
Abstract
Charge transport and extraction in polycrystalline perovskite films are often hindered by inefficient carrier transfer across grain domain boundaries (GDBs). Herein, we present a universal post-treatment strategy leveraging supramolecular crown ether-assisted slow release and precise delivery of Rb⁺ cations to GDBs, achieving in-situ GDB bridging. The solid-state nuclear magnetic resonance (NMR), transmission electron microscopic (TEM), and time-of-flight secondary ion mass spectrometry (ToF-SIMS) analyses confirm that Rb+ forms a non-perovskite phase, primarily localized at the surface and GDBs. Ultrafast time-resolved photoluminescence mapping revealed accelerated carrier diffusion across the grain boundaries for the Rb+-treated perovskite thin films which enables photo-generated charge carriers to travels over two grain domain boundaries before recombination. As a result, perovskite solar cells treated with this strategy achieved a champion efficiency of 26.02% (certified as 25.77%) and demonstrated remarkable stability, retaining 99.2% of their initial efficiency after 1300 h of continuous one-sun illumination under maximum power point tracking (ISOS-L-1I).
Similar content being viewed by others


Introduction
In recent years, perovskite solar cells (PSCs) have garnered considerable attention within the global academic and industrial sectors due to their enhanced performance while maintaining attributes, such as straightforward manufacturing processes and cost-effectiveness1,2,3,4,5,6,7,8. Despite these remarkable accomplishments, critical scientific and technological challenges, such as defect-induced structural degradation and defect-assisted carrier recombination, must still be urgently addressed to advance the large-scale industrial application of PSCs9,10,11,12. The polycrystalline nature and film-fabrication process of perovskites inherently lead to numerous grain domain boundaries (GDBs), regardless of the device performance, which serve as primary defect accumulation sites13,14,15.
Additive engineering is the primary strategy for passivating GDB defects16,17. However, additives inevitably influence crystallization and may introduce insulating moieties, which impede charge transport16,17,18. Post-treatment, which circumvents interference with the precursor composition and crystallization process, has emerged as an effective strategy for defect passivation in PSCs19,20,21,22. Nevertheless, post-treatment is generally confined to the top surface due to the fully crystallized perovskite structure, limiting its effectiveness in precisely targeting GDBs.
Herein, we propose a universal post-treatment strategy leveraging supramolecular crown ether (CE)-assisted slow release and precise delivery of Rb⁺ cations to GDBs, where they form in situ one-dimensional (1D) Rb-based non-perovskite phase bridge that facilitates defect passivation and carrier diffusion. The CE was judiciously selected as a suitable carrier molecule due to its multi-ring structure through rational optimization of the screening process23. The Rb+ cations were released successfully from the complex and formed a non-perovskite phase, located at the surface and the GDBs. This was confirmed by solid-state nuclear magnetic resonance (NMR), transmission electron microscopic (TEM), and time-of-flight secondary ion mass spectrometry (ToF-SIMS). Results from ultrafast time-resolved photoluminescence (TRPL) mapping indicated a cross-grain domain (GD) carrier diffusion in the Rb+-treated perovskite thin films, extending over several GDBs. The photoluminescence quantum yield (PLQY) exceeded 42% with an enhancement of over 2 mA cm−2 in short-circuit current (JSC) compared to the control sample. PSCs treated in this manner achieved a champion efficiency of 26.02% (certified as 25.77%) and demonstrated remarkable stability by retaining 99.2% of their initial efficiency after 1300 h of continuous one-sun illumination under maximum power point (MPP) tracking (ISOS-L-1I).
Results
Precise cation delivery and in situ inter-grain bridging
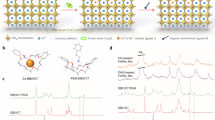
The low-temperature and layer-by-layer stacking preparation process results in the distribution of defects over various locations in the film, especially on the surface and at GDBs (Fig. 1a). Cs+ cations are often used to stabilize the FA+-rich perovskite phase. However, it is difficult to target specific locations of defects for repair since Cs+ occupies regular A cation sites in the perovskite lattice (Fig. 1b). Therefore, we selected Rb⁺ cations which cannot reside in A cation site due to their small size. We used a supramolecular host-guest strategy via CE complexation to control the delivery of Rb+ and achieve in situ formation of a 1D Rb-based non-perovskite phase at films’ surface and GDB (Fig. 1c). We also introduced an additional chemical rinsing step to remove the excess CE molecules. Considering the compatibility between the atomic radius of the cations and the cavity size of the CE, dibenzo-21-crown-7 ether (referred to as DB21-7) and 18-crown-6 ether (referred to as 18-6) were selected as carrier molecules to form host–guest supramolecular complexes with Cs+ (CE-Cs) and Rb+ (CE-Rb) cations, respectively24,25. The experiments were subsequently conducted, as shown in Supplementary Fig. 1.

a Defects are distributed at various locations in the film. b Cs+ cations captured by the lattice and distributed throughout the film. c Non-lattice-incorporated Rb⁺ cations were delivered precisely to A sites and achieve in situ formation of 1D Rb-based non-perovskite phase at films’ surface and GDB. d Room temperature 87Rb NMR spectra of CE-Rb-treated perovskite films rinsed and unrinsed by mixed solvent (respectively referred to as rinsed and unrinsed CE-Rb), δ-RbPbI3 and δ-FAyRb1-yPbI3 samples acquired with central transition selective (CT-selective) Hahn echo and simulated spectrum of CE-Rb with the parameters from ref. 40. e 1H → 13C CP spectra of CE-Cs-treated and CE-Rb-treated perovskite films rinsed and unrinsed by mixed solvent, respectively, as well as the α-FAPbI3 (containing MA+ as an additive) perovskite films at 100 K. f ToF-SIMS depth profiles for CE-Rb, Pb, and Sn cations. The inset is a schematic diagram of the CE-Rb-treated perovskite film sputtered by Ar+ cluster ion beam at different depths.
Multinuclear magic angle spinning (MAS) solid-state NMR experiments were performed to determine whether Cs+ or Rb+ cations have been released from the supramolecular complexes and, if so, have been incorporated into the perovskite lattice. The spectrum of the CE-Rb complex displays a characteristic second-order quadrupolar line shape at a low frequency (δ87Rb = 16 ppm) (Fig. 1d), indicating that most of the Rb+ ions were released from the complex. In CE-Rb-treated perovskites, the broad 87Rb signal corresponds to a non-perovskite phase, which is further corroborated by 207Pb solid-state NMR experiments (detailed discussion can be found in Supplementary Note 1 and Supplementary Fig. 2)26,27,28. The CE washing process was further investigated through 1H → 13C cross-polarization MAS experiments, as shown in Fig. 1e. Upon washing, the 13C signal intensities for both DB21-7 and 18-6 significantly decreased but did not completely disappear, suggesting that a few CEs remain in the film as passivators23,29. Overall, solid-state NMR results confirm that Cs+ cation incorporates into the perovskite phase, while Rb+ cations remain in the Rb-based non-perovskite phase, likely located on the surface and in the GDBs. The results can be proved by the theoretical calculations (detailed discussion seen in Supplementary Note 2 and Supplementary Fig. 3) showing that Rb+ cations are more easily released and form in situ 1D Rb-based non-perovskite phase. Additionally, ToF-SIMS was performed and showed that the Rb+ ions exhibited significant accumulation at top and bottom surfaces, as well as a substantial presence within the film, thus indicating that Rb+ ions were not incorporated into the lattice but instead expelled to facilitate in situ GDB bridging (Fig. 1f) (detailed discussion seen in Supplementary Note 3 and Supplementary Figs. 4–7).
Multi-grain-domain carrier transport in perovskite films
The carrier transport behavior in the polycrystalline perovskite films was investigated by photoluminescence (PL)-scanned imaging microscopy30 and the cumulative photon counts for different GDs were analyzed, as shown in Fig. 2a–d. In the control film, no PL signal was detected from GDs adjacent to the excited one, indicating that carriers’ diffusion was primarily confined within individual GDs (Fig. 2a). In the CE-Cs-treated film, a PL signal appeared in the adjacent GDs, suggesting that carriers were able to diffuse across one GDB (Fig. 2b). In contrast, the CE-Rb-treated film exhibited a strong PL signal at sub-adjacent GDs (Fig. 2c), demonstrating a multi-grain-domain carrier transportation, which is attributed to the in situ formation of 1D Rb-based non-perovskite phase at the GBDs. These results suggest that the 1D Rb-based non-perovskite phase bridges the GBDs and facilitates carrier transport, thereby enabling carriers to diffuse sufficiently far for charge accumulation to be detected in the sub-adjacent GD (Fig. 2d).

PL intensity images of individual GDs obtained by the PL-scanned imaging microscopy with the focused laser beam fixed at the indicated positions (Exc.) for a control, post-treated by b CE-Cs and c CE-Rb. The white dashed lines are the approximate boundary lines of the GDs. Insets: optical images of the films. d The cumulative number of photons at different positions. Comparison of the carrier transport kinetics extracted at a position with a transport distance of e 1.0 μm and f 2.0 μm in the control, CE-Cs, and CE-Rb-based films. g Schematic illustration of the excited carrier transport process. The “c” represents carrier, the 0, 1, and 2 represent the GD at the excitation point, the adjacent and sub-adjacent GDs, respectively.
We further extracted the kinetics at 1.0 and 2.0 µm from the excitation point, corresponding to the first and second neighbor GDs, respectively. After subtracting the wave-guiding components30,31,32, the PL intensity of the films treated with CE-Cs and CE-Rb is much higher than that of control after the carriers crossed to the first and second neighbor GDB, as shown in Fig. 2e, f. The detailed methodology for subtracting waveguide contributions to accurately attribute fluorescence signals is specifically discussed in the Supplementary Fig. 8. This phenomenon indicates that carriers did not escape from the GD where they were generated for the control sample, while the target samples, especially the CE-Rb-based films, exhibited carrier transport across multiple GDBs. Moreover, the faster rising process of CE-Rb iterated films indicates faster carrier transport, highlighting its superior potential for rapid carrier transport across multiple GDBs. Figure 2g provides an intuitive illustration of carrier transport across GDBs in perovskite films.
Long-range carrier diffusion via GDB bridging
We then employed a home-built TRPL imaging microscope to investigate carrier diffusivity in the perovskite films. The excitation laser beam was focused to a near diffraction-limited spot using an objective lens to excite the perovskite films at a fixed position. Simultaneously, the PL intensity image as a function of delay time was obtained by scanning the PL collection pathway using a pair of galvanometer mirrors. The PL intensity image and decay kinetics from each pixel were recorded using a time-correlated single-photon counting of the PL intensity image reflects changes in the carrier density distribution resulting from carrier transport, as illustrated in Fig. 3a. The spread of the PL intensity image indicates that the carriers transport away from the initial excitation site. The signal intensity of the control sample decays very quickly after 50 ns without any diffusion. A roughly triangle shape displayed in the CE-Cs sample after 100 ns, which is most likely caused by the hindering of GDB when the carrier diffuses18. The CE-Rb-based perovskite film exhibits excellent isotropic diffusion, with the light spot continuously expanding in a circular fashion. Then, the cross-sectional profiles of the PL spots at different delay times were extracted to quantitatively estimate the diffusion coefficient (D)33,34, as shown in Supplementary Fig. 9 and the converted Gaussian variance (σ2x(t)) was given in Fig. 3b. The intra-grain diffusion coefficient (D1) was obtained from a linear fit of the first stage in the Fig. 3b, about 0–20 ns (Supplementary Fig. 10). The fitted D1 of CE-Rb-based sample was more than twice as large as the other two, indicating that Rb+ also has a repairing effect on the intra-grain boundaries. Carrier diffusion becomes slow and the diffusion coefficient rapidly decreases when they are scattered or trapped by defects at GD edges or boundaries18. The light spot will continue to increase once the carriers diffused to the adjacent GD. From this data, we derived the cross-GD boundary diffusion coefficient (D2), which refers to carrier transfer from the excited GD to the adjacent GD. Results are summarized in Fig. 3c. No linear trend was observed for the control sample, which indicates that the carriers cannot diffuse to adjacent GD. The fitted D2 was 0.0037 and 0.0197 cm2 s−1 for the perovskite films treated with CE-Cs and CE-Rb-, respectively. The outer-grain diffusion coefficient was reduced by over 20 times for the CE-Cs-treated perovskite film, indicating that the carriers suffer significant losses at GDB. In contrast, less carrier loss (D2 was reduced by less than 10 times) when passing through GDB and arrived to adjacent or even sub-adjacent GD in CE-Rb-treated perovskite film.

a Two-dimensional carrier diffusion images of perovskite thin films at time frames of 0, 50, 100, 150, 200, and 250 ns. The PL intensity is normalized to its peak value. b All converted Gaussian variance σ2x(t) as a function of delay time from 0 to 260 ns every 10 ns. c The summarized intra and outer diffusion coefficient for the perovskite films before and after CE-Cs and CE-Rb post-treated. d The TRPL decay of control, CE-Cs, and CE-Rb-based perovskite films. e Statistical distribution of PLQYs for three perovskite films, the perovskite films prepared on glass (3 samples for each condition and measure 2 points for every sample). The box represents values within the range of 25–75%, the dots represent the median and the line segment represents the value range within 1.5 times the interquartile range.
Subsequently, the carrier diffusion length (LD) was calculated by the Eq. (1):
where t1 is 20 ns and τ is the carrier lifetime calculated from the TRPL kinetics, as shown in Fig. 3d. The calculated LD were 0.35, 1.02 and 2.71 μm for the control, CE-Cs, and CE-Rb samples, respectively, representing remarkably long LD for perovskite films of this composition and processing route35,36,37. The LD of CE-Rb-treated perovskite film is more than twice the average GD size (Supplementary Fig. 11), indicating that carriers could diffuse across adjacent GDs to sub-adjacent GDs. This observation is consistent with the cross-domain transport behavior revealed by PL mapping. These results provide strong evidence that the precise delivery of Rb+ induces effective in situ GDB bridging, thereby enhancing carrier diffusion across the perovskite film.
Additionally, the PLQY, stabilized open-circuit voltage (VOC), and quasi-Fermi level splitting ΔEF/q (QFLS/q) were measured for perovskite films based on control, CE-Cs, and CE-Rb treatments, as shown in Fig. 3e and Supplementary Fig. 12. The average PLQY values were 11.2 ± 2.1%, 33.7 ± 2.8%, and 38.8 ± 3.5% for control, CE-Cs and CE-Rb films, respectively, corresponding to a quasi-Fermi level splitting of 1.18, 1.23, and 1.25 eV. The notable increase in PLQY upon treatment indicates a suppression of non-radiative recombination. Moreover, the in situ GDB bridging may also alter the surface potential. To probe this effect, Kelvin Probe Force Microscopy (KPFM) was conducted on the perovskite films. The top-view and cross-section KPFM images for the three perovskite films are shown in Supplementary Figs. 13 and 14. Notably, a clear increase in contact potential difference was observed in the CE-Rb-treated film, whereas no such change was detected in the control or CE-Cs-treated films. This result suggests that the formation of 1D Rb-based non-perovskite phase influences surface energy level alignment at GDB. The effectiveness of the GDB bridging strategy was also confirmed by the increased current observed in the conductive atomic force microscopy (c-AFM) (Supplementary Fig. 15).
The morphology of the perovskite films was observed by scanning electron microscope (SEM), as shown in Supplementary Figs. 11 and 16. The GD size increased after modification, which may be due to secondary crystallization caused by the annealing process38. It is worth noting that the amount of lead iodide (PbI2) on the film surface decreased (consistent with the weakened PbI2 peak obtained in X-ray diffraction (XRD) pattern, shown in Supplementary Fig. 17), which is mainly due to the reaction with Cs+ or Rb+ cations and in situ reconstruction. The interaction between the target molecules and defects was confirmed by the shift of the Pb 4f orbital electrons, measured from X-ray photoelectron spectroscopy (XPS), as shown in Supplementary Fig. 18. The greater shift amplitude of CE-Cs compared to CE-Rb indicates stronger interactions of Cs+ with the A sites on the perovskite surface, consistent with the theoretical calculation. Moreover, the absorption, steady-state PL spectra and ultraviolet photoelectron spectroscopy (UPS) of perovskite films for control and post-treated films were examined, as shown in Supplementary Fig. 19. The closely overlapping absorption curves indicate that the introduction of cations did not cause a significant bandgap change. The PL intensity increased by 1.7 and 6.8 times compared to the control film for the CE-Cs and CE-Rb, respectively. The PL intensity and TRPL with sub-micrometer spatial resolution were measured to assess the homogeneity of post-treated perovskite films, as shown in Supplementary Figs. 20 and 21. It is evident that both PL intensity and homogeneity were enhanced in the treated perovskite films, especially in the CE-Rb sample. The above improved optoelectronic properties demonstrate the enhanced transport of charge carriers at the interface. As shown in Supplementary Fig. 22, the significant reduction in extraction time obtained from the perovskite films with hole transport layer (HTL) also confirms the above conclusion.
Devices performance and stability
We fabricated PSCs to explore the impact of the CE-Cs and CE-Rb additives on the photovoltaic metrics. Figure 4a and Supplementary Table 2 show the current density–voltage (J–V) curves under reverse and forward scanning and photovoltaic parameters of the three PSCs. The device based on CE-Rb displayed outstanding performance, with a VOC of 1.181 V, a JSC of 26.12 mA cm−2 and a fill factor (FF) of 84.32%, attaining a high power conversion efficiency (PCE) of 26.02% (certified 25.77%) The certification report is shown in Supplementary Fig. 23), For the control device we obtained a substantially lower performance with a VOC of 1.150 V, a JSC of 24.50 mA cm−2, a FF of 81.43%, yielding PCE of 22.94%. The statistics of parameters were shown in Fig. 4b and Supplementary Table 3, which further verified the remarkable improved performance. The same trend was reflected in the J–V curves and photovoltaic parameters of the PSCs based on wide-bandgap, narrow-bandgap and inverted devices, fully demonstrating the universal applicability of our approach (Supplementary Figs. 24–26). Moreover, a micro module PSCs based on CE-Rb with an area of 64.8 cm2 was successfully manufactured and achieved a PCE of 20.54% (Fig. 4c). The external quantum efficiency (EQE) spectra and corresponding integrated JSC were given in Fig. 4d. The integrated JSC were calculated to be 24.14, 25.48 and 26.12 mA cm−2 for the PSCs based on control, CE-Cs and CE-Rb-based perovskite films, respectively. These values were consistent with the JSC measured from J–V curves. The increased JSC can be reflected in the photocurrent imaging measurement results under illumination based on the rounded PSCs, as shown in Supplementary Fig. 27. This suggests that the non-radiative recombination is significantly reduced in the samples with the precise delivery strategy compared to the control. The long-term stability of three PSCs without any encapsulation (in an N2 atmosphere at ~40 °C) was tracked at MPP under continuous 1-sun illumination (shown in Fig. 4e). The CE-Rb-based device exhibited a negligible PCE decline after 1300 h. In addition, the CE-based device also showed a good thermal stability as it could retain over 90% of its initial PCE after 768 h storage at 85 °C in dark condition, as shown in Fig. 4f (ISOS-D-2I).

a J–V curves, b statistics for photovoltaic parameters including the JSC, VOC, PCE, and FF distribution collected from 20 devices for PSCs based on the control, CE-Cs and CE-Rb perovskite films. The box represents values within the range of 25–75%, the dots represent the median and the line segment represents the value range within 1.5 times of interquartile range. c J–V curve of large-area PSC (area, 64.8 cm2) based on CE-Rb. d The EQE spectra and corresponding integrated JSC, e operational stability and f long-term thermal stability of the three PSCs.
Discussion
In summary, this study introduces a host–guest supramolecular strategy using CEs for the precise delivery of Rb⁺ cations to GDBs, addressing key limitations of conventional post-treatment methods, such as random distribution of passivating agents and the introduction of nonconductive impurities. A systematic comparison of Cs+ and Rb+ highlights that the slow release of non-lattice-incorporated Rb+ ions enable precise in situ GDB bridging, effectively reducing potential barriers and facilitating multi-grain-domain carrier diffusion. This strategy achieves a carrier diffusion over two GDBs, a JSC density enhancement of over 2 mA cm−2, and a champion PCE of 26.02% (certified as 25.77%). Additionally, Rb-treated devices demonstrate exceptional stability, retaining over 99% of their initial PCE after 1300 h of continuous operation under MPP tracking and one-sun illumination. These findings establish a versatile and efficient post-treatment strategy, providing a robust pathway for the scalable fabrication of high-performance and stable PSCs.
Methods
Fabrications
The substrate of the device is fluorine-doped tin oxide (FTO) glass (purchased from Shangyang Company), which is sequentially cleaned by detergent, deionized water, ethanol, and 2-propanol (IPA, 99.5%, procured from Sigma-Aldrich) for 20 min, respectively. Subsequently, the FTO substrate was dried in an oven and treated by UV-ozone for 20 min before use.
Tin (IV) oxide colloidal dispersion (SnO2, 15 wt% in H2O colloidal dispersion, sourced from Alfa Aesar) was diluted three times with deionized water and spin-coated onto the treated FTO substrate at 4000 rpm for 30 s, followed by annealing at 170 °C for 30 min in ambient air. The prepared SnO2/FTO/glass substrate requires an additional 10-min UV-ozone treatment and transfers into a nitrogen-filled glove box to prepare perovskite film.
1.5 M of lead iodide (PbI2, 99.99%, obtained from You Xuan) was dissolved in a mixed solvent system of dimethylformamide (99.8%, procured from Sigma-Aldrich) and dimethyl sulfoxide (99.9%, procured from Sigma-Aldrich) (volume ratio of 9:1) and stirred for 10 min at 80 °C, followed by over 40 min at 30 °C. The organic salt solution was prepared by dissolving 90 mg of formamidinium iodide (99.8%, acquired from Greatcell Solar) and 18 mg of methylammonium chloride (99.5%, purchased from Xi’an Polymer Light Technology) in 1 mL of IPA and stirring for over 1 h at room temperature. For the first-step, the PbI2 precursor solution was spin-coated onto the SnO2/FTO/glass substrate at 1500 rpm for 30 s followed by annealing at 70 °C for 40 s. For the second-step, the organic salt was spin-coated onto the PbI2 at 1700 rpm for 25 s and annealed at 90 °C for 20 s. The samples were then annealed outside of the glove box at 150 °C for 15 min under approximately 35% humidity.
The target samples need an additional post-treatment step. The cesium iodide (99.5%, acquired from Alorich) mixed with dibenzo-21-crown-7 (acquired from Chemie Brunschwig) or rubidium chloride (99.5%, acquired from Sigma-Aldrich) mixed with 18-crown-6 (acquired from Apollo Scientific) with 1:1.15 molar ratio was dissolved in chlorobenzene (CB, 99.9%, procured from Sigma-Aldrich. The molar concentration of the CE is 0.027 M) and stirred vigorously at 70 °C for over 5 days in nitrogen glove box. The mixed solution was spin-coated onto the perovskite/SnO2/FTO/glass substrate at 4000 rpm for 30 s, annealed at 120 °C for 20 s and rinsed with 200 µL of solvent (mix IPA and CB with volume ratio of 1:1) three times at 3000 rpm for 30 s. The control samples were also rinsed three times with the mixed solvent at the same condition. The IPA is used to remove residual organic salts on the surface of the perovskite film, which further reduces the defects of the perovskite film (Supplementary Figs. 28–30).
The HTL solution was prepared by mixing 72.3 mg of 2,2′,7,7′-tetrakis-(N,N-di-4-methoxyphenylamino)-9,9′-spirobifluorene (Spiro-OMeTAD, 99.8%, obtained from Borun New Material Technology), 28.8 µL of 4-tert-butyl pyridine (96%, purchased from Tokyo Chemical Industry), 17.5 µL of bis(trifluoromethane) sulfonimide lithium salt (99.95% trace metals basis, procured from Sigma-Aldrich) solution (520 mg mL−1 in acetonitrile (99.8%, procured from Sigma-Aldrich), and 1 mL of CB. This HTL solution was spin-coated onto the perovskite or post-treated film at 2500 rpm for 25 s.
Finally, 80 nm thick layer of gold or silver was evaporated onto the HTL layer in a vacuum chamber (base pressure, 5 × 10−5 Pa) at a rate of 0.5 Å s−1. All chemicals were used as received without any additional treatment. The fabrication of large area module was similar to the small device39.
Characterizations
All solid-state NMR experiments were performed on a Bruker Avance III spectrometer operating at 11.7 T using a 3.2 mm 1H/X/Y low-temperature MAS probe. Thin films were scraped, tightly packed and sealed into 3.2 mm zirconia rotors in a nitrogen-filled glovebox. Before and between NMR experiments, samples were stored in Schlenk tubes filled with nitrogen. Samples were quickly inserted into the probe and spun at the magic angle using dry nitrogen (more details can be found in Supplementary Note 4).
Morphology and elements distribution of samples were characterized by scanning electron microscopy (SEM, SU82200304070201), TEM (Tecnai G2F30 STWIN03040101, FEI), Spherical Aberration-Corrected Transmission Electron Microscope (JEM-ARM300F2) and Focused Ion/Electron Dual Beam System (Helios G4 UX). The c-AFM and KPFM images were performed on JPK Nanowizard 4XP (Bruker).
Steady-state PL were obtained by a Fluorolog 322 spectrometer (Horiba Jobin Yvon iHr320 and a CCD) and excitation at 450 nm (Xenon lamp). TRPL was measured by a LifeSpec II (Edinburgh Instruments) fluorescence spectrometer with a picosecond pulsed diode laser (EPL-510, Edinburgh Instruments). UV–visible measurements were carried out using a Shimadzu UV-2600 spectrophotometer.
The PL and photocurrent imaging measurements are performed on a home-built laser-scanned and time-resolved PL microscopy coupled with a photocurrent detection module and a time-correlated single photon counting module. A CW laser with a wavelength of 405 nm (LSR405CP-80-FC, LASEVER INC, China) is coupled to the measurement system, and a Keithley 6485 picoammeter was used to detect the photocurrent. The photocurrent imaging was obtained by fast scanning the laser spot and recording the photocurrent during scanning of each point (256 × 256 pixels) on the perovskite solar cell. For PL and TRPL mapping measurements, a pulse laser with a wavelength of 460 nm (Superk fianium FIU-15 pp, NKT) was focused on the sample through a 100X air objective lens (NA = 0.95, Olympus SLMPlan N). By parking the excitation laser spot at a specific location in the sample, fast scanning of the galvanometer mirror ensures that photon emission was collected from the sample. A high-speed detector and a 510 nm long-pass filter were used to collect the fluorescence signal.
XRD was performed using X-ray diffractometer (D8 Advance, Bruker). The XPS and UPS were performed on a Thermo Scientific ESCA LAB250Xi system. An Al Kα 1486.8 eV X-ray radiation source was used for XPS. PLQY was measured by a constructed system, including an integrating sphere (Gigahertz-Optik), an Andor Kymera 193i spectrograph and a 660 nm continuous-wave laser (OBIS, Coherent) set at 1 Sun equivalent photon flux (elliptical beam profile with diameters (FWHM): 1.910 mm/2.330 mm, 2.35 mW), the PL signal was collected at normal incidence using an optical fiber.
ToF-SIMS measurements were conducted using a M6 Hybrid SIMS instrument (Model ION-TOF GmbH, Germany) equipped with Bi cluster, argon gas cluster and Cs ion sources. The analysis source was a 30-keV Bi3+ with a target current of 0.35 pA over an analysis area of 200 × 200 μm2. The sputter ion source was a 10-keV Ar+ cluster ion beam with a target current of 11 nA inside the sputtering raster area of 800 × 800 μm2.
The J–V curves of the PSCs (under simulated AM 1.5 G 1-sun illumination solar simulator (Sol3A Class AAA, Oriel, Newport, USA)) were measured by Keithley (2400, USA) in ambient atmosphere. The silicon cell (91150V-KG3, Newport, USA) was used to calibrate the light intensity. The effective area of the PSCs is 0.049 cm2, which was determined by a metal mask. The scan rate is 0.1 V s−1 and 0.2 s for every point from −0.1 to 1.2 V and 1.2 to −0.1 V under ambient atmosphere (20–25 °C, RH ~ 40%). The J–V curve of large area module was conducted without mask and under ≈400 mm diameter 1-sun illumination solar simulator. The module area was all areas except for electrodes and edges, including P1, P2, and P3. The EQE was recorded under a constant white light. The excitation beam coming from a 300 W Xenon lamp (ILC Technology) was focused through a Gemini-180 double monochromator (Jobin Yvon Ltd) and chopped at ≈2 Hz. The signal was recorded using a Model SR830 DSP Lock-In Amplifier (Stanford Research Systems).
A candlelight system (Switzerland) was used in a nitrogen glove box for operational long-term stability testing of unencapsulated PSCs. Under the room temperature and continuous white LED illumination (1 sun) (LED spectrum was provided in Supplementary Fig. 31), the performance of PSCs is measured every 12 h. The thermal stability was measured unencapsulated PSCs, which were placed on a hot plate (85 °C) open-circuit condition in nitrogen glove box.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the main data are available in the main text, the Supplementary Information, and the Source data file. All other data of this study are available from the corresponding authors on request. Source data are provided with this paper.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Shi, D. et al. Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science 347, 519–522 (2015).
Stranks, S. D. et al. Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 342, 341–344 (2013).
Dong, Q. et al. Electron-hole diffusion lengths >175 μm in solution-grown CH3NH3PbI3 single crystals. Science 347, 967–970 (2015).
Rong, Y. et al. Challenges for commercializing perovskite solar cells. Science 361, eaat8235 (2018).
Lin, R. et al. All-perovskite tandem solar cells with 3D/3D bilayer perovskite heterojunction. Nature 620, 994–1000 (2023).
Kim, H. J., Han, G. S. & Jung, H. S. Managing the lifecycle of perovskite solar cells: addressing stability and environmental concerns from utilization to end-of-life. eScience 4, 100243 (2024).
National Renewable Energy Laboratory, best research-cell efficiencies chart. https://www.nrel.gov/pv/cellefficiency.html (2025).
Domanski, K. et al. Migration of cations induces reversible performance losses over day/night cycling in perovskite solar cells. Energy Environ. Sci. 10, 604–613 (2017).
Bai, S. et al. Planar perovskite solar cells with long-term stability using ionic liquid additives. Nature 571, 245–250 (2019).
Zhang, J., et al. Inhibiting ion migration through chemical polymerization and chemical chelation toward stable perovskite solar cells. Angew. Chem. Int. Ed. 135, e202314106 (2023).
Divitini, G. et al. In situ observation of heat-induced degradation of perovskite solar cells. Nat. Energy 1, 15012 (2016).
Pan, Y. et al. Surface chemical polishing and passivation minimize non-radiative recombination for all-perovskite tandem solar cells. Nat. Commun. 15, 7335 (2024).
Wen, Y. et al. Amorphous (lysine)2PbI2 layer enhanced perovskite photovoltaics. Nat. Commun. 15, 7085 (2024).
Li, X. et al. Bifunctional ligand-induced preferred crystal orientation enables highly efficient perovskite solar cells. Joule 8, 3169–3185 (2024).
Alharbi, E. A. et al. Cooperative passivation of perovskite solar cells by alkyldimethylammonium halide amphiphiles. Joule 7, 183–200 (2023).
Frohna, K. et al. The impact of interfacial quality and nanoscale performance disorder on the stability of alloyed perovskite solar cells. Nat. Energy 10, 66–76 (2025).
Ni, Z. et al. High grain boundary recombination velocity in polycrystalline metal halide perovskites. Sci. Adv. 8, eabq8345 (2022).
Wang, W.-T. et al. Water- and heat-activated dynamic passivation for perovskite photovoltaics. Nature 632, 294–300 (2024).
Chen, J. et al. Surface molecular engineering for fully textured perovskite/silicon tandem solar cells. Angew. Chem. Int. Ed. 63, e202407151 (2024).
Zhang, S. et al. Surface passivation with tailoring organic potassium salt for efficient FAPbI3 perovskite solar cells and modules. Adv. Funct. Mater. 34, 2401945 (2024).
Zhang, X. et al. Machine learning for screening small molecules as passivation materials for enhanced perovskite solar cells. Adv. Funct. Mater. 34, 2314529 (2024).
Zhao, C. et al. Stabilization of highly efficient perovskite solar cells with a tailored supramolecular interface. Nat. Commun. 15, 7139 (2024).
Xiao, T. et al. Preparation of a fixed-tetraphenylethylene motif bridged ditopic benzo-21-crown-7 and its application for constructing AIE supramolecular polymers. Chin. Chem. Lett. 32, 1377–1380 (2021).
Dang, L. X. Mechanism and thermodynamics of ion selectivity in aqueous solutions of 18-crown-6 ether: a molecular dynamics study. J. Am. Chem. Soc. 117, 6954–6960 (1995).
Wang, Z. et al. Suppressed phase segregation for triple-junction perovskite solar cells. Nature 618, 74–79 (2023).
Kubicki, D. J. et al. Phase segregation in Cs-, Rb- and K-doped mixed-cation (MA)x(FA)1-xPbI3 hybrid perovskites from solid-state NMR. J. Am. Chem. Soc. 139, 14173–14180 (2017).
Gunes, U. et al. Formamidinium incorporates into Rb-based non-perovskite phases in solar cell formulations. Angew. Chem. Int. Ed. 64, e202416938 (2025).
Su, T. S. et al. Crown ether modulation enables over 23% efficient formamidinium-based perovskite solar cells. J. Am. Chem. Soc. 142, 19980–19991 (2020).
Tian, W. et al. Limiting perovskite solar cell performance by heterogeneous carrier extraction. Angew. Chem. Int. Ed. 55, 13067–13071 (2016).
Zhao, Q. et al. Oxygen vacancy mediation in SnO2 electron transport layers enables efficient, stable, and scalable perovskite solar cells. J. Am. Chem. Soc. 146, 19108–19117 (2024).
Tian, W., Zhao, C., Leng, J., Cui, R. & Jin, S. Visualizing carrier diffusion in individual single-crystal organolead halide perovskite nanowires and nanoplates. J. Am. Chem. Soc. 137, 12458–12461 (2015).
Yin, Y., Tian, W., Leng, J., Bian, J. & Jin, S. Carrier transport limited by trap state in Cs2AgBiBr6 double perovskites. J. Phys. Chem. Lett. 11, 6956–6963 (2020).
Delor, M., Weaver, H., Yu, Q. & Ginsberg, N. Imaging material functionality through three-dimensional nanoscale tracking of energy flow. Nat. Mater. 19, 56–62 (2020).
Wang, X. et al. PbI6 octahedra stabilization strategy based on π-π stacking small molecule toward highly efficient and stable perovskite solar cells. Adv. Energy Mater. 13, 2203635 (2023).
Liu, D. et al. Polymerization strategies to construct a 3D polymer passivation network toward high performance perovskite solar cells. Angew. Chem. Int. Ed. 62, e202301574 (2023).
Zheng, Y. et al. Managing interfacial hot-carrier cooling and extraction kinetics for inverted Ma-free perovskite solar cells over 23% efficiency via Dion-Jacobson 2D capping layer. Adv. Funct. Mater. 33, 2300576 (2023).
Wang, Y. et al. Grain boundary elimination via recrystallization-assisted vapor deposition for efficient and stable perovskite solar cells and modules. Adv. Mater. 35, 2304625 (2023).
Wang, P. et al. Precise anchoring of Pb-based defects for efficient perovskite solar cells: a universal strategy from lab-scale small-area devices to large-area modules. J. Phys. Chem. Lett. 16, 2695–2704 (2025).
Kim, J. et al. 87Rb, 85Rb, and 39K NMR studies of alkalides, electrides, and related compounds. J. Phys. Chem. 100, 2885–2891 (1996).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (12104081 for M.W., 52272193 for Y.S., 22122307 for W.T., 22403093 for Y.Y.). M.W. appreciates the Natural Science Foundation of Liaoning (2023-BS-063), the China Scholarship Council, and the open fund of the state key laboratory of molecular reaction dynamics in DICP, CAS. W.T. appreciates the Natural Science Foundation of Liaoning (2024JH3/50100010) and the Dalian Science and Technology Innovation Fund (2024RJ006). Y.S. appreciates the Fundamental Research Funds for the Central Universities (DUT22LAB602), and Y.Y. appreciates the Natural Science Innovation Team of Anhui Provincial Universities (2023AH010065).
Author information
Authors and Affiliations
Contributions
M.W., Y.S., W.T., L.Z., and M.G. conceived the idea and designed the experiments. M.W., L.Z., P.W., J.C., and J.B. supervised the experimental investigation and manuscript preparation. M.W., P.W., Y.Y., and K.H. fabricated and characterized perovskite films and devices. W.T., Y.Y., P.X., and S.J. contributed to the tracking and analysis of carrier dynamics process. Y.H. contributed to the preparation and testing of large-area PSC. P.W. and J.G. contributed to the morphology characterization. M.W., W.S., and T.F. participated in the DFT calculation. U.G., T.G., and L.E. contributed to the NMR measurement and analysis. Z.Y., R.C., and J.X. contributed to the ToF-SIMS, optical and electrical characterization of perovskite films. S.Z., S.J., and X.J. participated in the data analysis and result discussions. All authors contributed to suggestions for the manuscript and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kenji Katayama, Ji-youn Seo, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, M., Yin, Y., Wang, P. et al. In-situ boundary bridging unlocks multi-grain-domain carrier diffusion in polycrystalline metal halide perovskites. Nat Commun 16, 8755 (2025). https://doi.org/10.1038/s41467-025-63777-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63777-5
