
Abstract
Integrating distinct functional reaction sites within a single photocatalyst offers a promising approach for enhancing the photocatalytic H2 evolution by water splitting. However, the synergy between the dual active sites is hindered by suboptimal electronic states arising from the uniform coordination environments. Here we demonstrate a strategy for enhancing the synergy between Pt single atoms and nanoparticles by modulating the coordination environment. The optimal boron doped catalyst with B-Pt-O asymmetric coordination achieves a H2 evolution rate of 627.6 mmol g-1h-1, with an apparent quantum efficiency of 98.4%. Experimental and theoretical analysis reveal that the asymmetric coordination structure redistributes the electron density of Pt cocatalysts, promoting charge carrier separation, optimizing the dissociation and adsorption-desorption of the intermediate H2O* and H* on the dual sites. The findings highlight the importance of asymmetric coordination facilitates the photogenerated carrier transfer and surface reactions for efficient photocatalytic H2 evolution.
Similar content being viewed by others

Introduction
The efficient generation and utilization of hydrogen (H2) remains a core objective in catalysis research, driven by the global pursuit of sustainable energy solutions1,2,3. Photocatalysis presents a promising pathway to convert solar energy into green H2 via water splitting4,5,6. However, the current photocatalytic performance is constrained by poor light harvesting, high photogenerated carrier recombination, slow proton reduction kinetics, and the high energy barrier for activating the water reactants7,8. Particularly, charge-separation transfer represents a kinetically suppressive yet crucial step that determines the efficiency of subsequent surface redox reactions9. Overcoming these obstacles is vital to unlocking the potential of photocatalysts. Modifying the semiconductor surface with cocatalysts is an effective strategy for enhancing photocatalytic performance, which improves the separation and transfer of charge carriers, introduces additional active sites for the redox reactions and lowers the activation energy10,11. Moreover, the size of the cocatalyst significantly affects the activation of reactant molecules and the desorption of product molecules, both of which are crucial for maintaining photocatalytic efficiency12.
In the photocatalytic H2 evolution reaction (HER), single atoms (SAs) cocatalysts have demonstrated high intrinsic activity, attributed to their nearly 100% atomic utilization efficiency and defined electronic and structural properties. Nevertheless, SAs often struggle to activate water molecules13,14,15, a challenging and potentially rate-determining step in HER16,17. Recent studies have increasingly focused on the role of nanoclusters/nanoparticles (NCs/NPs) in catalysis18,19,20, which combine the high atomic utilization efficiency with metallic properties for facilitating the activation of H2O molecules. A promising approach is integrating distinct functional reaction sites within a single photocatalyst to address the limitations of SAs cocatalysts, particularly for multi-step reactions such as photocatalytic water splitting. Some studies have highlighted the synergy between Pt SAs and Pt NC/NPs on oxide semiconductor supports in the photocatalytic H2 evolution (PHE) by water splitting14,21,22,23. In this system, the Pt SAs serve as H* reduction sites, while the adjacent Pt NC/NPs are the activation sites of H2O molecules. This spatially coupled heterogeneity creates dual active sites that complement the distinct functions, boosting the overall photocatalytic performance24. Besides, the electronic states of the NPs and SAs sites are crucial for the adsorption-desorption processes and the selectivity of reactants25,26,27. To further enhance the synergistic effect of the NP and SA sites in promoting the photocatalytic activity of SA&NP-based catalysts, it is essential to employ appropriate methods for modulating the electronic state of the dual sites.
The modulation of the local coordination environment of the active sites, encompassing the regulation of coordination atom type and coordination number, can optimize the electronic state and enhance the metal-support interactions (MSI)28,29, thereby improving the activity. In particular, introducing heteroatoms into the first coordination shell of the metal single-atom active site forms an asymmetric coordination structure, typically represented by the Ha-M-Oa bonding configuration (Ha: Heteroatom; M: metal centre atom; Original coordination atom)25,30. This structural modification induces a redistribution of electron density due to the disparity in electronegativity between the heteroatoms and original coordination atoms, optimizing the desorption and conversion of reactants in electron deficient regions31. Moreover, the hybridization of the d-p orbital between the non-metallic heteroatoms and metallic active centre atoms can significantly alter the spin polarization of the active centre and further affect the catalytic activity32,33. Hence, the electronic states of dual active sites can be optimized to achieve enhanced photocatalytic efficiency by strategically incorporating heteroatoms asymmetric coordination.
Here, we developed an SAs&NPs-based photocatalyst featuring an asymmetric coordination structure. Titanium dioxide (TiO2) was selected as the substrate for modification due to its stability and wide availability33,34. Meanwhile, Pt served as the cocatalyst for its intrinsic activity and electronic properties35,36. To construct the asymmetric coordination environment, boron (B) was introduced as a heteroatom, capitalizing on its well-documented role as an electron-modulating dopant with trivalent electronic characteristics(2s22p1)37, which can fine-tune the nucleophilicity of Pt for facilitating HER kinetics38,39,40. In the Pt-TiO2-based catalysts, Pt atoms typically coordinate with O atoms24,35. The substitution of O with B creates a B-Pt-O coordination structure, which is anticipated to enhance photocatalytic performance by modulating electronic properties. Thus, a specialized metal-organic framework (MOF) precursor featuring Ti vacancies (MIL-125(TiV)) was utilized to anchor Pt atoms. The B species were introduced by a ligand substitution strategy, and the B-Pt-O coordination was eventually formed in the TiO2 support by heat treatment. The resultant B doped and Pt-SAs&NPs loaded photocatalyst (PtSA/NP-TiO2-xBx) exhibits notable photocatalytic performance with a PHE rate of 627.6 mmol g−1h−1, and an apparent quantum efficiency (AQE) of 98.4% at 365 nm and a turnover frequency (TOF) of 33090 h−1, outperforming the B-free counterpart. X-ray absorption fine structure (XAFS) analysis provides direct evidence for the B-Pt-O asymmetric coordination structure. Detailed investigations reveal that the asymmetric coordination structure leads to a redistribution of electron density between Pt SAs and NPs for optimizing the dissociation of H2O* and adsorption-desorption of H* at the dual sites, and disrupts the balanced spin electrons to increasing spin polarization, for promoting the charge carrier separation and further enhancing the PHE reaction. The findings underscore the pivotal role of asymmetric coordination in optimizing dual-site catalysis and advancing PHE performance, providing a framework for developing cocatalyst semiconductor photocatalytic systems with high activity and stability.
Results and discussion
Morphology and structure characterizations of the photocatalysts
The PtSA/NP-TiO2-xBx photocatalyst was synthesized via a multistep process, as illustrated in Fig. 1a. Initially, the MOF precursor with TiV was prepared using a previously reported hydrothermal method10. B species was introduced into the MOF structure through a ligand exchange strategy41. Subsequently, Pt was incorporated into the framework by an ion exchange process to fill the TiV. A final thermal treatment collapsed the MOF structure, thus B and Pt were uniformly dispersed within the TiO2 lattice, yielding a B and Pt co-modified TiO2 photocatalyst (PtSA/NP-TiO2-xBx). A control sample, PtSA/NP-TiO2 (without B doping), was prepared following the same method, excluding the B ligand exchange step (refer to the Methods section for details).

a Synthetic process of PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx, PTA denotes 1,4-benzenedicarboxylic acid, TBOT denotes Titanium butoxide, 4-PBA denotes 4-Carboxyphenylboronic acid, H2PtCl6 denotes Chloroplatinic acid. b FT-IR spectra of MIL-125, B-MIL-125 and B-MIL-125(Pt). c XRD images of the photocatalysts. d Raman spectra of the photocatalysts. SEM images of e PtSA/NP-TiO2 and f PtSA/NP-TiO2-xBx. g TEM images of PtSA/NP-TiO2-xBx. h High-resolution TEM images of PtSA/NP-TiO2-xBx. Source data are provided as a Source Data file.
X-ray diffraction (XRD) analysis confirms the successful synthesis of the MOF precursor, with the diffraction patterns of MIL-125 aligning with previous reports10 (Supplementary Fig. 1a). The retention of characteristic peaks in the B-MIL-125(Pt) matches those of MIL-125 (Supplementary Fig. 1a), verifying the preserved MOF crystal structure. Fourier-transform infrared (FTIR) spectroscopy demonstrates the successful preparation of the B-modified MOF, as B-MIL-125 and B-MIL-125(Pt) exhibit three additional peaks (Fig. 1b), corresponding to B-O stretching vibrations (~1100 and ~1400 cm−1) and B-C antisymmetric stretching vibrations (~1600 cm−1)41,42. Thermogravimetric analysis (TGA) reveals that the mass loss of B-MIL-125(Pt) ends at 450 °C (Supplementary Fig. 1b), representing the removal of organic components and the incorporation of thermally stable B and Pt species in the TiO2. The XRD pattern (Fig. 1c) of the photocatalysts shows that pristine TiO2 consists of anatase and rutile phases, while all the modified samples (TiO2-xBx, PtSA/NP-TiO2, and PtSA/NP-TiO2-xBx) exhibit only anatase phases. This indicates that B and Pt atoms migrate into the TiO2 lattice during the synthesis, modifying the local and short-range crystal structure and resulting in phase alterations, as evidenced by the weakened characteristic peaks. Samples with various contents of B and Pt were also analyzed (Supplementary Fig. 2a and b, XRD patterns), and no discernible peaks correspond to B2O3 and Pt species, confirming the high dispersion of B and Pt. Raman spectroscopy further confirms the substitutional B doping within the TiO2 lattice (Fig. 1d), evidenced by the anatase characteristic peaks of TiO2-xBx and PtSA/NP-TiO2-xBx exhibiting a positive shift (~ 3 cm−1) compared to TiO2 and PtSA/NP-TiO2, with no impurity peaks detected. The absence of the peak at ~ 820 cm−1 rules out the substitution of Ti by B, consistent with the previous conclusion that B preferentially substitutes O in the TiO2 lattice43, as its atomic radius closely matches that of O and requires minimal energy compared to substituting Ti or occupying interstitial sites41,43. Notably, the characteristic peaks of PtSA/NP-TiO2 remain unchanged, consistent with the low content of Pt.
The morphology of the photocatalysts, as observed through scanning electron microscope (SEM) and transmission electron microscopy (TEM) (Fig. 1e and f and Supplementary Fig. 3a-d), shows that TiO2 and PtSA/NP-TiO2 retained the typical cake-like morphology of MIL-125, while TiO2-xBx and PtSA/NP-TiO2-xBx exhibit rough, square-like structures, which attributed to the atomic reconstruction during the high-temperature collapse of the MOF framework caused by B introduction. Brunauer-Emmett-Teller (BET) measurements reveal that PtSA/NP-TiO2-xBx exhibits a larger specific surface area (SSA, 121.3 m2g−1, Supplementary Fig. 4c) and higher porosity (0.30 ml g−1, Supplementary Fig. 4d) than that of PtSA/NP-TiO2 (SSA, 88.7 m2g−1; porosity, 0.18 ml g−1, Supplementary Fig. 4a and b), thanks to the rough morphology induced by B doping. Besides, the rough surface morphology improves the hydrophilicity of the catalysts, as evidenced by the lower water contact angles of TiO2-xBx and PtSA/NP-TiO2-xBx (Supplementary Fig. 5a-d), which can facilitate the H2O molecule adsorption for photocatalytic water splitting. TEM image (Fig. 1g) reveals that the TiO2 consists of nanoscale crystalline grains and the photocatalysts exhibit regular cake-like morphology (Supplementary Fig. 6). PtSA/NP-TiO2-xBx (Supplementary Fig. 8) shows a more porous and loosely packed structure compared to PtSA/NP-TiO2-x (Supplementary Fig. 7), without observable aggregation. High-resolution TEM identifies the lattice fringes with a spacing of ~0.35 nm (Fig. 1h), corresponding to the anatase phase of TiO2.
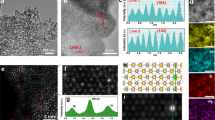
The aberration-corrected high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) was used to investigate the atomic structure of Pt species on the photocatalyst surface. As shown in Fig. 2a and Supplementary Fig. 9, Pt SAs and small Pt NPs are uniformly dispersed on both PtSA/NP-TiO2-xBx and PtSA/NP-TiO2 photocatalysts. The bright spots in the micrographs, attributed to the higher atomic number of Pt, correspond to Pt species. Specifically, Pt SAs and NPs are marked within dashed pink circles and boxes (Fig. 2a), respectively, confirming the coexistence of Pt SAs and NPs. Magnified views further illustrate the high dispersion of Pt SAs. The observed lattice fringes correspond to the arrangement of Ti atoms, and the individual bright spots along these fringes clearly represent Pt SAs (Fig. 2b), indicating that Pt atoms occupy the Ti sites. Atomic intensity profiles along the representative line corroborate this (Fig. 2d, Line 1 includes Ti and Pt atoms), the high-intensity signal of Pt contrasts with the lower intensity of Ti, confirming the presence of the Pt-O-Ti structural motif. The overall distribution and size statistics of Pt NPs are presented in Fig. 2c and Supplementary Fig. 10, showing a uniform dispersion with average grain sizes of 2.08 nm and 2.37 nm for PtSA/NP-TiO2-xBx and PtSA/NP-TiO2, respectively. The high-resolution image reveals a lattice spacing of 0.21 nm (Fig. 2c), consistent with the Pt (111) face, indicative of well-crystallized Pt NPs.

a, b HAADF-STEM images of PtSA/NP-TiO2-xBx. c HAADF-STEM, high-resolution image (inset), and the corresponding histogram of PtNP particle size distribution (inset) of the PtSA/NP-TiO2-xBx photocatalysts, the NPs size ranges from 1 to 3.8 nm, with ~ 80% of the NPs falling between 1.5 and 2.5 nm, yielding an average size of 2.08 nm. d The corresponding atomic intensity profiles of Line 1 marked in b. e DRIFTS spectra of CO adsorption on PtSA/NP-TiO2-xBx. f CO-DRIFTS spectra of PtSA-TiO2-xBx, PtSA/NP-TiO2-xBx and PtNP-TiO2-xBx, the peak at ~ 2096 cm⁻¹ is assigned to linear CO adsorption on Pt SAs of PtSA-TiO2-xBx, and the peaks at ~ 2055 cm⁻¹ and ~ 1825 cm⁻¹ are attributed to linear and bridged CO adsorption on Pt NPs of PtNP-TiO2-xBx, respectively. g Ti, O, B and Pt element mappings of PtSA/NP-TiO2-xBx. Source data are provided as a Source Data file.
The CO adsorption behavior of Pt species was investigated using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) to further confirm their dispersion properties and geometric configurations. As control samples, PtSA-TiO2-xBx and PtNP-TiO2-xBx containing only Pt SA and NP were synthesized, HAADF-STEM and XPS analyses verified the exclusive presence of SA and NP in the respective samples (refer to Supplementary Fig. 11 and 12). In the CO-DRIFTS spectrum of PtSA/NP-TiO2-xBx (Fig. 2e), the peaks at ~ 2172 cm⁻¹ and ~ 2117 cm−¹ correspond to the vibrational modes of saturated gas-phase CO molecules14. After adequate purging, three distinct peaks associated with stable CO adsorption on Pt species were observed. The peak at ~ 2103 cm⁻¹ is assigned to linear CO adsorption on Pt SAs44, consistent with the characteristic signal observed in the PtSA-TiO2-xBx (Fig. 2f). In contrast, the peaks at ~ 2063 cm⁻¹ and ~ 1829 cm⁻¹ are attributed to linear and bridged CO adsorption on Pt NPs45,46, respectively, in agreement with the spectral features of the PtNP-TiO2-xBx sample. The PtSA/NP-TiO2 exhibits similar three peaks with PtSA/NP-TiO2-xBx (Supplementary Fig. 13), further confirming the coexistence of SA and NP, which align with HAADF-STEM observations. Moreover, based on the deconvoluted peak areas in the CO-DRIFTS spectra (Supplementary Fig. 14) and the CO adsorption modes on Pt sites (Supplementary Fig. 15, CO adsorption on SA is directly proportional to the total SAs whereas NP is related only to the surface atoms44,47), the estimated atomic ratios of SA to NP in the PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx are approximately 1:16 and 1:13, respectively. (for detailed discussion, refer to Supplementary Figs. 14 and 15). Elemental mapping (Fig. 2g) confirms the homogeneous distribution of B and Pt across the surface of the PtSA/NP-TiO2-xBx.
Atomic coordination structure analysis
To better elucidate the chemical states and the local coordination environment of Pt cocatalysts, X-ray Photoelectron Spectroscopy (XPS) and X-ray Absorption Fine Structure (XAFS) analyses were conducted. The full spectrum of the photocatalysts (Supplementary Fig. 16a) verifies the presence of B, O, and Ti in the samples, with no detectable impurity peaks. The absence of Pt signal in the full spectrum can be attributed to its low content. Inductively coupled plasma (ICP) analysis of the optimal PtSA/NP-TiO2-xBx photocatalyst reveals that the contents of B and Pt are 1.56 wt.% and 0.37 wt.% (Supplementary Fig. 16b), respectively. XPS fine spectra were used to investigate the charge transfer dynamics between B and Pt. As shown in Fig. 3a, the B signal on TiO2-xBx and PtSA/NP-TiO2-xBx is directly detected. The binding energy of B 1 s appears at 192 eV, suggesting that B exists in a doped substitutional state without other impurity species37,43, consistent with the XRD results.

XPS spectra of a B 1s for TiO2-xBx and PtSA/NP-TiO2-xBx. b Pt 4f for PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx. c normalized Ti K-edge XANES spectra of TiO2, TiO2-xBx and reference Ti foil and TiB2. d normalized Pt L3-edge XANES spectra and e FT-EXAFS spectra of PtSA/NP-TiO2-xBx and reference Pt foil and PtO2. f Pt L3-edge fitted EXAFS spectra of R space for PtSA/NP-TiO2-xBx. g WT EXAFS contour plots for the k3-weighted Pt L3-edge EXAFS of Pt foil, PtO2 and PtSA/NP-TiO2-xBx. R denotes the radial distance (Å) and k denotes the photoelectron wave vector (Å−1) in XAFS analysis. Source data are provided as a Source Data file.
The Pt 4f fine spectrum (Fig. 3b) indicates that Ti 3s satellite peaks (~ 75 eV) overlap with Pt 4f peaks, complicating the deconvolution of Pt 4f curves for PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx48. During catalyst preparation, the Pt precursor (Pt4+) is partially reduced to form Ptδ+ species (Pt SA coordinated with the surface lattice O) and fully reduced to Pt0 (metallic Pt NP)49. The peak area ratio of Pt 4f5/2 to 4f7/2 was strictly fixed at 3:4, and the full width at half maximum (FWHM) of all Pt0 and Ptδ+ peaks was set uniformly during deconvolution48,49. In Fig. 3b, peaks at 70.84 and 74.41 eV are attributed to Pt0 4f7/2 and 4f5/2 signals of PtSA/NP-TiO2, respectively, while peaks at 72.45 and 76.65 eV correspond to Ptδ+ 4f7/2 and 4f5/2 signals. According to the principles of XPS binding energy measurements (Supplementary Fig. 17a), changes in the local valence electron density can affect the binding energy shift through two opposing mechanisms: (1) through variations in screening effect of the nuclear charge50 (Supplementary Fig. 17b, Mechanism 1), and (2) through Fermi level shifts associated with changes in carrier density51 (Supplementary Fig. 17c, Mechanism 2). Mechanism 1, which reflects the direct shielding of the nucleus by valence electrons, exerts a more dominant influence on both the direction and magnitude of binding energy shifts than the Fermi-level related band position changes described in Mechanism 2, particularly in metal-oxide catalysts49,50 (For more detailed discussion, refer to Supplementary Fig. 17). Based on this understanding, the observed increase in O 1s and Ti 2p binding energies of PtSA/NP-TiO2 relative to pristine TiO2 upon Pt loading (Supplementary Fig. 18) indicates a reduction in surface electron density, this electron depletion near the surface leads to the formation of upward band bending52 (For more detailed discussion, refer to Supplementary Fig. 19).
The reduced binding energy of Pt 4f (Pt0′: 70.63 and 74.07 eV; Ptδ′+: 72.24 and 76.37 eV) and further enhanced binding energy of the substrate elements (Ti, O, and B, Fig. 3a and Supplementary Fig. 18) for PtSA/NP-TiO2-xBx, as corroborated by the XPS results with different Pt and B contents (Supplementary Figs. 20 and 21), reflects more electrons transfer from the TiO2-xBx substrate to Pt cocatalysts by B doping, resulting in the formation of Ptδ′+ and Pt0′ species that are electron-rich and have a slightly lower valence state (For more detailed discussion on the valence state changes of the Pt species, refer to Supplementary Fig. 21), which further enhances electron depletion near the PtSA/NP-TiO2-xBx surface, leading to a more pronounced upward band bending (Supplementary Fig. 19). Moreover, this electron enrichment in Pt sites is expected to increase the 5d orbital occupation53, which is considered to play a critical role in determining the catalytic activity, thereby influencing the binding interactions of Pt sites and reaction intermediates54.
To further investigate the local coordination environment of Pt species in PtSA/NP-TiO2-xBx, element-selective XAFS measurements were performed. The Ti K-edge X-ray absorption near-edge structure (XANES) spectra show the absorption edge of TiO2-xBx is located between that of TiO2 and TiB2 (Fig. 3c), and the k2 weighted Fourier transform extended X-ray absorption fine structure (FT-EXAFS) reveals the longer bond length of TiO2-xBx than TiO2 (Supplementary Fig. 22), which further confirms that the B replaces O in TiO2-xBx since the length of Ti-B bond is longer than Ti-O bond43. Notably, the Ti K-edge FT-EXAFS spectra show the shorter bond length of TiO2-xBx than that of TiB2 (Supplementary Fig. 22). This is reasonable, as only a small fraction of O atoms in TiO2-xBx are replaced by B (contents of B is 1.56 wt.%), resulting in localized structural changes rather than the extensive bonding observed in TiB2, which means the overall lattice integrity maintains well while introducing the targeted modifications.
The Pt L3-edge XANES spectra reveal that PtSA/NP-TiO2-xBx exhibits a lower white line intensity than PtO2 but higher than Pt foil (Fig. 3d). This suggests that the Pt species in PtSA/NP-TiO2-xBx possess a valence range between 0 to +4, consistent with the coexistence of Pt0 and Ptδ+ states observed in the XPS results. FT-EXAFS spectral (Fig. 3e) analysis highlights the atomic dispersion of the Pt cocatalyst. Distinct features in the first and second coordination shells are observed: the signal around 2.2 Å in the second coordination shells corresponds to Pt-Pt bonds of Pt NPs. Notably, In the first coordination shell, the peak corresponding to Pt SAs resembles that of PtO2 but with a bond length elongated by 0.08 Å, which can be attributed to the partial substitution of O by B to form P-B/O (B-P-O) bonds as the length of Pt-B bond (2.18 Å) is longer than Pt-O bond (2.02 Å)38. In addition, Supplementary Fig. 23a illustrates that the k-space scattering pathways of PtSA/NP-TiO2-xBx diverge from those of PtO2, reflecting a distinct Pt coordination environment in PtSA/NP-TiO2-xBx relative to Pt-O. Based on the combined analysis of Raman, HAADF-STEM, XPS, and XAFS results, it can be inferred that Pt and B have respectively substituted for Ti and O in the TiO2 lattice, leading to the formation of B-Pt bonds. The proposed formation mechanism of the asymmetric B-Pt-O coordination structure is illustrated in Supplementary Fig. 24.
EXAFS fitting (Fig. 3f, Supplementary Table 2) reveals a coordination model involving Pt-O, Pt-B, and Pt-Pt structures. The fitted Pt-B coordination number of 1.5 suggests that one or two O atoms coordinated with Pt are replaced by B atoms, forming B-P-O motifs. Wavelet transform (WT) analysis provides further evidence for the Pt-B/O configuration. The WT intensity maxima for PtSA/NP-TiO2-xBx appears at R+α = 1.6 Å, shifting to lower k-values compared to PtO2 standards, indicating coordination between Pt and lighter atoms55, specifically B. Additionally, an intensity maximum at R+α = 2.2 Å can be observed, corresponding to the Pt-Pt coordination of the Pt NPs. The substantial electronegativity disparity between B and O facilitates the electron redistribution between B-Pt and O-Pt bonds within the B-P-O coordination, resulting in asymmetric electron density distribution. This further facilitates the electron transfer from the TiO2 to Pt cocatalysts, consistent with the XPS results.
Evaluation of photocatalytic activity
To evaluate the performance of the photocatalyst in water-splitting reactions, methanol served as a sacrificial agent, and the H2 evolution rate was monitored under 365 nm. As shown in Fig. 4a, pristine TiO2 generates H2 of 2.73 mmol g−1h−1, while TiO2-xBx exhibits a roughly sevenfold enhancement in PHE (10.31 mmol g−1h−1). Furthermore, the modification of Pt SAs and NPs yielded PtSA/NP-TiO2 photocatalyst with an enhanced PHE rate of 214.3 mmol g−1h−1 and AQE of 54.7%, which can be attributed to the high intrinsic activity of Pt cocatalysts and the synergistic effect between Pt SAs and NPs for HER15. Notably, B doping further elevates the PHE rate of PtSA/NP-TiO2-xBx to 627.6 mmol g−1h−1, representing 60.1-fold and 2.9-fold improvements over TiO2-x and PtSA/NP-TiO2, respectively, with an AQE of 98.4 % at 365 nm, nearing the theoretical maximum of unity (Fig. 4b and Supplementary Table 3). The TOF of PtSA/NP-TiO2-xBx is calculated to be 33090 h−1 (Supplementary Table 3), which is three times higher than that of PtSA/NP-TiO2 (10197 h−1). This notable photocatalytic performance is also evidenced by the continuous H2 bubble formation on the PtSA/NP-TiO2-xBx catalysts surface under irradiation (Supplementary Movie 1). To exclude the differences in Pt content due to B doping contributed to the observed PHE variations, the PHE rates of PtSA/NP-TiO2-xBx and PtSA/NP-TiO2 were compared with different Pt content. As shown in Fig. 4c, PtSA/NP-TiO2-xBx consistently exhibits higher PHE than PtSA/NP-TiO2 across all levels of Pt content and varying wavelengths (Supplementary Fig. 25), and the optimal PHE rate of PtSA/NP-TiO2-xBx is achieved with 0.4 wt.% Pt and 3 wt.% B (Supplementary Figs. 26 and 27). Coupled with the competitive performance evidenced in Fig. 4d, these results verify that B doping is the primary factor driving the enhancement in PHE activity. To confirm the synergy of SA and NP in the PHE reaction, PtSA-TiO2-xBx and PtNP-TiO2-xBx photocatalysts containing only SA and only NP were synthesized (Fig.2f, Supplementary Figs. 11 and 12) and evaluated the photocatalytic performance, respectively. The optimal PtSA-TiO2-xBx and PtNP-TiO2-xBx samples exhibit PHE rates of 136.7 and 302.7 mmol g−1h−1 (Supplementary Fig. 28), respectively, with corresponding TOF values of 14815 and 5789 h−1 (Supplementary Fig. 29), both of which were lower than those of PtSA/NP-TiO2-xBx, confirming the synergistic effect between SA and NP in enhancing HER performance.

a Photocatalytic H2 evolution rates and b AQE for H2 evolution of the photocatalysts, the error bars are defined as standard deviation, and the center of each error bar represents the mean of three independent measurements (n = 3). c Photocatalytic H2 evolution rates of PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx with different Pt content. d The PHE summary of the previously reported Pt-modified-photocatalysts and M-TiO2-based (M stands for metal) photocatalysts (reaction temperature ≥ 40 °C), the numbered data of photocatalysts are as follows: 1) Pt(1.0%)/TiO2, 2) Cu(0.01%)/Pt(1.0%)/TiO2, 3) Ni(0.01%)/Pt(1.0%)/TiO2, 4) Pd(0.01%)/Pt(1.0%)/TiO2, 5) PtCu-TiO2, 6) PtSA-TiO2, 7) Pt/TiO2-101, 8) 1 wt %Pt-TiO2, 9) 1 wt% Pt-g-C3N4, 10) 1%Pt/ZIS, 11) 1%Pt-TiO2, 12) 1%Ag/TiO2, 13) CuSA-TiO2, 14) 1%Au-1%Ag/TiO2, 15) Ag@SiO2@TiO2NS/Au, 16) Rh/TiO2, 17) 1%Au/TiO2, 18) InSA/TiO2. (summarized in detail in Supplementary Table 6). e PHE cycling stability of PtSA/NP-TiO2-xBx. Source data are provided as a Source Data file.
The PHE rates of PtSA/NP-TiO2-xBx with different methanol/water ratios are shown in Supplementary Fig. 30, highlighting the advantage of a 66.7% methanol proportion in enhancing photocatalytic H2 evolution. Isotope experiments were conducted by replacing water and methanol with heavy water (D2O) and deuterated methanol (CD3OD) with same ratio, respectively, proving water is the primary hydrogen source of photocatalytic H2 evolution (for more detailed discussion, refer to Supplementary Fig. 31). In addition, infrared thermograms during the photocatalytic reaction process and the PHE rates at different temperatures (20–70 °C) are presented in Supplementary Figs. 28 and 29, revealing the advantages of photocatalytic reaction under 40 °C. (For more detailed discussion, refer to Supplementary Figs. 32 and 33).
Furthermore, to verify the importance of B-Pt-O coordination for photocatalytic H2 evolution, a control sample PtSA/NP@TiO2-xBx (contains B doping and P loading but without B-P coordination) was synthesized and compared with PtSA/NP-TiO2-xBx. HAADF-STEM (Supplementary Fig. 34) imaging confirmed the coexistence of Pt SA and NP in PtSA/NP@TiO2-xBx, while XPS (Supplementary Fig. 35) and XAFS (Supplementary Fig. 36) analyses verified the exclusive presence of Pt-O coordination without Pt-B bonding (for more details, please refer to Supplementary Figs. 35 and 36, and Supplementary Table 4). The optimized PtSA/NP@TiO2-xBx sample exhibits a lower PHE rate of 335.4 mmol g−1h−1 (Supplementary Fig. 37) compared to PtSA/NP-TiO2-xBx, further confirming the critical role of B-Pt-O coordination in promoting photocatalytic H2 evolution.
Further comparisons with various metal cocatalysts of equivalent content reveal that B-doped M-TiO2 (M: Ag, Ru, Ir, Cu, Fe, Co, Ni, Zn) photocatalysts exhibit discrepant PHE rates (Supplementary Fig. 38), with Pt cocatalysts standing out due to its high intrinsic activity and the enhanced synergistic effects between Pt SAs and NPs facilitated by B doping. Long-term stability measurements conducted during cyclic water-splitting experiments show that the PHE rate remains stable over 40 hours (Fig. 4e). Post-reaction structural analysis using XRD, TEM, and HAADF-STEM further verify the stability of PtSA/NP-TiO2-xBx (Supplementary Fig. 39). The crystalline structure and surface lattice morphology of PtSA/NP-TiO2-xBx show no observable changes compared to the pre-reaction samples (Supplementary Fig. 39a-c). Line scan profiles and HAADF-STEM analysis confirm that Pt SAs remain well-dispersed on TiO2 (Supplementary Fig. 39d-e), with no observable agglomeration of Pt SAs or changes in the grain size of Pt NPs (Supplementary Fig. 39f). The element mapping also confirms the remaining dispersion of B and Pt after the photocatalytic reaction (Supplementary Fig. 39g). These analyses demonstrate the stability and strong anchoring of Pt SAs with B-Pt-O coordination on the TiO2 support, attributable to the porosity and unsaturated bonding of the MIL-125(TiV) framework, which effectively stabilizes Pt cocatalysts.
Charge transfer-separation efficiency of the photocatalysts
Photocatalysts Ultraviolet-visible (UV-vis) spectroscopy was employed to analyze the light absorption properties of the photocatalysts. As shown in Fig. 5a, PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx exhibit stronger absorption intensities beyond 400 nm compared to TiO2 and TiO2-xBx, attributed to the darkened colouration of the catalysts induced by Pt NPs. Furthermore, the red-shifted absorption edges observed for TiO2-xBx and PtSA/NP-TiO2-xBx, compared to TiO2 and PtSA/NP-TiO2, suggest that B doping effectively narrows the bandgap of TiO2. The Tauc plots further corroborate this (Fig. 5b). The calculated energy band positions from XPS valence band spectra (Supplementary Fig. 40), as depicted in Supplementary Fig. 41, indicate that TiO2-xBx and PtSA/NP-TiO2-xBx exhibit narrower bandgaps, and the estimated positions of the conduction band minimum (CBM) and valence band maximum (VBM) of PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx both moved upward, which is consistent with the XPS analysis of the upward band bending (Supplementary Fig. 19). These findings conform to the result that PtSA/NP-TiO2-xBx exhibits higher PHE efficiency under various wavelengths than PtSA/NP-TiO2.

a UV-vis spectra and b Tauc diagrams converted from UV-vis of TiO2, TiO2-xBx, PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx. c M-H hysteresis of TiO2, PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx. d Transient photocurrent responses and e EIS plots of TiO2, TiO2-xBx, PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx. f PL and g TRPL spectra of the photocatalysts. Source data are provided as a Source Data file.
Figure 5c presents the magnetic moment versus magnetic field (M-H) curves for TiO2, PtSA/NP-TiO2, and PtSA/NP-TiO2-xBx catalysts, measured in a magnetic field range from −3 to 3 T at 300 K. It can be seen that TiO2 exhibits a typical M-H curve with a saturation magnetization of 0.011 emu g−1, which is consistent with the previous reports56. In contrast, both PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx show significantly improved saturation magnetization compared to TiO2, attributed to the changes in electronic structure induced by the two-dimensional quantum confinement effect from Pt species, particularly the enhanced spin polarization57,58. Furthermore, PtSA/NP-TiO2-xBx exhibits a higher saturation magnetization (0.024 emu g−1) than PtSA/NP-TiO2 (0.020 emu g−1), suggesting that the B-Pt-O asymmetric coordination further strengthens the spin polarization of Pt. The disparity between spin-up and spin-down charge densities caused by spin polarization can enhance the charge separation59.
To investigate the carrier dynamics associated with the potential B-Pt-O interfacial sites, the photocurrent response and electrochemical impedance spectroscopy (EIS) were performed. The results indicate that the PtSA/NP-TiO2-xBx photocatalyst demonstrates the highest photocurrent density (Fig. 5d) and the smallest EIS semicircular radius (Fig. 5e) in the four samples, which can be attributed to the improved charge separation efficiency and reduced interfacial charge transfer resistance. Additionally, the PtSA/NP-TiO2-xBx photocatalyst shows notable stability and reproducibility, maintaining consistent photocurrent response over 10 on-off cycles. Photoluminescence (PL) and Time-Resolved PL (TRPL) results further substantiate the role of B in promoting charge carrier separation and transfer. As illustrated in Fig. 5f, pristine TiO2 exhibits a high PL intensity due to the significant charge carrier recombination and slow charge transfer. Upon introduction of B and Pt, PtSA/NP-TiO2-xBx exhibits a significant reduction in PL intensity, signifying the suppression of photogenerated electron-hole recombination. TRPL lifetime directly reflects the overall decay rate of singlet excitons and involves two competing processes: radiative recombination (fluorescence emission) or non-radiative transfer (exciton dissociation)60. Based on the fact of effective carrier separation confirmed by photocurrent and EIS results, the shortened lifetime of PtSA/NP-TiO2-xBx (Fig. 5g and Supplementary Table 5) suggests enhanced surface-mediated exciton dissociation of non-radiative processes, where photogenerated electrons and holes are rapidly captured by Pt species and the TiO2-xBx substrate61,62, respectively, which can be attributed to the B-Pt-O interfacial electric field. These findings highlight the role of B-Pt-O units in enhancing photocatalytic performance by promoting efficient charge carrier dynamics.
The separation and transfer of photogenerated carriers is a complex process of multiple time and space scales63, thus, electron transfer mechanisms of PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx during photocatalysis were studied using surface photovoltage (SPV), in-situ irradiated XPS (ISI-XPS) and in-situ irradiated Electron Paramagnetic Resonance (EPR) analysis. The SPV of the samples was measured using a Kelvin probe system (Supplementary Fig. 42), which is rigidly defined as the illumination-induced changes in the surface potential (Vs), and it is directly related to the initial transient separation of photogenerated charge carriers by the built-in electric field in the space charge region (SCR)52,64. Under dark conditions, PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx exhibit Vs of ~ −30 mV and ~ −71 mV, respectively, indicating the PtSA/NP-TiO2-xBx sample shows a more pronounced band bending. Upon illumination, as the light intensity increases, the Vs eventually stabilizes at a constant positive value (PtSA/NP-TiO2 for ~54 mV and PtSA/NP-TiO2-xBx for ~43 mV), indicating that the surface band bending has approached complete flattening64 (For more detailed discussion, refer to Supplementary Figs. 43 and 44 in Supplementary Information). This steady-state value corresponds to the saturated surface photovoltage (SPVsat)52. Previous studies have established that a positive SPV value corresponds to upward band bending, and the magnitude of the SPVsat value reflects the extent of band bending65. As shown in Supplementary Fig. 42, PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx exhibit SPVsat of ~ 84.4 mV and ~ 114.6 mV, respectively. This indicates that the upward surface band bending for the two samples, and that the band bending is more pronounced in the PtSA/NP-TiO2-xBx. These observations are consistent with the XPS analysis presented above.
ISI-XPS offers an approach for probing the electron transfer pathways during the photocatalytic process, which reflects the steady-state electronic structure that emerges after prolonged light exposure66. Figure 6a–c and Supplementary Fig. 45 exhibit a decreased binding energy in the Ptδ+ 4f and an increase in the substrate elements (Ti 2p, O 1s, and B 1s) under 30 minutes of continuous illumination, indicating that photogenerated electrons transfer to Pt sites while photogenerated holes migrate toward the TiO2 substrate. The shift in the Ptδ+ 4f binding energy under illumination is greater for PtSA/NP-TiO2-xBx (0.32–0.41 eV) than for PtSA/NP-TiO2 (0.19–0.24 eV). This further confirms that B-Pt-O coordination structure promots more efficient photogenerated electron transfer from the TiO2-xBx substrate to the Pt active sites under light irradiation, which further disrupts the equilibrium charge distribution within SCR, inducing stronger upward band bending.

ISI-XPS for a Pt 4f of PtSA/NP-TiO2, b Pt 4f of PtSA/NP-TiO2-xBx and c B 1s of PtSA/NP-TiO2-xBx. d Free energy diagrams for the water-dissociation process for Pt NPs and e Hydrogen adsorption for Pt SAs over the PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx. f Charge density difference maps of PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx. Electron accumulation is purple, and depletion is gray, and the green, cyan, white and yellow spheres depict the B, Pt, Ti and O atoms, respectively. g Project density of states for PtSA/NP-TiO2-xBx. h Mechanism of spin polarization for improved charge separation and synergistic PHE over the PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx photocatalysts. Source data are provided as a Source Data file.
Therefore, by combining SPV and ISI-XPS analyses, the mechanism of carrier separation and transfer under illumination can be effectively elucidated, as well as the corresponding changes in the surface band structure (Supplementary Fig. 46). At the onset of illumination, photogenerated electron-hole pairs are generated within the SCR, leading to a temporary flattening of the upward band bending, as revealed by SPV measurements. As illumination continues, photogenerated electrons are transferred to the Pt active sites while photogenerated holes accumulate on the TiO2 substrate, further disrupting the equilibrium charge distribution within SCR. This process leads to the re-establishment of a more pronounced upward band bending, as evidenced by ISI-XPS.
In-situ irradiated EPR measurements provide further evidence for this. As shown in Supplementary Fig. 47, PtSA/NP-TiO2 exhibits a prominent signal for the O vacancy (OV) at g = 2.002 under dark conditions. In contrast, the signal shifts to g = 2.005 in PtSA/NP-TiO2-xBx, which is attributed to the defect states introduced by B doping, including more OV and the covalent [BTi3]• structure43. Upon irradiation, both PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx exhibit a weak EPR signal at g = 2.01, corresponding to the Ti4+O2-Ti4+O•- units67. This structure acts as a hole-trapping site, stabilizing photogenerated holes and reducing their free transfer, thus slowing the recombination between photogenerated holes and electrons at the interface68. Notably, the EPR signal at g = 2.005 in PtSA/NP-TiO2-xBx is enhanced during irradiation and returns to its original state once the light source is removed. This demonstrates an increase in the number of unpaired electrons during illumination, further verifying that the transfer of photogenerated electrons can be promoted by B doping.
Mechanistic insights into the asymmetric coordination
Density functional theory (DFT) calculations have been performed to gain detailed and deep insights into the roles of the B-Pt-O coordination on the Pt-SA/NP dual sites. Both Pt SA and NP were placed in adjacent positions on the TiO2 surface, with B atoms substituted for O atoms and identified as stable structure (Supplementary Fig. 48). B atoms coordinated with Pt SA to form a stable 2-coordinate structure, as confirmed by the XAFS fitting results. Pt NP and SA were identified as the H2O activation and H2 desorption sites during model optimization, respectively, which aligns with ISI-XPS results and previous reports14,21. Therefore, the role of B can be determined by directly comparing the dissociation of H2O* at the Pt NP sites, along with the adsorption-desorption of H* at the Pt SA sites in PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx catalysts. In the water-splitting reactions, the dissociation of H2O molecules through the cleavage of the covalent HO-H bond is a critical and rate-limiting step. Figure 6d shows the optimized configurations of H2O, H2O*, and OH- + H* at the NP sites. H2O molecules are stably adsorbed at the Pt NP sites of both PtSA/NP-TiO2 and PtSA/NP-TiO2-xBx, forming a transition state of H2O*. However, the dissociation of H2O* into OH- + H* shows a more pronounced uphill process on PtSA/NP-TiO2 compared to PtSA/NP-TiO2-xBx. Specifically, at the Pt NP site, the energy barrier for H2O* dissociation is 0.72 eV for PtSA/NP-TiO2 and 0.23 eV for PtSA/NP-TiO2-xBx, indicating that B doping efficiently enhances the dissociation of H2O molecules. Additionally, the Gibbs free energy of H* adsorption/desorption is −0.67 eV for Pt-SA in PtSA/NP-TiO2-xBx and −0.04 eV in PtSA/NP-TiO2-xBx (Fig. 6e), suggesting the easier H* desorption on Pt-SA of PtSA/NP-TiO2-xBx than on PtSA/NP-TiO2, where the adsorption/desorption process is closer to equilibrium. These results demonstrate that B enhances the synergy between the dual sites, facilitating both the H2O* dissociation at the Pt NP and H* desorption at the Pt SA.
According to molecular orbital theory, the interaction between the H* intermediate and the Pt active site is governed by orbital coupling between the H 1s orbital and the Pt 5d orbital (Pt-Hads), leading to the formation of bonding and antibonding states. The electronic occupation of the Pt site determines the anti-bonding orbital filling of Pt-Hads, thereby influencing the catalytic activity53. However, due to strong interactions between Pt atoms and the TiO2, electrons tend to transfer from the Pt sites to the TiO2 substrate, creating electron-deficient Pt centers, which strengthens the Pt-Hads bond69, hindering the desorption of adsorbed H* and ultimately suppressing photocatalytic H2 evolution (Supplementary Fig. 49). This issue was alleviated by constructing the B-Pt-O asymmetric coordination through B doping, the charge density difference maps (Fig. 6f) reveal an asymmetric distribution of electron density on the Pt species, with more localized electron density at Pt sites of PtSA/NP-TiO2-xBx, suggesting the enhanced MSI for promoting the electron transfer from TiO2-xBx substrate to Pt sites and enriches the Pt 5d orbitals filling (consistent with the XPS results), increasing the occupation of the antibonding orbitals in the Pt-Hads. Therefore, the Pt-Hads bond strength is weakened, lowering the H* adsorption energy and facilitating the desorption of H* from the Pt active sites53,70 (Supplementary Fig. 49). This modulation of the electronic structure thus enhances the photocatalytic H2 evolution performance.
To understand the factors influencing the synergy, the project density of states (PDOS) of catalysts were analyzed. Figure 6g and Supplementary Fig. 50 illustrate the PDOS of PtSA/NP-TiO2-xBx and PtSA/NP-TiO2 photocatalysts. Obviously, the conduction band (CB) and valence band (VB) are primarily derived from Ti 3 d and O 2p orbitals, respectively. The introduction of B narrows the band gap of TiO2 (2.60 eV of PtSA/NP-TiO2 and 2.47 eV of PtSA/NP-TiO2-xBx), consistent with the above UV-Vis results (Fig. 5a). Additionally, a spin-up density of states of Pt-d up near the Fermi level in PtSA/NP-TiO2 can be observed (Supplementary Fig. 50), primarily ascribed to the hybridization of the d-p orbitals of Pt and O, which leads to a spin-polarized electron alignment, consistent with the M-H results. Notably, the spin-up state density of Pt-d up in PtSA/NP-TiO2-xBx (Fig. 6g) is much higher than PtSA/NP-TiO2 (Supplementary Fig. 50) due to the changed d-p orbital hybridization of Pt and B/O. This suggests the asymmetric B-Pt-O coordination disturbs the balance between spin-up and spin-down electrons of Pt, resulting in electron polarization and increasing the availability of spin-up electrons, disclosing its efficient charge separation. The associated mechanism is illuminated (Fig. 6h) to elucidate the impact of spin polarization on charge separation. Under light irradiation, spin-up electrons are excited into the CBM while the corresponding spin-up holes remain in the VBM. Due to the spin-orbit coupling (SOC) and hyperfine interactions (HFI), the excited spin-up electrons flip to spin-down while the spin-up holes maintain their orientation59. However, some of the holes with the opposite spin state can still recombine spontaneously with the flipped electrons. Stemming from the asymmetric coordination structure disturbs the balance between spin-up and spin-down electrons to increase the ratio of spin-up electrons, resulting in more spin-mismatched electrons and holes after the spin flipping, which helps suppress charge recombination71. This illustrates that higher spin polarization caused by the B-Pt-O asymmetric coordination enhances charge separation, which is consistent with the above PL results (Fig. 5f).
Therefore, the introduction of B enhances the efficiency of PHE through two main mechanisms: surface reactions and charge separation. First, B doping strengthens the MSI, facilitating the transfer of electrons from TiO2 to the Pt species. The B-Pt-O asymmetric coordination structure leads to a redistribution of electron density between Pt SAs and NPs, influencing the dissociation and desorption of reactants at the dual sites, thereby optimizing the H2 evolution reaction by water splitting. Second, the B-Pt-O asymmetric coordination disrupts the balance between spin-up and spin-down electrons on the Pt species, leading to increased spin polarization and more spin-up electrons. This promotes the charge carrier separation and suppresses the recombination. As a result, the synergy between the Pt SAs and NPs is enhanced to boost the PHE reaction (Fig. 6h).
In summary, the B-doped photocatalysts featuring the co-existence of Pt SAs and NPs were successfully synthesized by anchoring Pt species in TiV-containing MOF precursors and combining with a B-containing ligand substitution strategy. The optimal PtSA/NP-TiO2-xBx photocatalyst exhibits notable photocatalytic performance (PHE rate for 627.6 mmol g−1h−1, AQE for 98.4%, and TOF value for 33090 h−1), all of which are higher than that of the B-free counterpart. The experimental results confirmed that B doping modified the morphology and enhanced the light absorption of the photocatalyst. XAFS spectroscopy confirms the presence of B-P-O asymmetric coordination in PtSA/NP-TiO2-xBx, which enhances the MSI and promotes charge carrier separation and transport. In-situ characterization and theoretical analysis revealed that the B-Pt-O asymmetric coordination structure leads to a redistribution of electron density between Pt SAs and NPs, optimizing the adsorption-dissociation of the key intermediate H2O* by Pt NPs, as well as the adsorption-desorption of H* by Pt SAs, thus enhances the synergistic effect between Pt SAs and NPs. The findings highlight the importance of constructing asymmetric coordination through the introduction of heteroatoms to facilitate photogenerated carrier transfer and surface reactions. This design approach may be applied to other cocatalyst-semiconductor photocatalytic systems.
Methods
Materials and reagents
4-Carboxyphenylboronic acid (4-PBA acid, ≥97%), Titanium butoxide (TBOT, ≥99%), 1,4-benzenedicarboxylic acid (PTA, ≥99%), N, N-Dimethylformamide (DMF, ≥99.5%), Chloroplatinic acid (H2PtCl6, ≥99%), Na2SO4 (≥99.0%), methanol (≥99.5%) and ethanol (≥99.5%) were provided by Alad. Chem. RT Co. Ltd, (China).
Preparation of MOF B-MIL-125(TiV) precursor
The Ti-vacancy-containing MOF precursor, MIL-125(TiV), was synthesized via a modified solvothermal which adjustments to the titanium precursor amount to introduce Ti vacancies10. In a typical preparation, PTA (3.0 g) was dispersed in DMF (54 mL) and magnetically stirred for 15 min at ambient temperature. Methanol (6 mL) was then introduced into the suspension, followed by the dropwise addition of TBOT (1.25 mL, reduced from 1.56 mL reported in the literature10) under continuous stirring for 10 min. The resulting mixture was transferred into a 100 mL Schlenk tube, sealed, and heated at 130 °C for 20 h. After naturally cooling to room temperature, the solid product was collected by centrifugation, sequentially washed with DMF and methanol, and then dried under vacuum to remove residual solvents.
To fabricate B-MIL-125(TiV), a ligand substitution strategy was employed. Specifically, 0.5 g of 4-Carboxyphenylboronic acid was dissolved in 45 mL of DMF and stirred for 10 minutes. Subsequently, 5 mL of methanol and 1 g of MIL-125(TiV) were added to the solution, and ultrasonic treatment was applied to ensure thorough dispersion of the MIL-125(TiV). The mixture underwent a hydrothermal treatment at 130 °C for 20 hours, facilitating the ligand substitution process. The final product was then washed and dried to obtain the B-MIL-125(TiV).
Preparation of PtSA/NP-TiO2-xBx and PtSA/NP-TiO2
Typically, 0.3 g of B-MIL-125(TiV) was dispersed in 30 mL of deionized (DI) water. A specific amount of H2PtCl6 (e.g., 2.76 mg for 0.35 wt.% Pt) was then added to the suspension and stirred for 12 hours to facilitate the synthesis of the encapsulated precursor. The resulting B-MIL-125(Pt) was centrifuged, washed with water, and dried at 60 °C. To obtain the PtSA/NP-TiO2-xBx photocatalyst, the sample was annealed in air at 450 °C for 4 hours. The PtSA/NP-TiO2 was synthesized using the same procedure as PtSA/NP-TiO2-xBx, with the only difference being the use of MIL-125(TiV) without B as the precursor for encapsulation.
Preparation of blank TiO2 and TiO2-xBx
Blank TiO2 and TiO2-xBx were prepared by calcining the MIL-125 and B-MIL-125 at 450 °C for 4 hours, respectively.
Preparation of PtSA-TiO2-xBx
Typically, 4.5 mL of methanol, 40 mL of DMF, and 2.3 g of PTA were mixed under stirring. Subsequently, 0.96 mL of TBOT and a predetermined amount of H2PtCl6 (e.g., 3.7 mg for 0.2 wt.% Pt) were added to the solution. After stirring for 10 minutes, the mixture was transferred to a 100 mL Teflon-lined autoclave and heated at 130 °C for 20 hours. Upon completion of the hydrothermal reaction and natural cooling, the PtSA-MIL-125 was obtained. And B/PtSA-MIL-125 was then synthesized by introducing B via the same ligand exchange strategy. The resulting B/PtSA-MIL-125 was collected by centrifugation, washed with water, and dried at 60 °C. Finally, the sample was annealed in air at 450 °C for 4 hours to yield PtSA-TiO2-xBx.
Preparation of PtNP-TiO2-xBx
100 mg TiO2-xBx were dispersed in 30 mL of DI water. A specific amount of H2PtCl6 (e.g., 2.64 mg for 1 wt.% Pt) was then added to the suspension. The container was then sealed, and the mixture was stirred under a 365 LED illumination for 12 h. After the reaction, the product was washed with water and then dried at 60 °C to obtain PtNP-TiO2-xBx.
Preparation of PtSA/NP@TiO2-xBx
To obtain the PtSA/NP@TiO2-xBx photocatalyst (without B-Pt coordination), 50 mg of TiO2-xBx was dispersed in 200 mL of DI water. A specific amount of H2PtCl6 (e.g., 1.32 mg for 0.6 wt.% Pt) was then added to the suspension, and the mixture was stirred in a dark environment for 1 hour, the resultant sample was washed with water and then dried at 60 °C to yield PtSA/NP@TiO2-xBx.
Characterizations
The structural, morphological, and surface properties of the catalysts were comprehensively characterized using a combination of techniques. Thermogravimetric analysis (TG-DTA, ZCT-B, Beijing) was employed to determine the thermal stability. X-ray diffraction (Rigaku, Cu Kα radiation) was employed to determine the crystallinity, and Raman spectra were collected with a LabRAM HR Evolution spectrometer (HORIBA, 532 nm laser). Morphology and microstructure were observed by scanning electron microscopy (SEM, Nova Nano SEM 450) and transmission electron microscopy (TEM, JEM-2100), with aberration-corrected HAADF-STEM imaging performed on a Titan Cubed Themis G2 300 (Thermo Fisher).
Optical absorption was analyzed by UV-vis diffuse reflectance spectroscopy (Metash UV-9000S, BaSO4 as reference), and surface area and porosity were measured from N2 adsorption-desorption isotherms at 77 K using a BET analyzer (3H-2000PS2). Surface chemical states were investigated by X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250Xi, Al Kα), and elemental composition was determined by ICP-OES (Agilent 7700X). Magnetic properties were probed by a PPMS (DynaCool, Quantum Design), and paramagnetic centers were examined by electron paramagnetic resonance (EPR, Bruker EMXnano).
Synchrotron-based X-ray absorption spectroscopy was carried out to analyze Ti K-edge (RapidXAFS 2 M, Ge(311) analyzer) and Pt L3-edge (BL11B beamline, SSRF, Si(311) monochromator) EXAFS and XANES spectra. In situ XPS was conducted under 300 W Xe lamp irradiation (Perfectlight PLS-SXE300C), and in situ EPR spectra were recorded in Ar-filled quartz tubes (Bruker ESR500, X-band).
The charge-carrier dynamics were further investigated by steady-state photoluminescence (PL, Shimadzu FL-4500, excitation 365 nm) and time-resolved PL (FLS 1000, Edinburgh Instruments, excitation 365 nm, emission 430 nm). The TRPL average lifetime (τavg) were fitted using equation:
and the τave is calculated by following equation:
where A₁ and A₂ are the normalized amplitudes of the two decay components, and τ₁ and τ₂ are the corresponding lifetimes.
The characterization procedures were adapted from our previous reports10,41.
Photocatalytic property evaluation
A multichannel photochemical system (PCX-50C, Discover, Perfect Light Ltd.) was used for conducting photocatalytic H2 evolution (PHE) experiments. Typically, 5 mg of catalyst was dispersed in 30 mL of a water/methanol mixture (1:2, v/v) in a sealed quartz reaction vessel, followed by ultrasonic treatment for 3 min. The system was purged with Ar (10 cycles) to remove air before illumination. A 365 nm LED lamp (5 W) served as the light source, and the suspension was continuously stirred at 40 °C using a circulating water bath. The evolved H2 was quantified every 10 min by gas chromatography (GC7900, TCD detector, Ar carrier gas). After each 1 h cycle, accumulated H2 was purged with Ar, and the experiment was repeated three times to calculate the average H2 evolution rate. For long-term stability tests, the catalyst loading was reduced to 2 mg to avoid excessive pressure from continuous H2 generation.
The apparent quantum efficiency (AQE) was measured under monochromatic LED irradiation at 365, 385, and 420 nm with intensities of 62.4, 48.3, and 36.7 mW cm−2, respectively. In a typical test, 10 mg of catalyst was used under the same conditions as the PHE experiment. The AQE was calculated using Eq. (3):
where M is the amount of evolved H2 (mol), NA is Avogadro’s constant, h is Planck’s constant, c is the speed of light, I is the incident light intensity, A is the irradiation area, t is the reaction time, and λ is the irradiation wavelength. Each value was averaged from three independent measurements.
The turnover frequency (TOF) was determined using Eq. (4):
where nH2 is the number of moles of H2 generated, nPt is the number of moles of Pt in the catalyst, and t is the reaction time.
The photocatalytic testing protocols were adapted from our previous report41 with minor modifications.
Photo-electrochemical measurements
Photoelectrodes were fabricated by dispersing 5 mg of catalyst in 5 wt.% Nafion ethanol solution and drop-casting the suspension onto fluorine-doped tin oxide (FTO) glass (2.0 × 2.5 cm2), followed by drying at 60 °C for 3 h. Photoelectrochemical (PEC) tests were performed on a CHI-760E electrochemical workstation in a standard three-electrode configuration, with the prepared electrode as the working electrode, Ag/AgCl (saturated KCl) as the reference electrode, and a Pt foil as the counter electrode.
The electrolyte was 0.20 M Na2SO4 (pH = 6.98 ± 0.13), prepared within 24 h prior to use by dissolving anhydrous Na2SO4 (5.68 g) in 200 mL of deionized water under magnetic stirring until fully dissolved. The solution was stored in a sealed amber polypropylene container at room temperature in the dark. The pH was measured at 25 °C with a three-point calibrated benchtop pH meter (buffers at pH 4.00, 7.00, and 10.00), and values are reported as mean ± SD from three independent measurements (n = 3).
Electrochemical impedance spectroscopy (EIS) was conducted with an AC perturbation of 5.0 mV in the frequency range of 105–0.1 Hz. Photocurrent responses under chopped illumination (150 W Xe lamp, 94.7 mW cm−2) were recorded using the amperometric I-t method. Transient photocurrent measurements were collected at 1.0 kHz with an applied AC voltage of 5.0 mV.
The PEC measurement procedures were adapted from our previous work41 with minor modifications.
CO adsorption infrared spectrum measurements
CO chemisorption measurements were conducted using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) on a Bruker VERTEX 80 v spectrometer. To collect the CO-DRIFTS spectra, the sample was first reduced under a 10% H2/Ar gas flow at 150 °C for 0.5 hour after being loaded into the reflectance cell. Following the pretreatment, the sample was cooled to 30 °C under an Ar atmosphere, and a background spectrum was recorded. The sample was then exposed to a 10% CO/Ar gas flow at 20 mL min−1 for approximately 1 h until adsorption saturation was reached. Subsequently, the sample was purged with Ar at a flow rate of 20 mL min−1 for an additional 40 min to remove gas-phase CO.
Isotopic experiment
The isotopic experiment was conducted using a multichannel photochemical reactor under 365 nm LED irradiation. Typically, 15 mg of sample was added to 30 ml reaction solution, followed by irradiation. Two parallel experiments were performed under identical photocatalytic conditions, replacing water and methanol with heavy water (D2O) and deuterated methanol (CD3OD) with same ratio, respectively. The evolved products were subsequently analyzed by mass spectrometry (MS, Hiden HPR-40).
Surface photovoltage (SPV) experiments
Samples were uniaxially pressed into dense pellets (diameter: 10 mm; thickness: 1 mm). Surface potentials (Vs) before and during illumination were performed using the SKP5050 Kelvin probe with a calibrated LED light source (wavelength: 365 nm). The measurement area of samples is a circular region with a diameter of 2 mm. The surface photovoltage (SPV) was calculated as:
where Vₛ(illumination) and Vₛ(darkness) are the measured surface potentials before and during illumination, respectively.
In-situ irradiated XPS measurements
In-situ irradiated XPS measurements were performed by a photoelectron spectrometer (ESCALAB 250Xi) with an Al Kα radiation source (hv = 1486.68 eV).
Survey spectra were acquired with a pass energy of 100 eV and a step size of 1 eV, while high-resolution spectra were collected at 20 eV with 0.05 eV steps. During the measurement, the non-irradiated XPS spectra were collected first, and then the irradiated XPS spectra were collected after irradiation for 30 minutes. The light source (300 W Xe-lamp) is installed on the outside of the XPS instrument, and the irradiated XPS data is collected while it is irradiated.
Computational methods and details
Density functional theory (DFT) calculations were performed with the Vienna Ab initio Simulation Package (VASP). The generalized gradient approximation (GGA) within the Perdew-Burke-Ernzerhof (PBE) scheme was adopted for exchange–correlation. A plane-wave basis set was used with an energy cutoff of 500 eV. Structural optimizations were converged to a precision of 1×10−5 eV and 0.02 eV·Å−1. Electron states were sampled using a 1×2×1 k-point mesh centred at Γ. For Ti and Pt atoms, the U correction was applied with U values of 4.2 eV and 2.4 eV, respectively. A vacuum layer of 15 Å was employed in the surface model. After structure optimization, catalytic properties were evaluated by calculating the Gibbs free energy for H* adsorption.
where ΔE denotes the reaction energy difference, ΔZPE the zero-point energy correction, and ΔS the vibrational entropy contribution at temperature T. The atomic coordinates of all computational models are provided in Supplementary Data 1.
Data availability
The source data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Zhou, P. et al. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 613, 66–70 (2023).
Li, Y. Y. et al. Electrolyte-assisted polarization leading to enhanced charge separation and solar-to-hydrogen conversion efficiency of seawater splitting. Nat. Catal. 7, 77–88 (2024).
Lee, W. H. et al. Floatable photocatalytic hydrogel nanocomposites for large-scale solar hydrogen production. Nat. Nanotechnol. 18, 754–762 (2023).
Wan, S. et al. A superlattice interface and S-scheme heterojunction for ultrafast charge separation and transfer in photocatalytic H2 evolution. Nat. Commun. 15, 9612 (2024).
Kosco, J. et al. Generation of long-lived charges in organic semiconductor heterojunction nanoparticles for efficient photocatalytic hydrogen evolution. Nat. Energy 7, 340–351 (2022).
Sheng, H. X. et al. Strong synergy between gold nanoparticles and cobalt porphyrin induces highly efficient photocatalytic hydrogen evolution. Nat. Commun. 14, 1528 (2023).
Ruan, X. W. et al. A Twin S-Scheme Artificial Photosynthetic System with Self-Assembled Heterojunctions Yields Superior Photocatalytic Hydrogen Evolution Rate. Adv. Mater. 35, 2209141 (2023).
Song, S. et al. A selective Au-ZnO/TiO2 hybrid photocatalyst for oxidative coupling of methane to ethane with dioxygen. Nat. Catal. 4, 1032–1042 (2021).
Liu, B. X. et al. Regulating the Transfer of Photogenerated Carriers for Photocatalytic Hydrogen Evolution Coupled with Furfural Synthesis. ACS Nano 18, 17939–17949 (2024).
Zhang, Y. M. et al. Single-atom Cu anchored catalysts for photocatalytic renewable H2 production with a quantum efficiency of 56%. Nat. Commun. 13, 58 (2022).
You, X. M. et al. Ultrahigh Bifunctional Photocatalytic CO2 Reduction and H2 Evolution by Synergistic Interaction of Heteroatomic Pt-Ru Dimerization Sites. ACS Nano 18, 9403–9412 (2024).
Tang, M. H. et al. Upcycling of Polyamide Wastes to Tertiary Amines Using Mo Single Atoms and Rh Nanoparticles. Angew. Chem. -Int. Ed. 64, e202416436 (2024).
Zhu, Y. H. et al. Synergistic Effect of Platinum Single Atoms and Nanoclusters Boosting Electrocatalytic Hydrogen Evolution. CCS Chem. 3, 2539–2547 (2021).
Zhang, J. K. et al. Mechanistic Insight into the Synergy between Platinum Single Atom and Cluster Dual Active Sites Boosting Photocatalytic Hydrogen Evolution. Adv. Mater. 35, 2300902 (2023).
Liu, P. G. et al. Synergy between Palladium Single Atoms and Nanoparticles via Hydrogen Spillover for Enhancing CO2 Photoreduction to CH4. Adv. Mater. 34, 2200057 (2022).
Zhang, J. K., Zheng, X. H., Yu, W. L., Feng, X. & Qin, Y. Unravelling the synergy in platinum-nickel bimetal catalysts designed by atomic layer deposition for efficient hydrolytic dehydrogenation of ammonia borane. Appl. Catal. B-Environ. Energy 306, 121116 (2022).
Xi, S. B., Zhang, J. & Xie, K. Low-temperature Water-gas Shift Reaction Enhanced by Oxygen Vacancies in Pt-loaded Porous Single-crystalline Oxide Monoliths. Angew. Chem. -Int. Ed. 61, e202209851 (2022).
Lv, T. P. et al. Insights into synergistic effect of Pd single atoms and sub-nanoclusters on TiO2 for enhanced photocatalytic H2 evolution. Chem. Eng. J. 450, 137873 (2022).
Yu, Y. Y. et al. Synergistic Effect of Cu Single Atoms and Au-Cu Alloy Nanoparticles on TiO2 for Efficient CO2 Photoreduction. ACS Nano 15, 14453–14464 (2021).
Gao, Z. Y. et al. Photocatalytic Methanol Dehydrogenation Promoted Synergistically by Atomically Dispersed Pd and Clustered Pd. J. Am. Chem. Soc. 146, 24440–24449 (2024).
Zhang, J. K. et al. Species Heterogeneity and Synergy to Boost Photocatalytic Hydrogen Evolution. ACS Catal. 14, 15699–15712 (2024).
Ren, M. et al. Synergy between palladium single atoms and small nanoparticles co-anchored on carbon atom self-doped graphitic carbon nitride boosting photocatalytic H2 generation. Appl. Catal. B-Environ. Energy 345, 123680 (2024).
Wang, H. et al. High quantum efficiency of hydrogen production from methanol aqueous solution with PtCu-TiO2 photocatalysts. Nat. Mater. 22, 619–626 (2023).
Wang, W. Y. et al. Catalytic Refining Lignin-Derived Monomers: Seesaw Effect between Nanoparticle and Single-Atom Pt. Angew. Chem. -Int. Ed. 63, e202404683 (2024).
Chen, C. L. et al. Asymmetrically Coordinated Cu Dual-Atom-Sites Enables Selective CO2 Electroreduction to Ethanol. Adv. Mater. 36, 2409797 (2024).
Niu, X. D., Wei, J., Xu, D. Y., Pei, J. J. & Sui, R. Charge-asymmetry Fe1Cu single-atom alloy catalyst for efficient oxygen reduction reaction. Nano Res 17, 4702–4710 (2024).
Zhang, W. J. et al. Regulating Hydrogen/Oxygen Species Adsorption via Built-in Electric Field-Driven Electron Transfer Behavior at the Heterointerface for Efficient Water Splitting. Angew. Chem. -Int. Ed. 63, e202400888 (2024).
Zhai, H. H. et al. Electronic Regulation of Pt Single-Atom Catalysts via Local Coordination State Adjustment for Enhanced Photocatalytic Performance. ACS Catal. 13, 8063–8072 (2023).
Wei, J. et al. Site-specific metal-support interaction to switch the activity of Ir single atoms for oxygen evolution reaction. Nat. Commun. 15, 559 (2024).
Liu, W. J. et al. Regulating Local Electron Distribution of Cu Electrocatalyst via Boron Doping for Boosting Rapid Absorption and Conversion of Nitrate to Ammonia. Adv. Funct. Mater. 34, 2408732 (2024).
Huang, L. et al. Precisely Regulating Asymmetric Charge Distribution by Single-Atom Central Doped Ag-Based Series Clusters for Enhanced Photoreduction of CO2 to Alcohol Fuels. Angew. Chem. -Int. Ed. 63, e202412964 (2024).
Song, G. X. et al. High-spin state Fe(III) doped TiO2 for electrocatalytic nitrogen fixation induced by surface F modification. Appl. Catal. B-Environ. Energy 301, 120809 (2022).
Gao, Y. X. et al. Regulating the electron spin orbital by sulfur doping of Ti vacancies to manipulate spin flip for enhancing PEC water splitting performance. Energy Environ. Sci. 17, 6268–6278 (2024).
Zhang, R. X. et al. Direct Photocatalytic Methane Oxidation to Formaldehyde by N Doping Co-Decorated Mixed Crystal TiO2. ACS Nano 18, 12994–13005 (2024).
Zi, B. Y. et al. Pr doping promotes the formation of Pt single atoms by regulating metal-support interaction for remarkable photocatalytic hydrogen production. J. Colloid Interface Sci. 680, 298–306 (2025).
Kerketta, U., Kim, H., Denisov, N. & Schmuki, P. Grätzel-Type TiO2 Anatase Layers as Host for Pt Single Atoms: Highly Efficient and Stable Photocatalytic Hydrogen Production. Adv. Energy Mater. 14, 2302998 (2024).
Liu, C. W. et al. Knowledge-Driven Design and Lab-Based Evaluation of B-doped TiO2 Photocatalysts for Ammonia Synthesis. Adv. Energy Mater. 13, 2204126 (2023).
Cheng, X. et al. Ligand Charge Donation-Acquisition Balance: A Unique Strategy to Boost Single Pt Atom Catalyst Mass Activity toward the Hydrogen Evolution Reaction. ACS Catal. 12, 5970–5978 (2022).
Li, H., Qin, X. X., Zhang, X. G., Jiang, K. & Cai, W. B. Boron-Doped Platinum-Group Metals in Electrocatalysis: A Perspective. ACS Catal. 12, 12750–12764 (2022).
Mao, Z. J. et al. Interstitial B-Doping in Pt Lattice to Upgrade Oxygen Electroreduction Performance. ACS Catal. 12, 8848–8856 (2022).
Li, B. et al. Revealing the synergistic effect of bulk and surface co-doped boron on TiO2 for enhanced photocatalytic H2 evolution. Chem. Eng. J. 497, 154726 (2024).
Li, Y. J. et al. Boron containing metal-organic framework for highly selective photocatalytic production of H2O2 by promoting two-electron O2 reduction. Mater. Horiz. 8, 2842–2850 (2021).
Ma, K. W. et al. Revealing different depth boron substitution on interfacial charge transfer in TiO2 for enhanced visible-light H2 production. Appl. Catal. B-Environ. 315, 121570 (2022).
Guo, M. C. et al. Single-Atom Pt Loaded on MOF-Derived Porous TiO2 with Maxim-Ized Pt Atom Utilization for Selective Hydrogenation of Halonitro-benzene. Angew. Chem. -Int. Ed. 64, e202418964 (2025).
Jeong, H. et al. Controlling the Oxidation State of Pt Single Atoms for Maximizing Catalytic Activity. Angew. Chem. -Int. Ed. 59, 20691–20696 (2020).
Si, Y. et al. Fully exposed Pt clusters for efficient catalysis of multi-step hydrogenation reactions. Nat. Commun. 15, 4887 (2024).
Kim, T. S., O’Connor, C. R. & Reece, C. Interrogating site dependent kinetics over SiO2-supported Pt nanoparticles. Nat. Commun. 15, 2074 (2024).
Macino, M. et al. Tuning of catalytic sites in Pt/TiO2 catalysts for the chemoselective hydrogenation of 3-nitrostyrene. Nat. Catal. 2, 873–881 (2019).
Chen, J. J. et al. Reverse oxygen spillover triggered by CO adsorption on Sn-doped Pt/TiO2 for low-temperature CO oxidation. Nat. Commun. 14, 3477 (2023).
Zhang, J. J., Zhang, L. Y., Wang, W. & Yu, J. G. In Situ Irradiated X-ray Photoelectron Spectroscopy Investigation on Electron Transfer Mechanism in S-Scheme Photocatalyst. J. Phys. Chem. Lett. 13, 8462–8469 (2022).
Zhang, J. Y. et al. Electronic and transport properties of Li-doped NiO epitaxial thin films. J. Mater. Chem. C. 6, 2275–2282 (2018).
Zhang, Z. & Yates, J. T. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 112, 5520–5551 (2012).
Liu, D. J. et al. Modulating local coordination structure over single Pt atom to optimize adsorption behavior for high-efficiency photocatalytic H2 production. Appl. Catal. B-Environ. Energy 361, 124655 (2025).
Da, Y. M. et al. Single-Atom Pt Doping Induced p-Type to n-Type Transition in NiO Nanosheets toward Self-Gating Modulated Electrocatalytic Hydrogen Evolution Reaction. ACS Nano 17, 18539–18547 (2023).
Zhao, Y. L. et al. Vacancy Defects Inductive Effect of Asymmetrically Coordinated Single-Atom Fe-N3S1 Active Sites for Robust Electrocatalytic Oxygen Reduction with High Turnover Frequency and Mass Activity. Adv. Mater. 36, 2308243 (2024).
Pan, L. et al. Manipulating spin polarization of titanium dioxide for efficient photocatalysis. Nat. Commun. 11, 418 (2020).
Wu, H. et al. Strong intrinsic room-temperature ferromagnetism in freestanding non-van der Waals ultrathin 2D crystals. Nat. Commun. 12, 5688 (2021).
Sun, T. et al. Ferromagnetic single-atom spin catalyst for boosting water splitting. Nat. Nanotechnol. 18, 763–771 (2023).
Sun, K. et al. Manipulating the Spin State of Co Sites in Metal-Organic Frameworks for Boosting CO2 Photoreduction. J. Am. Chem. Soc. 146, 3241–3249 (2024).
Chen, F., Ma, T. Y., Zhang, T. R., Zhang, Y. H. & Huang, H. W. Atomic-Level Charge Separation Strategies in Semiconductor-Based Photocatalysts. Adv. Mater. 33, 2005256 (2021).
Jiang, X. H. et al. Silver Single Atom in Carbon Nitride Catalyst for Highly Efficient Photocatalytic Hydrogen Evolution. Angew. Chem. -Int. Ed. 59, 23112–23116 (2020).
Luo, H. et al. Pt single-atoms supported on nitrogen-doped carbon dots for highly efficient photocatalytic hydrogen generation. J. Mater. Chem. A 8, 14690–14696 (2020).
Chen, R. T. et al. Spatiotemporal imaging of charge transfer in photocatalyst particles. Nature 610, 296–301 (2022).
Chen, R. T., Fan, F. T., Dittrich, T. & Li, C. Imaging photogenerated charge carriers on surfaces and interfaces of photocatalysts with surface photovoltage microscopy. Chem. Soc. Rev. 47, 8238–8262 (2018).
Tousek, J. & Tousková, J. A novel approach to the surface photovoltage method. Sol. Energy Mater. Sol. Cells 92, 1020–1024 (2008).
Zhang, L. Y., Zhang, J. J., Yu, J. G. & Garcia, H. Charge-transfer dynamics in S-scheme photocatalyst. Nat. Rev. Chem. 9, 328–342 (2025).
Kumar, C. P., Gopal, N. O., Wang, T. C., Wong, M. S. & Ke, S. C. EPR investigation of TiO2 nanoparticles with temperature-dependent properties. J. Phys. Chem. B 110, 5223–5229 (2006).
Berger, T. et al. Light-induced charge separation in anatase TiO2 particles. J. Phys. Chem. B 109, 6061–6068 (2005).
Li, J. J. et al. Highly Active and Stable Metal Single-Atom Catalysts Achieved by Strong Electronic Metal-Support Interactions. J. Am. Chem. Soc. 141, 14515–14519 (2019).
Long, H. Y. et al. Fine-tuning d-p hybridization in Ni-Bx cocatalyst for enhanced photocatalytic H2 production. Nat. Commun. 16, 946 (2025).
Li, M. Y. et al. Engineering Spatially Adjacent Redox Sites with Synergistic Spin Polarization Effect to Boost Photocatalytic CO2 Methanation. J. Am. Chem. Soc. 146, 15538–15548 (2024).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB3803600 (Q.L.1,2*)), the National Natural Science Foundation of China (22378346, 22368050 (Q.L.1,2*)), the Key Research and Development Program of Yunnan Province (202302AF080002 (Q.L.1,2*)), and Scientific Research and Innovation Project of Postgraduate Students in the Academic Degree of Yunnan University (KC-24248466 (B.L.)). Authors thank the Electron Microscopy Center, the Advanced Computing Center, the Advanced Analysis and Measurement Center of Yunnan University for the sample testing and computations service.
Author information
Authors and Affiliations
Contributions
B.L., T.Z., and Q.L.1,2* conceived and designed the study. H.Z. and T.H. carried out the synthesis theory calculations of the study. Q.L.1, D.L., and B.Z. carried out the synthesis and characterizations of the materials. B.L., and M.Z. carried out the photocatalytic test. M.C, Y.Z1., H.S, Y.Z3, J.Z., Z.Z., G.Z., and J.H.Z. contributed to data analysis. T.Z. and Q.L.1,2* supervised the project. B.L. wrote the manuscript. B.L., T.Z., and Q.L.1,2* revised and reviewed the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kenichi Ozawa and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, B., Zheng, H., Zhou, T. et al. Asymmetric coordination enhances the synergy of Pt species dual active sites for efficient photocatalytic H2 evolution. Nat Commun 16, 8276 (2025). https://doi.org/10.1038/s41467-025-63637-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63637-2
