
Abstract
Current electrosynthesis catalysts typically rely on nanomaterial-based engineering with multi-dimensional structural modifications. However, such approaches may not always be necessary, especially for underexplored industrial electrochemical conversions. Here, we demonstrate that commercial platinum (Pt) foil catalysts excel in the electrochemical co-oxidation of waste polyethylene terephthalate (PET)-derived ethylene glycol (EG) and ammonia (NH3) into formamide (HCONH2), a process traditionally reliant on energy-intensive methods. This approach achieves a high Faradaic efficiency (FE) of 55.87 ± 1.4% and a productivity of 1003.63 ± 23.72 µmol cm−2 h−1 at industrially relevant current densities without any degradation for durable operation (more than 500 h and 300 h for H-cell and membrane electrode assembly (MEA) reactor, respectively). In situ spectroscopy, supported by theoretical calculations, suggests that *CH2O and *NH2 are likely key intermediates. Furthermore, the product sustainability index (ProdSI) and techno-economic analysis (TEA) underscore the cost-effectiveness and sustainability of noble Pt foil in this scenario, challenging the conventional reliance on complex electrocatalysts. This work provides distinctive insights into catalyst screening and demonstrates a viable strategy for upcycling waste plastics.
Similar content being viewed by others


Introduction
Renewable electrochemical conversion protocols are emerging with strong potential as an alternative to energy-intensive industrial chemical reactions1,2,3,4,5. Attributed to controllable rate and pathways of electrochemical reactions, waste resources can be converted into value-added products with reduced carbon footprint (Fig. 1a)6,7,8,9,10. Catalysts, as the engines for electrosynthesis, attract much attention from researchers, and enormous efforts have been put into downscaling the dimensions of catalysts for increased specific area11,12,13,14,15. Hence, catalyst nano-engineering is deemed as the holy grail in most recently developed electrocatalysis systems16,17. Nevertheless, latent uncertainty regarding the stability and cost-effectiveness exists behind the prosperity in catalyst nano-engineering. Attributed to aggregation of nanoparticles (resulting from weakened selectivity, Ostwald ripening, etc.), structure transformation, corrosion, unstable substrate, and many other unexpected factors18,19,20, nano-catalyst stability can hardly be ensured, especially for industrial-scale electrosynthesis21,22. More importantly, the lack of quantitative analysis makes the cost-effective comparison between non-noble metal catalysts and noble-metal counterparts ambiguous.

Schematic of (a) an integrating clean energy for sustainable production. b The traditional route, and (c) our plastic waste upcycling strategy for HCONH2 synthesis.
Herein, as a proof of concept, we systematically investigate the performance of non-noble metal nanocatalysts and mature noble metal electrodes during electrochemical synthesis with the production of formamide (HCONH2) as the representative. Traditionally, HCONH2 is produced industrially through the reaction of carbon monoxide (CO) and ammonia (NH3) under high temperature and pressure (Fig. 1b), which is energy-intensive and results in tremendous emissions of greenhouse gases23,24. Thus, electro/photocatalytic synthesis of HCONH2 becomes a promising solution to tackle the aforementioned challenges25,26,27,28,29,30,31. Studies have yielded a favorable Faradaic efficiency (FE) of 40.39% and a productivity of 515 µmol cm−2 h−1 through electrochemical upgrading of methanol (CH3OH) to HCONH2 via coupled NH3 co-oxidation over platinum (Pt)-based catalysts under ambient conditions. (Supplementary Fig. 1)29, but the high cost of CH3OH makes this method impractical. Others, like electrochemical upgrading of ethylene glycol (EG, (CH2OH)2) to HCONH2, is handicapped with side reactions like the formation of ammonium sulfate ((NH4)2SO4), and the unfavorable stability32.
In contrast, we focus on commercial catalysts, harnessing mature preparation and employment cases, and facilitating the implementation of the electrochemical reaction that has not been well investigated. Among them, Pt foil is chosen based on its advantageous adsorption/desorption energies, and EG from plastic degradation coupled with NH3 in potassium bicarbonate (KHCO3) solution is chosen as the sample electrosynthesis processes (Fig. 1c)33. Thereinto, by avoiding (NH4)2SO4 through the application of a weakly alkaline environment, the system achieves an FE of 55.87 ± 1.4% and a productivity of 694.9 ± 17.46 µmol cm−2 h−1 at industrially relevant current densities without any degradation for durable operation (> 500 h) in an H-type cell34,35,36,37. A production rate of 1003.63 ± 23.72 µmol cm−2 h−1 is obtained at a current density of 150 mA cm−2, which is double that of reported systems32. For the practical membrane electrode assembly (MEA) reactor configuration, the FE still consistently exceeds 40% with stable cycling at 100 mA cm−2 for more than 300 h. Mechanistic studies using in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), in situ differential electrochemical mass spectrometry (DEMS), and supported by density functional theory (DFT) calculations suggest that *CH2O and *NH2 are likely key electrophilic intermediates involved in the formation of HCONH2. Product sustainability index (ProdSI) and techno-economic analysis (TEA) further highlight the high cost-effectiveness and strong sustainability for noble Pt foil catalysts, which is opposite to the common perception of the selection of electrocatalysts. This study adds one dimension for future catalyst design and selection, and also paves the way for underlying electrochemical conversions originating from energy-intensive industrial reactions.
Results
Initial evaluation for the electrochemical synthesis of HCONH2
The electrochemical synthesis of HCONH2 is initially explored in an H-type cell equipped with a proton exchange membrane (PEM, Supplementary Fig. 2). The adopted electrolyte is 10.0 mL, affording 4.0 M EG, 1.0 M NH3, and 0.5 M KHCO3. EG and NH3 are chosen as C and N sources, respectively, while KHCO3 serves to stabilize pH, improve ionic conductivity, and suppress the formation of formic acid (HCOOH). A commercial Pt foil is employed as the cathode to facilitate the hydrogen evolution reaction (HER). Ten commercial anode catalysts—PtRu/C, Pt/C, Pt foil, Pd foil, TiO2, Ni(OH)2, NiO, Co3O4, MoS2, and Ag—each with an active area of 1.0 cm2, are systematically evaluated using galvanostatic methods. The carbonaceous liquid products are analyzed and quantified via 1H-nuclear magnetic resonance (1H-NMR, Supplementary Fig. 3).
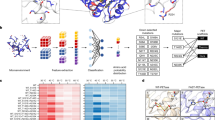
Electrolysis experiments are conducted at current densities of 20, 50, and 100 mA cm−2 for durations of 5 h, 2 h, and 1 h, respectively (Supplementary Figs. 4–6 and Fig. 2a). Among the catalysts evaluated, Pt foil showed favorable performance for HCONH2 production, achieving FE of 34.54%, 44.71%, and 56.86% under the conditions mentioned above. To validate the experimental results, DFT calculations are performed to evaluate the adsorption energies (Eads) of EG, NH3, and HCONH2 on various catalyst surfaces38. Accurate theoretical modeling necessitated the identification of the active catalyst phase during the reaction. Operando Raman spectroscopy is performed on all commercial catalysts under a constant current density of 50 mA cm−2, with spectra collected at 0 s, 30 s, 1 min, 2 min, 5 min, 8 min, and 10 min. The results revealed that most catalysts experienced varying degrees of surface reconstruction under anodic conditions. These observations are used to guide the selection of representative surface models for DFT calculations.

a Catalysts screening of HCONH2 electrosynthesis after 1 h at 100 mA cm−2 based on galvanostatic methods (4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3. pH: 9.44 ± 0.032). b Eads of EG, NH3, and HCONH2 on various catalyst surfaces. c I–V curves (with 20% iR compensation versus RHE) under varying conditions (1.0 M EG + 1.0 M NH3 + 0.5 M KHCO3, 1.0 M NH3 + 0.5 M KHCO3, 1.0 M EG + 0.5 M KHCO3, and 0.5 M KHCO3. pH: 10.00 ± 0.030, 10.00 ± 0.040, 8.30 ± 0.012, and 8.00 ± 0.028; Resistance: 5.47 ± 0.332, 4.63 ± 0.189, 7.86 ± 0.269, and 6.17 ± 0.435 ohms). d 1H NMR spectra after 1 h of continuous electrolysis at 100 mA cm−2 under the four conditions. e FEs of HCONH2 under each condition. The error bars represent the standard deviation of three independent measurements. Source data are provided as a Source Data file.
In particular, Pt-based systems (PtRu/C, Pt/C, and Pt foil; Supplementary Fig. 7a–c) exhibited a characteristic band at ~ 531–543 cm−1, attributed to α-PtO2, confirming the oxidative surface reconstruction29,39,40. However, to rule out possible contributions from other Pt oxides such as β-PtO2, we further conducted X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) analyses. The high-resolution XPS analysis for the Pt foil after electrolysis (Supplementary Fig. 8a) revealed a peak at ~ 79.6 eV, confirming the presence of oxidized Pt4+ 4f5/2 species41. Yet due to the nearly identical binding energies of α- and β-PtO2, XPS alone cannot discriminate between the two phases. Therefore, XRD is performed to determine the crystal structure: the pristine Pt foil displayed sharp reflections consistent with metallic Pt (PDF#04-0802), whereas the post-electrolysis sample showed additional peaks matching well with α-PtO2 (PDF#37-1087), with no signals assignable to β-PtO2 (Supplementary Fig. 8b, c). These results collectively confirm that α-PtO2 is the dominant surface oxide phase formed during electrolysis.
Additional Ru–O signals (~ 485 cm−1) observed in PtRu/C further support dual-metal surface evolution39. Pd foil (Supplementary Fig. 7d) exhibited bands at ~ 483 and ~ 528 cm−1, corresponding to PdO formation42. Ni(OH)2 and NiO (Supplementary Fig. 7f, g) showed bands at ~ 504 cm−1, indicative of β-NiOOH formation29,43,44. MoS2 (Supplementary Fig. 7i) displayed a distinct band at ~ 864 cm−1, assigned to Mo = O stretching vibrations, suggesting surface oxidation45. These reconstructed states are adopted for subsequent Eads analyses. In contrast, TiO2 (Supplementary Fig. 7e), Co3O4 (Supplementary Fig. 7h), and Ag (Supplementary Fig. 7j) exhibited no noticeable spectral changes, suggesting structural stability under the reaction conditions44,46,47. These operando insights provided an experimental basis for rational catalyst model construction in the theoretical calculations.
In Fig. 2b, the horizontal and vertical axes represent the Eads of NH3 and EG on various catalyst surfaces, respectively. The color intensity indicates the Eads of HCONH2, while the adsorption configurations are illustrated in Supplementary Fig. 9. Notably, α-PtO2-(110) crystal plane demonstrates the lower Eads for NH3 and EG, which are conducive to their adsorption for subsequent reactions. Simultaneously, this crystallography plane shows favorable Eads for HCONH2, facilitating its surfacial desorption while preventing product accumulation on active sites. Similarly, other crystal planes of α-PtO2, such as (100) and (001) planes, along with MoS2 and Ni(OH)2 catalysts, exhibit low Eads for NH3 and EG and high Eads for HCONH2, validating their catalytic efficacy in converting EG to HCONH2. These findings align well with electrochemical screening, demonstrating the feasibility of Pt foil in HCONH2 formation, which affords in situ surfacial conversion.
To elucidate the roles of C and N sources in the formation of HCONH2, current-voltage (I–V) curves (with 20% iR compensation versus RHE) and solution resistance are measured using various electrolyte compositions in 0.5 M KHCO3 (Fig. 2c and Supplementary Fig. 10). In the presence of 1.0 M NH3 (without EG), the onset potential is lower. The addition of 1.0 M EG increases the voltage significantly at a current density above 50 mA cm−248,49,50. Conversely, the presence of 1.0 M EG (without NH3) leads to a higher onset potential and voltage compared to the case with 0.5 M KHCO3 alone. The results suggest that NH3 oxidation is energetically favorable and occurs prior to EG oxidation, harnessing a higher decomposition energy barrier51,52,53. 1H NMR spectra confirm the sole formation of HCONH2 when both EG and NH3 are presented (Fig. 2d). At varying current densities (5–200 mA cm−2), an FE of 45.29 ± 1.93% is observed at 100 mA cm−2, along with a production rate of 563.3 ± 24.05 µmol cm−2 h−1 (Fig. 2e). At 200 mA cm−2, the FE can be maintained at 39.54 ± 2.02%, with a production rate of 983.56 ± 50.16 µmol cm−2 h−1 (Supplementary Fig. 11).
Investigation on the reaction mechanism for electrochemical HCONH2 synthesis
Identifying key intermediates is essential to the understanding of mechanisms for the electrosynthesis of HCONH2 from EG. The analysis of substantial reaction pathways is conducive to the optimization of reaction conditions and the extension of the electrochemical methodology to other applications, such as waste treatment. To this end, four high-performing catalysts: Pt foil, Pt/C, MoS2, and Ni(OH)2 are selected from the initial screening of ten materials. DFT calculations are performed to explore the reaction pathways on three crystal planes ((110), (100), and (001) planes) of α-PtO2, β-Ni(OH)2, and 1T-MoS2 (Fig. 3a and Supplementary Figs. 12–15), and the configurations are illustrated in Supplementary Data 1. The reaction begins with the oxidation of NH3 to form the *NH2 intermediate, which is consistent with the experimental result as shown in Fig. 2c. Simultaneously, EG interacts with the catalyst surface, that the system energy is reduced, indicating the stability of such a process. Subsequently, the cleavage of the C–C bond in EG generates two hydroxymethyl (*CH2OH) fragments, causing a moderate increase in free energy, which is consistent with reported results54. The pathway is observed for all four catalysts, with free energy changes (ΔG) calculated to be 0.70 eV, 1.38 eV, 1.98 eV, 2.10 eV, and 1.21 eV for α-PtO2-(110), -(100), and -(001), β-Ni(OH)2, and 1T-MoS2, respectively. Notably, α-PtO2-(110) exhibits the lower ΔG, indicating that it is more reactive and has superior catalytic performance, which is consistent with the results shown in Fig. 2b.

a Free energy diagrams for proposed reaction pathways on α-PtO2-(110). b–d Schematic diagram of pathway 1, 2, and 3 for HCONH2 synthesis during the co-oxidation of EG and NH3 over the surface of α-PtO2-(110). e In situ ATR-SEIRAS spectra during the reaction for the formation of HCONH2 in 4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3 electrolyte over Pt foil catalyst. f In situ DEMS signals for m/z = 46 (H2NCH2O), 45 (H2NCHO), 30 (CH2O), and 16 (NH2) species. Source data are provided as a Source Data file.
Subsequent analyses on the formation of HCONH2 on α-PtO2-(110) are carried out, with three primary pathways proposed (Fig. 3a and Supplementary Fig. 16). In the reaction coordinate diagram, the vertical axis represents free energy changes, while the horizontal axis denotes the reaction progress. The desorption of *CH2OH results in a significant free energy decrease (– 0.32 eV), followed by its partial oxidation in generating the aldehyde intermediate *CH2O, which induces a modest decrease in free energy (– 1.92 eV). Pathway 1 (red dashed line) involves the reaction between *CH2O and NH3-derived intermediate *NH2, which produces hemiaminal (*H2NCH2O) as accompanied by a decrease in free energy to – 5.65 eV. This intermediate undergoes further dehydrogenation that yields HCONH2, with the system’s free energy decreased to – 11.44 eV (Fig. 3b)55; Pathway 2 (black dashed line) involves the subsequent dehydrogenation of *CH2O to *CHO, which then couples with *NH2 to form HCONH2 (Fig. 3c); Pathway 3 (yellow dashed line) indicates that *CH2O could react directly with NH3 to form *H2NCH2OH. This intermediate is hydrolyzed to *H2NCH, which subsequently undergoes two dehydrogenation steps, yielding the nitrile intermediate *HNC and ultimately HCONH2 (Fig. 3d)56. Among these pathways, the crucial dehydration step of *NH2CH2OH is thermodynamically viable only over α-PtO2, with Gibbs free energy of – 1.08 eV. The results suggest that α-PtO2 facilitates HCONH2 formation predominantly by going through pathway 1, where pathway 3 is an alternative.
In contrast, pathway 2 involves the C–N coupling of two adjacent reactive intermediates, *CHO and *NH2, both of which are prone to oxidation, leading to the formation of C/N-containing by-products57. Consequently, the direct nucleophilic attack of NH3 on the in situ formed aldehyde *CH2O, as proposed in pathways 1 and 3, appears to be more feasible. During the formation of the *CHO intermediate in pathway 2, the system undergoes a free energy decrease of – 3.06 eV, resulting in a total free energy of – 5.4 eV. Upon the formation of HCONH2, the coupling of *CHO and *NH2 leads to a further energy reduction to – 7.6 eV, with an energy change of – 3.84 eV. Comparatively, the energy change from *NH2CH2O to HCONH2 in pathway 1 is – 5.79 eV, while the transition from *HNC to HCONH2 in pathway 3 involves the energy change of – 1.44 eV. These findings underscore that among the three reaction pathways for producing HCONH2 from EG, pathway 1 is the most energetically favorable and thus becomes the primary route.
To elucidate the mechanistic pathway for HCONH2 formation, in situ ATR-SEIRAS is employed to monitor the real-time evolution of reaction intermediates and ionic species at the catalyst-electrolyte interface. Measurements are conducted in an electrolyte comprising 0.5 M KHCO3, 4.0 M EG, and 1.0 M NH3 under galvanostatic conditions at a current density of 50 mA cm−2. Spectra are recorded at time intervals of 0 s, 30 s, 1 min, 2 min, 5 min, 8 min, and 10 min (Fig. 3e). The time-dependent growth of multiple infrared bands revealed the stepwise formation and accumulation of adsorbed intermediates on the catalyst surface.
According to established vibrational assignments, the band at 1241 cm−1 corresponds to the C–N stretching vibration of HCONH2. Additional bands at 1339 and 1360 cm−1 correspond to formate (HCOO−) and the asymmetric C–N stretching mode of HCONH2, respectively. The peak at 1652 cm−1 is assigned to the asymmetric bending vibration (δasNH2) of adsorbed *NH2 as well as the N–H bending in HCONH2. A strong feature at ~ 1745 cm−1 is indicative of *CH2O species, consistent with earlier reports. To ensure the fidelity of these assignments, reference IR spectra are acquired for a set of standard compounds, including HCONH2, HCOOH, EG, NH3, KHCO3, and K2CO3 (Supplementary Fig. 17), showing good agreement with the in situ data and supporting the molecular origins of the observed features28,32,58,59,60,61,62.
To further corroborate these findings, in situ DEMS is conducted under alternating potentials. The detection of mass-to-charge (m/z) signals at 46 (H2NCH2O), 45 (H2NCHO), 30 (CH2O), and 16 (NH2) provided direct evidence of key reactive intermediates and final products (Fig. 3f). These mass fragments align with the infrared bands at 1241, 1360, 1652 and 1749 cm−1, respectively, supporting a mechanistic pathway involving the coupling of *CH2O and *NH2 in the formation of HCONH2. Taken together, DFT calculations and spectroscopic measurements suggest that pathway 1 on the α-PtO2-(110) surface is likely involved in the electrocatalytic synthesis of HCONH2.
Optimization of electrochemical performance for HCONH2 synthesis
To elucidate the effect of reactant concentration on reactivity, initial tests with various concentrations of EG are conducted. As the EG concentration increases, both current density and resistance increase, likely due to the formation of more active electrophilic intermediates through EG oxidation (Fig. 4a and Supplementary Figs. 18, 19). With the increases in EG concentration from 1.0 M to 8.0 M, the FE of HCONH2 initially increases and then decreases (Fig. 4b, and Supplementary Fig. 20). In contrast, the FE of HCOOH increases with increasing EG concentration (Supplementary Fig. 21). The conversion ratio remains stable with all concentrations (Supplementary Fig. 22). At 100 mA cm−2, the FE for HCONH2 is 55.87 ± 1.4% with an EG concentration of 4.0 M. At 150 mA cm−2, using 4.0 M EG, the FE remains at 53.8 ± 1.27%, with a production rate of 1003.63 ± 23.72 µmol cm−2 h−1 (Fig. 4c). The NH3 concentration also influences product selectivity, with an FE of 55.87 ± 1.4% observed at 1.0 M NH3 (Supplementary Fig. 23).

a I–V curves (with 20% iR compensation versus RHE), (b) FEs, and (c) production rates with varying EG concentrations (1.0–8.0 M) in 1.0 M NH3 + 0.5 M KHCO3 (pH: 9.38 ± 0.016, 9.52 ± 0.029, 9.44 ± 0.032, 9.54 ± 0.035, and 9.52 ± 0.012; Resistance: 5.235 ± 0.151, 5.235 ± 0.491, 8.416 ± 0.264, 12.20 ± 0.02, and 15.219 ± 0.287 ohms). d I–V curves (with 20% iR compensation versus RHE), and (e) FEs in 4.0 M EG + 1.0 M NH3 with different electrolytes (0.5 M KOH, 0.5 M KHCO3, 0.25 M H2SO4, and 0.25 M K2SO4 (pH: 12.51 ± 0.045, 9.44 ± 0.032, 9.58 ± 0.017, 11.17 ± 0.048; Resistance: 4.62 ± 0.24, 8.416 ± 0.264, 8.03 ± 0.301, and 11.173 ± 0.442 ohms) (f) 13C NMR spectra for the electrolyte solution of 4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3 after 1 h at 100 mA cm−2. g Chronopotentiometry experiment profile showing the cell voltage (with 20% iR compensation versus RHE) and HCONH2 production at 100 mA cm−2 over 500 h in an H-type cell (4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3, electrolyte refreshed every 24 h. pH: 9.44 ± 0.032; Resistance: 8.416 ± 0.264 ohms). h Comparison on the performance against those reported H-type cell systems. The error bars in (b), (c), and (e) represent the standard deviation of three independent measurements. Source data are provided as a Source Data file.
In addition, the impact of electrolyte composition on reaction reactivity is examined using various electrolytes. The I–V curves (with 20% iR compensation versus RHE) and resistances of a solution containing 4.0 M EG and 1.0 M NH3 are measured in the electrolytes of 0.5 M potassium hydroxide (KOH), 0.5 M KHCO3, 0.25 M sulfuric acid (H2SO4) and 0.25 M potassium sulfate (K2SO4) (Fig. 4d and Supplementary Fig. 24), ensuring a consistent number of cations. The reaction demonstrates the low onset potential in a 0.5 M KHCO3 solution. In the 0.5 M KOH solution, a lower voltage is observed at the same current density after 20 mA cm−2 as compared to that in the 0.5 M KHCO3 solution. At this stage, the only carbonaceous liquid product is HCOOH, with HCONH2 being completely suppressed in the 0.5 M KOH solution (Supplementary Fig. 25). Such suppression may result from the reaction between the intermediate *CHO resulting from EG oxidation and OH−, producing HCOOH63,64. Upon reducing the alkalinity, the production of HCONH2 can be observed, further supporting the hypothesis regarding the reaction. In the 0.25 M H2SO4 electrolyte, the yield of HCONH2 is slightly higher than that in 0.5 M KOH, but remains significantly lower than that in KHCO3. The result supports the hypothesis that acidic conditions protonate NH3 into NH4+, thereby reducing its nucleophilicity and promoting the formation of byproducts such as (NH4)2SO4, which hinder C–N coupling65,66. In contrast, KHCO3 provides a weakly alkaline buffering environment that suppresses undesired ammonium salt formation and facilitates selective *NH2–*CH2O coupling toward HCONH2. At various current densities (5–200 mA cm−2), the FE of HCONH2 in 0.5 M KHCO3 electrolyte reaches 55.87 ± 1.4% at 100 mA cm−2, which is comparatively higher than under other conditions (Fig. 4e). At 150 mA cm−2, the conversion ratio remains approximately 90%, and the FE reaches 53.8 ± 1.27%, corresponding to a production rate of 1003.63 ± 23.72 µmol cm−2 h−1 (Supplementary Figs. 26, 27).
To evaluate thermal effects, the FE for the system with 4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3 is tested at various temperatures (20–60 °C) at 100 mA cm−2. The FE observed under laboratory conditions at room temperature (20 °C) is relatively higher (Supplementary Fig. 28). Under these conditions (4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3 at 20 °C), we quantified FEs for all detectable products: HCONH2, HCOOH, NO2, NO, N2, CO2, CO, and O2 over 5–200 mA cm−2. Across this current-density range, HCONH2 is the dominant carbonaceous product, oxygen evolution reaction (OER) is the main competing pathway, producing O2 as the principal by-product, its FE is 15.37% at 100 mA cm−2, and all other products appear only in minor amounts (Supplementary Fig. 29). The formation of HCONH2 is further confirmed by a distinct signal at 164.2 ppm in the 13C NMR spectra (Fig. 4f). The stability of the H-cell for HCONH2 electrosynthesis is tested in a solution of 4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3 at 100 mA cm−2. The cell voltage and HCONH2 production rate remain stable for over 500 hours, with the electrolyte refreshed every 24 hours (Fig. 4g). After cycling, the extent of O2 increases on the Pt foil, suggesting the occurrence of self-activation in the electrosynthesis process (Supplementary Fig. 30). It is also worth noting that no Pt would be consumed except for the self-activation surficial reaction. This system demonstrates superior performance compared to other reported electrochemical methods for HCONH2 production (Fig. 4h)29,32. In terms of product selectivity, the electrosynthesis system achieves a higher FE of 55.87 ± 1.4% at an industrial-level current density of 100 mA cm−2. The production rate of 1003.63 ± 23.72 µmol cm−2 h−1 with a high FE of 53.8 ± 1.27% is double the reported values, which is obtained at 150 mA cm−2. Additional comparisons in Supplementary Table 1 highlight substantial improvements in key performance metrics, underscoring the advantages of this electrosynthesis strategy.
Continuous production of HCONH2 in a membrane electrode assembly reactor
To achieve continuous production of HCONH2, an MEA reactor is developed to replace the traditional H-type cell used in the electrosynthesis (Supplementary Fig. 31). In this setup, titanium plates act as anode current collectors, while stainless steel plates serve as cathode current collectors. Commercial Pt foils (1.0 cm × 1.0 cm) are used as catalysts at both the anode and cathode, taking advantage of their high catalytic activity and stability. Based on the optimized conditions, the anolyte consists of a solution of 4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3, with H2O acting as the catholyte. The anode and cathode compartments are separated by a PEM (Nafion-117), which facilitates proton transport while maintaining the separation of the reactants. The effective geometric electrode area is restricted to 1.0 cm2. The liquid flow rates on both the anode and cathode sides are maintained at 0.92 mL min−1 using syringe pumps. The anode reaction continuously produces HCONH2, while the cathode reaction generates hydrogen (H2) gas (Fig. 5a), with a low impedance of 3.87 ohms during the reaction (Supplementary Fig. 32). This innovative design maintains a constant reactant concentration, simplifies the transport of reactants and the removal of products, and minimizes ohmic losses67,68.

a Schematic of the MEA reactor using Pt foil electrodes separated by Nafion-117 for HCONH2 synthesis through the co-oxidation reaction of EG and NH3. b I–V curves (no iR compensation), (c) FEs, and (d) production rates of electrochemical HCONH2 synthesis at different current densities. e Chronopotentiometry experiment profile showing the cell voltage (no iR compensation) and FEs of electrochemical HCONH2 synthesis at 100 mA cm−2 over 300 h in the MEA reactor (4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3, electrolyte refreshed every 30 h. pH: 9.44 ± 0.032; Resistance: 3.87 ohms). f Radar plot comparing this work with other reported MEA-based systems. Source data are provided as a Source Data file.
The I–V curve of the MEA system, covering the current density range of 1–300 mA cm−2, is shown in Fig. 5b. The FE of HCONH2 is changed in a pattern similar to that observed for the H-type cell, though it is slightly decreased, peaking at 43.3% at 100 mA cm−2 and maintaining at 41.42% at 150 mA cm−2 (Fig. 5c). The corresponding production rates are 538.42 μmol cm−2 h−1 and 772.8 μmol cm−2 h−1, respectively (Fig. 5d). It is worth noting that the FE of HCOOH for the MEA system is higher than that for the H-type cell. Notably, the system maintains continuous and stable HCONH2 production, sustaining an FE of approximately 40% for 300 h at 100 mA cm−2 without significant fluctuations in the full-cell voltage, which remained around 4.65 V. (electrolyte refreshed every 30 hours, Fig. 5e). Such stability is essential to meet the requirements of industrial-scale production, since it ensures consistent output and reduces the need for frequent maintenance or adjustments on the systems. Notably, the radar plot underscores that the system possesses superior stability compared to the reported electrochemical systems for HCONH2 production (Fig. 5f). Further performance comparisons, as provided in Supplementary Table 2, reveal that the system developed in this work for the electrosynthesis has substantial improvements in key metrics over other electrochemical synthesis methods. These enhancements demonstrate the distinct advantages of our MEA reactor design, including higher efficiency, reduced costs, and scalability for large-scale applications.
Upcycling plastic waste to HCONH2 as achieved at industrial-level current densities with ProdSI and TEA
Based on the successful conversion of EG and NH3 into HCONH2, the upcycling of wasted plastic bottles to HCONH2 through the electrochemical synthesis route is implemented (Fig. 6a). As confirmed by the 1H NMR spectra (Supplementary Figs. 33 and 34), EG and terephthalic acid (TPA) are the primary monomers produced during the initial step of PET depolymerization. TPA could be separated and recycled by the H2SO4 neutralization. The recovery and repurposing of TPA for the resynthesis of PET not only promotes a recycling economy but also adds value to the waste management process by creating a closed-loop system where materials are continuously reused. The step described above ensures that the presence of TPA does not interfere with the efficiency of the subsequent electrochemical reactions.

a Waste plastic upcycling process for HCONH2 production. b I–V curves (with 20% iR compensation versus RHE), and (c) FEs of HCONH2 synthesis at different current densities using EG derived from waste PET (1.34 M EG + 1.0 M NH3 + 0.5 M KHCO3. pH: 10.14 ± 0.02; Resistance: 6.541 ± 0.289 ohms). The error bars represent the standard deviation of three independent measurements. d Proposed integration of waste valorization into a circular energy system. e ProdSI-based sustainability assessment result of four catalysts. f Estimating production costs, and (g) projected yearly profits at different current densities. Source data are provided as a Source Data file.
Following the neutralization and separation processes, the resulting EG solution is co-oxidized with NH3 to form HCONH2. The electrochemical performance of the electrosynthesis system is evaluated, with the I–V curve (with 20% iR compensation versus RHE) measured over a current density range of 1–200 mA cm−2, as presented in Fig. 6b. The conversion ratio remains over 80%, indicating efficient utilization of reactants (Supplementary Fig. 35). The FE for HCONH2 synthesis reaches 47.68 ± 1.02% at a current density of 100 mA cm−2 (Fig. 6c). At the higher current density of 200 mA cm−2, a production rate of 879.4 ± 97.16 μmol cm−2 h−1 is obtained (Supplementary Fig. 36), highlighting the great potential of the system for high-throughput production and the scalability of the process. The proposed route not only facilitates the synthesis of valuable chemicals from plastic waste but also contributes to resolving the imminent environmental challenges posed by plastic waste. By combining the burgeoning renewable energy technologies, it is anticipated to generate more valuable materials from the wastes towards a net-zero society (Fig. 6d).
To this end, cost and sustainability evaluation are conducted using the ProdSI and TEA (Supplementary Note 1, and 2) methods to assess potential environmental and economic benefits. Akin to the depreciation of fixed assets, the cyclic decay rate exerts a strong impact on pragmatic employment, which is of equal importance to productivity and efficiency. Even tiny decay per cycle would be indefinitely magnified, especially at the industrial level. Under this occasion, the noble Pt foil is anticipated to afford low cost and better sustainability. An intuitionistic comparison in ProdSI is drawn between Pt foil and the other three catalysts, harnessing good initial performance. As illustrated in Fig. 6e, Pt foil catalyst demonstrates significant advantages over other catalyst counterparts among all sustainability considerations.
To further compare the advantages of Pt foil, we conduct two consecutive cycling tests on Pt/C, Pt foil, MoS2 and Ni(OH)2 catalysts in H-type cells at a current density of 100 mA cm−2, and evaluated their cell voltage, FE and HCONH2 generation rate (Supplementary Fig. 37). Pt foil exhibits stable performance, with negligible variation across cycles. In contrast, the other three catalysts show significant voltage decay, particularly in the second cycle, and a downward trend in both FE and yield. Notably, only Pt foil displays enhanced FE and productivity in the second cycle, which corresponds well with the increased surface O signal observed after long-term electrolysis (Supplementary Fig. 30), indicating a self-activation process involving the formation of Pt–O species. Importantly, this surficial oxidation does not lead to Pt loss, affirming the structural robustness of Pt foil under operating conditions.
We then conduct TEA on the synthesis of HCONH2 over Pt foil. Detailed cost analysis shows that the plastic waste upcycling process can reduce the cost of HCONH2 production to around $1500 per ton at a current density varying between 50 and 150 mA cm−2, which is nearly $200 less than that of traditional methods (Fig. 6f). Moreover, the projected annual profit across different current densities remains relatively high and consistent, with an estimated profit of $115.50 million at a current density of 100 mA cm−2 (Fig. 6g). At current densities above 50 mA cm−2, the estimated annual profit exceeds $114 million, suggesting promising applicability and industrial potential of our system for electrosynthesis. The net present value (NPV) analysis indicates that by the end of Year 3, the proposed factory will produce benefits, the cumulative present value will be over zero ($33.39 million). Over a 20-year operational period, the system is projected to generate a total profit exceeding $717.85 million under a current density of 100 mA cm−2. These findings underscore the economic potential and environmental benefits of our upcycling approach for the production of HCONH2 on a large scale, offering a sustainable and profitable solution for upcycling plastic waste to valuable products and tackling some imminent challenges in chemical synthesis. Collectively, Pt foil catalyst, which might be labeled as ‘high cost’, should be qualified as a good catalyst in the scenario of HCONH2 electrosynthesis. It is suggested that there should be a valuable comparison between the application of mature Pt foil processing technologies and the ongoing development of suitable nano-catalysts for catalytic electrosynthesis. Such concept could be extended to other unexplored industrial reactions (Supplementary Fig. 38).
Discussion
A sustainable and efficient strategy has been developed for the electrochemical synthesis of HCONH2 via the co-oxidation of EG derived from PET waste and NH3 over commercially Pt foil catalysts. Catalyst selection was guided by a combination of experimental screening and theoretical calculations. Employing KHCO3 as the electrolyte establishes a mildly alkaline environment, which suppresses the formation of (NH4)2SO4 by-products while preserving PET degradation efficiency. Under industrially relevant conditions, the system achieves an FE of 55.87 ± 1.4% and a production rate of 1003.63 ± 23.72 µmol cm−2 h−1. Long-term electrolysis confirms operational durability, with stable performance maintained over more than 500 h and 300 h for the H-cell and MEA reactor, respectively. The reaction mechanism is proposed based on in situ ATR-SEIRAS, DEMS, and supported by DFT calculations. These analyzes suggest *CH2O and *NH2 as key surface-bound intermediates that promote HCONH2 formation. Furthermore, TEA and ProdSI reveal that the use of noble metal catalysts, when properly evaluated, may offer competitive economic and environmental benefits compared to conventional energy-intensive processes. These findings provide a broader framework for reassessing the role of precious metal catalysts in sustainable electrosynthesis.
Methods
Chemicals and materials
Ethylene glycol (C2H6O2, 99.5%, Aladdin), ammonia solution (NH3.H2O, 25-28%, Aladdin), potassium bicarbonate (KHCO3, 99.5%, Aladdin), potassium hydroxide (KOH, 95%, Aladdin), potassium sulfate (K2SO4, 99%, Aladdin), sulfuric acid (H2SO4, 98%, RCI Labscan), formamide (CH3NO, 99%, Aladdin), formic acid (CH2O2, 99%, Aladdin), polyethylene terephthalate (PETTMCB-102, 300 mesh, Dupont), p-Phthalic acid (C8H6O4, 99%, Aladdin), Nafion (5 wt.% in a mixture of lower aliphatic alcohols and water, contains 45% water, Aladdin), 2-proponal (C3H8O, Aladdin), deuterium oxide (D2O, 99.9% atom% D, contains 0.05 wt% TMSP, Aladdin), dimethyl sulfoxide (C2H6OS, ≥ 99.9%, Sigma Aldrich) were used in the experiments. Platinum–ruthenium nanoparticles supported on carbon (PtRu/C), platinum nanoparticles supported on carbon (Pt/C), titanium dioxide (TiO2), nickel hydroxide (Ni(OH)2), nickel(II) oxide (NiO), cobalt(II,III) oxide (Co3O4), and silver (Ag) were purchased from Aladdin Company. Molybdenum disulfide (MoS2) was obtained from Xianfeng Nano Company. Platinum (Pt) foil and palladium (Pd) foil were obtained from Shanghai Yueci Electronics Company. The DI water (18.25 MΩ·cm−1) was used in all experiments.
Material characterizations
The morphologies of the samples were examined using a Hitachi SU-8230 field emission scanning electron microscope (FESEM). Operando Raman spectroscopy was conducted using a LabRAM HR800 Raman Spectroscopy System (Supplementary Fig. 39a). In situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) measurements were conducted using a BRUKER Invenio Fourier-transform infrared (FTIR) spectrometer equipped with a diamond ATR window (Supplementary Fig. 39b). Spectra were collected in ATR-SEIRAS mode with 64 scans and a spectral resolution better than 4 cm−1. X-ray photoelectron spectroscopy (XPS) was conducted on a Kratos AXIS UltraDLD with Al-Kα radiation (1486 eV) to gain the chemical composition and state of samples. X-ray diffraction (XRD) analysis was performed using a Rigaku Smart Lab 9 kW X-ray Diffractometer.
In situ differential electrochemical mass spectrometry (DEMS) measurements
In situ DEMS measurements were performed using an HPR-40 quadrupole mass spectrometer (Hiden Analytical, UK) equipped with a gas-permeable PTFE membrane inlet (thickness: 60 μm, pore size: 0.22 μm, porosity: 50%). The catalyst ink was drop-cast onto the PTFE membrane, which served as the working electrode. A saturated calomel electrode (Hg/Hg2Cl2) and a Pt wire were used as the reference and counter electrodes, respectively (Supplementary Fig. 39c). The electrolyte consisted of 4.0 M EG, 1.0 M NH3, and 0.5 M KHCO3 in aqueous solution.
Prior to electrolysis, the system was stabilized under open-circuit conditions for 30 min to establish the baseline signal. Subsequently, alternating potential cycling was applied between 1.2 V and 2.8 V vs Hg/Hg2Cl2 for 6 consecutive cycles, lasting a total of 3000 s. During the entire process, the mass spectrometer continuously monitored the gaseous products in real-time. The characteristic mass-to-charge (m/z) ratios of 46 (H2NCH2O), 45 (HCONH2), 30 (CH2O), and 16 (NH2) were tracked, and the corresponding signal intensities were baseline-corrected using the open-circuit background. No isotope labeling was used in this study.
Preparation of electrodes
For Pt foil and Pd foil experiments, commercial Pt foil and Pd foil (99.99%, 1.0 cm2, 0.1 mm thickness) were used directly as the working electrode after ultrasonic cleaning in ethanol and deionized water for 5 min each.
For all other catalysts were prepared as follows: 40 mg of catalyst powder was dispersed in 4.0 mL of isopropanol (Aladdin) and 160 μL of Nafion solution (Aladdin). The ink was ultrasonicated in an ice-water bath for 30 min to ensure homogeneity, and then airbrushed onto a 5 × 5 cm2 hydrophilic carbon paper (TGP-H060H(M), Toray, purchased via Science Materials Station). The resulting electrode was dried under vacuum at room temperature (20 °C) for 24 h before use with a geometric area of 1.0 cm2.
Activation of the Nafion-117 membrane
The proton exchange membrane (PEM, Nafion-117) was purchased from Fuel Cell Store. It was pretreated in 5% H2O2 and 10% H2SO4 solutions at 80 °C for 1 h each before use. The membrane thickness is ~ 183 μm, and the size was cut to match a 1.0 cm2 active area.
Electrochemical measurements
All electrochemical measurements were conducted on a BioLogic VSP-300 electrochemical workstation at room temperature (20 °C). All data were collected with EC-Lab software, and post-processing was performed using Origin.
Electrochemical measurements were conducted in a dual-chamber H-type electrolytic cell (C007-10, Gaoss Union) with each compartment containing 10.0 mL of electrolyte. A three-electrode configuration was used, with Pt foil acting as the counter and reference electrodes. We used a three-electrode configuration in an H-type cell, with Pt foil acting as the counter and reference electrodes. For all the reactions, Hg/Hg2Cl2 were used as reference electrodes. All potentials measured were calibrated to the reversible hydrogen electrode (RHE) by the Eq. (1):
The Hg/Hg2Cl2 reference electrode was calibrated against a reversible hydrogen electrode (RHE) in 0.5 M H2SO4 by recording the hydrogen evolution/oxidation potential on a Pt black electrode under continuous H2 purging. The zero-current potential was identified as 0 V vs. RHE, allowing us to determine the offset potential of the reference electrode. The reference electrode was calibrated regularly to ensure accuracy. All potentials in the manuscript have been converted accordingly. The compartments were separated by a proton exchange membrane (PEM, Nafion-117 purchased from Fuel Cell Store).
Electrochemical impedance spectroscopy measurements were carried out in a frequency range of 0.01-100,000 Hz with an amplitude of 6.0 mV. The uncompensated cell resistance was measured via EIS before each experiment. Linear sweep voltammetry and cyclic voltammetry of anodic reactions was measured at a scan rate of 5.0 mV s−1. The electrolyte solution consisting of 4.0 M EG, 1.0 M NH3, and 0.5 M KHCO3 was freshly prepared before each experiment. For 500 mL of electrolyte: Ethylene glycol (C2H6O2, 99.5%, Aladdin): 111.5 mL, ammonia solution (NH3.H2O, 25-28%, Aladdin): 34.08 mL, potassium bicarbonate (KHCO3, 99.5%, Aladdin): 25.03 g. These components were sequentially added into deionized water under continuous magnetic stirring until fully dissolved. The mixture was then diluted to 500 mL with deionized water. The electrolyte was used immediately or stored in a sealed container at room temperature (20 °C) for no more than 24 h. The pH of the electrolyte (4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3) was also measured at room temperature (20 °C) using a calibrated pH meter, yielding a value of 9.44 ± 0.032 (n = 3)
Membrane electrode assembly (MEA) reactor
The Pt foil electrode was used as the anode and cathode for formamide synthesis. The two electrodes were positioned on opposite sides and separated by a proton exchange membrane (PEM, Nafion-117 purchased from Fuel Cell Store). The geometric surface area of the catalyst is 1.0 cm2. On the cathode side, a stainless-steel gas flow chamber supplied DI water while the anode was supply with 4.0 M EG + 1.0 M NH3 + 0.5 M KHCO3. The flow rate was 0.92 ml min−1 in both chambers, controlled by a syringe pump. Impedance (IR) compensation was not performed unless specifically stated. The MEA device maintains continuous and stable HCONH2 production, sustaining an FE of approximately 40% for 300 hours at 100 mA cm−2 without significant fluctuations in the full-cell voltage, which remained around 4.65 V (electrolyte refreshed every 30 hours).
Product analysis
One-dimensional 1H NMR spectra were collected on a JEOL ECZ500R 500 MHz Solid-State nuclear magnetic resonance (NMR) spectrometer to quantify the liquid products at the anode. Typically, 500 μl of the electrolyte after electrolysis was mixed with 49.0 μl of D2O (Aladdin, 99.9 at% D) and 1.0 μl dimethyl sulfoxide (Sigma Aldrich, ≥ 99.9%) as internal standard. Gas products were analyzed online using an Agilent 7890B gas chromatograph (GC) equipped with a high-sensitivity flame ionization detector (FID) and a thermal conductivity detector (TCD). The FID was used for hydrocarbons and other carbon-containing organics, while the TCD provided a strong response for inorganic gases (e.g., O2 and N2). High-purity helium served as the carrier gas at 1.0 mL product identities and concentrations were determined by min−1. Comparison with certified gas standards. The detected gas were O2, CO2, CO, N2, NO, and NO2.
The corresponding FE was calculated as Eq. (2):
where ni is the number of electrons transferred in one product molecule, whereas the value is 3 for formamide and 4 for formic acid formation, respectively. F is the Faraday constant (96485 C mol−1); C is the concentration of the formamide and formic acid calculated by the calibration curve, and V is the volume of the electrolyte.
The production rate and conversion ratio were calculated based on the Eq. (3)−(4):
Data availability
The data generated in this study are provided in the Supplementary Information and Source Data file. All data supporting the findings of this study are available within the paper and its Supplementary Information or from the corresponding authors upon request. Source data are provided in this paper.
Code availability
The central code used in this paper is VASP. Detailed information related to the license and user guide are available at https://www.vasp.at. Other codes used in the study are available from the corresponding authors upon request.
References
Yang, H. et al. Amine-impregnated polymeric resin with high CO2 adsorption capacity for biogas upgrading. Chem. Eng. J. 430, https://doi.org/10.1016/j.cej.2021.132899 (2022).
Xia, Q. et al. Efficient synthesis of organosulfur compounds via electrochemical biomass conversion. Nat. Synth. 4, 765–775 (2025).
Tang, M. et al. High-energy-density aqueous zinc-ion batteries: recent progress, design strategies, challenges, and perspectives. Adv. Mater. e2501361 https://doi.org/10.1002/adma.202501361 (2025).
Zou, X. et al. Carbon-based electrocatalysts for rechargeable Zn-air batteries: design concepts, recent progress and future perspectives. Energy Environ. Sci. 17, 386–424 (2024).
Wu, J. et al. Immobilizing ordered oxophilic indium sites on platinum enabling efficient hydrogen oxidation in alkaline electrolyte. J. Am. Chem. Soc. 146, 20323–20332 (2024).
Zhu, P. et al. Continuous carbon capture in an electrochemical solid-electrolyte reactor. Nature 618, 959–966 (2023).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Li, Y. et al. Sequential co-reduction of nitrate and carbon dioxide enables selective urea electrosynthesis. Nat. Commun. 15, 176 (2024).
Gong, S. et al. Paired electrolysis for efficient coproduction of CO and S8 with techno-economic analysis. Chem. Eng. J. 507, 160286 (2025).
Wang, X. et al. Reaction and reactor designs for improving electrochemical CO2 capture. Device 3, 100837 (2025).
Corma, A. Heterogeneous catalysis: Understanding for designing, and designing for applications. Angew. Chem. Int. Ed. Engl. 55, 6112–6113 (2016).
Trindell, J. A., Duan, Z., Henkelman, G. & Crooks, R. M. Well-defined nanoparticle electrocatalysts for the refinement of theory. Chem. Rev. 120, 814–850 (2020).
Zhang, Y. et al. Design and regulation of defective electrocatalysts. Chem. Soc. Rev. 53, 10620–10659 (2024).
Peljo, P. & Kallio, T. Electrocatalyst nanoparticles go with the flow. Nat. Catal. 4, 445–446 (2021).
Sun, L., Reddu, V. & Wang, X. Multi-atom cluster catalysts for efficient electrocatalysis. Chem. Soc. Rev. 51, 8923–8956 (2022).
Duchesne, P. N. et al. Golden single-atomic-site platinum electrocatalysts. Nat. Mater. 17, 1033–1039 (2018).
Xu, H., Cheng, D., Cao, D. & Zeng, X. C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 1, 339–348 (2018).
Minguzzi, A. How to improve the lifetime of an electrocatalyst. Nat. Catal. 3, 687–689 (2020).
Sorsa, O. et al. Optimization and aging of Pt nanowires supported on single-walled carbon nanotubes as a cathode catalyst in polymer electrolyte membrane water electrolyser. Int. J. Hydrog. Energy 45, 19121–19132 (2020).
Liu, L. & Corma, A. Structural transformations of solid electrocatalysts and photocatalysts. Nat. Rev. Chem. 5, 256–276 (2021).
Akbashev, A. R. Electrocatalysis goes nuts. ACS Catal. 12, 4296–4301 (2022).
Xie, C., Chen, W., Wang, Y., Yang, Y. & Wang, S. Dynamic evolution processes in electrocatalysis: structure evolution, characterization and regulation. Chem. Soc. Rev. 53, 10852–10877 (2024).
Beyazay, T., Martin, W. F. & Tuysuz, H. Direct synthesis of formamide from CO2 and H2O with nickel-iron nitride heterostructures under mild hydrothermal conditions. J. Am. Chem. Soc. 145, 19768–19779 (2023).
Massolo, E., Pirola, M. & Benaglia, M. Amide bond formation strategies: Latest advances on a dateless transformation. Eur. J. Org. Chem. 2020, 4641–4651 (2020).
Guo, C. et al. Electrochemical upgrading of formic acid to formamide via coupling nitrite co-reduction. J. Am. Chem. Soc. 144, 16006–16011 (2022).
Lu, Y. et al. Anodic electrosynthesis of amide from alcohol and ammonia. CCS Chem. 6, 125–136 (2024).
Lan, J. et al. Efficient electrosynthesis of formamide from carbon monoxide and nitrite on a Ru-dispersed Cu nanocluster catalyst. Nat. Commun. 14, 2870 (2023).
Ramadhany, P. et al. Triggering C‒N coupling on metal oxide nanocomposite for the electrochemical reduction of CO2 and NOx‒ to formamide. Adv. Energy Mater. 14, 2401786 (2024).
Meng, N. et al. Electrosynthesis of formamide from methanol and ammonia under ambient conditions. Nat. Commun. 13, 5452 (2022).
Yang, W. et al. Photocatalytic formamide synthesis via coupling of electrophilic and nucleophilic radicals over atomically dispersed Bi sites. Angew. Chem. Int. Ed. Engl. 63, e202408379 (2024).
Mou, S., Liu, S., Dai, W., Sun, Y. & Dong, F. Fluorine-induced D-band center shift in Nb2O5 nanosheets for efficient electrochemical formamide synthesis. Appl. Catal. B Environ. 365, 124963 (2025).
Shi, Q. et al. Electrocatalytic upgrading of plastic and biomass-derived polyols to formamide under ambient conditions. Angew. Chem., Int. Ed. Engl. 63, e202407580 (2024).
Li, X. Y., Zhang, Z. M., Zhuang, X. X., Jia, Z. T. & Wang, T. Electrolyte effects in electrocatalytic kinetics. Chin. J. Chem. 42, 3533–3552 (2024).
Yan, Y. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144–6155 (2023).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 12, 4679 (2021).
Abedsoltan, H. A focused review on recycling and hydrolysis techniques of polyethylene terephthalate. Polym. Eng. Sci. 63, 2651–2674 (2023).
Yu, H. et al. Biobased dimethyl isosorbide as an efficient solvent for alkaline hydrolysis of waste polyethylene terephthalate to terephthalic acid. Green. Chem. 25, 7807–7816 (2023).
Chen, J. et al. Accelerating discovery of next-generation power electronics materials via high-throughput ab initio screening. Npj Comput. Mater. 11, 249 (2025).
Lin, X.-M. et al. In situ probe of the hydrogen oxidation reaction intermediates on PtRu a bimetallic catalyst surface by core-shell nanoparticle-enhanced Raman spectroscopy. Nano Lett. 22, 5544–5552 (2022).
Huang, Y. F., Kooyman, P. J. & Koper, M. T. Intermediate stages of electrochemical oxidation of single-crystalline platinum revealed by in situ Raman spectroscopy. Nat. Commun. 7, 12440 (2016).
Wang, Z. et al. Ultrafine PtO2 nanoparticles coupled with a Co(OH)F nanowire array for enhanced hydrogen evolution. Chem. Commun. 54, 810–813 (2018).
Sun, Y. L. et al. Visualization of electrooxidation on palladium single crystal surfaces via in situ Raman spectroscopy. Angew. Chem. Int. Ed. Engl. 63, e202408736 (2024).
Louie, M. W. & Bell, A. T. An investigation of thin-film Ni-Fe oxide catalysts for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 135, 12329–12337 (2013).
Joya, K. S. & Sala, X. In situ Raman and surface-enhanced Raman spectroscopy on working electrodes: spectroelectrochemical characterization of water oxidation electrocatalysts. Phys. Chem. Chem. Phys. 17, 21094–21103 (2015).
Huang, J. et al. In situ monitoring of the electrochemically induced phase transition of thermodynamically metastable 1T-MoS2 at nanoscale. Nanoscale 12, 9246–9254 (2020).
Doan, T. L., Nguyen, T. N., Hong, H. & Kim, T. Development of an oxygen evolution reaction electrocatalyst of TiO2 supported IrO2 (IrO2@TiO2) for enhancing performance and durability of polymer electrolyte membrane water electrolysis. Int. J. Hydrog. Energy 110, 754–763 (2024).
Luo, H., Ji, X. & Cheng, S. Investigation into the electrochemical behaviour of silver in alkaline solution and the influence of Au-decoration using operando Raman spectroscopy. RSC Adv. 10, 8453–8459 (2020).
Iriawan, H. et al. Methods for nitrogen activation by reduction and oxidation. Nat. Rev. Methods Prim. 1, 1–26 (2021).
Dann, E. K. et al. Structural selectivity of supported Pd nanoparticles for catalytic NH3 oxidation resolved using combined operando spectroscopy. Nat. Catal. 2, 157–163 (2019).
Pignet, T. & Schmidt, L. Kinetics of NH3 oxidation on Pt, Rh, and Pd. J. Catal. 40, 212–225 (1975).
Hong, W., Shang, C., Wang, J. & Wang, E. Bimetallic PdPt nanowire networks with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 8, 2910–2915 (2015).
Yue, H., Zhao, Y., Ma, X. & Gong, J. Ethylene glycol: properties, synthesis, and applications. Chem. Soc. Rev. 41, 4218–4244 (2012).
Xin, L., Zhang, Z., Qi, J., Chadderdon, D. & Li, W. Electrocatalytic oxidation of ethylene glycol (EG) on supported Pt and Au catalysts in alkaline media: Reaction pathway investigation in three-electrode cell and fuel cell reactors. Appl. Catal. B 125, 85–94 (2012).
Kang, H. et al. Mining the carbon intermediates in plastic waste upcycling for constructing C-S bond. J. Am. Chem. Soc. 146, 18639–18649 (2024).
Gunanathan, C., Ben-David, Y. & Milstein, D. Direct synthesis of amides from alcohols and amines with liberation of H2. Science 317, 790–792 (2007).
Yamaguchi, K., Kobayashi, H., Oishi, T. & Mizuno, N. Heterogeneously catalyzed synthesis of primary amides directly from primary alcohols and aqueous ammonia. Angew. Chem. Int. Ed. Engl. 51, 544–547 (2011).
Gong, L. et al. Recent development of methanol electrooxidation catalysts for direct methanol fuel cell. J. Energy Chem. 27, 1618–1628 (2018).
Nagaraj, R. et al. Fructose and methylglyoxal-induced glycation alters structural and functional properties of salivary proteins, albumin and lysozyme. Plos ONE 17, e0262369 (2022).
Li, X. et al. Photocatalytic C‒N coupling from stable and transient intermediates for gram-scale acetamide synthesis. Nat. Commun. 16, 3590 (2025).
McNaughton, D., Evans, C. J., Lane, S. & Nielsen, C. J. The high-resolution FTIR far-infrared spectrum of formamide. J. Mol. Spectrosc. 193, 104–117 (1999).
Sugawara, Y., Hamada, Y. & Tsuboi, M. Vibration-rotation spectra of formamides. Bull. Chem. Soc. Jpn. 56, 1045–1050 (1983).
Brummel, C. L., Shen, M., Hewett, K. B. & Philips, L. A. High-resolution infrared spectroscopy of formamide and deuterated formamide in a molecular beam. J. Opt. Soc. Am. B 11, 176–183 (1994).
Shi, Y. et al. Unveiling hydrocerussite as an electrochemically stable active phase for efficient carbon dioxide electroreduction to formate. Nat. Commun. 11, 3415 (2020).
Xia, Q. et al. Methanol-facilitated surface reconstruction catalysts for near 200% faradaic efficiency in a coupled system. Adv. Funct. Mater. 34, 2314596 (2024).
Bavarella, S. et al. Chemically reactive membrane crystallisation reactor for CO2–NH3 absorption and ammonium bicarbonate crystallisation: Kinetics of heterogeneous crystal growth. J. Membr. Sci. 599, 117682 (2020).
Leaist, D. Proton-coupled transport of ammonia in aqueous hydrochloric acid. Aust. J. Chem. 38, 249–260 (1985).
Xu, Q. et al. Identifying and alleviating the durability challenges in membrane-electrode-assembly devices for high-rate CO electrolysis. Nat. Catal. 6, 1042–1051 (2023).
Wan, L. et al. Oriented intergrowth of the catalyst layer in membrane electrode assembly for alkaline water electrolysis. Nat. Commun. 13, 7956 (2022).
Acknowledgements
G.P.Z. acknowledges the support from the Innovative and Technology Fund (#ITS-005-22MS) from the Innovative and Technology Commission of the Hong Kong Special Administrative Region, and the Research Institute for Advanced Manufacturing (RIAM) of The Hong Kong Polytechnic University (project code: 1-CDJV). J.Y.S. acknowledges the support from China and National Natural Science Foundation of China (22179089). S.X.D. acknowledges the support from the grants include national science foundation overseas senior scientist team program ID 22379097 and Australian Research Council DP220102215.
Author information
Authors and Affiliations
Contributions
X.Z.W., G.P.Z., S.X.D., X.Z., and J.Y.S. conceived the idea. X.Z.W. designed the experiments, performed the electrochemical tests, and explored the mechanisms. J.S.C., X.Z.W., and Y.W.S. contributed to the theoretical calculation. X.Z.W., E.H.Y., and W.S.Y. calculated the ProdSI and TEA of this study. X.Z.Y. assisted in gas product tests. Y.Y.Y., J.J.W., and F.J.X. assisted in SEM. X.Z.W. wrote the paper. X.Z.W., Y.W.S., M.C.T., Y.B.M., Q.X., J.W., and S.H.G. revised the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks Yongwen Tan and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Su, Y., Chen, J. et al. Revisiting Pt foil catalysts for formamide electrosynthesis achieved at industrial-level current densities. Nat Commun 16, 8040 (2025). https://doi.org/10.1038/s41467-025-63313-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63313-5
