
Abstract
The practical applications of enzymes often require their immobilization for multiple recycling or long-term running. However, practically efficient enzyme immobilization methods are lacking. Herein, we present an enzyme immobilization approach by engineering a porous “interphase” between water and oil around the surfaces of Pickering emulsion droplets. The designed “interphase” consists of a porous, nanometer-thick silica shell serving as a scaffold to incorporate enzymes. Within this “interphase”, enzymes can simultaneously be in contact with enzyme-preferred aqueous microenvironment and the oil phase containing organic reactants. The porous “interphase” with its tunable structure and properties allows modulation of transport of reactants, crudely akin to a cell membrane, and of local concentration of reactants. As a proof of the concept, we showcase that our “interphase” strategy is very effective in immobilization of Candida antarctica lipase B (CALB) for continuous-flow olefin epoxidation. Long-term stabilization (800 h), 16-fold increase in catalysis efficiency relative to batch reactions, and 99% H2O2 utilization efficiency are achieved. The integration of unique microenvironment and hydrophobic pores of the “interphase” is found to be crucial for such excellent performances, practically providing the most efficient enzymatic epoxidation system. This strategy opens an avenue for the design of efficient and sustainable biocatalytic processes.
Similar content being viewed by others

Introduction
Enzymatic reactions are becoming increasingly important for the synthesis of fine chemicals due to their high activity and excellent selectivity under mild reaction conditions1,2,3,4. However, their practical applications are often hampered by two challenges: limited reusability and rapid deactivation of enzymes caused by concentrated reactants or toxic molecules5,6. For instance, Candida antarctica lipase B (CALB) has exhibited considerable catalytic potentials in olefin epoxidation, generating high-value epoxides7,8,9,10. Unfortunately, hydrogen peroxide (H2O2) as an oxidant needed in this reaction can frequently lead to enzyme deactivation, thus impeding the practical applications of this enzymatic reaction7.
To address the challenge of enzyme recycling, various immobilization methods have been extensively explored. These include physical adsorption11,12, covalent bonding13,14, cross-linking15,16, and encapsulation17,18,19. Despite significant advances, these methods occasionally suffer from either enzyme leaching or enzyme deactivation, resulting in limited success of their use in industrial processes20,21,22. One of key reason for the loss of catalytic efficacy is that the immobilized enzymes lose their preferred native microenvironment, where the presence of surrounding physiological water is necessary in maintaining the 3D structure of enzyme and therefore its functioinality23,24. To preserve the aqueous environment, researchers have designed micelles25,26, vesicles27,28,29, emulsion droplets30,31, and colloidosomes32,33 to accommodate enzymes. These approaches aim to host free enzymes within their native aqueous environments, simulating cells or cell membranes34,35,36,37,38,39. However, the inherent thermodynamic and mechanical instability of these soft structures often poses a challenge to their efficient recycling, particularly in relation to the continuous-flow biocatalysis processes desired by industry40,41.
Pickering emulsions, renowned for their exceptional stability and versatile design potentials, have proven to be an excellent platform for constructing cell-mimicking structures42,43,44,45,46,47,48. For example, Wu et al. demonstrated a method to grow silica-polymer shells around Pickering emulsion droplets, achieving improved mechanical stability and enabling continuous-flow enzymatic catalysis49. Very recently, our group also developed a straightforward approach to grow porous silica or metal-organic framework (MOF) shells around Pickering emulsion droplet surfaces. These shells mimick the selective permeability and protective functions in a somewhat analogous manner to cell membranes50,51. Despite these advances, achieving effective control over the transport of reactive and potentially harmful molecules such as H2O2, remains a significant challenge. To the best of our knowledge, up to now there has been no practically effective methods to immobilize enzymes for H2O2-involed epoxidation or oxidation.
Herein, inspired by the structure and function of cell membranes that can host enzymes and regulate the accessibility of enzyme towards poisonous molecules, we develop an enzyme immobilization strategy. This involves growing a porous, hydrophobicity-tunable, nanometer-thick silica shell at the water‒oil interface of Pickering emulsion droplets (Fig. 1a). The technique leads to the formation of a porous “interphase” between water and oil where enzymes are incorporated, referred to as enzyme@IP. Somewhat similar to enzymes residing in living cell-membranes, the enzymes incorporated within enzyme@IP enable control of the diffusion rate and local substrate concentrations due to the tunable thickness of “interphase” and hydrophobicity of nanopores. Additional three other aims of this design are: (i) allowing enzymes to remain in contact with physiological environment for maintaining their structure and thus native activity; (ii) The enzymes can facilely be accessed by the organic substrates in oil phase; And (iii) the enzyme@IP in form of robust cell-like capsules can be directly packed into a column reactor to achieve continuous-flow reactions. The effectiveness of our strategy is demonstrated by continuous-flow olefin epoxidation which often suffers from enzyme deactivation caused by H2O2. Long-term operational stabilization (800 h), 16-fold increase in catalysis efficiency relative to batch reaction, and 99% H2O2 utilization efficiency are achieved. The interesting catalytic effects arising from the “interphase” are studied here and unraveled.

a Schematic illustration of the fabrication of the enzyme@IP and the enzymatic olefin epoxidation process. b SEM image of enzyme@IP. c Magnified SEM image of a single enzyme@IP. d SEM image of a single deliberately broken enzyme@IP showing the hollow interior, within which water previously resided. e SEM image of the “interphase”. f Elemental mapping of “interphase” for the N element. g CLSM image of enzyme@IP with the enzyme labeled by FITC-I. h 3D CLSM image for a single enzyme@IP. i Fluorescence intensity profiles showing the FITC-labeled enzyme distribution within enzyme@IP.
Results
Fabrication and characterization of enzyme@IP
The fabrication of the targeted enzyme@IP mainly involves two steps: (i) a water-in-oil Pickering emulsion was produced by shearing a mixture comprising an enzyme-contained aqueous solution and n-octane, in the presence of partially hydrophobic silica nanospheres serving as solid emulsifiers (Supplementary Figs. 1, 2 present the characterizations of the solid emulsifier and the type of Pickering emulsions); (ii) An interfacial sol-gel procedure was conducted by adding an organosilane to form a porous shell around the Pickering emulsion droplet, generating a porous “interphase” between water and oil. Since enzyme molecules have a high affinity to the water‒oil interface, they are incorporated within the “interphase”.
After removing the external oil and internal water, the scanning electron microscopy (SEM) images of enzyme@IP show the formation of discrete cell-like capsules with a perfectly spherical structure and an average size of 45 ± 5 µm (Fig. 1b, c), which closely corresponds to the size of the Pickering emulsion droplets before solid shell formation (Supplementary Fig. 3). The porosity of enzyme@IP was determined with nitrogen sorption analyzer, where the pore size and specific surface area were measured to be ca.2.2 nm and 167 m2 g−1, respectively (Supplementary Fig. 4a). Since no additional surfactant templates were introduced during the synthesis process, the disordered nanopores of the solid shell were generated due to the sol-gel process50, and were slightly influenced by the surface coverage of solid emulsifiers on the emulsion droplets (Supplementary Fig. 4b). In the SEM image of a deliberately crushed sample, the hollow structure within which water previously resided is clearly seen (Fig. 1d). Based on the cross-sectional image, the shell thickness was estimated to be ~170 nm (Fig. 1e). Lipase CALB tends to adsorb onto the interface of water-in-oil Pickering emulsion droplets due to its amphiphilic nature and the adsorption effects of the hydrophobic solid emulsifier52,53. Therefore, once the porous hydrophobic silica shell layer is formed around the emulsion droplet, the enzyme incorporates itself within the enzyme@IP. This is also evidenced by elemental mapping analysis, since the N element from the enzyme was distributed in the porous shell region, as seen in the micrograph of the cross-section of an enzyme@IP (Fig. 1f). To better visualize the positioning of CALB at the enzyme@IP, we labeled CALB with fluorescein isothiocyanate I (FITC-I). The confocal laser scanning microscopy (CLSM) images showed that nearly all of the enzyme was localized within the enzyme@IP. No fluorescent signals were detected outside enzyme@IP (Figs. 1g and 1h). The fluorescence signal is much stronger at the edges of the enzyme@IP, further indicated that enzyme was predominantly located within the “interphase” (Fig. 1i).
The obtained enzyme@IP exhibited a good mechanical stability. After treatment with heating at 110 oC for 3 h, the enzyme@IP retained its original morphology without any discernible damage, whereas the Pickering emulsion droplets underwent destruction under comparable conditions (Supplementary Fig. 5). Furthermore, when the enzyme@IP was packed into a column reactor and was then subjected to continuous pumping of various polar organic solvents, such as ethyl acetate, alcohol, and N, N-dimethylformamide (DMF), at a flow rate of 5 mL h−1, the enzyme@IP preserved its structural integrity over a period of 30 min. This was notably different from the severe breakage observed in the case of Pickering emulsion droplets without the formation of the “interphase” (Supplementary Fig. 6). Notably, even under a higher flow pressure of 1 MPa and a flow rate of 50 mL h−1 (with ethyl acetate as the mobile phase), the morphology of enzyme@IP remained intact. There was no change in the height of the packed column even after a continuous-flow operation for 48 h (Supplementary Fig. 7). These findings provide a number of compelling evidences for the robust structural stability of the fabricated enzyme@IP.
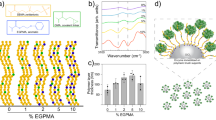
Interestingly, we also found that the partition coefficient of organosilane between the oil and water phases, together with the hydrolysis rate of organosilane jointly dictate the formation of “interphase”. Four organosilanes, trimethoxysilane (TMS), tetramethoxysilane (TMOS), trimethoxymethylsilane (MTMS), and trimethoxyoctylsilane (OTMS), with different log P values (P represents the partitioning coefficient of a compound between oil and water) were separately used to grow water‒oil “interphase”. In this process, hexylamine was acting as catalyst used at a the molar ratio of organosilane to hexylamine of 1:3. The hydrolytic rate of these organosilanes gradually decreases due to the change in molecular structure54. With the log P value being <0, TMS which is partially soluble in water is inclined to partition in the aqueous phase, diffusing from the oil phase into the water droplets. Due to the high hydrolytic activity of Si-H, TMS is rapidly hydrolyzed and the condensation of the resultant hydrolyzed species occurs within the water droplets. This generates numerous irregular silica nanoparticles inside the droplets (Fig. 2a1 and 2b). When the log P value of the organosilane increases to about 0 (e.g., TOMS), the organosilane is less soluble in water, and its hydrolysis and the subsequent condensation mainly take place at the oil-water interfaces, leading to the formation of so-called “interphase” between water and oil (enzyme@IP). However, owing to the high reactivity of TOMS with water55, the condensation rate cannot keep up with this rapid hydrolysis rate. The “interphase” was relatively fragile because of a lack of efficient linkages between hydrolyzed species, making this unamenable to the design of practical catalysis (Fig. 2a2 and 2b). Much more promisingly, when hydrophobic MTMS (containing a hydrophobic group ‒CH3, with a log P value between 0 and 1) was used, a well-defined enzyme@IP was generated (Fig. 2a3 and 2b). This is because the moderately hydrophobic methyl group of MTMS leads to a decreased hydrolysis rate. In this case, the hydrolysis can progress at a rate more comparable to that of the condensation rate at the interface. The elemental mappings revealed a uniform distribution of C elements throughout the “interphase” (Supplementary Fig. 8). However, when the hydrophobicity of organosilane increased even further, as with OTMS (with a log P value considerably >1), almost no “interphase” was formed (Fig. 2a4 and 2b). This can be explained by the fact that, at high log P values, organosilane was primarily distributed in the oil phase. In such a situation, it was not so easy for hydrolysis to take place at the water‒oil interface, thus making it more difficult to form an “interphase”.
Moreover, the thickness of the “interphase” was largely influenced by the amount of the applied organosilane. As the MTMS dosage was progressively increased from 0.15 to 0.30, 0.45, and 0.60 g mL−1 (with respect to the water phase), the “interphase” thickness increased correspondingly from 120 to 170, 320, and 620 nm, while the enzyme@IP continued to maintain a comparable shape and diameter (Fig. 2c). These results demonstrate that our approach allows controllable adjustment of the interphase’ thickness. We employed fluorescent dye Rhodamine B (RB) to assess the transport property of the enzyme@IP (Supplementary Fig. 9). The CLSM images demonstrated rapid diffusion of RB molecules toward the interior of the enzyme@IP, accompanied by an increase in fluorescence intensity with time, in the interior (Fig. 2d). The findings revealed a correlation between the molecular exchange rate and “interphase” thickness. Notably, an increase in “interphase” thickness led to a decrease in the rate of molecular exchange, primarily due to the increased tortuosity and extended diffusion paths. These findings can serve as a valuable guide for the design of enzyme@IP featuring controllable diffusion of molecules.

a SEM images of enzyme@IP synthesized with various organosilanes: a1 trimethoxysilane (TMS), a2 tetramethoxysilane (TMOS), a3 trimethoxymethylsilane (MTMS), and a4 trimethoxyoctylsilane (OTMS). b Log P values of various organosilanes (P represents the partition coefficient of compound between oil and water) and the corresponding SEM images of enzyme@IP synthesized using each organosilane. The shading represents the region where 0 <Log P <1. c SEM images showing the different shell thickness of enzyme@IP at various MTMS concentrations: c1 0.15 g mL−1, c2 0.3 g mL−1, c3 0.45 g mL−1, and c4 0.6 g mL−1. d Fluorescence intensity of rhodamine B diffusion into the interior of enzyme@IP over time at different shell thicknesses.
Enhanced CE in olefin epoxidations
We examined the catalytic performance of our enzyme@IP in the enzymatic epoxidation of olefins. Lipase CALB was selected as a catalyst to produce peracids in situ by catalyzing oxidation of carboxylic acids with H2O2 (Fig. 3a). Subsequently, based on the Prileshajev reaction mechanism, the peracids directly oxide olefin into epoxide. The generated carboxylic acid could be reused as an acyl donor in the next catalytic cycle7. Before testing continuous-flow reactions, we performed this enzymatic olefin epoxidation in a batch system. A conventional biphasic reaction involving an ethyl acetate phase containing cyclooctene and H2O2, and a phosphate buffered saline (PBS) phase containing CALB was examined. Ethyl acetate was chosen because it can be hydrolyzed by lipase CALB to generate a small amount of acetic acid, which then acts as a precursor for the in situ formation of peroxyacetic acid. Additionally, ethyl acetate is capable of dissolving both the olefin substrates and H2O2. After 10 h of reaction, the cyclooctene conversion reached only 20%, and the conversion remained virtually unchanged even with an extended reaction time (Fig. 3b). In the subsequent cycles, the conversion decreased down to 4% in the second cycle and then to 1% in the third cycle, indicating a significant decline in the enzyme activity (Supplementary Fig. 10). The reason for this is that the free enzyme rapidly loses activity at high H2O2 concentrations, as has been demonstrated previously7. Next, we replaced the batch biphasic reaction with an enzyme@IP-based continuous-flow system. Here, enzyme@IP was acting as solid catalyst which had been directly packed into a column reactor (equipped with a sand filter at the bottom of column). The reactant solution was continuously pumped into the inlet of the column reactor, and the product-rich stream was collected from the outlet. The setup and operational details are shown in Supplementary Fig. 11. Surprisingly, the cyclooctene conversion gradually increased, reaching up to 98%, while no by-products were detected (Fig. 3c). Based on the conversions obtained in the batch and flow systems, we estimated the CE values of CALB (CE, defined as μmol of substrate converted per milligram of enzyme per min, Fig. 3d). The continuous-flow system resulted in a CE value of 76 mol mg−1 min−1, which was ~16 times higher than that obtained for the batch reaction (4.8 mol mg−1 min−1). Furthermore, the H2O2 utilization efficiency (defined as the molar ratio of product to actual consumed H2O2) in the continuous-flow system was about 99%, exhibiting a 1.5-fold increase relative to the batch system (65.6 %, Fig. 3e). The method for determining residual H2O2 content after the reactions is detailed in Supplementary Method 3. The remarkable increase in CE in the continuous-flow system is partially attributed to the continuous-flow effect56,57,58,59. Essentially this effect maintains high reaction efficiency by making available a constant supply fresh reactants while reducing the accumulation of H2O2 at the “interphase”. This was further confirmed by an experiment, where suspending the flow for 48 h led to a remarkable decrease in olefin conversion to 65% upon restarting (Supplementary Fig. 12).

a Reaction network of olefin epoxidation using enzymatically in situ-generated peracids. b Conversion of cyclooctene over time in batch system. c Conversion of cyclooctene over time in the continuous-flow system. d Comparison of catalytic efficiency (CE) of Candida antarctica lipase B (CALB) in batch and flow systems. CE is defined as μmol of substrate converted per milligram of enzyme per min (i.e., μmol mg−1 min−1). e Comparison of H2O2 utilization efficiency in batch and flow systems. f Comparison of CE of CALB in flow systems with various “interphase” thicknesses. g Comparison of CE of CALB in flow systems involving enzyme@IP of various sizes. Batch reaction conditions: 6 mL aqueous enzyme solution (1.2 mg mL−1 of protein; pH = 7.4, 0.05 M), 3 mL ethyl acetate containing cyclooctene (0.5 mol L−1) and H2O2 (0.5 mol L−1), 35 oC, and 500 rpm. Continuous flow reaction conditions: 9 mL enzyme@IP (containing 7.2 mg of CALB), 0.5 mol L−1 cyclooctene, and 0.5 mol L−1 H2O2 in ethyl acetate as the mobile phase, with a flow rate of 1 mL h−1 for Fig. 3c and that of 3 mL h−1 for Fig. 3f, g. Residence time: 3 h (Fig. 3c); 1 h (Fig. 3f, g).
Next, we examined how the “interphase” thickness and the size of the enzyme@IP influenced its catalysis efficiency. Under the same reaction conditions, the catalytic performance of CALB in the continuous-flow system differed considerably for “interphase” systems with various differing thicknesses (Fig. 3f and Supplementary Fig. 13). With an increase in the “interphase” thickness from 120 to 170, 320, and then 620 nm, the cyclooctene conversions at steady state conditions decreased from 99% to 85%, 82%, and finally 72%, respectively. Concurrently, the CEs of CALB was estimated to decrease from 118 to 101, 97, and 85 mol mg−1 min−1, respectively. This decreasing trend was attributed to the reduced permeability resulting from the increased “interphase” thickness, which was consistent with our aforementioned fluorescent experimental results (Supplementary Fig. 9). Moreover, the size of enzyme@IP also affects the observed CE. As the diameter of enzyme@IP increased from 36 to 52, 72, and 103 µm (Supplementary Fig. 14), the cyclooctene conversions at steady state decreased from 89% to 85%, 77%, and 65%, respectively. Similarly, the CEs of CALB decreased from 106 to 101, 91, and 77 mol mg−1 min−1, respectively (Fig. 3g and Supplementary Fig. 15). The CE observed as a function of the enzyme@IP size can be explained by the fact that increasing the size reduces the “interphase” surface area of the enzyme@IP.
Enhanced enzyme stability at the water‒oil “interphase”
To further showcase the advantages of enzyme@IP, we conducted a comparative analysis of the catalytic performance among three distinct immobilized enzyme catalysts: water-enriched enzyme@IP (water is referred to as physiological water, pH=7.4, 0.05 M PBS), water-free enzyme@IP, and the traditional immobilized enzyme catalyst where CALB is adsorbed into the pores of porous silica microspheres (Fig. 4a). With the same enzyme loading in the three systems, the cyclooctene conversion in the water-enriched enzyme@IP remained >99% after 200 h of continuous-flow reaction (Fig. 4b). However, the water-free enzyme@IP retained its catalytic activity only in the first 20 h, but the conversion then significantly declined as the reaction progressed further. After 200 h of continuous flow, the cyclooctene conversion had decreased down to 58%. Similarly, for the system where the enzyme was adsorbed in the pores of silica microspheres (in the absence of physiological water environment), the cyclooctene conversion was only maintained at ~43% after 200 h. Based on the conversions seen after 200 h of continuous flow, the CEs of CALB in these three systems were estimated to be 76, 45, and 33 mol mg−1 min−1, respectively (Fig. 4c). These results revealed that the presence of physiological water within enzyme@IP plays an important role in enzyme stability and activity.

a Schematic of CALB in various environments: I water-enriched enzyme@IP, II water-free enzyme@IP, and III CALB adsorbed onto microspheres. b Conversion of enzymatic epoxidation over time in different flow systems. The reaction conditions are the same as those in Fig. 3c. c CE of CALB in different continuous-flow systems. d Fluorescence spectra of CALB in phosphate buffered saline (PBS) and ethyl acetate (EA), both containing 0.5 mol L−1 H2O2, before and after standing for 24 h. e Circular dichroism (CD) spectra of CALB after 24 h treatment in different PBS/EA mixtures, including PBS, a mixture of 33% (v/v) PBS in EA, and pure EA (all containing 0.5 mol L−1 H2O2). f Secondary structure changes of CALB after 24 h treatment in various PBS/EA mixtures, including α-helices, β-sheets, β-turns, and random coils. g Raman spectral intensity profiles showing the H2O2 concentration distributions from the exterior to interior. h Schematic illustration showing H2O2 diffusion through the hydrophobic pores of the “interphase”. i CE comparison of CALB with different “interphase” hydrophobicity. j Comparation of residual enzyme activity between hydrophilic and hydrophobic “interphase” after 12 h incubation in H2O2 solutions with varying concentrations from 0.5 to 1, 2 and 4 mol L−1. k Comparison of kinetic parameters (Km, Vmax and kcat) for enzymes incorporated in “interphase” with varying levels of hydrophobicity.
In order to demonstrate the promoting effect of the aqueous environment on enzyme stability, we compared the structural changes of enzymes after 24 h of treatment in a PBS and in an organic phase (ethyl acetate, EA), with both media containing 0.5 mol L−1 H2O2. It has previously been demonstrated that tertiary structural changes of proteins can be determined by monitoring the intrinsic fluorescence at 280 nm60. The fluorescence spectra of the CALB in the PBS and EA differed noticeably (Fig. 4d). The maximum emission wavelength remained virtually unchanged after being left in PBS for 24 h, whereas CALB exhibited a considerable red shift when exposed to the EA for the same duration. This observation was consistent with previous results that in an aqueous environment, enzymes maintain their conformation through interactions with water molecules23,24,61,62. In contrast, under identical high H2O2 concentration conditions, the absence of a sufficient protective hydration shell around enzymes in organic solvents renders them more susceptible to structural perturbation and deactivation. An additional experiment was conducted to further confirm this point by examining changes in circular dichroism (CD) spectra of CALB in three different solutions: PBS, a mixture containing 33% volume fractions of PBS in EA, and pure EA, respectively (all solutions contained 0.5 mol L−1 H2O2). Figure 4e shows the CD spectra of CALB after 24 h in these treatments. As the organic solvent content increased, the CD spectrum changed more dramatically. Furthermore, the secondary structure elements of CALB were analyzed (Fig. 4f, the method is provided in the Supplementary Method 2). Initially, in PBS, the secondary structure of CALB consisted of 4% α-helices, 49% β-sheets, 23% β-turns, and 26% random coils. However, after treatment with 33% (v/v) PBS in EA, significant alterations were observed in all secondary structure elements of CALB. Specifically, after treatment in pure EA, the structural composition shifted to 14% α-helices, 32% β-sheets, 21% β-turns, and 33% random coils. This evidence further underscores the importance of aqueous environments in maintaining enzyme structure and reaction activities.
In addition to the positive effect of the aqueous environment within the enzyme@IP on the enzyme stability, the hydrophobic pores in the “interphase” also play a crucial role in regulating H2O2 diffusion. Raman spectra were used to investigate the diffusion process of H2O2 from the oil phase to the “interphase” and water phase. In this experiment, H2O2, initially present in the oil phase, was observed to spontaneously enter the water phase. Within a short period of 45 s, H2O2 was detected at both the “interphase” and in the water phase (Supplementary Fig. 16). At a subsequent time of 60 s, the Raman intensity of H2O2 in the water phase gradually increased, indicating an accumulation of H2O2 within the enzyme@IP. Notably, after 75 s, the Raman intensity of H2O2 in the water phase (~42) consistently surpassed that observed at the “interphase” and in the oil phase region (~14 and ~17, respectively), and in particular with “interphase” showing the lowest intensity (Fig. 4g). These results demonstrate that the hydrophobic pores at the “interphase”, combined with the water‒oil microenvironment, significantly impact enzyme performance in olefin epoxidations. The hydrophobic methyl groups inside the pore channels make it difficult for H2O2 molecules to stay within them while diffusing from the oil phase (Fig. 4h). However, owing to the high solubility in the internal aqueous environment, H2O2 molecules easily pass through the channels and accumulated in the water environment. These effects ensure a lower local concentration of H2O2 at the “interphase”, protecting the enzyme from deactivation, despite ensuring a relatively higher concentration of H2O2 in the aqueous interior.
To further investigate the role of “interphase” hydrophobicity, four different “interphase” systems were prepared using various organosilanes: TMOS, a 1:1 molar mixture of TMOS and MTMS, MTMS, and a 1:1 molar mixture of MTMS and OTMS. The corresponding water contact angles of these “interphase” systems were measured to be 46o, 86o, 117o, and 129o, respectively (Fig. 4i). As the “interphase” hydrophobicity increased, the relative water content within enzyme@IP progressively decreased, from 64% to 45%, 15%, and 13%, respectively (Supplementary Fig. 17). This result suggests that a more hydrophobic interphase effectively reduces water retention within the porous of “interphase”, which not only maintains a suitable aqueous microenvironment for enzyme activity but also helps regulate the local concentration of H2O2, thereby minimizing undesired enzyme inactivation. We next examine how “interphase” hydrophobicity affects enzymatic catalytic performance. The enzyme@IP prepared with the more hydrophilic TMOS showed an average substrate conversion of only 31% during the first 30 h of continuous flow, which progressively decreased to <10% after 60 h of continuous operation (Supplementary Fig. 18), indicating poor enzyme stability likely due to higher local water content, resulting in excessive exposure to H2O2. In contrast, incorporation of MTMS into TMOS (1:1 molar ratio) increased the “interphase” hydrophobicity and lowered water content, allowing the conversion to remain around 86% after 60 h of continuous operation, demonstrating a marked improvement in enzyme performance. Notably, enzyme@IP formed with MTMS alone or with an MTMS/OTMS mixture maintained conversions above 98% over the same period. Based on the conversions after 60 h of continuous flow, the enzyme CEs were estimated to be 7, 66, 76 and 77 mol mg−1 min−1, respectively (Fig. 4i). These results highlight the critical role played by the hydrophobic “interphase” in retaining enzyme activity and promoting catalytic stability.
To further validate the protective effect of hydrophobic “interphases”, we compared the residual activity of enzymes immobilized within hydrophilic and hydrophobic “interphases”, which were prepared using TMOS and MTMS as the respective organosilanes. Both systems were pre-incubated in varying concentrations of H2O2 for 12 h and subsequently transferred to column reactors for continuous-flow enzymatic olefin epoxidation under the same reaction conditions to assess residual activity. As displayed in Fig. 4j and Supplementary Fig. 19, as the H2O2 concentration increased from 0.5 to 1, 2, and 4 mol L⁻¹, residual enzyme activity in the hydrophilic “interphase” decreased significantly from 52% to 45%, 35% and 15%, respectively. In contrast, enzymes in the hydrophobic “interphase” retained over 99% residual activity even at 4 mol L⁻¹ H2O2. These results highlight the critical role of “interphase” hydrophobicity in enhancing enzyme stability against H2O2-induced deactivation. In addition, to investigate how “interphase” hydrophobicity influences the interaction between enzyme active sites and substrates, we evaluated the kinetic parameters, including the apparent Michaelis−Menten constant (Km), maximal reaction rate (Vmax, representing the reaction rate when the enzyme active sites are saturated with substrate) and the apparent turnover number (kcat), under continuous-flow conditions, obtained at various levels of hydrophobicity of the “interphase”. Figure 4k and Supplementary Fig. 20 show that increasing the water contact angle of “interphase” from 46o to 86o and further to 117o and 129o resulted in a progressive decrease in Km (from 0.21 to 0.18, 0.13, and 0.12 M). This suggests an enhanced substrate affinity. Simultaneously, Vmax increased from 0.017 to 0.046, 0.074 and 0.091 M min⁻¹, and kcat rose from 0.47 × 103 to 1.28 × 103, 2.06 × 103, and 2.53 × 103 min⁻¹, indicating improved catalytic efficiency. These results confirm that the hydrophobic “interphase” not only protects enzyme stability but it also facilitates substrate accessibility and binding, thereby enhancing the overall enzymatic performance in olefin epoxidation.
Substrate scope and durability
Various olefins were examined, including cyclooctene, cyclohexene, styrene, 1-heptane, 1-octene, and α-pinene (Fig. 5). For all of the investigated substrates, the conversion gradually increased to >80% with time and then leveled off, reaching a steady state. In the case of cyclooctene, the continuous-flow system showed good stability after 800 h without substantial catalyst deactivation. Over this long period, the cyclooctene conversion was maintained at ~98.5% with a flow rate of 1 mL h−1 (Fig. 5a). The productivity of CALB, defined as the grams of accumulated epoxide produced per gram of enzyme, is calculated to be 6.9 × 103 g g−1, far exceeding the best results currently possible with the existing methods. Moreover, the enzyme@IP demonstrates superior H2O2 tolerance compared to these established methods (Supplementary Table 1)63,64,65,66,67. Furthermore, the morphology and size of the enzyme@IP remained virtually unchanged, as shown in the SEM image (Supplementary Fig. 21). The retention of CALB within the water–oil “interphase” after 120 h of continuous flow was determined to be 98%, based on N content measurements (Supplementary Table 2). Cyclohexene and styrene were also converted into the corresponding epoxide, with conversions exceeding 98% and 80%, respectively, maintained over a runtime period of 400 h (Fig. 5b, c). Due to the lower epoxidation reactivity of long-chain alkenes with peracids8, additional n-octanoic acid was introduced into the reaction feed to enhance the epoxidation performance. During 400 h of continuous operation, the conversions of 1-heptene and 1-octene reached 90–99% and 79–83%, respectively (Fig. 5d, e). Moreover, α-pinene was also efficiently converted into the corresponding products with conversions >99% over 400 h of stream time (Fig. 5f). For α-pinene, in addition to the introduction of n-octanoic acid, a saturated Na2HPO4 solution was used instead of PBS in the enzyme@IP to maintain an appropriate pH and prevent epoxide ring-opening, thereby ensuring high conversion and product stability. The products were easily obtained after the evaporation of ethyl acetate and were subsequently confirmed using gas chromatography/mass (GC-MS) spectrometry analysis and 1H NMR spectrometry (Supplementary Figs. 22, 23). These findings highlight the durability and high level of productivity of enzyme@IP. In addition, the scalability of enzyme@IP up to a 200 mL preparation scale and its storage stability were investigated, further demonstrating its suitability for practical applications (Supplementary Fig. 24). We next extended the enzyme@IP to a different type of enzymatic reaction, using aldo-keto reductase (AKR) to catalyze the enantioselective reduction of ketones (Supplementary Fig. 25). The reaction maintained an enantiomeric excess (e.e.) greater than 99% and a conversion above 85% over 450 h of continuous operation. The results further demonstrate the general applicability of the enzyme@IP system.

a cyclooctene, b cyclohexene, c styrene, d 1-heptane, e 1-octene, and f α-pinene. For Fig. 5a−c, the reaction was performed using enzyme@IP prepared with 5.1 mL PBS (0.05 M, pH 7.4) and 0.9 mL CALB solution (8 mg mL−1 of protein). The mobile phase consists of 0.5 mol L−1 olefin and 0.75 mol L−1 H2O2 in ethyl acetate, flow rate = 0.8 − 1 mL h−1, 35 oC. For Fig. 5d−f, the reaction conditions were the same as in Fig. 5a, except that the volume of enzyme@IP was doubled and 0.75 mol L⁻¹ n-octanoic acid was added to the mobile phase. Additionally, for Fig. 5f (α-pinene epoxidation), 0.5 g of Na2HPO4 was added into internal aqueous phase of enzyme@IP. Residence time: 3–3.8 h (Fig. 5a–c); 6–7.5 h (Fig. 5d–f).
Discussion
We have successfully developed an efficient method for immobilizing enzymes while maintaining high reactivity and long-term stability even under harsh reaction conditions. The key to the success of the method is the construction of a porous, hydrophobicity-tunable, and enzyme-incorporated “interphase” between water and oil phases, based on utilizing Pickering emulsions. On a crude level, this design mimics the cell membrane to some extent, where enzymes not only maintain their native activity due to contact with a physiological aqueous environment but also are able to easily access the substrates residing in the oil phase. The hydrophobic pores in the “interphase” allow for control over the local concentration of H2O2 at the “interphase” and thereby protect the enzyme from deactivation. More importantly, this “interphase” reaction system is well suited to use in continuous-flow catalysis, particularly in enzymatic olefin epoxidation. Employed as such, the method achieves CE values up to 16-fold higher than those of free enzymes in batch reactions. With demonstrated durability of 800 h under continuous operation and 99% H2O2 utilization efficiency, the technique represents a significantly more efficient and yet practically feasible enzymatic epoxidation method. Furthermore, by engineering different sizes and thicknesses of “interphase”, we were able to regulate their catalytic efficiency, even enabling controllable reactions at microscale. We believe our approach here establishes a robust platform for advancing the use of continuous-flow enzymatic reactions.
Methods
General procedure for preparing enzyme@IP
Typically, 1 mL of oil phase (n-octane) containing silica emulsifier (40 mg) was added into 2 mL enzyme solution (1.2 mg mL−1 of protein; pH = 7.4, 0.05 M). After vigorous shearing of the mixture at a speed of 3500 rpm for 1 min by a homogenizer, a W/O Pickering emulsion was generated. Afterwards, the resulting Pickering emulsion was dispersed into 16 mL n-octane containing a given amount of organosilane (e.g., MTMS, 4.4 mmol), as well as 13.2 mmol CH3(CH2)5NH2. The mixture was rotated on a rolling apparatus at 50 oC for 12 h. Then, the above n-octane phase was removed. The solid materials were washed with n-octane for several times. The materials so obtained were re-dispersed in n-octane for the subsequent packing into a column reactor.
Continuous-flow enzymatic olefin epoxidations
Typically, a suspension of enzyme@IP in n-octane was poured into a column reactor with an inner tube diameter of 2 cm. The column was equipped with a sand filter at its bottom, having a pore diameter ranging from 4.5 to 9 µm. Subsequently, a mobile phase solution consisting of 0.5 mol L−1 of olefins, 0.5 mol L−1 of H2O2, and 0.5 mol L−1 of n-octanoic acid (if required) dissolved in ethyl acetate was prepared. This solution was pumped through the inlet of the column reactor at a predetermined flow rate and allowed to pass through the reactor. The reactor column was maintained at a constant temperature of 35 oC. During the reaction, samples of the outflow were collected at suitable intervals for GC analysis. The presence of the product was further confirmed with GC-MS and 1H NMR.
Data availability
All data underlying this study are available from the corresponding author upon request. The data for Figs. 2–5 generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Bornscheuer, U. T. et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012).
Benkovic, S. J. & Hammes-Schiffer, S. A perspective on enzyme catalysis. Science 301, 1196–1202 (2003).
Sheldon, R. A. & Woodley, J. M. Role of biocatalysis in sustainable chemistry. Chem. Rev. 118, 801–838 (2018).
Devine, P. N. et al. Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem. 2, 409–421 (2018).
Bommarius, A. S. & Paye, M. F. Stabilizing biocatalysts. Chem. Soc. Rev. 42, 6534–6565 (2013).
Wu, S. K., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 60, 88–119 (2021).
Aouf, C. et al. The use of lipases as biocatalysts for the epoxidation of fatty acids and phenolic compounds. Green. Chem. 16, 1740–1754 (2014).
Li, K., Zou, H. B., Ettelaie, R., Zhang, J. X. & Yang, H. Q. Spatial localization of two enzymes at Pickering emulsion droplet interfaces for cascade reactions. Angew. Chem. Int. Ed. 62, e202300794 (2023).
Xu, Y., Khaw, N. R. B. J. & Li, Z. Efficient epoxidation of alkenes with hydrogen peroxide, lactone, and lipase. Green. Chem. 11, 2047–2051 (2009).
Biermann, U. et al. New syntheses with oils and fats as renewable raw materials for the chemical industry. Angew. Chem. Int. Ed. 39, 2206–2224 (2000).
Sánchez-Morán, H., Kaar, J. L. & Schwartz, D. K. Supra-biological performance of immobilized enzymes enabled by chaperone-like specific non-covalent interactions. Nat. Commun. 15, 2299 (2024).
Jin, Q. R., Jia, G. Q., Zhang, Y. M., Yang, Q. H. & Li, C. Hydrophobic surface induced activation of pseudomonas cepacia lipase immobilized into mesoporous silica. Langmuir 27, 12016–12024 (2011).
Xing, C. Y. et al. Enhancing enzyme activity by the modulation of covalent interactions in the confined channels of covalent organic frameworks. Angew. Chem. Int. Ed. 61, e202201378 (2022).
Zou, X. Q. et al. Investigating the effect of two-point surface attachment on enzyme stability and activity. J. Am. Chem. Soc. 140, 16560–16569 (2018).
Peschke, T. et al. Self-assembling all-enzyme hydrogels for flow biocatalysis. Angew. Chem. Int. Ed. 57, 17028–17032 (2018).
Lopez, S. et al. Cross-linked artificial enzyme crystals as heterogeneous catalysts for oxidation reactions. J. Am. Chem. Soc. 139, 17994–18002 (2017).
Chen, W. H., Vázquez-González, M., Zoabi, A., Abu-Reziq, R. & Willner, I. Biocatalytic cascades driven by enzymes encapsulated in metal–organic framework nanoparticles. Nat. Catal. 1, 689–695 (2018).
Chen, G. S., Huang, S. M., Ma, X. M., He, R. W. & Ouyang, G. Encapsulating and stabilizing enzymes using hydrogen-bonded organic frameworks. Nat. Protoc. 18, 2032–2050 (2023).
Zhang, Y. F. et al. Harnessing self-repairing and crystallization processes for effective enzyme encapsulation in covalent organic frameworks. J. Am. Chem. Soc. 145, 13469–13475 (2023).
Wang, K. Y. et al. Bioinspired framework catalysts: from enzyme immobilization to biomimetic catalysis. Chem. Rev. 123, 5347–5420 (2023).
DiCosimo, R., McAuliffe, J., Poulose, A. J. & Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 42, 6437–6474 (2013).
Maghraby, Y. R., El-Shabasy, R. M., Ibrahim, A. H. & Azzazy, H. M. E. Enzyme immobilization technologies and industrial applications. ACS Omega 8, 5184–5196 (2023).
Bellissent-Funel, M. C. et al. Water determines the structure and dynamics of proteins. Chem. Rev. 116, 7673–7697 (2016).
Ball, P. Water as an active constituent in cell biology. Chem. Rev. 108, 74–108 (2008).
Cortes-Clerget, M. et al. Bridging the gap between transition metal- and bio-catalysis via aqueous micellar catalysis. Nat. Commun. 10, 2169 (2019).
Zhang, N., Trépout, S., Chen, H. & Li, M. H. AIE polymer micelle/vesicle photocatalysts combined with native enzymes for aerobic photobiocatalysis. J. Am. Chem. Soc. 145, 288–299 (2023).
Che, H. L., Cao, S. P. & van Hest, J. C. M. Feedback-induced temporal control of “breathing” polymersomes to create self-adaptive nanoreactors. J. Am. Chem. Soc. 140, 5356–5359 (2018).
Wang, Y. X., Zhao, Q. C., Haag, R. & Wu, C. Z. Biocatalytic synthesis using self-assembled polymeric nano- and microreactors. Angew. Chem. Int. Ed. 61, e202213974 (2022).
Rifaie-Graham, O. et al. Photoswitchable gating of non-equilibrium enzymatic feedback in chemically communicating polymersome nanoreactors. Nat. Chem. 15, 110–118 (2023).
Ni, L., Yu, C., Wei, Q. B., Liu, D. M. & Qiu, J. S. Pickering emulsion catalysis: interfacial chemistry, catalyst design, challenges, and perspectives. Angew. Chem. Int. Ed. 61, e202115885 (2022).
Chang, F. Q., Vis, C. M., Ciptonugroho, W. & Bruijnincx, P. C. A. Recent developments in catalysis with Pickering emulsions. Green. Chem. 23, 2575–2594 (2021).
Tian, J. Q. et al. Interfacial energy-mediated bulk transport across artificial cell membranes. Nat. Chem. Eng. 1, 450–461 (2024).
Buddingh, B. C. & van Hest, J. C. M. Artificial cells: synthetic compartments with life-like functionality and adaptivity. Acc. Chem. Res. 50, 769–777 (2017).
Xu, C., Martin, N., Li, M. & Mann, S. Living material assembly of bacteriogenic protocells. Nature 609, 1029–1037 (2022).
Liu, X. L., Formanek, P., Voit, B. & Appelhans, D. Functional cellular mimics for the spatiotemporal control of multiple enzymatic cascade reactions. Angew. Chem. Int. Ed. 56, 16233–16238 (2017).
Guindani, C., da Silva, L. C., Cao, S. P., Ivanov, T. & Landfester, K. Synthetic cells: from simple bio-inspired modules to sophisticated integrated systems. Angew. Chem. Int. Ed. 61, e202110855 (2022).
Xu, Z., Hueckel, T., Irvine, W. T. M. & Sacanna, S. Transmembrane transport in inorganic colloidal cell-mimics. Nature 597, 220–224 (2021).
Podolsky, K. A. & Devaraj, N. K. Synthesis of lipid membranes for artificial cells. Nat. Rev. Chem. 5, 676–694 (2021).
Kurihara, K. et al. A recursive vesicle-based model protocell with a primitive model cell cycle. Nat. Commun. 6, 8352 (2015).
Amstad, E. Capsules: their past and opportunities for their future. ACS Macro Lett. 6, 841–847 (2017).
Vriezema, D. M. et al. Positional assembly of enzymes in polymersome nanoreactors for cascade reactions. Angew. Chem. Int. Ed. 46, 7378–7382 (2007).
Crossley, S., Faria, J., Shen, M. & Resasco, D. E. Solid nanoparticles that catalyze biofuel upgrade reactions at the water/oil interface. Science 327, 68–72 (2010).
Dedovets, D. et al. Multiphase microreactors based on liquid-liquid and gas-liquid dispersions stabilized by colloidal catalytic particles. Angew. Chem. Int. Ed. 61, e202107537 (2022).
Sun, Z. Y., Glebe, U., Charan, H., Böker, A. & Wu, C. Z. Enzyme-polymer conjugates as robust Pickering interfacial biocatalysts for efficient biotransformations and one-pot cascade reactions. Angew. Chem. Int. Ed. 57, 13810–13814 (2018).
Schoonen, L. & van Hest, J. C. M. Compartmentalization approaches in soft matter science: from nanoreactor development to organelle mimics. Adv. Mater. 28, 1109–1128 (2016).
Jiang, S., Caire da Silva, L., Ivanov, T., Mottola, M. & Landfester, K. Synthetic silica nano-organelles for regulation of cascade reactions in multi-compartmentalized systems. Angew. Chem. Int. Ed. 61, e202113784 (2022).
Deng, N. N., Yelleswarapu, M. & Huck, W. T. S. Monodisperse uni- and multicompartment liposomes. J. Am. Chem. Soc. 138, 7584–7591 (2016).
Jiang, H. et al. Engineering hybrid microgels as particulate emulsifiers for reversible Pickering emulsions. Chem. Sci. 13, 39–43 (2022).
Wu, H., Du, X. L., Meng, X. H., Qiu, D. & Qiao, Y. A three-tiered colloidosomal microreactor for continuous flow catalysis. Nat. Commun. 12, 6113 (2021).
Zhang, X. M. et al. Pickering emulsion-derived liquid−solid hybrid catalyst for bridging homogeneous and heterogeneous catalysis. J. Am. Chem. Soc. 141, 5220–5230 (2019).
Tian, D. P. et al. Pickering-droplet-derived MOF microreactors for continuous-flow biocatalysis with size selectivity. J. Am. Chem. Soc. 143, 16641–16652 (2021).
Wei, L. J., Zhang, M., Zhang, X. M., Xin, H. C. & Yang, H. Q. Pickering emulsion as an efficient platform for enzymatic reactions without stirring. ACS Sustain. Chem. Eng. 4, 6838–6843 (2016).
Plikat, C., Drews, A. & Ansorge-Schumacher, M. B. Effects of protein content on Pickering-assisted interfacial enzyme catalysis. ChemCatChem 14, e202200444 (2022).
Ciriminna, R., Sciortino, M., Alonzo, G., de Schrijver, A. & Pagliaro, M. From molecules to systems: sol-gel microencapsulation in silica-based materials. Chem. Rev. 111, 765–789 (2011).
Gonçalves, J. P. et al. Confining the sol-gel reaction at the water/oil interface: creating compartmentalized enzymatic nano-organelles for artificial cells. Angew. Chem. Int. Ed. 62, e202216966 (2023).
Zhang, M. et al. Compartmentalized droplets for continuous flow liquid-liquid interface catalysis. J. Am. Chem. Soc. 138, 10173–10183 (2016).
Zhang, M. et al. Ionic liquid droplet microreactor for catalysis reactions not at equilibrium. J. Am. Chem. Soc. 139, 17387–17396 (2017).
Zhang, M. et al. Pickering emulsion droplet-based biomimetic microreactors for continuous flow cascade reactions. Nat. Commun. 13, 475 (2022).
Zhang, M. et al. Pickering emulsion droplets and solid microspheres acting synergistically for continuous-flow cascade reactions. Nat. Catal. 7, 295–306 (2024).
Liu, Y., Chen, D. W. & Wang, S. H. Effect of sub- and super-critical CO2 pretreatment on conformation and catalytic properties evaluation of two commercial enzymes of CALB and lipase PS. J. Chem. Technol. Biotechnol. 88, 1750–1756 (2013).
Ingenbosch, K. N. et al. Effect of organic solvents on the structure and activity of a minimal lipase. J. Org. Chem. 87, 1669–1678 (2022).
Doukyu, N. & Ogino, H. Organic solvent-tolerant enzymes. Biochem. Eng. J. 48, 270–282 (2010).
Adomi, L. & Drews, A. Impact of Pickering emulsions composition on lipase catalyzed peroxyacetic acid synthesis. Chem. Ing. Tech. 96, 535–544 (2024).
Brandolese, A., Lamparelli, D. H., Pericàs, M. A. & Kleij, A. W. Synthesis of biorenewable terpene monomers using enzymatic epoxidation under heterogeneous batch and continuous flow conditions. ACS Sustain. Chem. Eng. 11, 4885–4893 (2023).
Su, W. et al. Improved efficiency of lipase-mediated epoxidation of α-pinene using H2O2 in single-phase systems. Mol. Catal. 508, 111585 (2021).
Meyer-Waßewitz, J., Holtmann, D., Ansorge-Schumacher, M. B., Kraume, M. & Drews, A. An organic-single-phase CSTR process for the chemo-enzymatic epoxidation of α-pinene enables high selectivity and productivity. Biochem. Eng. J. 126, 68–77 (2017).
Meyer, J. et al. A continuous single organic phase process for the lipase catalyzed synthesis of peroxy acids increases productivity. Eng. Life Sci. 17, 759–767 (2017).
Acknowledgements
This work is supported by the National Key Research and Development Program of China (2021YFC2101900, H.Q.Y.), the Natural Science Foundation of China (22332002, U24A20492, H.Q.Y.; 22472088, M.Z.), Fundamental Research Program of Shanxi Province (202303021211016, M.Z.), Young Scholars Development Program of Shanxi Research Institute of Huairou Laboratory (2023SY3007, M.Z.), Foundation of State Key Laboratory of Coal Conversion (grant number J24-25-909, M.Z.), and the Program for Sanjin Talent of Shanxi Province from M.Z.
Author information
Authors and Affiliations
Contributions
H.Q.Y. conceived and supervised the project; M.Z. executed the experiments and collected the data; M.R.W., Y.M.M., T.L., D.W.H., and H.M.Y. helped M.Z. to conduct part of the experiments; X.L.T. and H.T.L. helped to perform the Raman measurements; T.T.Z. helped to conduct CD spectroscopy testing. H.Q.Y. and M.Z. wrote the paper. All authors edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Wang, M., Mi, Y. et al. Enhanced enzyme stability at the interphase of water-oil for continuous-flow olefin epoxidation. Nat Commun 16, 8087 (2025). https://doi.org/10.1038/s41467-025-63476-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63476-1
