tumors were treated with anti-PD-1 antibody (Fig. 3o). As expected, SMS knockout plus anti-PD-1 antibody significantly inhibited tumor growth and extended mouse survival (Fig. 3p, q), along with high levels of CD8+ T cell infiltration and cytokine production (Fig. S6g), highlighting the importance of spermine in ICB efficacy.
In summary, these results indicate that spermine promotes tumor growth through an immune-dependent mechanism and creates a TME that suppresses immune responses, thereby reducing the effectiveness of ICB therapy.
Spermine inhibits calcium uptake and suppresses OXPHOS in CD8+ T cells
To elucidate the molecular mechanisms by which spermine inhibits the activity of cytotoxic CD8+ T cells, we first performed an energy metabolomics analysis on CD8+ T cells isolated from the spleens of C57BL/6 mice, both with and without spermine treatment. Spermine treatment significantly altered the metabolic profile of CD8+ T cells, notably downregulating oxidative phosphorylation (OXPHOS), glycolysis, and the tricarboxylic acid (TCA) cycle (Fig. 4a). RNA sequencing (RNA-seq) further revealed a marked downregulation of the mTORC1 and OXPHOS signaling pathway in CD8+ T cells treated with spermine (Fig. 4b). Key regulatory components of the mTORC1 pathway, including the phosphorylation of mTOR, S6K, and 4EBP-1, were notably suppressed (Fig. 4c, and Fig. S7a).

a Differential energy metabolism-related metabolites in CD8+ T cells treated with spermine (n = 3 biologically independent samples per group). b GSEA plots of mTORC1 and OXPHOS signaling based on RNA-seq of CD8+ T cells treated with spermine (n = 3 biologically independent samples per group). c Western blotting of mTOR, p-mTOR, S6K, pS6K, 4EBP1, and p-4EBP1 levels in CD8+ T cells treated with spermine (2 μM, 24 h). d Flow cytometric analysis of Ca²+ levels in the cytoplasm of CD8+ T cells. (n = 3 biologically independent samples). e Flow cytometric analysis of Ca²+ levels in the mitochondria of CD8+ T cells. (n = 3 biologically independent samples). f Representative images and quantification of mitochondria in CD8+ T cells (n = 5 biologically independent samples). Scale bars: 0.5 μm. g Representative images and quantification of MitoSOX in CD8+ T cells treated with spermine (n = 3 biologically independent samples). Scale bars: 8 μm. h Representative images and quantification of JC-1 in CD8+ T cells treated with spermine (n = 3 biologically independent samples). Scale bars: 8 μm. i Quantification of ATP levels in CD8+ T cells treated with spermine (n = 3 biologically independent samples). j OCR data of CD8+ T cells treated with spermine (n = 3 biologically independent samples). k ECAR data of CD8+ T cells treated with spermine (n = 3 biologically independent samples). l Representative images and quantification of apoptosis in CD8+ T cells treated with spermine (n = 3 biologically independent samples). m CFSE images and quantification of CD8+ T cells treated with spermine (n = 3 biologically independent samples). n Flow cytometric analysis of Ki67, PD-1, Granzyme B, IFN-γ, TNF-α, and Perforin in CD8+ T cells treated with spermine (n = 3 biologically independent samples). For c n = 3 biologically independent samples. For f, data were presented as mean ± S.D., n = 5 biologically independent samples. For d, e, and g–n data were presented as mean ± S.D., n = 3 biologically independent samples. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. Source data are provided as a Source Data file.
Polyamines have been known to act as antagonists at relatively high concentrations for calcium-sensing receptors, a family C G protein-coupled receptor31. Indeed spermine led to a reduction in Ca²+ levels in the cytoplasm and mitochondria of CD8+T cells, as measured using Fluo-4 AM and Rhod-2 AM, respectively, and analyzed by flow cytometry (Fig. 4d, e). Calcium is believed to play a crucial role in modulating mitochondrial function, with research indicating that low levels of mitochondrial calcium can reduce ATP production by influencing the activity of calcium-sensitive enzymes within the mitochondrial matrix32. Additionally, the OXPHOS pathway was downregulated in CD8+ T cells treated with spermine (Fig. S7b). Given that mitochondria, the cell’s powerhouse, are closely linked to T cell antitumor effectiveness33,34, and that the mTORC1 pathway is crucial for cellular metabolism—including glycolysis and OXPHOS35,36,37—the impact of spermine on mitochondrial function was investigated. We reasoned that spermine impairs mitochondrial biogenesis and function. Electron microscopy revealed that spermine treatment reduced the number of mitochondria while increasing their size in CD8+ T cells (Fig. 4f), a finding corroborated by MitoTracker flow cytometry analysis (Fig. S7c). Following spermine treatment of CD8+ T cells, qPCR analysis revealed a significant downregulation of Mfn1 and Mfn2, indicating a potential inhibitory effect of spermine on mitochondrial fusion. In contrast, the expression levels of fission-related genes Fis1 and Mff remained largely unchanged (Fig. S7d). These results suggest that spermine selectively modulates mitochondrial dynamics by suppressing fusion rather than enhancing fission in CD8+ T cells. Further assessment of mitochondrial function showed elevated levels of mitochondrial reactive oxygen species (mtROS, as indicated by MitoSOX staining) in spermine-treated T cells (Fig. 4g), along with a decreased mitochondrial membrane potential (measured by JC-1 staining, with aggregates shown in red and monomers in green) (Fig. 4h). Consequently, ATP production was reduced in spermine-treated T cells (Fig. 4i). Seahorse analysis confirmed that spermine decreased both basal and maximal oxygen consumption rates (OCR) as well as the extracellular acidification rate (ECAR) (Fig. 4j, k). While spermine did not significantly induce apoptosis in CD8+ T cells (Fig. 4l), it did reduce their proliferation, as evidenced by reduced carboxyfluorescein succinimidyl ester (CFSE) staining (Fig. 4m). Production of cytotoxic effectors, including GZMB, IFN-γ, tumor necrosis factor α (TNF-α), and perforin, was also decreased upon spermine treatment (Fig. 4n).
Collectively, spermine inhibits calcium uptake and mitochondrial function, leading to CD8+ T cell dysfunction by suppressing the mTOR pathway.
Spermine promotes TAM M2 polarization and exacerbates CD8+ T cell dysfunction
TAMs play a crucial role in creating an immunosuppressive TME that enables cancer to evade immune surveillance38,39. In our in vivo studies, beyond its effects on CD8+ T cells, spermine promotes M2 polarization of TAMs and increases Treg abundance (Fig. 3i). However, spermine does not directly induce Treg differentiation in vitro (Fig. S7e). Instead, spermine treatment led to a reduction in CD86 expression and an increase in CD206, PD-L1, and RELMa expression in bone marrow-derived macrophages (BMDMs) and Raw cell lines co-cultured with KPC tumor cell-conditioned medium (Fig. 5a). Additionally, spermine treatment induced the upregulation of Arg1 in BMDMs and Raw cell lines, as demonstrated by Western blotting analysis (Fig. S7f, g). Similar to the effect of spermine in CD8+ T cells, spermine-treated BMDMs showed a significant reduction in mitochondrial Ca²+ levels (Fig. 5b). Additionally, spermine led to decreased gene signature scores for the electron transport chain (ETC) and OXPHOS, while increasing the gene signature scores for purine metabolism (Fig. 5c, d). Previous studies have demonstrated that cells reprogram purine metabolism in response to mitochondrial respiratory deficiencies40. Key enzymes in the purine metabolism pathway, including ADA, GUK1, XDH, and IMPDH, were significantly upregulated, indicating that macrophages exhibited a protein signature characteristic of purine metabolism-driven macrophages (Fig. 5e and Fig. S7h). These purine metabolism macrophages mediate immunosuppression and may promote M2 polarization41.

a Flow cytometric analysis of CD86, CD206, PD-L1, and RELMa in BMDMs and Raw cells treated with spermine (n = 3 biologically independent samples). b Flow cytometric analysis of Ca²+ levels in the mitochondria of BMDMs (n = 3 biologically independent samples). c GSEA plot of ETC and OXPHOS based on proteomic sequencing of BMDMs treated with spermine. d GSEA plot of purine metabolism based on proteomic sequencing of BMDMs treated with spermine. e Western blotting of ADA, GUK1, XDH, and IMPDH levels in BMDMs treated with spermine (10 μM, 24 h). f KEGG enrichment analysis of differentially expressed proteins in BMDMs treated with spermine, highlighting the top ten pathways (n = 3 biologically independent samples per group). g Western blotting of Smad2, p-Samd2, Smad3, and p-Smad3 levels in BMDMs treated with spermine (10 μM, 24 h). h Extracellular TGF-β levels in BMDMs treated with spermine (n = 3 biologically independent samples). i Schematic diagram illustrating the interaction between BMDMs and CD8 + T cells under the influence of spermine. j Flow cytometric analysis of CD206, PD-L1, and RELMa in BMDMs (n = 3 biologically independent samples). k OCR data of CD8+ T cells (n = 3 biologically independent samples). l CFSE images and quantification of CD8+ T cells (n = 3 biologically independent samples). m Flow cytometric analysis of Ki67, PD-1, GZMB, IFN-γ, TNF-α, and Perforin in CD8+ T cells (n = 3 biologically independent samples). For a, b, h, j–m data were presented as mean ± S.D., n = 3 biologically independent samples. For e and g, n = 3 biologically independent samples. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. Panel i was created in part with BioRender. Hanshen, Y. (https://BioRender.com/rbvmrne). Source data are provided as a Source Data file.
Next, we conducted quantitative proteomics analysis on control and spermine-treated BMDMs, identifying 538 differentially expressed proteins (Fig. S7i). TGF-β signaling pathway stands out as one of the top-ranked pathways upregulated by spermine (Fig. 5f). As an indicator for TGF-β signaling activity, phosphorylated Smad2 and Smad3 levels were induced by spermine (Fig. 5g and Fig. S7j). TGF-β secretion was also elevated (Fig. 5h). Previous studies have demonstrated that TGF-β suppresses mTOR signaling in exhausted T cells and plays a crucial role in regulating the metabolism and function of CD8+ T cells42. To investigate the effect of spermine in this context, CD8+ T cells were co-cultured with TAMs and subsequently treated with spermine for 24 hours. Following treatment, CD8+ T cells and TAMs were isolated for further analyses (Fig. 5i). In the TAMs, spermine promoted M2 polarization (Fig. 5j). In CD8+ T cells, spermine inhibited their OCR to a greater extent when co-cultured with BMDMs compared to CD8+ T cells alone (Fig. 5k). Similarly, spermine significantly suppressed CD8+ T cell proliferation, increased exhaustion, and markedly reduced the production of cytotoxic effectors, including GZMB, IFN-γ, TNF-α, and Perforin in CD8 + T cells (Fig. 5l, m). Since spermine activates the TGF-β signaling pathway, which may contribute to the development of less-differentiated tumors, we performed IHC staining for GATA6 under different conditions to assess tumor differentiation status. Our results show that Serpinb9 knockdown led to an increase in GATA6 staining levels, suggesting a shift toward a more differentiated tumor phenotype (Fig. S7k). In contrast, Serpinb9 overexpression resulted in decreased GATA6 staining levels, indicating a transition to a less-differentiated tumor subtype. Notably, in SMS KO tumors, the changes in GATA6 staining levels induced by Serpinb9 OE were no longer observed, suggesting that SMS plays a critical role in mediating these effects (Fig. S7l). Additionally, we systematically analyzed Serpinb9 mRNA expression across the molecular subtypes defined by Collisson, Bailey, and Moffitt, as well as in the TCGA dataset43,44,45. Notably, Serpinb9 expression varied significantly among subtypes in the Bailey dataset, with higher expression observed in more aggressive subtypes. However, no statistically significant differences were found in the other three datasets (Fig. S7m). Furthermore, clinical data analysis revealed that plasma spermine levels were significantly higher in patients with poorly differentiated tumors compared to those with well-differentiated tumors, further supporting the role of altered spermine metabolism in promoting tumor aggressiveness and poor differentiation in PDAC (Fig. S7n).
In TAMs, spermine activates the TGF-β signaling pathway, inducing purine metabolism and driving macrophage polarization toward immunosuppressive, thereby further exacerbating CD8+ T cell dysfunction.
Depletion of Serpinb9 sensitizes pancreatic cancer to immunochemotherapy
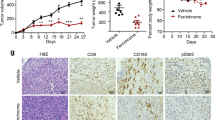
Given the essential role of Serpinb9 in spermine biosynthesis, we wanted to test whether inhibiting Serpinb9 level or activity suppresses pancreatic tumor growth and therapeutic sensitivity. Knockdown of Serpinb9 modestly inhibited PDAC cell proliferation in vitro (Fig. S8a). However, spermine concentrations in tumor interstitial fluid of Serpinb9-KD KPC tumors were markedly lower compared to control KPC tumors (Fig. 6a). To assess the role of Serpinb9 in pancreatic tumor growth, mouse KPC with altered levels of Serpinb9 (control, knockdown, or overexpression) were subcutaneously inoculated into both nude mice and C57BL/6 mice. Serpinb9 knockdown markedly decreased tumor volume and weight in C57BL/6 mice while Serpinb9 overexpression promoted tumor growth. The effects of Serpinb9 on tumor growth were modest and much weaker in the nude mice (Fig. 6b, c, and Fig. S8b–e). Similar results were obtained in Panc02 cell-derived tumors (Fig. S8f–i). In orthotopic KPC-derived tumor models, Serpinb9 knockdown extended survival significantly in immunocompetent C57BL/6 mice, but not in immunodeficient nude mice (Fig. 6d, e). In terms of tumor immune profiles, Serpinb9 knockdown significantly increased the infiltration and activity of CD8+ T cells, along with a decrease in Tregs and CD206+ TAMs, but an increase in CD86+ TAMs (Fig. S9a). Minimal effects were observed on other types of immune cells, such as B cells, NK cells, MDSCs, and dendritic cells (Fig. S9b). Multiplex IHC staining assays confirmed these results (Fig. S9c). To determine if CD8+ T cells mediate Serpinb9-associated anti-tumor immunity, we depleted CD8+ T cells from the C57BL/6 mice prior to KPC tumor cell inoculation (Fig. 6f). Serpinb9 knockdown, in combination with anti-PD-1 treatment, resulted in a significant reduction in tumor growth compared to the control group. Depletion of CD8+ T cells completely eliminated the difference in tumor growth between the control group and the combinational therapy group (Fig. 6g,h). Interestingly, Serpinb9-KD tumors exhibited a reduced number of cancer-associated fibroblasts (CAFs) and less collagen deposition, suggesting a microenvironment that may be more susceptible to therapy (Fig. 6i). We also treated orthotopic control and Serpinb9-KD tumors (expressing luciferase for in vivo imaging) with Gemcitabine and/or anti-PD-1 antibody (Fig. S10a). Analysis of tumor luminescence intensity demonstrated that the tumors gradually shrank with continued drug administration, with the most pronounced reduction in the Serpinb9-KD group with the combinational treatment of Gemcitabine and anti-PD-1 antibody (Fig. 6j, k, and Fig. S10b).

a Serpmine concentrations in TIF from Serpinb9 NC and Serpinb9 KD KPC tumors (n = 5 mice). b, c Tumor growth curves of Serpinb9 NC and Serpinb9 KD KPC tumors from immunocompetent (b) and immunodeficient mice (c) (n = 5 mice). d, e Survival of Serpinb9 NC and Serpinb9 KD KPC-bearing immunocompetent (d) and immunodeficient mice (d) (n = 10 mice). f Schematic of the protocol for CD8+ T cells depletion in orthotopic KPC (5 × 105)-bearing mice receiving combination therapy. g Tumor photographs and weights from each mouse group (n = 5 mice). h Flow cytometry and statistical analysis of CD8+ T cells in spleens (n = 5 mice). i Representative images and quantification of aSMA (upper) and Sirius red (lower) in Serpinb9 NC and Serpinb9 KD pancreatic cancer tissues (n = 5 mice). Scale bars: 100 μm (upper), 75 μm (lower). j Representative images of luminescence intensity change in tumors, with final day luminescence intensity (n = 3 mice). k Tumor weights from each mouse group (n = 5 mice). l Schematic of the protocol of Serpinb9 deficiency combined with αPD-1/αPD-L1 therapy in orthotopic KPC Serpinb9 NC and Serpinb9 KD tumor-bearing mice. m Tumor photographs and weights from each mouse group (n = 5 mice). n Survival of mice in each group (n = 7 mice). o Flow cytometric analysis of CD8+ T cells, Ki67+CD8+ T cells, GZMB+CD8+ T cells, IFN-γ+CD8+ T cells, and PD-1+CD8+ T cells (n = 5 mice). For a–c, g–k, m, and o, data were shown as the means ± SD., n = 5 biologically independent samples. For d, e, and n data were generated using Kaplan-Meier survival curves based on log-rank tests. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. NS, no significance. f and l were created in part with BioRender. Hanshen, Y. (https://BioRender.com/nfthfna). Source data are provided as a Source Data file.
Analysis of human cancer immunotherapy datasets revealed that high Serpinb9 levels are associated with a negative response to ICB in bladder cancer (BLCA), melanoma, and renal cell carcinoma (RCC) (Fig. S10c). Additionally, elevated Serpinb9 levels correlate with poor OS following ICB treatment (Fig. S10d). To determine the additive effect of Serpinb9 inhibition when combined with ICB, orthotopic control and Serpinb9-KD KPC tumors in C57BL/6 mice were treated with anti-PD-L1 or anti-PD-1 monoclonal antibodies (Fig. 6l). It was clear that combining ICB therapy with Serpinb9 depletion significantly enhances the efficacy of immunotherapy. While both ICB and Serpinb9 depletion alone inhibited tumor growth, the combination treatment showed additive effects (Fig. 6m). There were no significant changes in mouse body weight and spleen weight among the six groups (Fig. S10e, f), indicating the tolerability of single or combinational treatments. Notably, the longest mouse survival was achieved with Serpinb9 depletion combined with anti-PD-1 (Fig. 6n). Flow cytometry analyses revealed that the combinational therapy significantly increased the infiltration of CD8+ T cells and the subpopulations of GZMB+CD8+ T cells, IFN-γ+CD8+ T cells, Ki67+CD8+ T cells while decreasing the subpopulation of PD-1+ CD8+ T cells in the tumors compared to the control or single-agent treatments (Fig. 6o). Serpinb9 has been reported to potentially affect CAFs and collagen24, which are major barriers to the penetration of chemotherapeutic agents. We also found reduced CAFs and collagen deposition in the Serpinb9-deficient KPC pancreatic orthotopic tumor models compared to NC (Fig. 6i). Similar results were obtained for Panc02 tumors with the same types of treatments (Fig. S10g–i).
Taken together, these results suggest that Serpinb9 inhibition enhances the therapeutic efficacy of ICB in pancreatic cancer in a manner dependent on CD8+ T cells.
A peptide inhibitor targeting the Serpinb9-SMS interaction
Given the role of Serpinb9 in inhibiting GZMB activity, there has been an interest in developing an inhibitor against Serpinb9. A small-molecule inhibitor, named 3034, was designed to target the Serpinb9-GZMB interaction and showed efficacy in suppressing tumor progression in melanoma, lung cancer, and breast cancer24. We evaluated the anti-tumor efficacy of 3034 in orthotopic KPC tumor models, both as a single agent and in combination with ICB therapies (anti-PD-L1 or anti-PD-1). Although 3034 demonstrated considerable anti-tumor activity on its own, the combination with ICB did not provide any additional benefit over the ICB alone or 3034 alone treatments (Fig. S11a, b). Importantly, we confirmed that 3034 reduces the Serpinb9-Granzyme B interaction, but had no effect on the Serpinb9-SMS interaction (Fig. S11c, d). We reasoned that an inhibitor disrupting the Serpinb9-SMS interaction could be a more promising approach for treating PDAC. Based on the molecular interactions between Serpinb9 and SMS, we designed three human Serpinb9-mimicking peptides (Pep1, Pep2, and Pep3) and one derived from mouse Serpinb9 (Pep4). These peptides, including the residue F336 of Serpinb9, demonstrated stable binding to SMS in molecular dynamics simulations (Fig. 7a and Fig. S11e). All the peptides were actually N-terminally acetylated (neutralized) during synthesis to block the potential artifact from the charged free amine group. We found that Pep2 most effectively attenuated the Serpinb9-SMS interaction among the three human peptides (Fig. 7b and Fig. S11f). The IC50 value of Pep2 was 1661 μM in human BxPC-3 cells, and the IC50 of Pep4 was 1640 μM in mouse KPC cells (Fig. S11g). The ~1.6 mM IC50 concentration was for the inhibition of the ultimate tumor cell viability (the half-maximal inhibitory concentration). However, the IC50 concentration of these peptides to inhibit SMS levels and spermine secretion is at much lower concentrations (50–200 µM). Immunoblotting analysis revealed a decrease in SMS protein levels in PDAC cells following treatment with varying concentrations and durations of Pep2 and Pep4 (Fig. 7c, d, and Fig. S11h, i). Spermine production and secretion were significantly reduced following treatment with the inhibitory peptides: Pep2 (200 µM, 24 hours) for BxPC-3 cells and Pep4 (200 µM, 24 hours) for KPC cells (Fig. 7e). Moreover, the inhibitory peptides markedly decreased intracellular spermidine levels, while extracellular levels of both spermidine and putrescine remained unaffected (Fig. S11j). This observation also suggests that the peptides are safe, as they achieve their intended effects without significantly affecting cell viability at biologically relevant concentrations. They primarily function by inhibiting SMS levels and reducing spermine secretion at concentrations ranging from 50–200 µM. Through this mechanism, the peptides help alleviate the immunosuppressive tumor microenvironment rather than directly targeting tumor cell viability. Additionally, KPC tumor-bearing mice received intratumoral injections of Pep4 (30 mg/kg, once every other day) for 2 weeks. As a result, SMS levels and spermine concentrations in the tumor interstitial fluid of Pep4-treated KPC tumors were significantly reduced compared to those in control KPC tumors (Fig. 7f,g, and Fig. S11k).

a Computational design workflow for Serpinb9-mimic peptides targeting SMS. b Co-IP and western blotting analysis showing the interaction between HA-tagged SMS and Flag-tagged Serpinb9. c, d Heatmap of relative SMS levels in pancreatic cancer cell lines (BxPC-3, KPC) treated with a concentration and time gradient of Pep2 or Pep4. e Statistical analysis of relative extracellular spermine levels in BxPC-3 and KPC cells treated with Pep2 or Pep4 (n = 3 biologically independent samples). f Statistical analysis of relative SMS levels in KPC tumors treated with Pep4 (n = 5 mice). g Statistical analysis of relative spermine concentrations in TIF from KPC tumors treated with Pep4 (n = 5 mice). h, i Tumor growth curves and weights from mice treated with Pep4, αPD-1, or their combination (n = 5 mice). j Changes in body weights of mice in each group (n = 5 mice). k Representative t-SNE images and statistical results of CD8+ T cells by flow cytometry (n = 5 mice). l Representative mIHC staining and quantification of CD8, GZMB, CD206, and Foxp-3 (n = 5 mice). Scale bars = 20 μm. For b–d, n = 3 biologically independent samples. For e data were presented as mean ± S.D., n = 3 biologically independent samples. For f–l data were presented as mean ± S.D., n = 5 biologically independent samples. Statistical significance was determined using two-sided unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. Source data are provided as a Source Data file.
As a proof-of-concept study, we next assessed the effect of Pep4 on tumor progression and ICB response in vivo. The Pep4 was administered via intratumoral injection (30 mg/kg, once every other day) as a single agent or in combination with anti-PD-1 antibody to mice bearing subcutaneous KPC-derived tumors. The treatment of Pep4 significantly reduced tumor growth and had even greater anti-tumor activity when combined with anti-PD-1 antibody in comparison with the control group (Fig. 7h, i, and Fig. S12a). No notable toxicity was observed as indicated by minimal changes in body weight and spleen weight (Fig. 7j and Fig. S12b), as well as histological analysis of organ sections and blood biochemical tests (Fig. S12c, d) among the four treatment groups. To further assess Pep4-induced changes in the tumor microenvironment, we performed spectral flow cytometry (Cyteck) analysis of tumor-infiltrated immune cells. Unsupervised density-based clustering (t-SNE) revealed significant differences in CD8+ T cell proportions in tumors (Fig. 7k). Notably, Pep4 increased CD8+ T cell infiltration and cytotoxic activities (GZMB, IFN-γ, TNF-α, Perforin) and decreased PD-1+CD8+ T cell subpopulations. Additionally, the numbers of CD206+ macrophages and Tregs were significantly reduced, while CD86+ macrophages increased markedly in the combination group compared to control and single agent groups (anti-PD-1 or Pep4) (Fig. S12e, f). Multiplex IHC staining confirmed the flow cytometry results (Fig. 7l).
To assess whether immunological memory is induced by the combination therapy with Pep4 and anti-PD-1, we rechallenged KPC tumor-free mice from the initial subcutaneous model with the same KPC cells, without administering additional therapy (Fig. S13a). The combination therapy markedly reduced tumor size and volume compared to the control group (Fig. S13b, c). Notably, in some mice from the combination therapy group, rechallenge with KPC cells did not result in tumor formation (CR: 3/7; CR denotes tumors smaller than 60 mm3 that continued to regress until the end of the study). In contrast, all age-matched control mice developed tumors (Fig. S13d, e). Flow cytometry analysis revealed a significantly higher number of effector memory T cells (TEM, CD44hiCD62Llow) in the mouse spleen of the combination therapy group compared to the control group (Fig. S13f). These findings suggest that Pep4 and anti-PD-1 treatment induces an immune memory effect capable of inhibiting tumor growth.
In summary, developing inhibitors to block the Serpinb9-SMS interaction alters the spermine metabolism and reprograms the tumor immune microenvironment of PDAC, thereby improving the efficacy of ICB therapy. Combining a Serpinb9 inhibitor with PD-1 blockade presents an attractive therapeutic strategy for treating pancreatic cancer.
Discussion
Therapeutic resistance of ICB in PDAC is primarily due to immunosuppressive TME and metabolic dysregulation46,47. The findings presented in this study elucidate the critical role of spermine metabolism in shaping the immune landscape of pancreatic cancer and highlight its implications for therapeutic translation. Our study revealed that Serpinb9 interacts with SMS to stabilize its levels, thereby elevating spermine production and secretion within the TME. This interaction profoundly influences immune cell functions and ultimately impacts the efficacy of ICB in pancreatic cancer (Fig. 8).

Spermine levels are markedly elevated in the plasma of pancreatic cancer patients and correlate with poor prognosis. In tumor cells, Serpinb9 directly interacts with spermine synthase to hinder its lysosomal degradation, thereby augmenting the biosynthesis of spermine. Inhibiting Serpinb9 or its interaction with spermine synthase enhances the efficacy of pancreatic cancer immunotherapy. This Figure was created with BioRender. Hanshen, Y. (https://BioRender.com/dwim8mz).
Understanding metabolic interactions within the pancreatic tumor microenvironment enhances our insight into disease progression and reveals potential vulnerabilities for therapeutic intervention48,49,50. Recent research has focused on the role of polyamine metabolism in tumorigenesis and cancer immunity, given its frequent dysregulation in human cancers11,12. Specifically, polyamine biosynthesis, including spermine, has been linked to oncogenic signaling pathways51,52. Previous studies have demonstrated that spermidine supplementation can restore impaired immunotherapy efficacy in aged mice by enhancing mitochondrial function and activating CD8+ T cells53. In contrast, spermine has been shown to facilitate tumor progression by inducing PD-L1 expression and reducing CD8+ T cell infiltration in hepatocellular carcinoma54. However, the precise mechanisms by which spermine directly modulates CD8+ T cell function and shapes the macrophage phenotype in pancreatic cancer remain poorly understood. In this study, we observed that elevated plasma spermine levels in pancreatic cancer patients correlate negatively with survival. Additionally, spermine concentrations in tumor interstitial fluid from PDAC patient tissues negatively correlated with the presence of intratumoral CD8+ T cells. Intraperitoneal spermine injection promoted pancreatic tumor growth in immunocompetent mice, worsened the TME, and led to resistance to ICB treatment. Notably, spermine adversely affected CD8+ T cell function beyond merely inhibiting GMZB. Spermine treatment suppressed the mTORC1 signaling pathway in CD8+ T cells, decreased mitochondrial activity, and impaired OXPHOS, resulting in reduced proliferation, diminished cytotoxic effector production, and increased exhaustion of CD8+ T cells. This is in line with previous studies suggesting that polyamines influence CD8+ T metabolic fitness by modulating mitochondrial activity53. Furthermore, spermine-induced M2 polarization in TAMs enhanced TGF-β signaling, exacerbating CD8+ T cell dysfunction within the TME38,55. The observed upregulation of purine metabolism in TAMs following spermine treatment adds a dimension to our understanding of TAM dynamics in pancreatic cancer. The dual impact of spermine on both CD8 + T cells and TAMs underscores its role in creating an immunosuppressive environment that facilitates tumor immune evasion and progression.
While previous research has explored Serpinb9 primarily as an inhibitor of Granzyme B24,26, its functions extend beyond this single interaction, and its specific roles in modulating tumor immunometabolism are not yet fully understood. In this study, we observed that Serpinb9 is predominantly upregulated in cancer cells compared to other types of cells within pancreatic tumors. Furthermore, Serpinb9 levels correlate with tumor grade, TNM stage, CA19-9 levels, and overall patient survival in pancreatic cancer. Notably, our findings demonstrate that inhibiting Serpinb9 enhances T cell infiltration and improves tumor responsiveness to immune checkpoint blockade. Additionally, the identification of Serpinb9 as a crucial regulator of spermine biosynthesis and SMS stability unveils a previously unrecognized link between polyamine metabolism and the tumor immune microenvironment. This finding not only advances our understanding of Serpinb9 immunosuppressive mechanisms in cancer but also highlights potential avenues for therapeutic intervention.
From a therapeutic perspective2,56, targeting the Serpinb9-SMS-spermine axis represents a promising strategy for enhancing the sensitivity of pancreatic cancer to immunotherapy. Our research demonstrates that Serpinb9 depletion sensitizes pancreatic tumors to immune checkpoint blockade, leading to increased infiltration of CD8+ T cells and improved effector functions within the tumor microenvironment. Additionally, the development of the peptide inhibitor Pep4, which targets the Serpinb9-SMS interaction, provides an innovative approach for disrupting spermine production in pancreatic cancer. Pep4 effectively reduced SMS levels, lowered spermine concentrations in the tumor microenvironment, and significantly inhibited tumor progression. Combining Pep4 with anti-PD-1 therapy not only suppressed tumor growth but also fostered the development of immunological memory against tumor rechallenge, suggesting a sustained anti-tumor immune response. However, there are notable limitations with the current peptide inhibitor. The peptide functions primarily as a pro-drug in proof-of-concept studies, and its effective concentration is substantially higher than the desired nanomolar range. Advancing this approach to clinical application necessitates further refinement of the peptide’s design, optimization of its pharmacological properties, and development of efficient delivery methods to achieve therapeutic efficacy at clinically relevant doses. Although the biological activity of the peptides supports effective intracellular engagement, further studies are warranted to clarify their exact mode of cell entry and cross-species specificity.
In conclusion, our study advances the understanding of spermine-mediated immune evasion mechanisms in pancreatic cancer and introduces a previously uncharacterized therapeutic strategy targeting the Serpinb9-SMS interaction. By elucidating these mechanisms, we offer a rationale for therapeutic interventions designed to modulate polyamine metabolism and the tumor immune microenvironment. Future research has the potential to revolutionize treatment approaches and improve clinical outcomes for patients with pancreatic cancer receiving immunotherapy.
Methods
Ethical approval
All animal experiments were approved by the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (approval number: 2025-135).
All experiments involving human samples were approved by the Institutional Review Board of the First Affiliated Hospital, School of Medicine, Zhejiang University (approval number: 2025B IIT 0304), and written informed consent was obtained from all participants prior to sample collection.
Patient samples
Paired human pancreatic cancer tissue samples and plasma of PDAC patients were sourced from the Department of Hepatobiliary and Pancreatic Surgery at the First Affiliated Hospital, School of Medicine, Zhejiang University. Tumor tissue microarrays were prepared by Wuhan Service Bio-Technology. Plasma samples from PDAC patients used in Fig. 1a were obtained from participants enrolled in the clinical trial previously described27. All other human sample analyses were conducted using specimens from an independent cohort of pancreatic cancer patients. For selected assays, a subset of 40 patients was randomly chosen from this cohort based on sample availability. Plasma samples from 10 healthy donors (5 males, 5 females; aged 20–40 years) were collected during routine health check-ups at the First Affiliated Hospital, Zhejiang University School of Medicine. All donors provided written informed consent before sample collection. No compensation was provided.
Animals
Balb/c nude mice and C57Bl/6 J mice (aged 6-8 weeks) were obtained from the Model Animal Research Center of Nanjing University. The animals were maintained under specific-pathogen-free (SPF) conditions in the Experimental Animal Center at the First Affiliated Hospital, School of Medicine, Zhejiang University.
Cell culture
Pancreatic ductal adenocarcinoma (PDAC) cell lines, except KPC, were purchased from the American Type Culture Collection (ATCC). These cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; 11965092, Gibco) with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin (Pen-Strep). The KPC cell line, derived from a genetically engineered mouse model (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre), was cultured in modified McCoy’s 5 A Medium (16600108, Thermo Fisher Scientific) with 10% FBS and 1% Pen-Strep. All cell lines were routinely tested for mycoplasma contamination using PCR and found to be negative.
Establishment of mouse PDAC model
In vivo experiments were terminated at predefined endpoints, including a single tumor exceeding 20 mm in any dimension, a total tumor volume surpassing 4000 mm3 in cases of multiple tumors, body weight loss of more than 20%, or signs of severe distress such as lethargy or impaired mobility. Animals were randomly assigned to each group, and data collection and analysis were conducted by two independent investigators who were blinded to the group assignments. KPC cells (expressing a short hairpin RNA targeting Serpinb9 (shSerpinb9) and a negative control plasmid) were injected subcutaneously into the right flanks of mice (5 × 105 cells/mouse). Tumor size was measured using calipers, with the formula volume = length × width2 × 0.5. Mice were sacrificed after three weeks, and tumors were analyzed by flow cytometry, western blotting, and immunohistochemistry. For survival analysis, KPC Serpinb9 NC and KPC Serpinb9 KD cells were injected orthotopically into the pancreas of mice (5 × 105 cells/mouse), and the time of death was recorded. To evaluate the pro-tumorigenic function of Serpinb9 in an SMS-dependent manner, NC and SMS KO tumor cells were first generated using the pLV3-U6-MCS-sgRNA-Cas9-Flag-EGFP-Puro vector (either empty or containing sgRNA). Subsequently, Serpinb9 overexpression cell lines were established using the pLV3-CMV-MCS-HA-mCherry vector (either empty or containing Serpinb9 OE). These cells were then orthotopically implanted into C57BL/6 mice at a density of 5 × 105 cells per mouse. C57BL/6 or nude mice were implanted orthotopically with 5 × 105 KPC cells. Following implantation, mice were administered intraperitoneally with spermine (10 mg/kg, q.d. for 2 weeks). To assess the therapeutic effect of ICB on Serpinb9-deficient PDAC, C57BL/6 mice were implanted orthotopically with 5 × 105 Serpinb9 NC or Serpinb9 KD KPC cells, followed by treatment with dual ICB (anti-PD1 or anti-PD-L1 antibodies) or IgG isotype control (100 μg per mouse, six times every three days). To evaluate the therapeutic effect of Gemcitabine plus ICB, C57BL/6 mice were orthotopically implanted with 5 × 105 Serpinb9 KD or NC KPC-luc cells. Tumor size was monitored using in vivo imaging after intraperitoneal injection of D-Luciferin (Gold Biotechnology). For subcutaneous tumorigenesis experiments, KPC cells were injected into the right flanks of C57Bl/6 J mice pretreated or not with a Serpinb9 mimicking peptide (200 μM) for 24 h (1 × 105 cells/mouse). To evaluate the impact of Serpinb9 mimicking peptide on ICB therapy, mice were randomly divided into four groups: Pep4 treatment (30 mg/kg), αPD-L1 therapy (100 μg per mouse), Pep4-αPD-1 combination therapy, and control. Treatments were administered three times weekly for two weeks, after which tumors were harvested for further analysis.
Cell transfection
Human and mouse Serpinb9 shRNA plasmids, mouse SMS CRISPR-Cas9 plasmids, Human Serpinb9 expressing plasmid Flag-Serpinb9, and Human SMS expressing plasmid HA-Serpinb9 were purchased from Miaoling Bio. For lentivirus production, targeted plasmids, plasmid psPAX2, and plasmid pMD2.G were transfected into 293 T cells using jetPRIME reagent (Polyplus). The supernatant was filtered and concentrated after 48 h. KPC and BxPC-3 cells were stably transfected with lentiviral particles and selected with puromycin (10 μg/mL). Knockdown efficiency was confirmed by western blotting.
In vivo antibody depletion of CD8+ T cells
CD8+ T cells were depleted by intraperitoneal injection of anti-CD8 monoclonal antibodies (BP0061, Bio X Cell, 200 μg per mouse) three days before orthotopic inoculation with Serpinb9 NC or Serpinb9 KD KPC cells (5 × 105 cells). Anti-CD8 was administered every three days throughout the experiment.
Metabolic studies
The metabolic activity of CD8+ T cells was analyzed using a Seahorse XFe24 Analyzer (Agilent) to measure the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). For this, 1 × 105 CD8+ T cells were seeded in Seahorse 24-well plates, and the Seahorse XF Cell Mitochondria Stress assay was performed following the manufacturer’s protocol. For OCR measurement, cells were cultured in XF RPMI-1640 medium supplemented with 10 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate. Baseline OCR was recorded, followed by sequential injections of oligomycin (2 μM, to inhibit ATP synthesis), FCCP (1 μM, to uncouple mitochondrial oxidative phosphorylation), and a combination of rotenone and antimycin A (0.5 μM each, to inhibit complexes I and III of the electron transport chain). For ECAR measurement, cells were maintained in XF RPMI-1640 medium supplemented with 2 mM L-glutamine. Baseline ECAR was measured, followed by sequential additions of glucose (10 mM), oligomycin (1 μM), and 2-deoxyglucose (2-DG, 50 mM).
Western blotting and immunoprecipitation
Proteins were extracted using RIPA lysis buffer (P0013B, Beyotime Biotechnology) with phosphatase and protease inhibitors. Protein samples were quantified using bicinchoninic acid (BCA) (P0012, Beyotime Biotechnology) and subjected to SDS-PAGE and electrotransfer to PVDF membranes (Millipore). Membranes were blocked with 5% skim milk, incubated with primary antibodies overnight at 4 °C, and then with HRP-conjugated secondary antibodies. Immunoprecipitation was performed using IP/Western lysing solution (P0013, Beyotime Biotechnology) following the manufacturer’s instructions. Quantitative results were obtained using ImageJ software (NIH). The primary antibodies are listed in Table S3.
Immunohistochemistry (IHC) analysis
Tissue samples were fixed in neutral buffered formalin (10%), paraffin-embedded, and sectioned at 4 μm. PDAC tissue microarrays were immunostained with antibodies against Serpinb9 (ab233443, Abcam) and SMS (ab156879, Abcam). After overnight incubation with primary antibodies, sections were treated with HRP-conjugated secondary antibodies and visualized using a DAB chromogen kit (BDB2004, Biocare). Images were captured with Image Scope software (Leica Biosystems) and quantitative results were analyzed using ImageJ and GraphPad Prism. The primary antibodies are listed in Table S4.
Ex vivo treatment of CD8+ T cells and BMDMs with spermine
To investigate the effects of spermine on immune cells, CD8+ T cells and BMDMs were treated ex vivo under controlled conditions. For CD8+ T cells, freshly isolated cells were cultured in vitro and treated with 2 μM spermine for 24 hours. To prevent polyamine oxidation by serum amine oxidases, a serum-free medium was used prior to spermine treatment. For BMDMs, cells were first incubated with a KPC-conditioned medium containing FBS for 48 hours to simulate the tumor microenvironment. After this incubation, BMDMs were washed three times with PBS to remove the residual conditioned medium. Subsequently, cells were switched to a standard culture medium without FBS before spermine treatment (10 μM, 24 hours) in subsequent experiments.
RNA sequencing
mRNA sequencing was performed on Serpinb9 KD versus NC KPC cells and spermine versus PBS-treated CD8+ T cells. Total RNA was extracted using the RNA-Quick Purification Kit (RN001, YiShan Biotechnology). Library construction, sequencing (Illumina), and analysis were conducted at the Biomedical Big Data Center, First Affiliated Hospital, School of Medicine, Zhejiang University.
Metabolite analysis by LC–MS/MS
For spermine versus PBS-treated CD8+ T cells, metabolite extraction, detection, and quantitative analysis were performed by Wuhan Biotechnology.
Tumor interstitial fluid collection
Tumor interstitial fluid was obtained from either mouse xenograft tumors or human resected tumor tissues. The tumors were rinsed with PBS and then cut into small fragments (3–5 mm3). These fragments were placed on 70-μm filters within 50 mL centrifuge tubes. The samples were centrifuged at 400 × g for 15 minutes at 4 °C. The TIF was then collected and passed through 0.22-μm filters.
Quantitative proteomics analysis
Quantitative proteomics on Serpinb9 KD versus NC KPC cells was conducted by PTM-Biolabs (Hangzhou, China). Quantitative proteomics on spermine versus PBS-treated BMDMs was performed by the Mass Spectrometry Detection Platform at the First Affiliated Hospital, School of Medicine, Zhejiang University.
Cell proliferation
Cell proliferation was measured using a cell counting kit-8 (CCK-8; GK10001, GLPBIO). Two thousand cells per well were seeded in a 96-well plate, and optical density was measured daily at 450 nm for four days.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde, blocked with 3% BSA, and incubated with primary antibodies against Serpinb9 (ab233443, Abcam) and SMS (ab156879, Abcam) overnight at 4 °C, followed by secondary antibody incubation. Tissue sections followed similar steps. Fluorescent images were acquired using a TCS SP8 X confocal microscope (Leica).
GST pull-down assay
The GST pull-down assay was performed using a Pierce GST Protein Interaction Pull-Down Kit (21516, Thermo Scientific) with human SMS*His fusion protein (Ag8840, Proteintech), human Serpinb9-GST fusion protein (Ag6707, Proteintech), and recombinant human GST (ab70456, Abcam) following the manufacturer’s protocol.
Quantitative real-Time reverse transcription PCR (qRT-PCR)
Total RNA was isolated using Trizol LS Reagent (Invitrogen), quantified using a NanoDrop instrument, and reverse-transcribed using a PrimeScript RT reagent kit (RR047A, Takara). qRT-PCR was performed using an Applied Biosystems 7500 Fast Real-Time PCR System. Gene expression was normalized to β-Actin, and relative expression was calculated using the 2^-ΔΔCt method. The sequences of the primers were listed in Table S3.
Multiplex immunohistochemistry (mIHC)
Multiplex IHC was performed to visualize Serpinb9, CD3, CD68, Pan-Ck, and Asma in pancreatic cancer tissues using the Opal Polaris 7-color Manual IHC Kit (NEL861001KT, Akoya). After DAPI counterstaining, the stained slides were scanned using a Vectra 3.0 quantitative pathology imaging system (Akoya).
Duolink proximity ligation assay (PLA)
The interaction between Serpinb9 and SMS was assessed using the Duolink In Situ PLA kit (DUO92101, Sigma-Aldrich) following the manufacturer’s guidelines.
Flow cytometry analysis
Mouse tumors were mechanically dissociated and digested in RPMI-1640 medium supplemented with 2% FBS, 10 μg/mL DNase (D5025, Sigma-Aldrich), 1 mg/mL collagenase IV (17104019, Thermo Fisher Scientific), and 3 mM CaCl2 (21115, Sigma-Aldrich) at 37 °C for 1 hour. The dissociated tissues were filtered through 70 μm strainers to obtain single cells. Non-immune cells were removed using a 36% Percoll solution. Surface markers were analyzed by incubating the cells with specific antibodies in the dark at 4 °C for 30 minutes. For intracellular cytokine detection, cells were treated with a Cell Stimulation Cocktail (550583, BD Biosciences) for 4 hours. FOXP3 detection was performed using the FOXP3/Transcription Factor Staining Buffer Set (00-5523-00, Thermo Fisher Scientific). Data were acquired on a Fortessa flow cytometer (Becton Dickinson) or a Cytek Aurora (Cytek Biosciences) and analyzed with FlowJo software (Becton Dickinson, version 10.8.1). The primary antibodies are listed in Table S4.
Polyamine quantification
Polyamine levels were quantified by Hangzhou Cosmos Wisdom Biotechnology Co., Ltd. (Hangzhou, China) using a quantitative UPLC-MS/MS platform (ACQUITY UPLC-Xevo TQ-S) following established protocols. Polyamine standards were sourced from Sigma-Aldrich. The raw data generated by UPLC-MS/MS were processed with QuanMET software (v1.0, MetaboProfile) to perform peak integration, calibration, and quantification for spermine, spermidine, and putrescine.
Molecular dynamics (MD) simulations
The atomic binding modes of Serpinb9 and lamp2A to SMS were generated utilizing the AI-powered platform ColabFold29, which incorporates parameters from AlphaFold230. The input protein sequences were identified by their UniProt IDs: P50453 for Serpinb9, P52788 for SMS, and P13473 for lamp2A. In the peptide design section, the atomic models of peptide-bound SMS were derived from the dominant configuration of Serpinb9-SMS. All the complex models were visualized using ChimeraX57. Each system was parameterized using the CHARMM36 force field58 and solvated in a TIP3P59 water box. The water box was then neutralized and ionized to a concentration of 150 mM using Na+ and Cl-. After 5000 steps of energy minimization to stabilize the geometry and release any present strain, each system was equilibrated for 10 ns at 310 K using a 2 fs timestep. Finally, 500 ns production run was conducted for each system in triplicate, with a time step of 2 fs in the NPT ensemble at 310 K and 1 bar using the V-rescale thermostat60 and Parrinello-Rahman barostat61, respectively. The cutoff distance for van der Waals and electrostatics was set to 12 Å, while long-range electrostatic interactions were handled by the particle mesh Ewald method62. All simulation studies were conducted using GROMACS 2023.263.
The last 100 s of production run were used for analysis. The binding energies (ΔG) of Serpinb9 and lamp2A to SMS were assessed by end-point Molecular Mechanics Generalized Born Surface Area (MM/GBSA) using gmx_MMPBSA software64. Key parameters controlling the MM/GBSA analyses were used as follows: external dielectric constant ~78.5; internal dielectric constant ~10; and solvation model ~8. The RMSD was calculated using GROMACS tools.
Free energy perturbation
Binding affinity change of alanine scan was calculated via the free energy perturbation (FEP) method with Hamiltonian replica exchange65. The dominant configurations of the production MD simulations were selected for FEP calculations. We computed free energy perturbation calculations for the bound (Serpinb9 + SMS) and free states (Serpinb9 only) with 5 replicas for each calculation. The binding free energy change due to residue mutation was calculated as ΔΔG = ΔGbound − ΔGfree. For each mutation, 32 windows of sequential annihilation of electrostatics and van der Waals interactions were set up. Window exchange attempts at every 1000 steps between neighboring windows with an even-odd switch pattern. The production run of each FEP window was 1 ns, leading to 320 ns production run in total (5 replicas × 32 windows × 2 states (complex + free) × 1 ns). GROMACS 2023.2 with the CHARMM36 protein force field and TIP3P water model were used for FEP calculations, matching the MD work. The GROMACS bar analysis tool was used to estimate the binding free energy changes (ΔΔGs) and their statistical errors.
Design and synthesis of Serpinb9-mimicking peptides
In order to design peptides targeting SMS, we adopted a Serpinb9-mimicking strategy through the following process. First, the complex of Serpinb9 and SMS was constructed with AlphaFold2 and further refined through MD simulation protocol. Second, virtual alanine mutagenesis of key residues contributing to the binding of Serpinb9 to SMS was evaluated by FEP calculations. Pull-down experiments further validated the crucial role of residue F336 in the binding mode of Serpinb9 to SMS. Third, we designed three peptides, Pep1 (Human Sperbin9 residues 326-345), Pep2 (Human Sperbin9 residues 335-344) and Pep3 (Human Serpinb9 residues 326-334). The Pep1 and Pep2 included the F336, while Pep3, lacking the phenylalanine, served as a negative control. The Pep4 (Mouse Sperbin9 residues 335-344) was designed following the sequence of mouse Serpinb9, with the same truncation as Pep2, and prepared for mouse-associated animal experiments. Finally, the systems of these four peptides binding to SMS were built and analyzed by MD simulations following the protocols above. All peptides were chemically synthesized by Synthbio, achieving a purity above 95%.
Data acquisition and preprocessing
Bulk transcriptome sequencing data and clinical information of PDAC patients were sourced from the TCGA database (https://portal.gdc.cancer.gov/) and the GEO database (GSE15471, GSE28735, GSE71729). Transcriptome data from 167 normal pancreatic tissues were downloaded from the GTEx. Immunotherapy data were retrieved from the IMvigor210 cohort and the Tumor Immunotherapy Gene Expression Resource (TIGER) database (Melanoma-GSE78220, Melanoma-GSE91061, Melanoma-phs000452, Melanoma-PRJEB23709, RCC-Braun_2020 http://tiger.canceromics.org/). Single-cell data were obtained from the GEO database (GSE229413).
Statistical analyses
Statistical results are presented as means ± SD from at least three independent biological replicates. Differences between groups were assessed using Student’s t-test, while one-way ANOVA was used for comparisons among multiple groups. Overall survival curves were analyzed using the Kaplan-Meier method and compared with the log-rank test. Correlations between variables were assessed using Spearman’s rank correlation. Patients were stratified into high and low groups based on the median value, unless otherwise specified. This approach was used for survival and other statistical analyses. Analyses were performed with SPSS (V26, IBM Corp., Armonk, NY, USA) and GraphPad Prism 9.0 software, with statistical significance set at p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data are available on NCBI under the accession code PRJNA1152236. The Proteomics data have been deposited in the PRIDE database under the accession codes PXD058466 and PXD059411. The TCGA-PAAD dataset used in this study is publicly available from TCGA. The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Hu, Z. I. & O’Reilly, E. M. Therapeutic developments in pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 21, 7–24 (2024).
Topalian, S. L. et al. Neoadjuvant immune checkpoint blockade: a window of opportunity to advance cancer immunotherapy. Cancer Cell 41, 1551–1566 (2023).
Sun, Q. et al. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct. Target Ther. 8, 320 (2023).
O’Reilly, E. M. et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 5, 1431–1438 (2019).
DePeaux, K. & Delgoffe, G. M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 21, 785–797 (2021).
Gautam, S. K., Batra, S. K. & Jain, M. Molecular and metabolic regulation of immunosuppression in metastatic pancreatic ductal adenocarcinoma. Mol. Cancer 22, 118 (2023).
Martínez-Reyes, I. & Chandel, N. S. Cancer metabolism: looking forward. Nat. Rev. Cancer 21, 669–680 (2021).
Arner, E. N. & Rathmell, J. C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 41, 421–433 (2023).
Casero, R. A., Murray Stewart, T. & Pegg, A. E. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat. Rev. Cancer 18, 681–695 (2018).
Holbert, C. E., Cullen, M. T., Casero, R. A. & Stewart, T. M. Polyamines in cancer: integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 22, 467–480 (2022).
Zhang, T. et al. Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat. Metab. 2, 1062–1076 (2020).
Bi, G. et al. Polyamine-mediated ferroptosis amplification acts as a targetable vulnerability in cancer. Nat. Commun. 15, 2461 (2024).
Wolters-Eisfeld, G., Hackert, T. & Güngör, C. Unmasking metabolic dependencies in pancreatic cancer: aberrant polyamine synthesis as a promising new therapeutic target. Signal Transduct. Target. Ther. 8, 410 (2023).
Lee, M.-S. et al. Ornithine aminotransferase supports polyamine synthesis in pancreatic cancer. Nature 616, 339–347 (2023).
Murthy, D. et al. Cancer-associated fibroblast-derived acetate promotes pancreatic cancer development by altering polyamine metabolism via the ACSS2–SP1–SAT1 axis. Nat. Cell Biol. 26, 613–627 (2024).
Halbrook, C. J., Lyssiotis, C. A., Pasca di Magliano, M. & Maitra, A. Pancreatic cancer: Advances and challenges. Cell 186, 1729–1754 (2023).
Connor, A. A. & Gallinger, S. Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat. Rev. Cancer 22, 131–142 (2022).
Bird, C. H. et al. Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 18, 6387–6398 (1998).
Sun, J. et al. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem. 271, 27802–27809 (1996).
Kaiserman, D. & Bird, P. I. Control of granzymes by serpins. Cell Death Differ. 17, 586–595 (2010).
Medema, J. P. et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 98, 11515–11520 (2001).
Bots, M. & Medema, J. P. Serpins in T cell immunity. J. Leukoc. Biol. 84, 1238–1247 (2008).
Jiang, L. et al. Direct tumor killing and immunotherapy through anti-serpinB9 therapy. Cell 183, 1219–1233.e18 (2020).
Dervovic, D. et al. In vivo CRISPR screens reveal Serpinb9 and Adam2 as regulators of immune therapy response in lung cancer. Nat. Commun. 14, 3150 (2023).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med 24, 1550–1558 (2018).
Fu, Q. et al. Sintilimab plus modified FOLFIRINOX in metastatic or recurrent pancreatic cancer: the randomized phase II CISPD3 trial. Ann. Surg. Oncol. 30, 5071–5080 (2023).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784 (2017).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Tu, Y.-C. et al. Mechanisms of dual modulatory effects of spermine on the mitochondrial calcium uniporter complex. J Biol Chem 301, 108218 (2025).
Giorgi, C. et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium 52, 36–43 (2012).
Vardhana, S. A. et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 21, 1022–1033 (2020).
Nava Lauson, C. B. et al. Linoleic acid potentiates CD8+ T cell metabolic fitness and antitumor immunity. Cell Metab. 35, 633–650.e9 (2023).
Aj, V. & Bd, M. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab. 1, 321–333(2019).
Cunningham, J. T. et al. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740 (2007).
Morita, M. et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711 (2013).
Kersten K. et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer cell 40, 624–638 (2022).
Tharp, K. M. et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat. Cancer https://doi.org/10.1038/s43018-024-00775-4 (2024).
Wu, Z. et al. Electron transport chain inhibition increases cellular dependence on purine transport and salvage. Cell Metab. 36, 1504–1520 (2024).
Li, S. et al. Metabolism drives macrophage heterogeneity in the tumor microenvironment. Cell Rep. 39, 110609 (2022).
Gabriel, S. S. et al. Transforming growth factor-β-regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity 54, 1698–1714.e5 (2021).
Collisson, E. A. et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med 17, 500–503 (2011).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
Moffitt, R. A. et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet 47, 1168–1178 (2015).
Na, U., Pr, B., Rf, D. & Dc, L. Immunologic strategies in pancreatic cancer: making cold tumors hot. J. Clin. Oncol. 40, 2789–2805 (2022).
Liu, J. et al. QDPR deficiency drives immune suppression in pancreatic cancer. Cell Metab. 36, 984–999.e8 (2024).
Y, O., P, V. & Sp, H. The interactive role of inflammatory mediators and metabolic reprogramming in pancreatic cancer. Trends in cancer 8, 556–569 (2022).
Cm, S. et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–83 (2016).
Niu, N. et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell 42, 869–884.e9 (2024).
Guo, Y. et al. Spermine synthase and MYC cooperate to maintain colorectal cancer cell survival by repressing Bim expression. Nat. Commun. 11, 3243 (2020).
Novita Sari, I. et al. Metabolism and function of polyamines in cancer progression. Cancer Lett. 519, 91–104 (2021).
Al-Habsi, M. et al. Spermidine activates mitochondrial trifunctional protein and improves antitumor immunity in mice. Science 378, eabj3510 (2022).
Hx, S. et al. Elevation of spermine remodels immunosuppressive microenvironment through driving the modification of PD-L1 in hepatocellular carcinoma. Cell communication and signaling: CCS 20, 175 (2022).
Rodrigo Nalio, R. et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell 185, 1189–1207 (2022).
Neoptolemos, J. P. et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 15, 333–348 (2018).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput 8, 3257–3273 (2012).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A. & Moreno, E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput 17, 6281–6291 (2021).
Jiang, W. & Roux, B. Free energy perturbation hamiltonian replica-exchange molecular dynamics (FEP/H-REMD) for absolute ligand binding free energy calculations. J. Chem. Theory Comput 6, 2559–2565 (2010).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China [U20A20378 and 82188102 to TBL, U23A20462 and 82071867 to XLB, 82403852 to XZZ]; “Ling Yan” Research and Development Program of Department of Zhejiang Province Science and Technology [82071867 to XLB]; National Key Research and Development Program of China [2024YFA1306400 to TBL, XLB and XBL]; Noncommunicable Chronic Diseases-National Science and Technology Major Project [2024ZD0524900 to XBL]; China Postdoctoral Science Foundation [2024T170810 and 2024M752879 to XZZ]. We thank Minghua Sun, Lei Ni, Jianfeng Wang, and Junyu Qiu for their support during sample processing and assistance with the mouse experiments. We extend our gratitude to Pengda Zou, Ziqian Wan, and Yahong Wu from the Core Facility, Central Laboratory, First Affiliated Hospital, Zhejiang University School of Medicine, for their technical support with mass spectrometry. We also thank the Biomedical Big Data Center of the First Affiliated Hospital, Zhejiang University School of Medicine, for their assistance with RNA-seq analysis.
Author information
Authors and Affiliations
Contributions
T.L. and X.B. supervised this study; H.Y., X.Z., and X.L. conceived the idea, designed the experiments, and composed the paper; H.Y. and S.Z. conducted most of the animal and in vitro experiments; Y.Y., Y.J., and R.Z. performed computer simulation; Q.L. and Jinyan Huang (J.H.) assisted in bioinformatics analysis; Y.C., L.L., K.S., W.D. assisted in flow cytometry analysis; L.H. and M.L. analyzed the IHC staining tumor samples; J.S.(Shi), N.L., and X.L. (Liu) provided assistance in the mouse experiments; J.S.(Song) and Junming Huang (J.H.) provided technique supports; H.Y. analyzed the data and wrote the manuscript; H.Y., X.Z., X.L., and X.B. revised the manuscript. All authors revised and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ilaria Elia, Drazen Raucher, Tracy Stewart and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, H., Zhang, X., Zhang, S. et al. Targeting spermine metabolism to overcome immunotherapy resistance in pancreatic cancer. Nat Commun 16, 7827 (2025). https://doi.org/10.1038/s41467-025-63146-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63146-2
