
Abstract
The extensive dendrite formation and unstable interfacial chemical environment pose significant obstacles to operating lithium metal batteries under extreme conditions. Here, we develop an allyl ether electrolyte operated across a wide-temperature range. Leveraging the neighboring group participation effect of alkenyl groups, the designed electrolyte possesses a quasi-weak solvation structure with low desolvation energy. Moreover, this effect facilitates the anion decomposition to form a dual-layer solid electrolyte interface, suppressing dendrite formation and surface parasitic reactions. Therefore, the single-salt, single-solvent electrolyte enables reversible lithium plating/stripping with high Coulombic efficiencies from −40 °C to 60 °C. The assembled 50 μm lithium | |3.5 mAh cm−2 sulfurized polyacrylonitrile full cells achieve capacity retention of 93.1% after 150 stable cycles (0.2 C) at 25 °C, where the positive electrode could retain 78% of its room temperature capacity at −40 °C. Moreover, the pouch cells demonstrate promising cycling stabilities, with a capacity retention of 94.8% (0.5 C), 92.4% (0.2 C), and 72.7% (0.1 C) after 100 cycles at 60 °C, 25 °C, and −40 °C, respectively. This terminal group modification strategy offers perspectives for wide-temperature electrolyte design, representing a crucial advancement in enhancing the performance of lithium metal batteries.
Similar content being viewed by others

Introduction
Lithium (Li) metal is recognized as the ideal negative electrode material for high-energy-density batteries owing to its high theoretical specific capacity (3860 mAh g−1) and lowest operating potential (−3.04 V vs. standard hydrogen electrodes)1,2,3. Nevertheless, the practical application of Li metal anodes (LMAs, negative electrode) is significantly hindered by issues, e.g., limited plating/stripping Coulombic efficiency (CE) and the notorious dendrite formation, jeopardizing cycling stability and raising safety hazards4,5,6,7. These problems are exacerbated when operating under extreme conditions like subzero temperatures8,9,10. Notably, the increased Li+ desolvation energy and insufficient Li+ diffusion kinetics across the solid electrolyte interface (SEI) have been identified as primary kinetic challenges encountered obstacles at low temperatures (LT)11. Therefore, among the various approaches explored8,12,13,14,15,16,17,18,19,20,21,22,23, the design of functional electrolytes stands out as a highly accessible and promising method for regulating the Li+ solvation structure and modifying the SEI composition. Enhancing the performance of electrolytes to overcome these issues is widely recognized as crucial in enabling the reversible operation of LMAs at low temperatures4,20.
In contrast to the earlier approach of incorporating low-melting-point solvents24,25,26, an interfacial desolvation energy reduction strategy was proposed by introducing diethyl ether as a weakly solvated solvent27. Building upon this concept, a range of solvents with weak solvation properties, including BFE28, DMM29, N-mixTHF30, DDE31, and TFMP16, have been investigated to enhance low-temperature performance. Furthermore, these solvents promote the accumulation of anions (PF6−, FSI−, and TFSI−) within the solvation structure to form contact ion pairs (CIPs) and ion-pair aggregates (AGGs)32,33,34. The associated anions preferably decompose on the Li metal surface and generate inorganic-rich SEIs, which could effectively suppress electrolyte degradation and mitigate electron tunneling effects35. Among the diverse solvents studied, fluorinated solvents have garnered significant attention due to the electron-withdrawing capability of fluorine atoms. This property weakens the solvation ability of the electrolyte, facilitating the formation of a LiF-rich SEI layer9,28,36,37,38,39,40. However, individual LiF-rich SEI layers without organic species exhibit inadequate mechanical properties, e.g., high hardness, poor adhesion, and a tendency to fragment41. These drawbacks hinder the formation of stable SEIs and lead to a substantial increase in impedance. Additionally, the weakly solvated Li+ cations negatively affect the ionic conductivity, and the interaction between ion pairs may deteriorate the desolvation process at low temperatures42. More importantly, whether these weakly coordinated solvents participate in the anion decomposition process has not been fully explored, where the commonly used “cocktail strategy” complicates the investigation. Therefore, the rational design of solvent molecules with functional groups should consider an appropriate solvation structure and a multifunctional SEI layer to simultaneously ensure Li+ diffusion and stable interface chemical environments.
In this work, we introduced a non-fluorinated allyl ether (AE) electrolyte specifically designed for LMAs. Leveraging the neighboring group participation (NGP) effect, this electrolyte forms a quasi-weak solvation structure while maintaining relatively low desolvation energy. The NGP effect also induces lithophilic properties, promoting the decomposition of FSI− anions on the Li metal surface. This process generates a dual-layer SEI composed of a LiF-rich outer layer and a Li2O-rich inner layer, effectively suppressing further electrolyte decomposition, preventing SEI incrassation, and facilitating fast Li+ diffusion. Therefore, the single-salt, single-solvent 2 M LiFSI/AE electrolyte exhibits a dendrite-free Li deposition morphology and impressive Li plating/stripping Coulombic efficiencies (CEs) across a broad temperature range (−40 °C to 60 °C). The 50 μm Li | |3.5 mAh cm−2 sulfurized polyacrylonitrile (SPAN) full cells (negative electrode to positive capacity ratio, N/P = 2.9) with AE electrolyte maintain 93.1% of the initial capacity after 150 cycles at 25 °C. Moreover, the Li | |SPAN full cell demonstrates a specific capacity of 490 mAh g−1 at −40 °C, retaining 78% of room temperature capacity. Meanwhile, the full cell could also reversibly operate for 100 cycles at 60 °C. The Li | |SPAN pouch cells demonstrate promising cycling stabilities, with a capacity retention of 94.8%, 92.4%, and 72.7% after 100 cycles at 60 °C, 25 °C, and −40 °C, respectively. The alkenyl-functionalization design and NGP effect present a promising approach to developing non-fluorinated electrolytes for lithium metal batteries (LMBs) that operate effectively across a broad temperature range (Supplementary Table 1).
Results
NGP effect molecule design principle
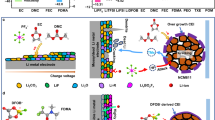
To design an effective solvent molecule for wide temperature LMAs, we focus on modifying 1,2-dimethoxyethane (DME), a commonly used ether solvent for LMBs. DME processes a high ionic conductivity due to the strong solvation effect of dioxygen atoms along the carbon chain26. However, this strong interaction between DME molecules and Li+ cations results in high desolvation energy, hindering efficient Li deposition and compromising low-temperature performance. Moreover, the solvent-separated ion pairs (SSIPs) solvation structure in DME electrolytes leads to the formation of unstable, highly porous, and thick organic-rich SEIs. Therefore, we proposed two design principles to address these issues: decreasing desolvation energy and forming multifunctional SEIs. These principles aim to ensure effective LMB operation at low temperatures with highly reversible Li plating/stripping capability (Fig. 1).

Molecular structures and chemical/electrochemical properties of three electrolytes, including electrostatic potential, solid electrolyte interface (SEI), Coulombic efficiency (CE), and low-temperature (LT) performance.
Firstly, we select a linear analog of the dioxygen DME, dipropyl ether (DPE) with a single oxygen atom, to reduce the chelation energy between Li+ cations and the solvent at low temperatures. Therefore, the 2 M LiFSI/DPE electrolyte could achieve improved desolvation kinetics, enhancing Li plating/stripping CE and mitigating dendrite formation. However, the as-formed thick and rigid inorganic-rich SEI may increase interfacial resistance and hinder Li+ diffusion at low temperatures. Secondly, we adopt a terminal group modification approach to construct a robust SEI with hierarchical structures, where the electron-withdrawing carbon-carbon double bond (C=C) reduces the electron cloud density of oxygen in the DPE. Leveraging the NGP effect, the π-bonds and Li+ cations undergo slight coordination to form a quasi-tridentate solvation structure. Furthermore, the NGP effect could promote the decomposition of FSI− anions to form a thin and robust hierarchical SEI layer for rapid Li+ migration. Notably, the AE electrolyte also exhibits comparable thermal stability and an expanded operational temperature window (Supplementary Fig. 1 and Table 2). Therefore, a non-fluorinated 2 M LiFSI/AE electrolyte is employed for low-temperature LMBs.
Solvation energy and structure of different electrolytes
Since the desolvation process significantly affects the electrochemical performance, we first evaluate the solvation structure of the 2 M LiFSI electrolytes and their corresponding solvents (DME, DPE, and AE). The electrolyte concentration is selected based on the electrochemical properties’ comparison of AE-based electrolytes (Supplementary Fig. 2 and Supplementary Note 1)43,44,45. The density functional theory calculated Li+-solvent binding energy and bond length demonstrate that the bidentate interaction of DME solvent exhibits the strongest binding affinity with Li+ across all temperature ranges (Fig. 2a and Supplementary Data 1). Interestingly, compared to DPE, the chelating effect of the π-bonds enables AE to possess a stronger binding energy with a longer bond length. Therefore, the Raman spectroscopy reveals that the S-N-S stretching vibration peak of FSI− anion (680-780 cm−1) undergoes a greater redshift in AE electrolyte than DPE electrolyte, where more solvent and less anion participate in the solvation sheath due to the NGP effect (Fig. 2b)39,46. Owing to the strong solvent coordination and augmented electron density around the Li nucleus, the strongly solvated DME electrolyte experiences a prominent redshift, and the 7Li nuclear magnetic resonance (NMR) also displays the most upfield shift (Fig. 2c)28. Intriguingly, a larger upfield shift in the 7Li NMR of the DPE electrolyte, compared to the AE electrolyte, might be attributed to more anion coordination in the Li+ solvation sheath, consistent with the Raman spectroscopy28,47,48.

a Binding energy and bond length of Li+-solvent coordination. b Raman spectrum of LiFSI salt, AE, DPE, and DME electrolytes. c 7Li NMR of AE, DPE, and DME electrolytes (1 M LiCl in D2O as the internal reference). d, e The 2D NMR spectrum of 1H-7Li HOESY (d) and 1H-19F HOESY (e) for AE electrolyte, respectively. f 13C NMR of AE solvent and AE electrolyte, (CDCl3 as the internal reference,77.5–76.5 ppm). g, h MD simulation snapshots and solvation structures for AE electrolytes at −40 °C. i Statistical results of different solvation structures in DME, DPE, and AE electrolytes at different temperatures. Source data are provided as a Source Data file.
Furthermore, we performed 2D 1H-7Li and 1H-19F heteronuclear Overhauser enhancement spectroscopy (HOESY) on the AE electrolyte to further elucidate these short-range (cation-solvent) and long-range (anion-solvent) interactions at the molecular level32,35,49. The -CH2-, -CH=, and =CH₂ groups in the AE molecule exhibit distinct and constant correlations for 1H and 7Li among different mixing times, indicating the NGP effect induces the coordination between the Li+ and C=C double bond (Fig. 2d, Supplementary Fig. 3a–c and Supplementary Note 2). Moreover, as the formation of a quasi-tridentate coordination structure, the inductive effect of Li+ results in =CH₂ carrying a more positive charge, attracting the FSI− into closer spatial proximity and causing electrostatic interactions between C3 of AE molecule and FSI− (Fig. 2e and Supplementary Fig. 3d–f). This phenomenon could also be observed in 13C NMR measurements, where the opposite chemical shifts of the C2 and C3 carbon signals suggest the involvement of the FSI− anions and C=C double bonds in the solvation structure through the NGP effect (Fig. 2f). In contrast, both the C2 and C3 carbon of DPE molecules experience the upfield shift with smaller \(\triangle \delta\) values due to the shielding effect from the weakly solvation induced Li+-FSI− interactions (Supplementary Fig. 4)50,51.
To gain a better understanding of the coordination environment, we conducted molecular dynamics (MD) simulations of the solvation structures for three electrolytes at −40 °C, 25 °C, and 60 °C (Fig. 2g, Supplementary Figs. 5–10, and Supplementary Note 3). Analysis of radial distribution functions (RDFs) and coordination numbers (CNs) further reveals that a significant presence of anions is observed in the primary solvation sheaths of DPE and AE, rather than solvent molecules accumulating in the DME electrolyte at all temperatures. Interestingly, due to the NGP effect, the alkenyl groups provide more coordination to Li+ at −40 °C, enhancing the Li salt dissociation degree with a reduced ion aggregate population (Fig. 2b and Supplementary Table 3). Therefore, the AE electrolyte possesses a higher ionic conductivity while maintaining a relatively low desolvation energy (Fig. 2g, Supplementary Fig. 11, and Table 4). The MD-calculated statistical results further reveal that the solvation structures in DPE and AE electrolytes are dominated by CIPs/AGGs, benefiting the higher Li+ transference number and formation of anion-derived inorganic-rich SEI layers (Fig. 2i, Supplementary Fig. 12, Supplementary Note 4, and Supplementary Data 2).
Reversible Li plating/stripping at different temperatures
To evaluate the compatibility of these electrolytes with the Li metal negative electrode, we assembled Li | |Cu cells to measure the Li plating/stripping CEs based on a modified Aurbach method52. At 25 °C, the AE electrolyte demonstrates an impressive Li plating/stripping CE of 99.64%, surpassing that of the DME electrolyte (99.31%) and DPE electrolyte (98.30%) (Fig. 3a). At −40 °C, the high desolvation energy and melting point of DME electrolyte result in an ineffective Li plating/stripping and significantly reduce the CE value (79.55%) (Fig. 3b). Due to the weak solvation modification, the DPE electrolyte exhibits an improved CE of 97.07%, yet remains unfavorable for LMB’s cycling performance. Supplemented with the NGP effect, the AE electrolyte achieves a promising CE of 98.86% at −40 °C, highlighting the synergistic effect of weak solvation and functional SEI layer in the realm of Li battery research. Moreover, the stable SEI layer further enables the AE electrolyte to maintain a high CE value (99.13%) even at an elevated temperature of 60 °C.

a–c Comparison of Li plating/stripping CE values of three electrolytes at different temperatures with modified Aurbach methods. d–f Li metal initial deposition morphology (0.5 mA cm−2, 5 mAh cm−2) at −40 °C for different electrolytes. g–i Morphology of Li metal after 100 cycles (0.5 mA cm−2, 0.5 mAh cm−2) in different electrolytes at −40 °C. j, k Li plating/stripping behavior of Li | |Li symmetric cells (0.5 mA cm−2, 0.5 mAh cm−2) at 25 °C and −40 °C, respectively. l Critical current density of three electrolytes at 25 °C in Li | |Li cell, with the current increasing by 0.2 mA cm−2 every cycle. Source data are provided as a Source Data file.
The substantial disparity in CE values may reflect the deposition morphology variations of LMAs in the three electrolytes at different temperatures. At −40 °C, the DME electrolyte could barely enable Li deposition on the Cu current collector, producing dendritic morphology (Fig. 3d). Due to the reduced desolvation energy, the DPE electrolyte enables effective Li deposition at −40 °C but generates numerous mossy Li dendrites (Fig. 3e). In contrast to DME and DPE electrolytes, AE electrolyte achieves more uniform, dense, and dendrite-free Li deposition across all temperature ranges (Fig. 3f and Supplementary Fig. 13). Furthermore, we employed Li | |Li symmetric cells to evaluate the cycle life of LMAs in three electrolytes at various temperatures, along with the morphology of LMAs after Li plating/stripping for 100 cycles. At −40 °C, the symmetric cell with DME electrolyte fails quickly due to the rampant dendrite formation, while the DPE electrolyte demonstrates a limited plating/stripping life with large voltage hysteresis and mossy surface morphology (Fig. 3g, h, and k). Conversely, AE electrolyte exhibits a more uniform deposition morphology and a prolonged Li plating/stripping lifespan across wide temperature ranges, even at −40 °C (Fig. 3i–k and Supplementary Figs. 14–16). Achieving uniform and compact Li deposition could suppress the formation of dead Li (i.e., Li metal regions which are electronically disconnected from the current collector) and parasitic reactions between the electrolyte and LMAs, improving the CE values and cyclability. Additionally, AE electrolyte exhibits an enhanced resilience to elevated current densities, attributed to the accelerated Li+ desolvation and migration process (Fig. 3l). Therefore, these observations precisely elucidate the high CE, extended cycle life of Li metal, and enhanced critical current densities achieved in the AE electrolyte within the temperature range from −40 to 60 °C.
The SEI layer and its formation mechanism
To explore the Li+ diffusion kinetic advantage of the AE electrolyte, the distribution of relaxation time (DRT) techniques is employed to analyze the impedance of the LMAs across the broad temperature range (Fig. 4a–c and Supplementary Fig. 17). Notably, both charge transfer resistance (Rct) and SEI resistance (RSEI) exhibit a pronounced temperature dependence, which is drastically enlarged at low temperatures53. According to the DRT results, the Li | |Li symmetric cell with DME electrolyte has high RSEI and Rct value at −40 °C, indicating that its ionic conductivity and desolvation process are greatly restricted at low temperatures. In contrast, the weakly solvated DPE electrolyte mitigates this phenomenon to some extent with a reduced Rct value of around 250 Ω, but the RSEI is still insufficient. Additionally, the bulk resistance (R0) of DPE electrolyte increases as the temperature decreases, due to the rapid drop in its ionic conductivity (Supplementary Fig. 11). Notably, the Li | |Li symmetric cell with AE electrolyte exhibits much lower Rct and RSEI values. Similarly, the charge transfer and SEI activation energies are calculated to be 36.1 kJ/mol and 35.1 kJ/mol for the AE, 41.3 kJ/mol and 45.7 kJ/mol for the DPE electrolyte, and 56.3 kJ/mol and 60.8 kJ/mol for the DME, respectively, highlighting the significant role of the AE electrolyte in promoting low-temperature LMAs performance (Supplementary Fig. 18). The smaller RSEI of the AE electrolyte represents the rapid migration of Li+ through the SEI layer, demonstrating the generation of a distinctive SEI composite and structure.

a–c Temperature-dependent DRT plot derived from electrochemical impedance spectroscopy (EIS) data analysis for Li | |Li symmetric cells after 10 cycles (0.5 mA cm−2, 0.5 mAh cm−2) for DME, DPE, and AE electrolytes. d XPS spectra of SEI on Li negative electrode of Li | |Li symmetric cells after 10 cycles (0.5 mA cm−2, 0.5 mAh cm−2) in AE electrolyte at −40 °C, with depth profiles of Li 1 s, C 1 s, O 1 s, and F 1 s. Source data are provided as a Source Data file.
To investigate the interfacial structure and chemical composition of the SEI layer formed with/without the NGP effect in different electrolytes at −40 °C, we conducted X-ray photoelectron spectroscopy (XPS) with depth-profiling analysis (Supplementary Fig. 19). The thick SEI generated in the DME electrolyte comprises more organic components, primarily attributed to the strong solvation effect of the DME solvent, impacting the transport dynamics of Li+ cations through the SEI with a large impedance (Supplementary Fig. 20). Meanwhile, a reduction in organic content on the Li metal surface is observed in the SEI layer generated in the DPE electrolyte, accompanied by a certain amount of LiF and Li2O derived from anion decomposition (Supplementary Fig. 21). However, no Li0 signal is detected until 500 s of sputtering time, indicating a substantial thickness of the SEI layer. In comparison, the SEI generated in the AE electrolyte reveals that aside from a minor organic influence in the outermost layer, the SEI in the sublayer contains a large amount of LiF (Fig. 4d and Supplementary Fig. 22). Notably, although the allyl group is electrochemically active towards polymerization, the Attenuated total internal reflectance Fourier transform infrared (ATR-FTIR) spectroscopy reveals that no significant polymerization occurs in the AE electrolyte or on the Li metal interface (Supplementary Fig. 23 and Supplementary Note 5). Along with Ar+ sputtering, LiF signals decrease and Li2O signals increase, indicating the formation of a dual-layer SEI structure composed of a LiF-rich outer layer and a Li2O-rich inner layer. The electronic insulation LiF-rich outer SEI layer passivates the surface to prevent parasitic reactions, while the Li+ conductive Li2O-rich inner layer promotes Li+ diffusion and fosters uniform Li deposition7. Additionally, the presence of Li0 signal at 300 s demonstrates that the AE electrolyte derived the thinnest SEI layer among the three electrolytes. Moreover, the SEI layers generated at 25 °C of three electrolytes follow a similar trend as that of −40 °C (Supplementary Figs. 24–27). Therefore, this thin and flexible dual-layer SEI derived from the NGP effect of the AE electrolyte possesses promising Li+ conductivity, mechanical properties, and adsorption affinity for Li metal, which is conducive to the stability of the SEI and the reduction of ion transport impedance (Supplementary Fig. 28 and Supplementary Note 6).
Furthermore, we utilized cryogenic transmission electron microscopy (cryo-TEM) techniques to explore the nanostructure and dominant components of this dual-layer SEI originating from the AE electrolyte. As shown in Fig. 5a and Supplementary Fig. 29, at 25 °C, the SEI layer presents a distinct and well-defined structure, primarily consisting of LiF (111), Li2O (111), and Li3N (110) with lattice spacings of 0.23, 0.26, and 0.39 nm, respectively. The microscopy image reveals a gradual transition from LiF to Li2O in the stacking sequence, which is in line with the XPS depth profiling and helps to prevent parasitic reactions and promote Li+ migration54,55. Moreover, the SEI generated at −40 °C possesses a similar nanostructure and dominant components with larger crystalline domains where the sluggish nucleation kinetics at low temperatures impede the formation of nanocrystals56 (Fig. 5b and Supplementary Figs. 30–31). The thickness of this SEI (~40 nm) is much thinner than the SEI formed in the DPE electrolyte at −40 °C, aligning with the Li 1 s XPS depth profiling (Supplementary Fig. 32). To obtain a more comprehensive vision of the SEI layer, we further employed the time-of-flight secondary ion mass spectrometry (TOF-SIMS) measurements in the negative ion mode (Fig. 5d–f). The depth profiles of the representative anion fragments indicate that the evolution of F− and O− fragments is consistent with the XPS depth-profiling results, composed of a LiF-rich outer layer and a Li2O-rich inner layer (Fig. 5d). Furthermore, the 3D variation images expound that the organic carbon species constitute the outermost layer and F− fragments are evenly distributed at the outer SEI layer, where LiF signal could be clearly detected from the mass spectra (Fig. 5e, f). The intensity of F− gradually decreases with a dominance of O− fragments, which homogenously fulfill the inner layer of the SEI. Therefore, by combining the XPS, cryo-TEM, and TOF-SIMS results, we could conclude the successful generation of a functional dual-layer SEI on the Li metal surface in the AE electrolyte at different temperatures (Supplementary Fig. 33).

a, b Cryo-TEM images of the SEI layer formed in the AE electrolyte at 25 °C and −40 °C, respectively. c Schematic presentation of the dual-layer SEI. d TOF-SIMS depth profiles of element fragments in SEI formed in AE electrolyte at −40 °C. e 3D variation of the TOF-SIMS intensity related to the corresponding element fragments. f The corresponding TOF-SIMS mass spectrum. g AIMD simulation of the dual-layer SEI formation process on the Li metal surface at −40 °C. Source data are provided as a Source Data file.
To gain a deeper understanding of the NGP effect on the SEI formation process, we carried out ab initio molecular dynamics (AIMD) simulations of the AE electrolyte decomposition on a Li metal surface at −40 °C (Fig. 5g and Supplementary Data 3). At the initial stage of 1.7 ps, the NGP effect of the AE molecules triggers the adsorption of complex ions onto the Li metal surface, thereby elucidating the decreased Rct value at low temperatures. Subsequently, FSI− anions undergo deoxidation, resulting in the formation of LiF on the Li metal surface. At 2.3 ps, the AE electrolyte is further drawn towards the Li metal surface, and FSI− anions are decomposed to generate LiF and Li2O. The C-O bond on the AE carbon chain also breaks at 2.3 ps and establishes a connection with the Li surface, facilitating the complete decomposition of anions at 3 ps to form LiF, Li2O, and Li3N (Supplementary Fig. 16). The simulation is prolonged for an additional 17 ps, during which no other reactions occur, indicating that the SEI layer effectively inhibits further parasitic reactions. It is noteworthy that the pronounced solvation capability of DME molecules effectively displaces FSI− anions from the Li⁺ solvation sheath, thereby reducing the coordination of FSI− to diminish its chemical reactivity. Under identical simulation conditions, the DME electrolyte exhibits a prolonged decomposition time of 12.5 ps, much slower than 3 ps for the AE electrolyte, impeding the formation of an inorganic-rich inner SEI on the Li metal surface (Supplementary Fig. 34 and Supplementary Note 7 and Supplementary Data 4). Thus, the NGP effect not only facilitates the interfacial Li+ diffusion but also promotes the formation of a stable, dense, and robust SEI layer.
Electrochemical performance of Li | |SPAN full cells
SPAN was selected as the positive electrode material due to its benefits in terms of low cost, high capacity, and suitability for the oxidation stability window of ether-based electrolytes (Supplementary Figs. 35–36). The electrochemical performance of the Li | |SPAN half cells was evaluated in three electrolytes at temperatures of 25 °C, −40 °C, and 60 °C (Supplementary Fig. 37). At 25 °C, AE electrolyte delivers a specific capacity of 650 mAh g−1 at 0.2 C with promising cycling stability, maintaining a capacity retention of 89% after 400 cycles. Notably, the capacity retention rate is calculated based on the stabilized discharge capacity (10th cycle). Even at high current rates of 1 C and 2 C, it maintains promising capacity and stable cycling (92% for 1 C and 83% for 2 C after 300 cycles) (Supplementary Fig. 38). Meanwhile, the performance of the DME electrolyte decays rapidly, while the DPE electrolyte shows some improvement but also suffers short-circuiting after 180 cycles. At −40 °C, the AE electrolyte maintains 78% of room temperature capacity and achieves an impressive capacity retention of 80% after 100 cycles (Supplementary Fig. 37b). Furthermore, when compared with its counterparts, the AE electrolyte displays superior cycling stability and capacity retention, particularly at 60 °C (Supplementary Fig. 37c). The 50 μm Li | |3 mAh cm−2 SPAN full cells were further assembled to verify their rate performance (Supplementary Fig. 39). The AE electrolyte offers stable cycling and high capacity retention at high current rates, courtesy of their weak coordination property and high ionic-conductive SEI. However, the Li | |SPAN cells with DPE and DME cells fail to complete the rate capability test. Especially for the DPE electrolyte, the kinetic limitation could be attributed to the low ionic conductivity in the electrolyte and the elevated desolvation energy at the positive electrode interface induced by the ion clusters in the DPE electrolyte (Supplementary Fig. 40 and Supplementary Note 8). Comparably, the cycling stability of the 50 μm Li | |SPAN full cell reveals that the AE electrolyte exhibits consistent cycling behavior, delivering an areal capacity of 3.5 mAh cm−2 and a specific capacity of 654 mAh g−1, with a higher capacity retention of 93.1% after 150 cycles at 25 °C (Fig. 6a). Additionally, at an elevated temperature of 60 °C, the full cell demonstrates an enhanced specific capacity of 718 mAh g−1 and maintains 83% of capacity after 100 cycles due to the suppression of the polysulfide shuttling (Fig. 6b and Supplementary Figs. 41 and 42). Noteworthy is the fact that even at −40 °C, the 50 μm Li | |2.2 mAh cm−2 SPAN full cell displays a specific capacity of 490 mAh g−1 with stable cycling performance over 100 cycles (Fig. 6c). Notably, the full cell could also demonstrate a similar performance with a high loading positive electrode (3.5 mAh cm−2), attributing to the sufficient Li plating/stripping CE and smooth Li plating morphology at low temperature (Supplementary Fig. 43). Therefore, the results highlight the AE electrolyte’s commendable electrochemical performance within a broad temperature range.

a–c Cycling performance at (a) 25 °C, (b) 60 °C, and (c) −40 °C. d–f Pouch cell cycling performance at (d) 25 °C, (e) 60 °C, and (f) −40 °C (Inset: digital images of the pouch cells). All the specific capacity refers to discharge specific capacity, where 1 C equals 700 mA g−1. Source data are provided as a Source Data file.
To evaluate the feasibility of the developed AE electrolyte, we subsequently fabricated single-layer 50 μm Li | |SPAN pouch cells and conducted electrochemical measurements under various temperatures. The results indicate that the AE electrolyte demonstrates promising cycling stability at 25 °C, with a capacity retention of 92.4% after 100 cycles and a specific energy of 613.62 Wh kg−1 based on the mass of electrode materials (Fig. 6d and Supplementary Table 5 and 6). Furthermore, the pouch cell could maintain 94.8% and 72.7% of stable capacity after stably operating for 100 cycles at 60 °C and −40 °C, respectively (Fig. 6e, f). These findings validate the efficacy of the design electrolyte for LMAs across the wide temperature range and highlight its significant potential for practical application in LMBs.
Discussion
In summary, we have developed a non-fluorinated allyl ether-based electrolyte with a tuned solvation property and dual-layer SEI for wide temperature LMBs. The NGP effect of the alkenyl group allows for a quasi-tridentate solvation structure, ensuring promising ionic conductivity and maintaining a weak solvation property to facilitate a rapid interfacial Li+ desolvation process. This unique solvation structure also promotes the decomposition of FSI− anions to generate a dual-layer SEI composed of an electron-insulated LiF-rich outer layer and an ionic-conductive Li2O-rich inner layer, suppressing surface parasitic reactions and accelerating fast Li+ diffusion. Therefore, the Li metal negative electrodes with 2 M LiFSI/AE electrolyte demonstrate promising Li plating/stripping CEs with dendrite-free deposition morphologies across a broad temperature range (−40 °C to 60 °C). For LMBs full cells with 50 μm Li | |3.5 mAh cm−2 SPAN (N/P = 2.9), the AE-based electrolyte could deliver a high area capacity (3.5 mAh cm−2) and stable cycling (93.1%, 150 cycles) at 25 °C. Moreover, the full cells operated at −40 °C maintain 78% of the initial room temperature capacity and stably cycle for 100 cycles. The pouch cells with the AE electrolyte also exhibit a stable cycling performance, retaining 94.8%, 92.4%, and 72.7% capacity after 100 cycles at 60 °C, 25 °C, and −40 °C, respectively, highlighting their practical potential for LMBs. This work mitigates the reliance on costly and toxic fluorinated solvents while highlighting the function of the NGP effect group in electrolyte solvent design for LMBs operating within a wide temperature range.
Methods
Materials
Lithium bis(fluorosulfonyl)imide (LiFSI, ≥99.9%), 1,2-dimethoxyethane (DME, ≥99.9%), and tetrahydrofuran (THF) were obtained from DodoChem. Dipropyl ether (DPE, >98%) and allyl ether (AE, >98%) were purchased from TCI. DPE and AE were both dehydrated with 4 Å molecular sieves for a week in the glovebox before use. Sulfur (S, ≥99.8%) was purchased from Sigma-Aldrich. Deuterium oxide (D2O, 99.9%), chloroform-d (CDCl3, 99.8%), 2,2’-Azobis(2-methylpropionitrile) (AIBN, ≥98%), and N-methyl-2-pyrrolidone (NMP) were purchased from Aladdin. Polyacrylonitrile (PAN, average Mw 150,000) was obtained from Macklin. Li chip (16 mm * 0.6 mm), Celgard 2325, carbon-coated Al foil, Cu foil, Super-P, and cell case (2032) purchased from Canrud. 50 μm Li foil was purchased from Changsha Xinkang New Material Co. Tuball binder slurry contained 0.4 wt.% single-walled carbon nanotubes (SWCNTs), 2 wt.% polyvinylidene difluoride (PVDF), and 97.6 wt.% N-Methylpyrrolidone (NMP), purchased from OCSiAl.
Synthesis of sulfurized polyacrylonitrile (SPAN)
S powder (2 g) and Polyacrylonitrile (PAN) (0.5 g) powder were mixed by grinding, then heated in N2 at 450 °C for 6 h with a ramping rate of 5 °C/min.
Preparation of the SPAN electrode
The SPAN positive electrode was fabricated by mixing SPAN powder (80 wt.%), Super P (8 wt.%), and a pre-formulated Tuball binder slurry (purchased from OCSiAl, containing 0.4 wt.% SWCNTs, 2 wt.% PVDF, and 97.6 wt.% NMP). This mixture was designed to achieve a final composition ratio of SPAN: SuperP: SWCNTs: PVDF = 80:8:2:10. The slurry was hand-milled for 30 min and coated onto carbon-coated aluminum foil, followed by drying overnight at 60 °C in a vacuum oven. For coin cells, the SPAN positive electrodes’ mass loadings are 5 mg cm−2 and 4.4 mg cm−2 for areal capacities of 3.5 mAh cm−2 and 3 mAh cm−2, respectively. For pouch cells, the SPAN electrodes’ mass loading is 5 mg cm−2, 4.4 mg cm−2, and 3.5 mg cm−2 for areal capacities of 3.5 mAh cm−2 (25 °C), 2.2 mAh cm−2 (−40 °C), and 2.7 mAh cm−2 (60 °C), respectively. Notably, all the areal capacities of SPAN positive cathodes are calculated based on the electrochemical results.
Preparation of the Li metal electrode
For Li | |SPAN full cell, 50 µm thick Li metal was punched into discs with a diameter of 1 cm and placed on a spacer for assembly with the positive electrode material. For the Li | |SPAN pouch cell, 50 µm Li metal was cut into 2.5 × 2.5 cm pieces, placed on a copper foil, and assembled with the positive electrode material. For the Li | |SPAN half-cell, Li | |Li, and Li | |Cu cells, Li chips were directly used for assembly without further processing.
Electrochemical testing
All electrolytes tested in this study were prepared inside an Ar-filled glovebox with O2 < 0.1 ppm and H2O < 0.1 ppm. The electrochemical tests at −40 °C, 25 °C, and 60 °C mentioned in the article were conducted in temperature-controlled chambers. CR-2032-type coin cells were chosen to assemble the testing cells (Land battery tester). Celgard 2325 (thickness, 25 μm) was used as the separator for all cells. To assess the Coulombic efficiency of lithium negative electrodes in different electrolytes, we applied the modified Aurbach method to determine the reversibility of the electrolytes under different conditions. Firstly, a current density of 0.5 mA cm−2 was deposited on copper foil for 10 h, which was then stripped to 1 V and cycled twice to form a stable SEI. Secondly, 5 mAh cm−2 was redeposited as a lithium source, followed by 10 cycles of 1 mAh cm−2 plating and stripping before finally stripping to 1 V. The CE was calculated by dividing the total capacity removed during stripping by the total capacity added during plating. Li deposition morphology studies at various temperatures were conducted on Cu working electrodes plated with 5 mAh cm−2 at 0.5 mA cm−2 at 60° C, 25 °C, and −40 °C after resting for 2 h to achieve temperature equilibration. Li | |Li symmetric cells were tested at 0.5 mA cm−2, 0.5 mAh cm−2, and Li plating/stripping morphology was investigated after 100 cycles at 60 °C, 25 °C, and −40 °C. The limiting current density test was performed using a Li | |Li symmetric cell, starting with a current density of 0.2 mA cm−2 for the first cycle and increasing by 0.2 mA cm−2 for each cycle until the cell failed. The oxidative stability of the electrolytes was determined by linear sweep voltammetry (LSV) from 0 V (vs. open circuit voltage) to 6 V (vs. Li/Li+) of Li | |Al cells at 0.1 mV S−1. Electrochemical impedance spectroscopy (EIS) (Biologic SAS, France) analyses were performed using a Li | |Li symmetric cell cycled for 10 cycles at −40 °C followed by EIS tests at different temperatures in the frequency range of 1 MHz to 3 mHz and alternating voltage amplitude of 5 mV on a VMP3 multichannel electrochemical station, with 15 points per decade. The SEI mechanical properties were tested for AE and DPE electrolytes with Li | |Cu cells, depositing 5 mAh cm−2 on copper foils at a current of 0.5 mA cm−2 at −40 °C. The galvanostatic intermittent titration technique (GITT) measurements were performed using a multichannel battery testing system. After activating the Li | |SPAN cell at 0.1 °C for one cycle, it was left to rest for 4 h. Then, it was discharged at a current rate of 0.2 C for 10 min, followed by another 4-h rest, until the voltage was discharged to 0.5 V. The GITT for the charging process was similar, with charging for 10 min followed by a 4-h rest, until the voltage reached above 3.5 V. The voltage range for all Li | |SPAN cell tests was 0.5 V to 3.5 V. Li | |SPAN pouch cells were prepared using SPAN positive electrode (2.5 cm * 2.5 cm) and a copper negative electrode with 50-μm Li rolled on the inside of the glovebox. A pressure of 100 kPa is applied during the resting and testing process. Notably, the areal capacities of pouch cells’ SPAN positive electrodes are 2.7, 3.5, and 2.2 mAh cm−2 for 60 °C, 25 °C, and −40 °C, respectively. The specific amount of electrolyte used in Li | |SPAN full cell and pouch cell is 10 g Ah−1 and 5 g Ah−1, respectively. After assembly, coin cells were rested at 25 °C for 6 h to ensure complete electrolyte infiltration, while pouch cells were rested for 12 h. Coulombic efficiency (CE) for all cells is defined as the ratio of the discharge capacity to the charge capacity in the preceding charge cycle.
Synthesis of poly-AE
For chemical polymerization, 490 mg of AE and 16 mg of AIBN were added to a 4 mL glass vial in the glovebox. The sealed glass vial was then transferred outside the glovebox and heated at 75 °C under stirring overnight (~8 h). The obtained reaction mixture was cooled to 25 °C before tetrahydrofuran (THF) was added to dissolve the obtained reaction mixture. Then, the obtained solution was dropped into hexane (~10–15 mL) to extract the poly-AE. The obtained suspension was centrifuged and dried at 60 °C under vacuum for 2 h to collect poly-AE (yellow solid).
Characterizations
All morphology images of Li chips were detected by a scanning electron microscope (SEM, SU8020, Hitachi, Japan). The XPS with Ar+ depth profiling (Thermo Scientific, NEXSA) was employed to investigate the interface chemistry of the Li negative electrode after 10 cycles of Li | |Li testing at varying temperatures. Prior to analysis, all Li negative electrodes were thoroughly washed with a solvent to remove any residual electrolyte, followed by vacuum drying for 10 min. To ensure analytical accuracy, the measurements were conducted using a vacuum transfer vessel to prevent air exposure, and the spectra were calibrated using the C 1 s peak at 284.8 eV. Depth-profiling experiments were performed using argon cluster ion sputtering with a beam energy of 0.5 keV, covering an analysis area of 2 × 2 mm2. The NMR spectra were tested by a Bruker AVANCE III 600 MHz liquid NMR instrument. The coaxial NMR tubes were used to avoid the impact on the solvation structure from the internal standard solution or the deuterated solvents. 7Li peaks calibrated using 1 M LiCl in D2O as the internal reference at chemical shift = 0 ppm. 13C NMR was used CDCl3 as deuterated solvents. Two-dimensional HOESY experiments (1H-7Li and 1H-19F) were performed on a Bruker AVANCE III 600 MHz with different variable mixing times (200 ms, 400 ms, and 900 ms). The solvation structure of the Li+ in different electrolytes was measured by a Raman spectrometer (MonoVista CRS). Cryo-TEM (Talos F200C) experiments were prepared by depositing Li on bare Cu grids, with a deposition capacity of 0.25 mAh cm−2 under a current density of 0.5 mA cm−2 in the corresponding electrolyte. The obtained Li samples on Cu grids were washed with the corresponding solvents and dried under vacuum for 10 min. Atomic force microscopy (AFM) measurements were conducted using a Bruker Dimension Icon system operating in Quantitative Nanomechanical Mapping (QNM) mode. To prevent oxidation, the entire measurement process was performed within a nitrogen-filled glovebox. The samples consisted of Li metal electrodeposited onto Cu foil at an areal capacity of 5 mAh cm−2. Prior to AFM analysis, the samples were transferred into the glovebox using a vacuum transfer module to ensure air-free conditions. The ATR-FTIR spectroscopy was examined via Bruker VERTEX 80/80 v. The TOF-SIMS was tested by FIB-SEM-Raman-TOF-SIMS All in One System (TESCAN C-TOF) for etching of cycled Li negative electrode (plating/stripping 10 cycles, 0.5 mA cm−2, 0.5 mAh cm−2) at a current of 10 nano-ampere and 30 KV, and the analysis area is 50 * 50 μm at the Li metal surface.
MD simulation
Classical molecular dynamics simulations were performed using the open-source Gromacs. The behavior of the solvents was described using the OPLS-AA/L force field, while the force field parameters for Li+ cations and FSI− anions were obtained from published literature57. The initial model was constructed using the open-source PACKMOL software58. The electrolyte system was first energy-minimized using the Steepest Descent (SD) method, with a simulation time step of 1 fs and a total duration of 10 ps. During each step of the minimization, the system was considered to have converged to the minimum energy state when the force on any atom in the system was less than or equal to 100.0 kJ/mol/nm. The optimized electrolyte system was first annealed to the desired temperature under constant volume conditions. Subsequently, the electrolyte system was simulated for 10 ns under constant temperature and pressure (NPT ensemble) using the V-rescale thermostat and Berendsen barista to achieve the appropriate density. Finally, the equilibrated system was simulated for 10 ns under constant temperature and volume (NVT ensemble) using the V-rescale thermostat. The radial distribution function (RDF) and coordination number were calculated using VMD software.
Quantum chemistry calculations
All quality control calculations were performed using the Gaussian 16 software package. The initial geometries were all extracted from molecular dynamics simulations. The B3LYP method with the 6-311 + + G (d, p) basis set was used to optimize the molecules and complexes. Frequency analysis was conducted to ensure that all structures correspond to minima on the potential energy surface.
AIMD simulation
We performed ab initio molecular dynamics (AIMD) simulations using the CP2K software package58. The Li metal negative electrode was modeled as a 7-layer Li metal slab exposing the lowest surface energy plane (100), with the middle two layers fixed to simulate the behavior of bulk lithium. The electrolyte system comprised 4 LiFSI and 16 AE, and its initial structure was equilibrated using classical molecular dynamics simulations to achieve an appropriate density. The Perdew-Burke-Ernzerhof (PBE) functional with GD3 (BJ) dispersion correction was employed to describe the exchange-correlation effects. Electronic orbitals were described using the DZVP-MOLOPT-SR-Goedecker-Teter-Hutter (GTH) basis set. Due to the large model size, only the gamma point was considered during calculations, with an energy convergence criterion of 1 * 10−5 and an energy cutoff of 400 Ry. AIMD simulations were conducted at 233.15 K using the Nose thermostat. The simulation duration was 20 ps after a 2 ps pre-equilibration with a time step of 1.0 fs.
Data availability
The experimental data that support the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Shi, Z. et al. Mechanically interlocked interphase with energy dissipation and fast Li-ion transport for high-capacity lithium metal batteries. Adv. Mater. 36, 2401711 (2024).
Yin, Y. et al. Fire-extinguishing, recyclable liquefied gas electrolytes for temperature-resilient lithium-metal batteries. Nat. Energy 7, 548–559 (2022).
Jin, Y. et al. An intermediate temperature garnet-type solid electrolyte-based molten lithium battery for grid energy storage. Nat. Energy 3, 732–738 (2018).
Hu, A. et al. Ion transport kinetics in low-temperature lithium metal batteries. Adv. Energy Mater. 12, 2202432 (2022).
Kim, S. C. et al. Potentiometric measurement to probe solvation energy and its correlation to lithium battery cyclability. J. Am. Chem. Soc. 143, 10301–10308 (2021).
Holoubek, J. et al. An all-fluorinated ester electrolyte for stable high-voltage li metal batteries capable of ultra-low-temperature operation. ACS Energy Lett. 5, 1438–1447 (2020).
Li, G.-X. et al. Enhancing lithium-metal battery longevity through minimized coordinating diluent. Nat. Energy 9, 817–827 (2024).
Gao, Y. et al. Low-temperature and high-rate-charging lithium metal batteries enabled by an electrochemically active monolayer-regulated interface. Nat. Energy 5, 534–542 (2020).
Fan, X. et al. All-temperature batteries enabled by fluorinated electrolytes with non-polar solvents. Nat. Energy 4, 882–890 (2019).
Zhang, N. et al. Critical review on low-temperature Li-ion/metal batteries. Adv. Mater. 34, e2107899 (2022).
Li, Z. et al. 40 Years of low-temperature electrolytes for rechargeable lithium batteries. Angew. Chem. Int. Ed. 62, e202303888 (2023).
Yang, Y. et al. Liquefied gas electrolytes for wide-temperature lithium metal batteries. Energy Environ. Sci. 13, 2209–2219 (2020).
Dong, X. et al. High-energy rechargeable metallic lithium battery at −70 °C enabled by a cosolvent electrolyte. Angew. Chem. Int. Ed. 58, 5623–5627 (2019).
Jiang, H. Z. et al. Electrophilically trapping water for preventing polymerization of cyclic ether towards low-temperature Li metal battery. Angew. Chem. Int. Ed. 62, e202300238 (2023).
Lin, Y. et al. Activating ultra-low temperature Li-metal batteries by tetrahydrofuran-based localized saturated electrolyte. Energy Storage Mater. 58, 184–194 (2023).
Shi, J. et al. An amphiphilic molecule-regulated core-shell-solvation electrolyte for Li-metal batteries at ultra-low temperature. Angew. Chem. Int. Ed. 135, e202218151 (2023).
Yu, L. et al. A localized high-concentration electrolyte with optimized solvents and lithium difluoro(oxalate)borate additive for stable lithium metal batteries. ACS Energy Lett. 3, 2059–2067 (2018).
Jin, C. B. et al. Taming solvent–solute interaction accelerates interfacial kinetics in low-temperature lithium-metal batteries. Adv. Mater. 35, 2208340 (2023).
Piao, Z. et al. Constructing a stable interface layer by tailoring solvation chemistry in carbonate electrolytes for high-performance lithium-metal batteries. Adv. Mater. 34, 2108400 (2022).
Xu, J. et al. Revealing the anion–solvent interaction for ultralow temperature lithium metal batteries. Adv. Mater. 36, 2306462 (2023).
Li, X. et al. Fast interfacial defluorination kinetics enables stable cycling of low-temperature lithium metal batteries. J. Am. Chem. Soc. 146, 17023–17031 (2024).
Li, Y. et al. Hybrid polymer-alloy-fluoride interphase enabling fast ion transport kinetics for low-temperature lithium metal batteries. ACS Nano 17, 19459–19469 (2023).
Li, N. et al. Customization nanoscale interfacial solvation structure for low-temperature lithium metal batteries. Energy Environ. Sci. 17, 5468–5479 (2024).
Yang, Y. et al. Electrolyte design principles for low-temperature lithium-ion batteries. eScience 3, 100170 (2023).
Selinis, P. & Farmakis, F. Review—A review on the anode and cathode materials for lithium-ion batteries with improved subzero temperature performance. J. Electrochem. Soc. 169, 010526 (2022).
Hou, R., Guo, S. & Zhou, H. Atomic insights into advances and issues in low-temperature electrolytes. Adv. Energy Mater. 13, 2300053 (2023).
Holoubek, J. et al. Tailoring electrolyte solvation for Li metal batteries cycled at ultra-low temperature. Nat. Energy 6, 303–313 (2021).
Zhang, G. et al. A monofluoride ether-based electrolyte solution for fast-charging and low-temperature non-aqueous lithium metal batteries. Nat. Commun. 14, 1081 (2023).
Ma, T. et al. Optimize lithium deposition at low temperature by weakly solvating power solvent. Angew. Chem. 134, e202207927 (2022).
Fang, H. et al. Regulating ion-dipole interactions in weakly solvating electrolyte towards ultra-low temperature sodium-ion batteries. Angew. Chem. 136, e202400539 (2024).
Liu, X. et al. Anchored weakly-solvated electrolytes for high-voltage and low-temperature lithium-ion batteries. Angew. Chem. Int. Ed. 63, e202406596 (2024).
Wang, Y. et al. Weak solvent–solvent interaction enables high stability of battery electrolyte. ACS Energy Lett. 8, 1477–1484 (2023).
Zheng, Y. et al. Achieving rapid ultralow-temperature ion transfer via constructing lithium-anion nanometric aggregates to eliminate Li+-dipole interactions. Nano Lett. 23, 3181–3188 (2023).
Yawei Chen, M. L. et al. Origin of dendrite-free lithium deposition in concentrated electrolytes. Nat. Commun. 14, 2655 (2023).
Li, A.-M. et al. Methylation enables the use of fluorine-free ether electrolytes in high-voltage lithium metal batteries. Nat. Chem. 16, 922–929 (2024).
Amanchukwu, C. V. et al. A new class of ionically conducting fluorinated ether electrolytes with high electrochemical stability. J. Am. Chem. Soc. 142, 7393–7403 (2020).
Yu, Z. et al. Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy 5, 526–533 (2020).
Zhang, D. et al. The fluorine-rich electrolyte as an interface modifier to stabilize lithium metal battery at ultra-low temperature. Adv. Funct. Mater. 32, 2112764 (2022).
Xu, J. et al. Electrolyte design for Li-ion batteries under extreme operating conditions. Nature 614, 694–700 (2023).
Yu, Z. et al. Rational solvent molecule tuning for high-performance lithium metal battery electrolytes. Nat. Energy 7, 94–106 (2022).
Jin, Y. et al. Self-healing SEI enables full-cell cycling of a silicon-majority anode with a coulombic efficiency exceeding 99.9%. Energy Environ. Sci. 10, 580–592 (2017).
Wan, H. et al. Designing electrolytes and interphases for high-energy lithium batteries. Nat. Rev. Chem. 8, 30–44 (2023).
Ravikumar, B. et al. Effect of salt concentration on properties of lithium ion battery electrolytes: a molecular dynamics study. J. Phys. Chem. C. 122, 8173–8181 (2018).
Yamada, Y. et al. Advances and issues in developing salt-concentrated battery electrolytes. Nat. Energy 4, 269–280 (2019).
Chang, Z. et al. A liquid electrolyte with de-solvated lithium ions for lithium-metal battery. Joule 4, 1776–1789 (2020).
Li, Z. et al. Non-polar ether-based electrolyte solutions for stable high-voltage non-aqueous lithium metal batteries. Nat. Commun. 14, 868 (2023).
Chen, Y. et al. Steric effect tuned ion solvation enabling stable cycling of high-voltage lithium metal battery. J. Am. Chem. Soc. 143, 18703–18713 (2021).
Gu, R. et al. An ether-based electrolyte solvation strategy for long-term stability and ultra-low temperature Li-metal batteries. Adv. Funct. Mater. 34, 2310747 (2024).
Liang, H. et al. Solvent–solvent interaction mediated lithium-ion (de)intercalation chemistry in propylene carbonate based electrolytes for lithium–sulfur batteries. ACS Nano 17, 18062–18073 (2023).
Ren, X. et al. Enabling high-voltage lithium-metal batteries under practical conditions. Joule 3, 1662–1676 (2019).
Wan, C. et al. Natural abundance 17O, 6Li NMR and molecular modeling studies of the solvation structures of lithium bis(fluorosulfonyl)imide/1,2-dimethoxyethane liquid electrolytes. J. Power Sources 307, 231–243 (2016).
Adams, B. D. et al. Accurate determination of coulombic efficiency for lithium metal anodes and lithium metal batteries. Adv. Energy Mater. 8, 1702097 (2017).
Chen, Y. et al. Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery. Nat. Commun. 14, 8326 (2023).
Guo, R. & Gallant, B. M. Li2O solid electrolyte interphase: probing transport properties at the chemical potential of lithium. Chem. Mater. 32, 5525–5533 (2020).
Zeng, H. et al. Beyond LiF: Tailoring Li2O-dominated solid electrolyte interphase for stable lithium metal batteries. ACS Nano 18, 1969–1981 (2024).
Lu, W. et al. Concentration-driven interfacial amorphization toward highly stable and high-rate Zn metal batteries. Nano Lett. 24, 2337–2344 (2024).
Canongia Lopes, J. N. et al. Potential energy landscape of bis(fluorosulfonyl)amide. J. Phys. Chem. B 112, 9449–9455 (2008).
Martínez, L. et al. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Acknowledgements
This work was supported by Major Research plan of the National Natural Science Foundation of China, Grant No. 92372207, (J.L.). It was also supported by the National Natural Science Foundation of China, Grant No. 22309059 (H.J.), 92372113 (F.D.), 12274176 (F.D.), 52202237 (Z.W.), and the Department of Science and Technology of Jilin Province (20250205069GH, H.J.). The authors thank Prof. Jun Ming, Liang Wang, Nailin Yue, and Chunyu Wang for their assistance with advanced physical characterization. The authors also appreciate the help provided by the instrumentation-sharing platform of the College of Physics, Jilin University.
Author information
Authors and Affiliations
Contributions
H.J. and J.T. conceived the idea and designed the experiments. Z.C. assisted in completing theoretical calculations and simulations. J.T. carried out the majority of the experiments and characterizations. H.J., Z.W., J.W., R.T., F.D., and J.L. analyzed the physical characterization and electrochemical data. J.T., H.J., F.D., and J.L. co-wrote the manuscript. All authors contributed to the manuscript discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Leonardo J. A. Siqueira, Steve Greenbaum and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, J., Wei, Z., Wu, J. et al. Neighboring alkenyl group participated ether-based electrolyte for wide-temperature lithium metal batteries. Nat Commun 16, 7917 (2025). https://doi.org/10.1038/s41467-025-63262-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63262-z
