
Abstract
The accumulation of intracellular aggregates of Tau protein is one main hallmark of Alzheimer’s disease (AD) and is the consequence of Tau conformational changes, increased phosphorylation, and self-association to form fibrillar aggregates. This pathological process prevents the physiological interaction of Tau with microtubules to the detriment of the structural integrity of neurons. In healthy cells, aberrant protein misfolding and aggregation are counteracted by chaperone proteins whose protective capacity decreases with age. The role of the chaperone Hsp90 and the mechanism by which it can prevent Tau aggregation are controversial. In this work, the strategy of mimicking Hsp90 through the design of the β-hairpin like peptidomimetic β-Hsp90, inspired by two Hsp90/Tau interaction sequences, is presented. β-Hsp90 inhibits Tau aggregation both in vitro and in cells, restoring Tau’s physiological interaction with microtubules. β-Hsp90, which interacts with the P1 region of Tau, is more effective than individual peptide sequences from the chaperone HSP90 and another β-hairpin mimic based on Tau sequences. Moreover, β-Hsp90 reduces AD-associated Aβ1-42 aggregation, offering the development of a dual inhibitor. This work paves the way for the design of new drugs targeting devastating untreated amyloid diseases, by mimicking physiological chaperones with small synthetic peptide drugs.
Similar content being viewed by others


Introduction
The accumulation of protein deposits in the brain is a common hallmark of many neurodegenerative pathologies, including Alzheimer’s (AD), Parkinson’s (PD), and Huntington’s diseases (HD), among others1. The misfolding and aggregation of these proteins (amyloid-β (Aβ) and Tau in AD, α-synuclein in PD, and Huntingtin in HD)2,3,4 lead either to the formation of toxic species or to the depletion of a functional protein which plays an important physiological role in the homeostasis of neuronal cells. The incidence of these pathologies, called amyloid diseases, increases with the aging of the population and constitutes a major threat to all health systems. AD is the most prevalent devastating neurodegenerative disorder and the most prevalent form of tauopathies5. Tau is a member of the microtubule-associated protein (MAP) family, and its physiological role is to promote the assembly and dynamics of microtubules (MTs), which are essential for axonal transport and maintenance of structural integrity of neurons6,7,8,9. Under pathological conditions, Tau monomers undergo conformational changes and increased phosphorylation at selected sites, which are associated with the development of abnormal soluble oligomers and filamentous aggregates, which are unanimously considered to cause degeneration of neuronal and glial cells5,8. AD is also characterized by the formation of other amyloid aggregates composed mainly of Aβ1-42. Many efforts have been made to develop a treatment for AD, mainly with single-target drugs (Tau or Aβ1-42), but unfortunately, most of them have failed10,11. Despite their controversial utility and high cost, three antibodies targeting Aβ1-42, Aducanumab (FDA approval in June 2021), Lecanemab (approved in July 2023 by the FDA and in November 2024 by the EMA)12, and Donanemab (FDA approval in July 2024)13, have recently arrived on the market, ushering in the last three decades of the amyloid hypothesis. However, to date, no drug targeting Tau has been approved for the therapeutic treatment of AD. Targeting cross-protein-protein interactions (PPIs) involving Aβ or Tau proteins is a recent and promising strategy to explore. PPIs are fundamental in maintaining cellular functions, but many PPIs are aberrant in the pathogenesis of numerous diseases, such as AD14. Deciphering the protein partners of Aβ and tau proteins and determining whether these PPIs are beneficial or harmful is still far from being understood and exploited for therapeutic strategies15,16,17,18,19. PPIs involving chaperone proteins are of major interest, and a hitherto unexplored strategy for combating AD, as well as other amyloidoses, consists in drawing inspiration from chaperone proteins to design effective inhibitors against pathological aggregation processes. Indeed, in healthy cells, aberrant protein misfolding and aggregation are counteracted by a group of proteins called molecular chaperones, considered to be part of the cellular quality control machinery20,21,22. Neurons rely on molecular chaperones to cope with misfolded protein levels throughout their lifespan. However, with age, the protective capacity of molecular chaperones decreases and becomes less responsive to stress signals, allowing misfolded protein species to accumulate and eventually aggregate23,24. Until now, how molecular chaperones defend neurons against the accumulation of misfolded proteins remains unclear25. Tau and Aβ (as well as many other amyloid proteins) are hybrid proteins that possess both structured domains and intrinsically disordered protein (IDP) regions26. Chaperones interacting with IDPs could exhibit a holdase mechanism, which involves capturing free unfolded proteins and maintaining them in soluble forms27. However, full-length physiological chaperones cannot be effectively used as drugs, mainly due to pharmacokinetic and cost issues. We considered the very little explored strategy to increase the proteostasis capacity by mimicking natural protein chaperones with small peptide drugs to combat tauopathies. To our knowledge, the strategy of designing small peptides derived from natural chaperone sequences has only been reported to prevent Aβ aggregation, one using a 19-residue peptide based on αA-crystallin28, and the other developed by some of us using diaza-nonapeptides based on Transthyretin29. However, this strategy has not been investigated for tauopathies and is the focus of this promising new study presented here.
The heat shock protein 90 kDa (Hsp90) chaperone particularly attracted our attention because the role of Hsp90 in neurodegeneration is rather controversial. In particular, the exact activities and molecular mechanism of the Hsp90-Tau complex remain enigmatic. Regarding the activity of Hsp90, its interaction with Tau appears to either promote or protect Tau against degradation and aggregation, depending in particular on the associated co-chaperone30,31,32,33,34. However, there is limited evidence that a direct interaction of HSP90 with tau affects its aggregation. Hsp90 alone has been shown to inhibit the formation of tau fibrils but promote the formation of small tau oligomers35,36. In complex with the cis–trans peptidyl-prolyl isomerase FKBP51, Hsp90 also acts to promote Tau oligomers formation37. Interestingly, the dihydropyridine derivative LA1011 was reported as interacting with HSP90 to disrupt the binding of FKBP51 (the increase of FKBP51 seems to be a major player in hyperphosphorylated Tau aggregation), explaining its activity to reduce tau pathology and amyloid plaque formation in transgenic AD mice38,39. On another hand, the Hsp70/Hsp90 machinery counteracts the aggregation of the highly affine phosphorylated Tau40. Regarding the binding sites of Tau to Hsp90, they are reported as highly polymorphic. A direct interaction of Hsp90 with Tau repeat domain (Tau-RD) has been mainly reported, while this region is also involved in the binding of Tau to MTs (Tau-RD is also called microtubule-binding region, MTBR, Fig. 1A) and in its self-aggregation41,42. However, in the ternary Hsp90/FKBP51/Tau complex, an interaction of the proline-rich region P1 of Tau with the Hsp90 N-domain was observed37. Phosphorylated Tau has been also reported to interact with the Hsp70/Hsp90 complex at its proline-rich region P2 and its MTBR flanking pseudo-repeat R’40. These observations suggest that Hsp90 can act by interacting through the MT-binding domain to favor aggregates that might be less toxic than oligomers and fibrils, and that this Hsp90/Tau complex might prevent deleterious interactions of Tau aggregates with cytoplasmic proteins. In multi-complex systems, Hsp90 rather interacts with the proline-rich region P1/P2 and R’ flanking Tau-RD to prevent or increase Tau oligomerization. The question of whether the Hsp90-Tau interaction allows to preserve the physiological interaction between Tau MTBR and MT has not yet been addressed.

A Structure of Tau: schematic representation of Tau longest CNS isoform; B Schematic representation of Tau folded structure when interacting with MT6; C Chemical representation of Tau-MTs β-hairpin mimic compound (β-Tau); D Crystal structure of the Hsp90 N-terminal domain in complex with EC44 (PDB = 3NMQ) and schematic representation of the spatial distributions of β-strands placed at the crystal structure of the Hsp90 N-terminus (M = middle domain, N = N-terminal domain); E Primary structure of the Hsp90 N-terminal region highlighting key interaction sites according to Karagöz et al.41 β-strands in blue, helical structures in gray, strong hydrophobic interactions in red, and significant mono-amino acid interactions in green. F Chemical depiction of the designed peptides (H1, S4, S6, S7, S4short, S7short) and β-hairpin mimics β-Hsp90 inspired by the Tau-NHsp90 interaction. Figure 1D was adapted from Fig. 4A of Karagoz et al. Hsp90-Tau Complex Reveals Molecular Basis for Specificity in Chaperone Action, 201441.
Interestingly, for AD, although the Hsp90/Aβ complex has not been studied much, Hsp90 has also been described to be able to inhibit Aβ aggregation43,44.
In this work, in light of these considerations, the strategy of mimicking Hsp90 by small peptidomimetics inspired by Hsp90-tau interaction sequences is presented to further explore the effects of HSP90-tau interaction on tau aggregation and tau-MT interaction. The interest in using peptide derivatives rather than using small non-peptide compounds to target specifically PPIs in order to either inhibit or mimic these PPIs is very well established. Their ability to mimic and modulate the large and relatively flexible surface areas of the hot-spots, i.e., peptide sequences located at PPIs, makes them more attractive than conventional small molecules14,45,46. Furthermore, they offer several advantages such as greater affinity, selectivity, and safety, owing to their intermediate size between small molecule drugs and protein therapeutics47. However, the major drawbacks of small peptides are their proteolytic instability and their low ability to adopt stable conformations, while PPI hot-spots generally involve well-defined secondary structures47,48. The introduction of non-natural amino acids into peptide derivatives, inspired by the sequences and structural features of PPIs, makes it possible to induce bioactive conformations and increase their stability towards proteolysis48,49. These new small peptidomimetics inspired by Hsp90-Tau interaction sequences have been employed here both as exploratory tools to provide some useful insights into the mechanism and physiological role of Hsp90 on Tau, and to achieve efficient inhibitors of Tau aggregation able to restore physiological Tau-MT interaction. A flexible β-hairpin mimic, β-Hsp90, built on a piperidine-pyrrolidine β-turn inducer and bearing defined sequences from Hsp90, demonstrated to be a strong inhibitor of both wild-type Tau (Wt-Tau) and pro-aggregative ΔK280 Tau mutant fibrillization, as assessed by Thioflavin-T (ThT) fluorescence spectroscopy. This design was compared to the canonical self-recognition elements (SREs) approach based on specific Tau sequences (compound β-Tau). Transmission electron microscopy (TEM) images gave information about the morphology of Tau species in the presence of these new Tau aggregation inhibitors. Then, through live cell imaging, FDAP (fluorescence decay after photoactivation), the anti-aggregative property of compound β-Hsp90 was confirmed in a cellular context, also highlighting its ability to restore Tau-MT interaction to physiological levels. The unprecedented involvement of the P1 region of Tau in the interaction with this efficient compound was proven thanks to 15N-HSQC titrations of 15N-Tau. Furthermore, the ability of β-Hsp90 to act as a dual inhibitor of Tau and Aβ1-42 aggregation was shown by ThT and TEM experiments conducted on Aβ1-42. These results open new prospects for future research in the field of Alzheimer’s therapy, considering that combination therapies and dual targeting molecules, such as dual inhibitors of Tau and Aβ aggregation, have already been suggested as promising therapeutic strategies, with respect to single-target approaches10.
Results
Designing β-hairpin mimics to interact with Tau and inhibit its aggregation
The effectiveness of rationally designed acyclic β-hairpin mimics, built on a piperidine-pyrrolidine β-turn inducer and bearing two small peptides based on SRE sequences involved in the misfolding of Aβ1-42 or hIAPP, in selectively inhibiting either Aβ1-42 or hIAPP aggregation processes, has already been shown by some of us50,51,52. For Tau, the aggregative SRE sequences are essentially localized in the 306-378 region located in the MTBR53, while in physiological conditions, this sequence actively participates in the binding of Tau with MTs. Thus, in a first approach, a selective peptidomimetic targeting Tau was designed taking inspiration from the secondary structure and self-interactions that Tau engages when it is bound to MTs6, with the objective to stabilize this conformation and delay the misfolding towards pro-aggregative conformations. However, we were aware of the risk that mimicking this interaction zone could compete and compromise the physiological interaction of Tau with MTs. By exploring the conformation that Tau adopts when binding to MTs using NMR analysis, Kadavath et al. noticed that the two hexapeptide sequences, PHF6* (275VQIINK280 at the beginning of R2) and PHF6 (306VQIVYK311 at the beginning of R3. Figure 1A), form a hairpin-like structure allowing the stabilization of the Tau-MT interaction (Fig. 1B)6. Noteworthy, these sequences are also known to be the “hot spots” of Tau self-association, spontaneously aggregating in solution into β-sheet structures and triggering both the oligomerization and the fibril growth54,55. Therefore, considering that the hairpin conformation is stabilized during the interaction with MTs and that these two sequences are possibly hidden into the globular form of the full-length protein, thereby reducing their solvent exposure and their aggregation propensity, the two PHF6 and PHF6* sequences have been inserted into the piperidine-pyrrolidine synthetic β-turn inducer to mimic this kind of β-hairpin. Our hypothesis is that this pre-organized conformation might increase the interaction with Tau and behave as a conformational pillar able to induce the full-length Tau (TauFL) protein to adopt and maintain its physiological shape for its interaction with MTs (compound named β-Tau, Strategy A, Fig. 1C).
Secondly, given the intriguing role of the chaperone machinery, notably Hsp90, in regulating Tau misfolding and aggregation, our second design approach focused on mimicking the Hsp90-Tau interaction. This novel synthetic chaperone was hypothesized to reduce Tau fibrillization, and it might help to clarify whether the reported promotion of oligomerization exerted by Hsp90 on Tau is detrimental or beneficial for its interaction with MTs in a cellular context. Thus, the sequences of Hsp90 suspected to be the recognition elements for its interaction with Tau were identified and inserted in a β-hairpin template to mimic the core element of the interaction. The choice of these sequences referred to a comprehensive study employing Small-Angle X-ray Scattering (SAXS) and Nuclear Magnetic Resonance (NMR) spectroscopy to elucidate structural details of the Tau-Hsp90 complex (Fig. 1D)41. It is, however, essential to emphasize that, although this study disclosed the most prominent interacting regions (MTBR for Tau and N-terminal and central domains of Hsp90), the exact interacting residues involved in both proteins remain elusive, limiting the design of Hsp90-based inhibitors. To bridge this gap, the aforementioned study41 has been combined to a mapping of the crystallographic structure of the N-terminal domain of Hsp90 (NHsp90; PDB = 3NMQ), highlighting key interacting regions (helices, β-sheets) and single amino acids hot spots (negative charges, hydrophobic sites and single interactions) within the Hsp90-Tau complex (Fig. 1E, Strategy B, Right side). This comprehensive analysis facilitated localizing the most relevant interacting regions and possible binding sites of NHsp90 with Tau (Fig. 1D, E), leading to the identification of four sequences suspected to play a pivotal role in the interaction (NHelix 1 as compound H1, NStrand 4 as compound S4, NStrand 6 as compound S6 and NStrand 7 as compound S7, Fig. 1E). Notably, two of the selected sequences, S4 and S7, exhibited the highest density of interacting sites and were spatially close, facing each other in an antiparallel orientation, albeit not directly connected. Consequently, the core section of these sequences (each consisting of 6 amino acids: 151TIVVVK146 and 183TKVILH188) was kept and incorporated into the piperidine-pyrrolidine β-turn inducer (compound β-Hsp90, Fig. 1F). Additionally, to further assess and underline the importance of the β-hairpin like structure of β-Hsp90 for an optimal interaction with the target protein, the shorter S4 and S7 hexapeptides, inserted into the β-hairpin mimic β-Hsp90, were also synthesized (compounds S4short and S7short, Fig. 1F).
Synthesis and conformational analysis
The preparation of the β-hairpin mimics β-Tau and β-Hsp90 and of the peptides H1, S4, S6, S7, S4short, S7short was performed with satisfactory yields using classical Fmoc-based solid-phase peptide synthesis (SPPS) on the Rink amide resin (more detailed information and characterization of the compounds in supporting information). The preparation of the piperidine-pyrrolidine β-turn scaffold was done accordingly to our previous publications50,51,52.
Conformational analysis of compounds β-Tau and β-Hsp90 was performed using Circular Dichroism spectroscopy (CD) in 20 mM phosphate buffer (pH 7.4) at 20 °C and 37 °C, showing for both compounds in aqueous medium a dynamic equilibrium between β-sheet conformation (negative Cotton effect at 218 nm) and random coil (negative band at 195 nm) structure as demonstrated by deconvolution56 (~30% β-sheet/60% random coil, see SI, Supplementary Fig. 12 and Fig. 13, and Supplementary Table 6). However, in MeOH, both compounds at 125 µM showed the typical bands of a β-sheet structure with β-Tau having a more pronounced tendency to aggregate and precipitate than β-Hsp90 (see the decrease of CD signal intensity for β-Tau in SI, Supplementary Fig. 14 and Supplementary Table 6 for deconvolution). These results are consistent with our previous studies using NMR, CD and molecular dynamics (MD) on this class of compounds based on the same piperidine-pyrrolidine β-turn inducer, that showed a distinct degree of flexibility according to the peptide sequences50,51,52. CD analysis of compound H1 (NHelix 1) was also performed to evaluate its ability to maintain its helical structure outside the protein context. However, due to its limited solubility in aqueous medium, the CD analysis was only conducted in MeOH at 37 °C. Remarkably, H1 exhibited a typical helical structure characterized by two consecutive negative minima at 218 and 209 nm and a positive maximum at 195 nm (SI supplementary Fig. 15). CD spectra for compounds S4, S6, S7, S4short and S7short were not registered, as their short peptide length was not expected to stabilize any significant secondary structure.
Mechanistic investigations by in vitro and cellular aggregation assays and by Tau-compounds interaction
Thioflavin-T (ThT) fluorescence spectroscopy is the standard technics used to study the aggregation kinetics of amyloidogenic proteins due to the ability of ThT to fluoresce upon binding to β-sheet rich structures57,58. The aggregation curve of an amyloid protein alone classically displays a sigmoidal pattern, characterized by 3 different portions: i) an initial lag phase also called primary nucleation phase, characterized by the absence of a fluorescent signal due to the lack of β-sheet rich structures of the initial nuclei and small “on-pathway” oligomers; ii) a growth phase, also called elongation phase corresponding to the fast increment of the fluorescent signal caused by the formation of bigger β-sheet rich species (secondary nucleation); iii) a final plateau reached when the maximal level of mature fibrils is reached59. The ability of the compounds to modulate the Ac-PHF6* hexapeptide model, Wt-Tau and Tau-ΔK280 mutant, as well as Aβ1-42and hIAPP (to evaluate the specificity of the compounds) aggregation was assessed by ThT fluorescence spectroscopy, considering the time of the half-aggregation (t1/2), giving insight into the overall kinetics of aggregation and the intensity of the experimental fluorescence plateau (F), assumed to be influenced by the β-sheet content, morphology and quantity of aggregates formed. TEM was used to confirm promising ThT results on Tau and Aβ1-42 and to give information on the morphology of Tau. The most promising peptide compounds were evaluated for their stability towards proteolysis, their ability to restaure Tau-MT interaction in cells, and to provide molecular information of their interaction with Tau by NMR.
β-Hairpin mimetics and NStrand 7 of Hsp90 inhibit the aggregation of Ac-PHF6* hexapeptide model
Among the many short peptide models reported to mimic Tau aggregation without using TauFL60,61,62, the Ac-PHF6* hexapeptide model is one of the latest proposed; it is cheap and easy to synthesize and to handle, robust and able to aggregate rapidly63. Furthermore, the VQIINK segment (PHF6*) has been described as the more powerful driver of tau aggregation by Seidler et al.64 Thus, this model was used to screen the activity of all the prepared compounds (β-Tau, β-Hsp90, H1, S4, S6, S7, S4short and S7short) at 5/1, 1/1 and 0.1/1 compound/Ac-PHF6* ratios. Due to the high aggregative propensities of Ac-PHF6*, the lag phase period was not observed, thus t1/2 could not be calculated (Supplementary Fig. 1), and only the relative maximum of fluorescence F with respect to the control curve was considered (Fig. 2A, Supplementary Fig. 1 and Table 1).

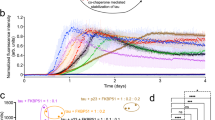
A ThT-fluorescence at 240 min of compounds β-Tau, β-Hsp90, H1, S4, S6, S7, S4short and S7short related to the maximum fluorescence obtained for the ThT-fluorescence control curve of Ac-PHF6* at 5/1 (green bars), 1/1 (yellow bars) and 0.1/1 (blue bars) compound/Ac-PHF6* ratios. Parameters are expressed as mean ± SE, n = 3 (technical replicates). Compounds were dissolved in water. The concentration of Ac-PHF6* was 25 µM. The fluorescence for Ac-PHF6* control was set at 100% for the maximal fluorescence and the relative fluorescence was calculated related to this value. B, C Effects of compounds β-Tau, β-Hsp90, S4, S7, S4short, S7short, MB and β-hIAPP on Wt-Tau:Hep fibrillization assessed by ThT-fluorescence spectroscopy at 5/1 (green bars) and 1/1 (yellow bars) ratios of compound/Wt-Tau. Parameters are expressed as mean ± SE, n = 2 or 3 (technical replicates). Compounds were dissolved in water. The concentration of Wt-Tau was 10 µM. B The relative t1/2 (%) was calculated related to the t1/2 of Wt-Tau control (see “Methods” for the detailed calculations). NA = No Aggregation over 30 h and (C) The relative F (%) was calculated at 30 h related to the fluorescence plateau of Wt-Tau control. D Transmission electron micrographs of Wt-Tau with inserted width distribution of the observed ordered structures. Fibrils of Wt-Tau: Hep (control); short nanotube structures of Wt-Tau: Hep in the presence of β-Hsp90 (1/1 and 1/5 ratios, respectively); amorphous aggregates of Wt-Tau in the presence of β-Tau (1/1 and 1/5 ratios, respectively) and S7 (1/1 and 1/5 ratios, respectively).
Compound H1 (NHelix 1) alone showed strong ThT fluorescence at 125 µM suggesting a self-aggregating behavior, which can be due to its hydrophobic character and its low solubility in aqueous medium (Supplementary Fig. 1). At 5/1 and 1/1 ratios (H1/Ac-PHF6*), the increase of F (+143% and +33% respectively) suggested a co-aggregation process of H1 with Ac-PHF6* or rather simply the addition of the two individual F values. On the contrary, the two β-hairpin mimics β-Tau and β-Hsp90, along with the Hsp90-derived peptides S4, S6, S7, S4short and S7short, displayed excellent activities at a 5/1 ratio, almost completely suppressing the fluorescence. At stoichiometric ratio, only the hairpin mimics β-Tau, β-Hsp90 and peptide S7 maintained excellent activities (respectively, F = −81%; −75%; −97%) while S4, S6 and S7short displayed a pro-aggregative effect with an overall increment in the detected fluorescent level up to +155%. Remarkably, at a sub-stoichiometric ratio (0.1/1), only the β-hairpin mimics β-Tau and β-Hsp90 exhibited activity, with β-Tau in particular giving comparable results to those observed at higher ratios (F = −87%). Interestingly, the simultaneous presence of both S4short and S7short at 2.5 + 2.5/1 ratio let to maintain a strong activity which is reduced at 0.5 + 0.5 /1 ratio while no major effect was observed in 0.05 + 0.05/1 ratio (Supplementary Fig. 1). These preliminary results on Ac-PHF6* hexapeptide model underly the relevance of the hairpin secondary structure and of the two rational design strategies based on Tau or Hsp90 sequences.
β-Hairpin mimetics inhibit the fibril formation of the full-length Wt-Tau in vitro
The addition of anionic co-factors like heparin is mandatory to induce the aggregation of TauFL. However, heparin-induced TauFL aggregates are different from those observed in diseases65,66. Thus, Tau aggregation assay was performed in 25 mM phosphate buffer at pH 6.6, as reported by Huvent et al.67 but with a decreased Tau/heparin ratio (160/1), to induce the Tau assembly in an almost co-factor-free environment68. The ThT-fluorescence curves were nicely reproducible and characterized by a sigmoidal shape with a lag phase of around 5 h followed by an elongation phase and a final plateau reached after 12 h. The two parameters t1/2 and F were used to evaluate the activity of compounds β-Tau, β-Hsp90, S4, S6, S7, S4short and S7short, which were compared to the activity of Methylene Blue (MB, methylthioninium chloride, considered as a reference Tau inhibitor, analog of the reduced form hydromethylthionine mesylate, LMTM or TRx0237, which has been in phase 3 clinical trials conducted by TauRx Therapeutics Ltd for mild to moderate AD (Fig. 2B,C, Supplementary Fig. 2 and Supplementary Table 2).
Only β-hairpin mimics β-Tau, β-Hsp90, peptide S7, and MB showed a complete inhibition of the aggregation of Wt-Tau at a 5/1 ratio. In contrast with MB, β-hairpin mimics β-Tau and β-Hsp90 and peptide S7 were still very active at 1/1 ratio to delay the aggregation (t1/2 = +55% for MB; +266% for β-Tau; + 249% for β-Hsp90 and N.A. for S7) and reduce F (F = − 86%; −29%; −84%; −94% respectively), with β-Hsp90 and peptide S7 being the most active compounds on both parameters (Figs. 2B,C, Supplementary Fig. 2 and Table 2). At the very low ratio 0.1/1, β-Tau, β-Hsp90, peptide S7 and MB showed no significant inhibitory effect (consistently with literature data69, MB did not delay the kinetics but decreased F by 50%). The rationale of the choice of the peptide sequences was also challenged by the evaluation of a similar β-hairpin mimic (β-hIAPP), recently reported by some of us52, based on human islet amyloid polypeptide (hIAPP, also known as amylin) sequences (A13N14F15L16V17 and N22F23G24A25I26L27) and whose aggregation is involved in type 2 Diabetes (T2D). As expected, β-hIAPP was much less active with no activity on F at any ratio, only a small ability to delay the aggregation kinetics at 5/1 ratio and even slightly speeding up the aggregation process at 1/1 and 0.1/1 ratios (Figs. 2B,C, Supplementary Fig. 2 and Supplementary Table 2).
The superior inhibitory effect of β-Hsp90 among our lead compounds was next confirmed by transmission electron microscopy (TEM) experiments of Wt-Tau at the end of the aggregation process (96 h). At the low ratio of heparin used for the ThT assays (Tau/heparin ratio 160/1), control Wt-Tau formed long amyloid fibrils with an average diameter of 12.6 ± 0.1 nm with various morphologies as previously reported (Fig. 2D)65.
In the presence of our lead compounds β-Tau, β-Hsp90, and S7 at Tau: compound ratios of 5:1 and 1:1, the characteristic Wt-Tau fibril formation was widely perturbed (Fig. 2D). Notably, in the presence of β-Hsp90, short rod-like nanostructures, that were undetectable by ThT assay, were observed. At Wt-Tau:Hep:β-Hsp90 ratio of 1:0.1:1, these rod-like nanostructures exhibited an average width of 18.5 ± 0.1 nm, with some structures partially open, revealing their internal morphology. Increasing the β-Hsp90 concentration fivefold resulted in sparsely populated grids, where only planar rod-like structures with an average width of 15.7 ± 0.2 nm were detected. These observations suggest that β-Hsp90 stabilizes intermediate structures, thereby inhibiting the formation of mature amyloid fibrils.
In contrast, in the presence of β-Tau and S7 at all tested concentrations, aggregates were observed. Given their positive ThT signals, these aggregates likely contain significant β-sheet content.
We further investigated the effect of the chaperone-mimicking hairpin compound β-Hsp90 at a higher heparin ratio (Tau/Hep 4/1). Under these conditions, Wt-Tau fibrils are formed more rapidly compared to the lower heparin ratio (Tau/Hep 160/1). Interestingly, in the presence of β-Hsp90, fibril formation was completely inhibited, and only amorphous structures—negative in the ThT assay—were observed (Supplementary Fig. 3).
Together, these results demonstrate that all tested compounds effectively inhibit Wt-Tau: Hep amyloid fibril formation.
Stability toward proteolysis
Before the cell evaluation of the most active compounds β-Tau, β-Hsp90 and S7, their stability towards proteolysis was assessed. The aim was to observe whether the incorporation of the unnatural piperidine-pyrrolidine β-turn inducer could limit enzymatic degradation by comparing the stability of β-Hsp90 with S4short, S7short. Stability studies are a fundamental phase of drug development, especially with peptide-based compounds, which are easily degraded by proteolytic enzymes ubiquitously found inside and outside cells. In the Pronase medium, which has a broad specificity (mixture of neutral protease, chymotrypsin, trypsin, carboxypeptidase, and aminopeptidase), natural peptide S7 and S7short were highly vulnerable to proteolysis, with S7 completely degraded and only 46% of S7short remaining after 30 min (Supplementary Table 3). Conversely, the shorter peptides S4short and β-Tau displayed more pronounced stability with 71% and 30% of peptides intact after 2 h, respectively. Remarkably, β-Hsp90 emerged as the most stable candidate, showing a remarkable resistance to proteolytic degradation with complete stability up to 1 h and 79% of the compound intact after 2 h (detailed cleavages are represented in Supplementary Fig. 4). Overall, the incorporation of the non-natural β-turn inducer notably enhanced the stability of β-Hsp90 and limited the degradation of β-Tau. Thus, in vitro aggregation and stability studies allowed us to identify the two hairpin structures, β-Tau and β-Hsp90, as the most promising candidates for further development, while peptide S7, exhibiting extreme instability towards proteolytic degradation, was discarded for cell evaluation.
β-hairpin mimetics also inhibit aggregation of the pro-aggregative Tau-ΔK280 mutant in vitro
As the assessment of the compounds to inhibit Tau aggregation and to restore physiological Tau-MT interaction in cells implies model neurons expressing Tau-ΔK280 (single amino acid deletion mutant, which has been reported in two cases of tauopathies70,71,72), an in vitro ThT assay on Tau-ΔK280 was carried out for β-Tau and β-Hsp90.
The ThT-fluorescence assays revealed that β-Hsp90 was notably more efficient than β-Tau (this superiority being much more pronounced than towards Wt-Tau, Fig. 3A). The activity of β-Hsp90 was similar to that of MB. In the presence of β-Hsp90, a complete inhibition of the aggregation of Tau-ΔK280 at both 5/1 and 1/1 ratios was observed (as in the presence of MB). An interesting activity in decreasing F at the very low 0.1/1 ratio was preserved (F = −66% and −61% for β-Hsp90 and MB respectively), with a slight aggregation of the aggregation kinetics observed, and more pronounced in the presence of MB (t1/2 = −44% for MB and −16% for β-Hsp90) (see Figs. 3A–C, Supplementary Table 4 and Supplementary Fig. 5). β-Tau suppressed the aggregation at 5/1 ratio, kept an interesting but lower activity at 1/1 ratio (t1/2 = +179%, F = -75%), and did not show any activity at 0.1/1 ratio (Figs. 3A, B, Supplementary Table 4 and Supplementary Fig. 5).

A, B Effects of compounds β-Tau, β-Hsp90 and MB on Tau-ΔK280 fibrillization assessed by ThT-fluorescence spectroscopy at 5/1 (green bars) 1/1 (yellow bars) and 0.1/1 (blue bars) ratios of compound/Tau-ΔK280. Parameters are expressed as mean ± SE, n = 2 or 3 (technical replicates). Compounds were dissolved in water. The concentration of TauΔK280 was 10 µM. A) The relative t1/2 (%) was calculated with respect to the t1/2 of Tau-ΔK280 control (see Methods for the detailed calculations). NA = No Aggregation over 30 h. B The relative F (%) was calculated at 30 h related to the fluorescence plateau of Wt-Tau control. C Representative curves of ThT fluorescence assays over time showing Tau-ΔK280 fibrillization (10 µM) in the absence (red curve) and in the presence of β-Hsp90 at compound/Tau-ΔK280 ratios of 5/1 (purple curves), 1/1 (green curves) and 0.1/1 (blue curves). The control curve of the compound is represented in an orange line and the buffer in gray.
Quantitative live cell imaging shows that β-hairpin based on Hsp90 restores the physiological microtubule interaction of aggregation-prone Tau in model neurons
β-Hairpin mimetics β-Tau and β-Hsp90 were then evaluated for their ability to inhibit Tau aggregation and to restore physiological Tau-MT interaction in axon-like processes of model neurons expressing Wt-Tau and aggregation-prone Tau mutant (Tau-ΔK280) (Fig. 4A). Assessment of neuronal cell toxicity using MTT and LDH assays73,74 on the model neurons revealed no cell membrane damage or metabolic impairment at concentrations up to 100 μM for β-Tau and β-Hsp90 (Supplementary Fig. 6)

A Schematic representation of the live cell imaging approach to determine Tau dynamics in axon-like processes of neuronally differentiated PC12 cells. The schematic shows that calculation of the effective diffusion constant (Deff) using a one-dimensional diffusion model allows determination of the fraction of microtubule-bound Tau75. Schematic representations of the expressed Tau constructs are shown on the right. The MT-binding repeat regions (RR1–RR4) are indicated by yellow boxes, the N-terminal PAGFP fusion by green. The lysine deletion of the aggregation-prone construct is located in the second repeat. B Effect of β-Hsp90 on the dynamics of aggregation-prone Tau in axon-like processes. FDAP diagrams, scatterplots of effective diffusion constants (Deff), bar graphs of the fraction of Tau bound to microtubules, and scatterplots of k*on and koff rate constants are shown. Mean ± SE of 12 (control), 16 (3.12 µM β-Hsp90), 17 (6.25 µM β-Hsp90), 18 (12.5 µM β-Hsp90) and 12 (25 µM β-Hsp90) cells are shown. Deff β-Hsp90 25 µM vs control p-value = 0.0074. MT bound Tau β-Hsp90 25 µM vs control p-value = 0.0340. C Effect of β-Hsp90 on wild-type Tau dynamics in axon-like processes. In contrast to aggregation-prone Tau, β-Hsp90 does not affect fluorescence decay, Deff, MT-bound fraction and k*on and koff values. This indicates that the increased binding of aggregation-prone Tau is caused by the inhibition of soluble Tau aggregate formation without affecting the physiological Tau-microtubule interaction. Shown are mean ± SE of n = 14 (control), n = 16 (6.25 µM β-Hsp90), n = 15 (12.5 µM β-Hsp90), n = 11 (25 µM β-Hsp90) cells. D Effect of β-Tau on the dynamics of aggregation-prone Tau in axon-like processes. β-Tau does not affect Deff and the MT-bound fraction of aggregation-prone Tau. However, β-Tau leads to a significant increase in the k*on and koff rates, suggesting that it increases the dynamics of the Tau-microtubule interaction. k*on β-Tau 25 µM vs control p-value = 0.0010. koff β-Tau 25 µM vs control p-value = 0.0285. Mean ± SE of 19 (control) and 16 (25 µM β-Tau) cells are shown. Statistically significant differences from control determined by one-way ANOVA with Dunnett’s post hoc test (B, C) or unpaired two-tailed Student’s t-tests (D) are indicated. *p < 0.05, ** p < 0.01. In all experiments, incubation with the compound lasted 24 h, and the final concentration of DMSO was 0.125%.
The quantitative live-cell imaging approach is based on the photoactivation of a defined population of Tau molecules, followed by fluorescence decay after photoactivation (FDAP) measurements to determine the dynamics of the interaction of Tau with MTs in axon-like processes (Fig. 4A)75,76. After focal photoactivation, Tau-ΔK280 showed dissipation from the region of photoactivation, which was significantly reduced in the presence of β-Hsp90 (Fig. 4B, left). Accordingly, calculating the effective diffusion constant (Deff) from the FDAP curves using a one-dimensional diffusion model resulted in lower values for β-Hsp90-treated cells, which became significant at 25 µM treatment (Fig. 4B, middle). This corresponds to an approximately 5% increased binding of Tau-ΔK280 to the microtubules. The application of a previously developed, refined reaction-diffusion model made it possible to estimate the binding constants (k*on and koff rates) of the Tau-MT interaction75,76. Although we did not observe a statistically significant change in k*on and koff rates, there was a trend toward increased kon. Estimation of the dose-response relationship of β-Hsp90 on Deff using a one-site nonlinear equation adjusted for competitive binding yielded an IC50 value of 24 μM.
To confirm that β-Hsp90 acts by reducing the aggregation of pathological Tau and does not directly affect the physiological Tau-MT interaction, we also performed similar FDAP assays with Wt-Tau expressing cells. As expected, Wt-Tau showed increased interaction with axonal microtubules compared to Tau-ΔK280 due to the lack of oligomerization under the cellular conditions. Interestingly, in the presence of Wt-Tau, β-Hsp90 did not induce any change in any of the parameters, indicating that β-Hsp90 does not affect the physiological interaction of Tau with MTs (Fig. 4C). Remarkably, the fraction of microtubule-bound Wt-Tau in the control sample (no compound) was very similar to the fraction of the aggregation-prone Tau-ΔK280 in the presence of 25 µM of β-Hsp90. This observation demonstrates that the β-hairpin mimetic based on the chaperone Hsp90 effectively inhibits Tau-ΔK280 aggregation in cells and thus fully restores the Tau-MT interaction back to physiological levels. This also suggests that the interaction of β-Hsp90 with Tau does not prevent the binding of Tau to MTs.
Conversely, β-Tau exhibited a smaller impact on most parameters (in particular, Deff and MT bound Tau), suggesting that β-Tau has a much lower activity to reduce Tau aggregation than β-Hsp90 in model neurons (Fig. 4D). Remarkably, treatment with β-Tau still resulted in a significantly increased k*on, indicating that the presence of β-Tau still increases the availability of aggregation-prone Tau-ΔK280 to axonal MTs. Interestingly, β-Tau also increased the koff rate of the Tau-MT interaction, indicating that it increases the dynamics of the Tau-MT interaction. This may indicate that, by mimicking Tau hot spots (see above the design), β-Tau competes with Tau to interact with MTs.
Titration of 15N-Tau with β-hairpin mimics β-Tau and β-Hsp90
In order to obtain molecular information about the interaction between Tau and our β-hairpin mimics, 2D 1H-15N HSQC titrations of 15N- Wt-Tau (20 μM) were conducted in the presence of β-Hsp90 or β-Tau at different concentrations (20 μM, 40 μM, and 100 μM). Conversely to β-Tau, in the presence of β-Hsp90, a global signal broadening of Tau was observed, suggesting that β-Hsp90 induces a partial oligomerization of Tau. Chemical shift perturbations (CSPs) induced by β-Hsp90 were significantly larger than those observed in the presence of β-Tau, suggesting that Tau has a higher affinity for β-Hsp90. Remarkably, in all molar ratios tested, the largest CSPs observed in the presence of β-Hsp90 were predominantly confined to the P1 region of Tau (residues ~175-187), as depicted in Fig. 5 (due to the large extent of broadening observed in titrations with Tau and β-Hsp90, it was not possible to determine the binding constant (KD) from CSP values). The observation that β-Hsp90 does not bind to the Tau MTBR surely explains the effectiveness of β-Hsp90 in inhibiting Tau aggregation without preventing Tau-MT interaction. On the other hand, the weaker CSPs (i.e., CSPs mostly corresponding to the noise level in Fig. 5) and lack of effect on 15N-Tau peak intensity in the presence of β-Tau suggest that this compound does not significantly interact with Tau protein or that this interaction is too transient at the NMR level (Supplementary Fig. 7). This might explain the lower efficiency of β-Tau compared to β-Hsp90 in decreasing Tau aggregation both in vitro and in cells.

Indicated in orange circles (A) and (B) are the locations of selected hydrophobic amino acids (F, Y, I, L, V, P) in the sequence of Wt-Tau. A Chemical shift perturbations (CSP) of Tau residues in the presence of β-Hsp90 (red) are more prominent than those of β-Tau (violet), particularly in the P1 region of Tau (residues ~175-187, within the region boxed in gray). Black triangles represent noise-level CSP values (B) I/I0 values of Tau residues (I represents NMR signal intensity in the presence of peptide, and I0 corresponds to conditions in the absence of compounds) showed a ~ 20% decrease upon addition of β-Hsp90 at the 1/5 mole ratio (also in Supplementary Fig. 8), however this was not observed for the same mole ratio upon addition of β-Tau.
β-Hairpin mimetic β-Hsp90 inhibits the aggregation of Aβ1-42 in vitro and acts as a dual and selective inhibitor for AD
As Tau and Aβ1-42 mutually interact during AD pathology causing a progressive worsening of symptoms18, and as Hsp90 was reported to inhibit Aβ1-42 aggregation even at a 50-fold lower ratio43,44, the potential effect of our β-hairpins on the aggregation process of Aβ1-42 was investigated, and compared with Resveratrol (classical control being very efficient in inhibiting the aggregation but showing clinical effectiveness due in particular to its low bioavailability77). We followed our classical aggregation protocol50,51, and the control curves were characterized by a lag phase of around 1–5 h, followed by a growth phase that reached the plateau after 10–15 h. β-hairpin mimics β-Tau and β-Hsp90 showed an effect on Aβ1-42 aggregation at a 10/1 (compound/Aβ1-42) ratio, being able to completely suppress the formation of the fibril, similarly to Resveratrol (Figs. 6A,B, Supplementary Table 5 and Supplementary Fig. 9). At the lower 1/1 ratio, only β-Hsp90 retained a strong inhibitory activity (t1/2 = +308%, F = -90%), and superior in decreasing the aggregation kinetics compared to control Resveratrol (t1/2 = +110%, F = -78%), while β-Tau induced a modest delay on the aggregation kinetics (t1/2 = +27%) and no effect on F. β-Hsp90 even kept a small activity at 0.1/1 ratio (t1/2 = +94%, F = -30%). Thus, we next investigated the activity of the isolated β-strand sequences of Hsp90 S4 and S7, and of the hexapeptides S4short and S7short present in β-Hsp90. Conversely to what was observed for Tau, only peptides S4 and S4short slightly delayed the aggregation at the high 10/1 ratio, while S7 and S7short were completely inefficient in decreasing Aβ1-42 aggregation.

A, B Effects of compounds β-Tau, β-Hsp90, S4, S7, S4short, S7short and Resveratrol on Aβ1-42 fibrillization assessed by ThT-fluorescence spectroscopy at 10/1 (green bars), 1/1 (yellow bars) and 0.1/1 (blue bars, only for β-Hsp90) ratios of compound/Aβ1-42. Parameters are expressed as mean ± SE, n = 2 (technical replicates). Compounds were dissolved in water. The concentration of Aβ1-42 was 10 µM. A The relative t1/2 (%) was calculated with respect to the t1/2 of Aβ1-42 control, see supporting information for the detailed calculations. NA = No Aggregation over 42 h, and (B) The relative F (%) was calculated at 42 h related to the fluorescence plateau of Aβ1-42 control. C Transmission electron micrographs, Aβ1-42 control and in the presence of β-Hsp90 (10/1 and 1/1 ratios) and β-Tau (10/1 ratio).
Notably, while β-Hsp90 and, to a lesser extent, β-Tau are promising dual aggregation inhibitors of the two proteins involved in AD, they did not show interesting activity on hIAPP fibrillization kinetics, an amyloid protein involved in T2D. In our ThT assays, both β-Hsp90 and β-Tau even increased the fluorescence plateau at 10/1 (compound/hIAPP) ratio (with no significant activity at 1/1 ratio, Supplementary Fig. 10).
The inhibitory results obtained with our β-hairpin mimics on Aβ1-42 aggregation were confirmed by TEM, demonstrating the absence of Aβ1-42 fibrils at the end of the aggregation process (24 h). In the case of β-Hsp90 and β-Tau at this 10/1 ratio, non-uniform structures were detected (Fig. 6C).
2D 1H-15N HSQC titrations of 15N-Aβ1-42 (50 μM) in the presence of β-Hsp90 (50 μM and 500 μM) were then performed. No CSP or broadening of Aβ1-42 1H-15N correlations could be observed at β-Hsp90/Aβ1-42 molar ratios of 1:1 or 10:1. A 50% decrease of all Aβ1-42 signals was nevertheless observed in the presence of a 10-fold excess of β-Hsp90, suggesting a co-aggregation process (Supplementary Fig. 11). The absence of CSP or signal broadening in the presence of an equimolar ratio of β-Hsp90 suggests that this compound does not significantly interact with Aβ1-42 monomers or that the interaction is too transient at the NMR scale. Therefore, this compound interacts rather with larger oligomers that are not visible by NMR.
Discussion
In this work, the possibility of inhibiting Tau aggregation was investigated by designing new β-hairpin mimics, β-Tau and β-Hsp90, containing a piperidine-pyrrolidine β-turn inducer bearing peptide sequences based on Tau SREs and inspired by the physiological interaction of Tau with MTs interactions (PHF6 and PHF6*) for β-Tau, and on two peptide sequences (NStrand 4 and NStrand 7) of the physiological chaperone Hsp90 for β-Hsp90. Four different peptides extracted from the crystal structure of Hsp90 N-terminal domain (PDB = 3NMQ) were synthesized and found to be the most relevant interacting regions and possible binding sites of NHsp90 in cross-interaction with Tau (NHelix 1 as compound H1, NStrand 4 as compound S4, NStrand 6 as compound S6 and NStrand 7 as compound S7). Their synthesis on the solid phase was efficient. The conformational analysis of β-Tau and β-Hsp90 by CD confirmed the presence of a flexible β-hairpin-like structure in aqueous medium (PBS), while a stabilized β-hairpin was observed in the organic protic solvent MeOH.
β-Tau and β-Hsp90 showed excellent ability to inhibit Wt-Tau fibril formation in vitro as demonstrated by ThT assays and TEM imaging, with superiority of the synthetic chaperone β-Hsp90. Among the four peptides H1, S4, S6 and S7, also evaluated by ThT, only S7 demonstrated activity against Wt-Tau aggregation. The need to use pre-organized β-hairpin structures to ensure inhibitory activity on Tau aggregation and the introduction of the non-natural piperidine-pyrrolidine dipeptide mimic to ensure stability against proteolysis were demonstrated. β-Hsp90 clearly proved to be the most stable candidate against proteolysis, showing a complete stability up to 1 h and only 21% degradation after 2 h. β-Tau showed a good stability up to 1 h (62% of intact β-Tau) but was more degraded after 2 h (64% of degradation). Despite the interesting in vitro activity of S7, its high sensitivity to proteolysis (76% and 100% degradation, after 10 and 30 min, respectively) led us to discard it from further cellular studies and to continue with our two β-hairpin mimics β-Tau, and β-Hsp90. The superiority of β-Hsp90 was confirmed on the aggregation-prone Tau mutant ΔK280-Tau, which is a single mutation that has been reported in two cases of tauopathies70,71,72. In the ThT assays conducted in the presence of β-Hsp90, a complete inhibition of the aggregation of Tau-ΔK280 at both 5/1 and 1/1 ratios was observed, and an interesting activity at the very low 0.1/1 ratio was preserved. Conversely, β-Tau suppressed the aggregation only at the 5/1 ratio, but it showed a lower activity at the 1/1 ratio and no activity at the 0.1/1 ratio.
This difference between β-Hsp90 and β-Tau was even more remarkable in the quantitative live cell imaging on model neurons, where only β-Hsp90 effectively inhibits Tau-ΔK280 aggregation and fully restores the Tau-MT interaction up to physiological levels (same levels as with Wt-Tau). This highly interesting result also suggests that the interaction of β-Hsp90 with Tau does not prevent the binding of Tau to MTs. Indeed, our NMR studies showed that β-Hsp90 was predominantly interacting with the P1 region of Tau (in particular, residues ~175-187), rather than with the region involved in the binding of Tau to MTs (MTBR). The reported observations in literature that Hsp90 alone could interact with Tau MTBR to increase oligomerization41,42, or in multi-complex systems with the proline-rich region P1 (in Hsp90/FKBP51/Tau complexe37), or P2 and R’ flanking Tau-RD (in Hsp70/Hsp90 complex40), to prevent Tau aggregation, might indicate that our small synthetic chaperone β-Hsp90 is capable of acting as multi-protein systems. Furthermore, the P1 region of Tau targeted by β-Hsp90 includes two major phosphorylation sites, Thr175 and Thr181 (in particular, the concentration of Tau phosphorylated at site 181 is a well-established biomarker of AD in the cerebrospinal fluid (CSF)78 and a studied plasma biomarker79). However, the slight oligomerization observed in our NMR and TEM analyzes of Tau in the presence of β-Hsp90 is in accordance with some literature data reporting that Hsp90 inhibits the formation of Tau fibrils but could promote the formation of small Tau oligomers35,36. Thus, mimicking HSP90 with small peptide derivatives is a valuable strategy to prevent the pathological aggregation of Tau and restaure its physiological conformation, allowing its interaction with MTs. The mechanism of action of β-Hsp90 is different from the one suspected for the dihydropyridine derivative LA1011 that is active to reduce tau pathology and amyloid plaque formation in transgenic AD mice, by interacting with HSP90 to disrupt the binding of FKBP51 and thus release HSP90. The increase of FKBP51 seems a major player in Tau aggregation, and blocking HSP90-FKBP51 allow HSP90 to play his beneficial role38,39. However, this study with LA1011 and our study point in the same direction, confirming that HSP90 is a crucial target for therapeutic strategies against AD and other Tauopathies.
Furthermore, while the question of whether the interaction Hsp90-Tau allows to preserve the physiological interaction between Tau MTBR and MT has not been addressed before, our cellular FDAP studies indicate that β-Hsp90 might induce non-toxic small off-pathway oligomers that can prevent deleterious interactions of Tau aggregates with cytoplasmic proteins (such as kinases) or that can be in equilibrium with monomers able to interact with MTs. It is important to understand that the increase in MT-bound fraction of Tau in the presence of β-Hsp90 could have several reasons. The first is that the amount of free tau is increased due to the anti-aggregative activity of the drug. It could, second, be due to the induction of a conformational change in tau or microtubules, which increases tau’s on-rate, promoting increased binding to microtubules. Or third, it could change the structure of microtubules, thereby decreasing the off-rate of tau. Or fourth, a combination of some of the previous effects could occur. Our modeling allows to distinguish between these possibilities. The data show, that β-Hsp90 increases the k*on rate. k*on is the pseudo-first-order on rate (kon[MT]eq (s-1)) and is therefore independent of the concentration of Tau. Thus, the effect must be due to either a conformational change in Tau induced by β-Hsp90 or in microtubules, which increases the affinity of tau to microtubules. Koff is unchanged. This means that β-Hsp90 does not affect microtubules with respect to changing the dwell time of Tau on the microtubule surface (the dwell time is 1/koff). However, kon and koff are not affected with Wt-Tau, as our further experiments in Fig. 4 show. This means that the effect of β-Hsp90 is specific for pathologic, aggregation-prone Tau and microtubules are not affected. Thus, the data show that β-Hsp90 is not just decreasing aggregation, but it also induces a conformational change in pathologic Tau that leads to a Tau conformer that binds better (with higher affinity) to microtubules thereby restoring the affinity back to Wt-Tau.
Overall, our results could indicate that HSP90 acts as β-Hsp90 by altering the pathological conformation of Tau, reducing the formation of pathological aggregates, inducing small off-pathway oligomers in equilibrium with monomers, and thereby increasing its ability to interact with microtubules. Remarkably, β-Tau based on PHF6 and PHF6* was also able to inhibit in vitro Wt-Tau and Tau-ΔK280 aggregation, albeit more weakly than β-Hsp90, and with reduced inhibitory activity in cells. This smaller activity could be due to its lower proteolysis stability or to its interaction with Tau MTBR that decreases the subsequent Tau-MT interaction. However, its lack of visible interaction with Tau in the NMR studies rather suggests that by mimicking Tau-MT hot spots, β-Tau might compete with Tau and interact with MTs, which is consistent with the increased koff observed in the FDAP assay in the presence of β-Tau.
Additional highly interesting results obtained in this work concern the selectivity of our designed compounds for AD. Indeed, a similar β-hairpin mimic (β-hIAPP), recently reported by us52 based on hIAPP sequences inhibiting hIAPP aggregation involved in T2D, was inactive towards Tau aggregation. Conversely, β-Hsp90 and β-Tau did not inhibit hIAPP aggregation and even significantly increased the aggregation in the ThT assays, at the high ratio of 10/1. A link between Tau and hIAPP was highlighted by the demonstration that these two proteins act in a coordinated manner to impair beta cell function and glucose homeostasis, linking AD and T2D80. The increased aggregation induced by β-Tau is in accordance with this report and indicates that PHF6 and PHF6* might be part of the hot-spots of Tau-hIAPP cross-interaction. Even if the ubiquitin–proteasomal system, which includes Hsp90, is important for hIAPP clearance81, to our knowledge, a direct link between Hsp90 and hIAPP has not yet been evidenced. Our results might suggest a potential but not beneficial Hsp90-hIAPP interaction.
Importantly, β-Hsp90 was also found to be highly efficient in inhibiting the aggregation of Aβ1-42, which is the second amyloid protein involved in AD, thus confirming the preliminary data found in the literature reporting the inhibition of Aβ1-42 aggregation by the physiological Hsp90 chaperone43,44. Our results with the isolated peptides indicate that, while NStrand 7 of Hsp90 (peptide S7) seems to be the key sequence of Hsp90 interacting with Tau, NStrand 4 (peptide S4) appears to be rather important for inhibiting Aβ1-42 aggregation. In contrast, β-Tau was much less efficient in inhibiting Aβ1-42 aggregation at low ratios of β-Tau/Aβ1-42. Tau and Aβ1-42 have been suspected to interact mutually during the AD pathology, through the cross-seeding of the amyloid core of Aβ1-42 and PHF6 and PHF6* of Tau18. Our data suggest that PHF6 and PHF6* are involved in this cross-interaction but may not be involved in the seeding of Aβ1-42.
To conclude, we demonstrate that mimicking natural protein chaperones with small peptides is a valuable strategy to explore the impact of chaperone-Tau interaction on Tau aggregation and on the physiological interaction of Tau with MTs. Small peptide drugs mimicking natural chaperones hold great promise for combating tauopathies. Furthermore, our lead β-hairpin mimic β-Hsp90-based on the physiological chaperone Hsp90, showing proteolytic stability and potent inhibitory activity on both Tau and Aβ1-42 aggregation, mutually implicated in AD, offers the possibility to develop a very efficient dual-inhibitor. In complex diseases, where single-target drugs do not achieve the desired results, drug combinations or multitarget drug treatment often result in higher effectiveness82,83. Dual-targeting compounds such as dual inhibitors of Tau and Aβ have been proposed as new strategies to achieve better therapeutic benefits for AD10.
To the best of our knowledge, there are no other Tau inhibitors (small molecules or peptides) that interact with the P1 region of Tau. This work paves the way to design new inhibitors of aggregation that do not affect the Tau-MTs physiological interaction. As proline-rich region appears to be a critical region both in regulating Tau interaction with MTs as well as Tau aggregation, it is also important to study the behavior of full-length Tau proteins rather than small peptide models that only contain the MT binding sequence.
Importantly, our β-hairpin mimics β-Hsp90 and β-Tau provide valuable explorative tools for further and deeper studies of Hsp90/amyloid proteins and amyloid/amyloid cross-interactions (such as Hsp90/Tau, Tau/Aβ1-42, Hsp90/Aβ1-42, Tau/hIAPP and Hsp90/hIAPP). We are confident that these new results in the amyloid field will open new ways for designing peptide drug candidates not only to combat AD, but also, by extending the design from other chaperones acting on different amyloid proteins, to target other untreated amyloid diseases such as PD, HD, T2D or Amyotrophic lateral sclerosis (ALS). We consider that this study may help to develop urgently needed therapies for neurodegeneration and other age-dependent pathologies.
Methods
General procedure for the Synthesis on Solid phase Peptide Synthesis Strategy (SPSS) for compounds β-Tau and β-Hsp90
All the reactions involved were agitated in plastic syringe tubes equipped with filters on an automated shaker on Rink Amide resin (0.2 mmol scale, loading 0.327 mmol/g, 600 mg). The coupling yields were monitored with the Fmoc-test procedure (reported below).
Removal of Fmoc group was performed in 20% (v/v) piperidine/DMF for 20 min twice. Capping steps were performed by treating the resin with the mixture of acetic anhydride (0.25 M) and NMM (0.25 M) in DMF solution for 20 min. After each reaction, the resin was washed with DMF (3 × 10 mL), MeOH (3 × 10 mL) and DCM (3 × 10 mL) successively.
Rink Amide resin (600 mg, 0.2 mmol/g) was swelled in DMF for 1 h before using. Natural amino acids and scaffold were coupled using different coupling reactive accordingly to their position:
First AA loaded, AA coupled after the scaffold and the two last AA
The resin was suspended in DMF (4 mL) and collidine (17 eq). Fmoc-AA-OH (5 eq), DIC (5 eq), and Oxyma pure (5 eq) were solubilized in DMF/DCM 33% (v/v) (3mL) and let under magnetic stirring for 7min. Afterward, the solution containing the amino acid was poured into the reactor containing the resin and the mixture shaked for 16 h at room temperature. (Positions 1, 7, 11 and 12)
Scaffold coupling
The resin was suspended in DMF (4 mL) and collidine (17 eq). The piperidine-pyrrolidine β-turn scaffold50 (1.5 eq), DIC (1.5 eq), and OXYMA (1.5 eq) were solubilized in DMF/DCM 33% (3 mL) and let under magnetic stirring for 7 min. Afterward, this solution was poured into the reactor containing the resin and the mixture was shaken for 16 h at room temperature.
All the other AA couplings
Fmoc-AA-OH (5 eq) and HCTU (5 eq) were solubilized in 5 mL of DMF/NMM 20% (V/V). The prepared solution is added to the resin and shaked for 20 min. Afterward, the solvent was evaporated and the resin washed with DMF (1 × 5 mL) and the coupling procedure repeated. (Positions from 2 to 6 and from 8 to 10).
At the end of the synthesis, the peptides were cleaved from the resin by shaking for 2 h in the presence of 5 mL of an acidic solution containing trifluoracetic acid/H2O/TIPS/Phenol/Thioanisole; 87.5%/5%/2.5%/2.5%/2.5%. The liquid was poured in dry cold Et2O (40 mL) in ice bath by filtering it over a cotton pad. The peptides precipitated and isolated by centrifugation at 6000 rounds/min for 7 min. The pad was resuspended with ether and centrifuged again to remove the remaining TFA. The hairpin mimics were lyophilized and purified over reverse-phase RP-HPLC.
Synthesis of natural peptides H1, S4, S6 and S7
Peptides were synthesized using microwave-assisted solid-phase peptide synthesis performed on a Liberty Blue Microwave Automated Peptide Synthesizer (CEM Corporation, Matthews, NC, USA), following the standard protocols for Fmoc/tBu strategy (0.1 mmol scale, 300 mg, 0.327 mmol/g). Fmoc-deprotection cycles were respectively of 15 s (75 °C, 155 W) and 30 s (90 °C, 30 W). Couplings of Arg residues were performed in 1500 s (25 °C, 0 W) and 300 s (75 °C, 30 W). Couplings of His were performed in 240 s (50 °C, 35 W). Couplings of other residues are performed in 15 s (90 °C, 170 W) and 110 s (90 °C, 30 W). Peptide cleavage (3 h at room temperature) from the resin and deprotection of the amino acid side chains were performed by using the reagent K (trifluoracetic acid/phenol/water/thioanisole/1.2-ethanedithiol; 82.5/5/5/5/2.5) for 180 min. The liquid was poured in dry cold Et2O (40 mL) in ice bath by filtering it over a cotton pad. The peptides precipitated and isolated by centrifugation at 6000 rounds/min for 7 min. The pad was resuspended with ether and centrifuged again to remove the remaining TFA. The peptides were lyophilized and purified over RP-HPLC if necessary.
Synthesis of natural peptides S4short and S7short via manual solid-phase peptide synthesis
All the reactions involved were agitated in plastic syringe tubes equipped with filters on an automated shaker, on Rink Amide resin (0.2 mmol scale, loading 0.327 mmol/g, 600 mg).
Removal of Fmoc group was performed in 20% (v/v) piperidine/DMF for 20 min twice. Capping steps were performed by treating the resin with the mixture of acetic anhydride (0.25 M) and NMM (0.25 M) in DMF solution for 20 min. After each reaction, the resin was washed with DMF (3 × 10 mL), MeOH (3 × 10 mL) and DCM (3 × 10 mL) successively.
Rink Amide resin (600 mg, 0.2 mmol/g) was swelled by DMF for 1 h before using. The loading was performed by suspending the resin with DMF (4 mL) and collidine (17 eq). Fmoc-AA-OH (5 eq), DIC (5 eq), and Oxyma pure (5 eq) were solubilized in DMF/DCM 33% (v/v) (3 mL) and let under magnetic stirring for 7 min. Afterward, the solution containing the amino acid was poured into the reactor containing the resin and the mixture shaked for 16 h at room temperature. Every following coupling using Fmoc-protected natural amino acid was carried out twice to get satisfactory yields using Fmoc-AA-OH/HCTU (2.5/2.5 eq) in NMM/DMF (20%V/V, 4 ml).
Peptides were cleaved from the resin by shaking for 2 h with an acidic mixture (TFA/water/TIPS/Thioanisole; 95%/2.5%/1.25%/1.25%) and repeated the same procedure explained in General procedure.
Fmoc loading Test phase (Fmoc-test)
From 5 to 10 mg of dry resin was shaken in DMF/Piperidine 20% solution (1 mL) for 30 min. The resin was sedimented by centrifugation and 1 mL of the supernatant was added to 9 mL of DMF and mixed well. This final solution was used for the UV analysis measuring the absorbance at 301 nm versus a DMF blank (triplicate analysis).
For the quantification of resin loading, we used the general formula: L = (A301 × V × d)/(Ec × w × M). Where: L = Resin loading; A301 = UV Absorbance at 301 nm; V = Volume of the cleavage solution =1 mL; d = Dilution = 10; Ec = Extinction coefficient = 7800 mL/mmol*cm; w = Width of the cuvette = 1 cm; M = Weight of the resin sample in g. Substituting the extinction coefficient, volume, dilution and cell width into the general formula results in this formula, which can be used to calculate the resin loading.
Circular Dichroism Spectroscopy
Peptidomimetics were dissolved in MQ water to a concentration of 500 μM as stock solutions. Before measurement, each compound was diluted to 125 µM concentration with 20 mM PB (pH 7.2) buffer or MeOH into a cuvette with a pathlength of 1 mm. The CD spectra were recorded on a J-815 spectropolarimeter (JASCO, Tokyo, Japan) from 190 to 260 nm at 20 and 37 °C and a scan rate of 50 nm/min (accumulation n = 3). Each CD spectrum was corrected by subtracting the corresponding baseline (PB buffer 20 mM or MeOH). Data processing was performed using Solver in Excel software (Microsoft). The deconvolution process was executed according to literature56.
Fluorescence-detected ThT binding assay (Ac-PHF6*-NH2)
Thioflavin-T and Heparin sodium salt (H-3149, average MW 18 kDa) were obtained from Sigma-Aldrich. Ac-PHF6*-NH2 was self-produced according to the general SPPS protocol of S4short and S7short and purified by HPLC. The purity was confirmed by LC-MS and HRMS. Ac-PHF6*-NH2 was dissolved in pure hexafluoro-isopropanol (HFIP) at a concentration of 1 mM and incubated for 10 min at room temperature to dissolve any preformed aggregates. Next, HFIP was evaporated under a stream of dry nitrogen gas, followed by vacuum desiccation for at least 3 h. The resulting thin film was dissolved in 20 mM MOPS buffer (pH 7.4), sonicated for 1 min and vortexed for 2 min to get fully disperse clean solution of AcPHF6*-NH2 (500 µM). Stock solutions of compounds to test were dissolved in water (10, 2 and 0.2 mM). Thioflavin-T binding assays were used to measure the formation of fibrils in solution using a plate reader (Fluostar Optima, BmgLabtech) and standard 96-wells flat-bottom black microtiter plates (final volume 200 µL) in combination with a 440 nm excitation and 480 nm emission filters. ThT assay was started by adding 2 µL of a 100 µM heparin solution (Heparin sodium salt H-3149, average MW 18 kDa) to a mixture containing 25 µM of Ac-PHF6*-NH2, 20 μM ThT in 20 mM MOPS pH 7.4 buffer. The concentration of Ac-PHF6*-NH2 was held constant at 25 μM for all experiments, and inhibitors were added to yield compound/ Ac-PHF6*-NH2 ratios of 5/1, 1/1 and 0.1/1. The ThT assays were performed in triplicate.
Production and purification of recombinant Wt-Tau and Tau-ΔK280
Prokaryotic expression plasmids were based on human adult Tau (Tau441wt) in a pET-3d vector84,85. pET-3d-tau plasmids expressing human Tau protein (Wt-Tau) and the ΔK280 deletion variant (Tau-ΔK280), were transformed into Escherichia coli BL21(DE3)pLysS cells for expression. Cells were grown, induced and harvested as previously described86. For TEM analysis, native Tau was purified from the cell extract by sequential anion exchange and phosphocellulose chromatography as previously described84. The Tau protein eluate was dialyzed against water and concentrated with Vivaspin® (15 R, 2000 MWCO, Sartorius, UK). Protein concentrations were determined by a bicinchoninic acid (BCA) assay (Thermo-Fisher Scientific, USA) using bovine serum albumin as a standard. For the ThT assays, Tau441wt protein was also purified using heat denaturation following established protocols85 and dialyzed against 50 mM ammonium bicarbonate buffer before being lyophilized.
Fluorescence-detected ThT binding assay (Wt-Tau441 and Tau441-ΔK280)
Thioflavin-T and Heparin sodium salt (H-3149, average MW 18 kDa) were obtained from Sigma-Aldrich. Lyophilized full-length Wt-Tau441 and Tau441 ΔK280 were diluted to 40 μM in NaPi 25 mM, NaCl 25 mM, and EDTA 2.5 mM pH 6.8. Stock solutions of compounds to test were dissolved in water. ThT fluorescence was measured to evaluate the development of Tau fibrils over time using a fluorescence plate reader (Fluostar Optima, BMG labtech) with 384-wells flat-bottom black plates (final volume in the wells of 40 μL). Experiments were started by adding Heparin (0.0625 μM) to each well containing the resulting Tau solution (final Tau concentration equal to 10 μM), thioflavin-T (25 μM) with and without the compounds to test at different concentrations (50, 10 and 1 μM, ratios compounds/Wt-Tau equal to 5/1, 1/1 and 0.1/1) in NaPi 25 mM, NaCl 25 mM, EDTA 2.5 mM pH 6.8. The ThT fluorescence intensity of each sample (performed in triplicate) was recorded over time with 440/480 nm excitation/emission filters set for 30 h under continuous agitation (orbital shaking) at 37 °C on plates sealed with a transparent film. The ability of compounds to inhibit Wt-Tau aggregation was assessed considering the time of the half-aggregation (t1/2) and the intensity of the experimental fluorescence plateau (F), both values were obtained by fitting the obtained kinetic data to a Boltzmann sigmoidal curve using GraphPad Prism 5. The relative t1/2 is defined as the experimental t1/2 in the presence of the tested compound relative to the one obtained without the compound and is evaluated as the following percentage: [t1/2 (Tau + compound)] / t1/2 (Tau) × 100. The relative F is defined as the intensity of experimental fluorescence plateau observed with the tested compound relative to the value obtained without the compound and is evaluated as the following percentage: (F(Tau + compound) / F(Tau) × 100. Curves of the tested compounds are fitted to a Boltzmann sigmoidal model and represented relative to the maximal fluorescence of the control experiment.
Fluorescence-detected ThT binding assay (Aβ1−42)
Aβ1−42 was purchased from Bachem, and Thioflavin-T was obtained from Sigma. The peptide was dissolved in a 1% aqueous ammonia solution to a concentration of 1 mM and then, just prior to use, was diluted to 0.2 mM with 10 mM Tris-HCl and 100 mM NaCl buffer (pH 7.4). Stock solutions of compounds to test were dissolved in water. ThT fluorescence was measured to evaluate the development of Aβ1−42 fibrils over time using a fluorescence plate reader (Fluostar Optima, BMG labtech) with standard 96-well black microtiter plates (final volume in the wells of 200 μL). Experiments were started by adding the peptide (final Aβ1−42 concentration equal to 10 μM) into a mixture containing 40 μM ThT in 10 mM Tris-HCl and 100 mM NaCl buffer (pH 7.4) with and without the compounds at different concentrations (100 and 10 μM, compound/ Aβ1−42 ratios of 10/1 and 1/1) at room temperature. The ThT fluorescence intensity of each sample (performed in duplicate) was recorded with 440/480 nm excitation/emission filters set for 42 h, performing a double orbital shaking of 10 s before the first cycle. The ability of compounds to inhibit Aβ1−42 aggregation was assessed considering the time of the half aggregation (t1/2) and the intensity of the experimental fluorescence plateau (F), both values were obtained by fitting the obtained kinetic data to a Boltzmann sigmoidal curve using GraphPad Prism 5. The relative t1/2 is defined as the experimental t1/2 in the presence of the tested compound relative to the one obtained without the compound and is evaluated as the following percentage: [t1/2 (Aβ + compound)] / t1/2 (Aβ) × 100. The relative F is defined as the intensity of the experimental fluorescence plateau observed with the tested compound relative to the value obtained without the compound and is evaluated as the following percentage: F(Aβ + compound) / F(Aβ) × 100. Curves of the tested compounds are fitted to a Boltzmann sigmoidal model and normalized to the maximal fluorescence of the control experiment.
Fluorescence-detected ThT binding assay (hIAPP)
Thioflavin-T was obtained from Sigma. hIAPP, purchased from Bachem, was dissolved in pure hexafluoro-isopropanol (HFIP) at a concentration of 1 mM and incubated for 1 h at room temperature to dissolve any preformed aggregates. Next, HFIP was evaporated under a stream of dry nitrogen gas, followed by vacuum desiccation for at least 3 h. The resulting peptide film was then dissolved in DMSO to obtain stock solutions of hIAPP (0.2 mM), and stock solutions of compounds to test were dissolved in water. Thioflavin-T binding assays were used to measure the formation of fibrils in solution or in the presence of membranes over time. A plate reader (Fluostar Optima, BmgLabtech) and standard 96-wells flat-bottom black microtiter plates in combination with a 440 nm excitation and 480 nm emission filters were used. The ThT assay was started by adding 5 μL of a 0.2 mM hIAPP stock solution to a mixture of 10 μM ThT and 10 mM Tris/HCl, 100 mM NaCl at pH 7.4, containing the compounds to test (50 and 5 μM, compound/ hIAPP ratios of 10/1 and 1/1) at room temperature. The ThT assays were performed once. The curves of the tested compounds are fitted to a Boltzmann sigmoidal model and normalized to the maximal fluorescence of the control experiment.
Transmission electron microscopy (TEM) for Wt-Tau441
Wt-Tau441 solubilization immediately before use: Lyophilized Tau was dissolved in buffer NaPi (Na2HPO4 and NaH2PO4 25 mM, NaCl 25 mM, EDTA 2.5 mM, pH 6.6) to a final concentration of 40 µM (stock solution).
Sample preparation: Experiments were started by adding 10 µL of Buffer, 10 µL of Tau solution (Final concentration of 10 µM), 10 µL of desired active compound (either 200 µM or 40 µM to evaluate 5:1 and 1:1 ratios), and 10 µL Heparin solution 0.25 µM solubilized in NaPi buffer (final concentration 0.0625 µM). In blank analysis, 10 µL of active compound was replaced by 10 µL MQ H2O. These solutions were maintained at 37 °C for 96 h at 1400 rpm.
The samples were prepared on carbon-coated copper grids (ECF200- Cu, 200 mesh, Science Services, Munich, Germany). The grids were pre-treated with argon in a plasma cleaner (Diener Electronics, Ebhausen, Germany). The sample (1.2 µL) was applied onto the grids, and after a sedimentation time of 5 min, the excess suspension was removed with filter paper. Grids were then stained with 1% acetate uranyl solution (1.2 µL) for 5 min, excess stain was removed with filter paper and washed thoroughly with 3 × 10 µL of Milli-Q water. The prepared grids were analyzed with a JEOL JEM-2200FS electron microscope (JEOL, Freising, Germany), a cold field emission electron gun, and an applied acceleration voltage of 200 kV. A bottom-mounted Gatan OneView camera (Gatan, Pleasanton, CA, USA) was used for digital recording. The images were processed using the image-processing system Digital Micrograph GMS3 (Gatan, Pleasanton, CA, USA) and the image editing software ImageJ.
Transmission electron microscopy (TEM) for Aβ1-42
Aβ1-42 Pre-treatment for storage: Aβ1-42 was dissolved in 1 mg/mL in NaOH 60 mM and sonicated for 5 min. Afterward the solution was centrifugated at 5000 rpm for 5 min and lyophilized87.
Aβ1-42 Pre-treatment immediately before use: pre-treated Aβ1-42 was dissolved in an HCl 60 mM obtaining a final concentration of 200 µM.
Sample preparation: Experiments were started by adding the peptide (final Aβ1−42 concentration equal to 10 μM) into a solution containing the active compound in the appropriate ratio (100 or 10 μM) into 20 mM phosphate buffer pH 7.4, containing 1 mM NaN3 and 200 µM EDTA. The mixture was then shacked for 24 h at 37 °C at 700 rpm.
Transmission electron microscopy. The samples were prepared on carbon-coated copper grids (ECF200- Cu, 200 mesh, Science Services, Munich, Germany). The grids were pre-treated with argon in a plasma cleaner (Diener Electronics, Ebhausen, Germany). The sample (2 µL) was applied onto the grids, and after a sedimentation time of 5 min, the excess suspension was removed with filter paper. Grids were then stained with 1% acetate uranyl solution (2 µL) for 5 min, excess stain was removed with filter paper and washed thoroughly with 3 × 10 µL of Milli-Q water. The prepared grids were analyzed with a JEOL JEM-2200FS electron microscope (JEOL, Freising, Germany), a cold field emission electron gun, and an applied acceleration voltage of 200 kV. A bottom-mounted Gatan OneView camera (Gatan, Pleasanton, CA, USA) was used for digital recording. The images were processed with the image-processing system Digital Micrograph GMS3 (Gatan, Pleasanton, CA, USA) and the image editing software ImageJ.
Cell lines
PC12 cells (originally obtained from J.A. Wagner, Harvard Medical School) were cultured in serum-DMEM (DMEM supplemented with 10% fetal bovine serum and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin)) at 37 °C with 10% CO2 in a humidified incubator. For live cell imaging, cells were transfected with the respective pRc/CMV expression vector using Lipofectamine 2000 (Thermo-Fisher Scientific, USA) as previously described88.
Metabolic activity and cytotoxicity profiling using a combined LDH and MTT assay
PC12 cells were cultured in 96-well plates at 1 × 104 cells/well in 50 µL of serum-reduced medium supplemented with 100 ng/mL 7S mouse NGF to induce neuronal differentiation. Cells were incubated for 48h, test compounds were added in an additional volume of 50 µL, and incubation was continued for 20 h. For the LDH assay, 50 µL medium from each well of the assay plate was transferred to a separate 96-well plate. To quantify LDH release, 50 µL of LDH reagent (4 mM iodonitrotetrazolium chloride (INT), 6.4 mM beta-nicotinamide adenine dinucleotide sodium salt (NAD), 320 mM lithium lactate, 150 mM of 1-methoxyphenazine methosulfate (MPMS) in 0.2 M Tris-HCl buffer, pH 8.2) was added. Absorbance was measured at 490 nm using a Thermomax Microplate Reader operated with SoftMaxPro Version 1.1 (Molecular Devices Corp., Sunnyvale, CA, U.S.A.). For the MTT assay, MTT reagent (3,(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide) was added to the wells of the remaining assay plate at a final concentration of 1 mg/ml MTT. Cells were incubated for 2 h before the reaction was stopped by adding 50 μl of lysis buffer (20% (wt/vol) sodium dodecyl sulfate in 1:1 (vol/vol) N, N-dimethylformamide/water, pH 4.7). After overnight incubation at 37 °C, optical densities of the formazan product were determined at 570 nm. MTT conversion measurements were normalized to the optical densities of negative control wells. All experiments were performed in triplicate on two independent plates.
Live-cell imaging and Fluorescence Decay After Photoactivation (FDAP)
FDAP experiments were performed essentially as previously described76,89,90. Briefly, cells expressing wild-type Tau or Tau-ΔK280 were plated on 35-mm glass-bottom culture dishes (MatTek, USA), transfected, and neuronally differentiated by medium exchange to serum-reduced DMEM containing 100 ng/mL 7S mouse NGF. After 3 days, the medium was exchanged to serum-reduced DMEM without phenol red with NGF, and the respective compound (or DMSO for carrier control) was added at the desired concentration. After 20 h, live cell imaging was performed using a laser scanning microscope (Nikon Eclipse Ti2-E (Nikon, Japan)) equipped with a LU-N4 laser unit with 488-nm and 405-nm lasers and a Fluor 60× ultraviolet-corrected objective lens (NA 1.4) and a C2+scanner enclosed in an incubation chamber at 37°C and 5% CO2. Photoactivation was performed with a 405-nm laser using the microscope software (NIS-Elements version AR 5.02.03 (Nikon, Japan)). The following steps were performed in a photoactivation experiment. A transfected cell with a suitable process (minimum length 30 µm) was selected in the field of view (50 µm × 50 µm), and a pre-activation image was saved at 488-nm excitation. The scan window was reduced to a 6-μm diameter activation spot (corresponding to 130 pixels), and photoactivation was performed in the center of the neurite using the 405-nm blue diode (laser intensity 1.0%; corresponding to an output of 30 µW) with two scans at a pixel dwell time of 4.08 microseconds (corresponding to a total activation time at the spot of 1.1 milliseconds). The scan window was enlarged to the initial size, and the first scan was performed after 1 s with the 488-nm laser at the lowest practical laser intensity. Subsequent images were acquired at a rate of 1 frame/second. 112 images with a resolution of 256 × 256 pixels were collected per activated neurite. To ensure that the imaging conditions did not affect cell viability and subsequent evaluation, neurites were excluded from analysis if they showed retraction or drifted out of focus. Effective diffusion constants were determined by fitting the fluorescence decay data from the photoactivation experiments using a one-dimensional diffusion model function for FDAP. A reaction-diffusion model was used to estimate the association rate k*on and the dissociation rate koff constant of Tau binding according to Igaev et al.75 The model is based on the assumptions that (1) PAGPF has no effect on the Tau-microtubule interaction, (2) the binding probabilities of Tau protein to microtubules are independent of the position, (3) the reactants are in dynamic equilibrium before photoactivation, (4) the number of the binding sites is not changed by photoactivation, but activation only affects the number of photoactivated molecules, (5) during an FDAP experiment, the transport of Tau along the neurite can be neglected, (6) binding of Tau protein to binding sites other than microtubules is negligible, and (7) The total concentration of tau proteins does not significantly exceed the concentration of available binding sites. All data sets were tested for normality using the D’Agostino-Pearson and Shapiro-Wilk tests. Statistical outliers were identified using the ROUT method. When necessary, data sets were logarithmically transformed to allow for further statistical testing. Equality was assessed using an F-test to compare variances. To compare two data sets, an unpaired two-tailed t-test was used. In case of unequal variances, Welch’s correction was applied. To compare different concentrations, statistically significant differences from control were determined by a one-way ANOVA with Dunnett’s post hoc test. All statistical analyzes were performed using the GraphPad Prism program.
Pronase stability assays
The assay buffer was prepared by combining 50 mM Tris, 10 mM CaCl2, 150 mM NaCl, and 0.05% (w/v) Brij 35, and by adjusting the pH to 7.5. The peptide was dissolved in 200 µL of assay buffer to achieve a concentration of 400 µM. The solution was placed in a thermomixer set at 37 °C with stirring. Pronase was diluted to a concentration of 5 µg/mL in 200 µL of assay buffer. The solution was placed in a thermomixer set at 37 °C with stirring. The peptide solution (200 µL) and the pronase solution (200 µL) were combined in an Eppendorf tube. The final concentration of pronase should be 2.5 µg/mL, and the final concentration of the peptide should be 200 µM. The reaction mixture was left in the thermomixer at 37 °C. The reaction progress was monitored by LC-MS at various time points: 0 min, 10 min, 30 min, 1 h, and 2 h. At each time point, 40 µL of the reaction solution was transferred into an HPLC vial containing 6 µL of 10% TFA (to deactivate the enzyme). This procedure was repeated for each time point.
Preparation of Wt (2N4R) 15N-Tau
The full-length isoform of Tau (2N4R, Uniprot ID P10636) enriched with the 15-Nitrogen stable isotope was prepared based on a previously published method91. Cloning was accomplished with a pNG2 vector (Novagen). The expression plasmid containing the gene encoding for Tau was subsequently used to transform BL21(DE3) Escherichia coli cells, which were grown to an optical density (at 600 nm) of 0.8 in Luria Broth at 37 °C. Cells were pelleted by centrifugation, washed, and resuspended in M9 minimal medium (6 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 4 g/L glucose, pH 7.0) containing 0.5 g ISOGRO-15N (Sigma Aldrich) and 2 g/L 15N-ammonium chloride as the nitrogen source. Induction of protein expression was accomplished by the addition of IPTG to 0.5 mM followed by overnight incubation at 37 °C.
The cell pellet was collected and subjected to lysis using a French cell press (lysis buffer contains 20 mM MES, 1 mM EGTA, 0.2 mM MgCl2, 5 mM DTT, 1 mM PMSF, 1 mg/mL lysozyme, 10 µg/mL DNAse, pH 6.8). Salt, 500 mM NaCl, was added to the lysate, which was then boiled to denature proteins. The lysate was cleared of cell debris, DNA, and precipitated proteins by high-speed centrifugation (12700 × g, 40 min, 4 °C). Residual nucleic acids were precipitated by incubating the supernatant with 20 mg/mL streptomycin. The precipitated nucleic acids were separated by centrifugation. The resulting supernatant was incubated with 0.36 g/mL ammonium sulfate then subjected to centrifugation to collect a protein pellet mostly containing Tau protein. The protein pellet was resuspended and dialyzed into a dialysis buffer (20 mM MES, 1 mM EDTA, 2 mM DTT, 50 mM NaCl, pH 6.8). Tau was purified from the mixture using cation exchange chromatography (MonoS 10/100, 2 mL/min with a buffer gradient of 50 mM to 1 M NaCl in 20 mM MES, 1 mM EGTA, 2 mM DTT, pH 6.8), and size exclusion chromatography (Superdex 75 26/600, 2 mL/min in phosphate buffered saline with additional 500 mM NaCl). The purified Tau protein was dialyzed overnight against 50 mM NaPi pH 6.8, concentrated and flash-frozen in aliquots, and stored at −80 °C until they were used in NMR experiments. The protein concentration was determined by bicinchoninic acid assay following the manufacturer’s instructions (Pierce).
NMR Spectroscopy: 2D 1H-15N HSQC titrations of 15N-Tau
The lyophilized compounds β-Tau and β-Hsp90 were resuspended in water to a concentration of 1000 µM, followed by dilution to a final buffer composition of 50 mM NaPi pH 6.8, 10 mM NaCl, 1 mM DTT and a working concentration of 400 µM compound. For each titration series, a 200 µL solution of 2N4R 15N-Tau (20 µM) was prepared in 50 mM NaP pH 6.8, 10 mM NaCl, 1 mM DTT, 10% D2O and incremented with 20 µM, 40 µM, and 100 µM of β-Tau and β-Hsp90.
Heteronuclear single quantum coherence (1H-15N HSQC) spectra of Tau with and without compounds were collected at 5 °C using 900 MHz and 800 MHz spectrometers equipped with cryoprobes. HSQC spectra were processed using TopSpin version 3.6.2 (Bruker) and analyzed using Sparky92. The Tau binding profiles were represented by peak intensity ratios (I/I0) and chemical shift perturbations (CSP). The I/I0 ratio was calculated by dividing the peak amplitude (I) of Tau amide peaks in the presence of the compound by the corresponding peak intensity (I0) in the absence of the compound. CSPs were calculated using the equation (1), \({{\rm{CSP}}}=\sqrt{0.5\times [{(\Delta {{\rm{\delta }}}{{\rm{H}}})}^{2}{+(\Delta {{\rm{\delta }}}{{\rm{N}}})}^{2}/25]}\), where ΔδH and ΔδN correspond to the chemical shift differences of H and N. CSPs were smoothed with a 3-residue averaging window. The noise level of the CSP values was determined by the collection of the 1H-15N HSQC spectra of Tau in the absence of compounds in duplicate and applying the CSP equation given above to compare chemical shift differences between the two spectra.
Preparation of 15N- Aβ1−42
Recombinant 15N-Aβ1 − 42 (rPeptide) was resuspended in 1% NH4OH at 1 mg/mL concentration, lyophilized overnight and stored at -20°C. Aliquots containing 70 µg 15N-Aβ1 − 42 were resuspended in 300 µL of 20 mM NaPi pH 7.2, 10% D2O buffer, to obtain a concentration of 50 µM. 15N-Aβ1 − 42 samples containing 50 µM or 500 µM β-Hsp90 were prepared by adding the appropriate volume of a 1 mM β-Hsp90 stock solution in the same buffer.
2D 1H-15N HSQC titrations of 15N-Aβ1-42
5 mm Shigemi tubes were used for NMR experiments. 2D 1H-15N HSQC spectra of 15N-Aβ1−42 in the absence and in the presence of β-Hsp90 at 1:1 and 10:1 ratios were collected at 5 °C using a 500 MHz spectrometer equipped with a cryoprobe. NMR spectra were processed with nmrPipe and analyzed using Sparky.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source Data are provided with this paper as a Source Data file. All data are available in the supporting information. Direct contact with the corresponding authors for any additional information is possible by email.
References
Lashuel, H. A. Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity? Curr. Opin. Chem. Biol. 64, 67–75 (2021).
Dobson, C. M. The amyloid phenomenon and its links with human disease. Cold Spring Harb. Perspect. Biol. 9, a023648 (2017).
Nguyen, P. H. et al. Amyloid oligomers: a joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, type II diabetes, and amyotrophic lateral sclerosis. Chem. Rev. 121, 2545–2647 (2021).
Willbold, D., Strodel, B., Schröder, G. F., Hoyer, W. & Heise, H. Amyloid-type protein aggregation and prion-like properties of amyloids. Chem. Rev. 121, 8285–8307 (2021).
Sexton, C. et al. Current directions in tau research: highlights from Tau 2020. Alzheimer's Dement 18, 988–1007 (2022).
Kadavath, H. et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl Acad. Sci. USA 112, 7501–7506 (2015).
Kellogg, E. H. et al. Near-atomic model of microtubule-tau interactions. Science 360, 1242–1246 (2018).
Wang, Y. & Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 (2016).
Barbier, P. Role of Tau as a microtubule-associated protein: structural and functional aspects. Front. Aging Neurosci 11, 204 (2019).
Ramesh, M. & Govindaraju, T. Multipronged diagnostic and therapeutic strategies for Alzheimer’s disease. Chem. Sci. 13, 13657–13689 (2022).
Zhang, J. et al. Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 9, 211 (2024).
Mullard, A. FDA approves second anti-amyloid antibody for Alzheimer's disease. Nat. Rev. Drug Discov. 22, 89–89 (2023).
Reardon S. Alzheimer’s drug with modest benefits wins backing of FDA advisers. Nature. https://doi.org/10.1038/d41586-024-01726-w (2024).
Lu, H. et al. Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct. Target. Ther. 5, 1–23 (2020).
Malhotra, A. et al. Exploring novel mechanistic insights in Alzheimer’s disease by assessing the reliability of protein interactions. Sci. Rep. 5, 13634 (2015).
Mao, Y., Fisher, D. W., Yang, S., Keszycki, R. M. & Dong, H. Protein-protein interactions underlying the behavioral and psychological symptoms of dementia (BPSD) and Alzheimer’s disease. PLOS ONE 15, e0226021 (2020).
Sinsky, J. et al. Tau protein interaction partners and their roles in alzheimer’s disease and other tauopathies. Int J. Mol. Sci. 22, 9207 (2021).
Ciccone, L. et al. The positive side of the Alzheimer’s disease amyloid cross-interactions: the case of the Aβ 1-42 peptide with Tau, TTR, CysC, and ApoA1. Molecules 25, 2439 (2020).
Di Lorenzo, D. Tau protein and tauopathies: exploring tau protein–protein and microtubule interactions, cross-interactions and therapeutic strategies. ChemMedChem 19, e202400180 (2024).
Altinok, S., Sanchez-Hodge, R., Stewart, M., Smith, K. & Schisler, J. C. With or without you: co-chaperones mediate health and disease by modifying chaperone function and protein triage. Cells 10, 3121 (2021).
Wentink, A., Nussbaum-Krammer, C. & Bukau, B. Modulation of amyloid states by molecular chaperones. Cold Spring Harb. Perspect. Biol. 11, a033969 (2019).
Tittelmeier, J., Nachman, E. & Nussbaum-Krammer, C. Molecular chaperones: a double-edged sword in neurodegenerative diseases. Front. Aging Neurosci. 12, 581374 (2020).
Klaips, C. L., Jayaraj, G. G. & Hartl, F. U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217, 51–63 (2018).
Hipp, M. S., Kasturi, P. & Hartl, F. U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 (2019).
Rutledge, B. S., Choy, W.-Y. & Duennwald, M. L. Folding or holding?—Hsp70 and Hsp90 chaperoning of misfolded proteins in neurodegenerative disease. J. Biol. Chem. 298, 101905 (2022).
Uversky, V. N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 7, 18 (2015).
Graff, A. M. de, Mosedale, D. E., Sharp, T., Dill, K. A. & Grainger, D. J. Proteostasis is adaptive: balancing chaperone holdases against foldases. PLOS Comput. Biol. 16, e1008460 (2020).
Santhoshkumar, P. & Sharma, K. K. Inhibition of amyloid fibrillogenesis and toxicity by a peptide chaperone. Mol. Cell. Biochem. 267, 147–155 (2004).
Shi, C. et al. Proteolytically stable diaza-peptide foldamers mimic helical hot spots of protein-protein interactions and act as natural chaperones. J. Med. Chem. 66, 12005–12017 (2023).
Lackie, R. E. The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front. Neurosci. 11, 254 (2017).
Gupta, A., Bansal, A. & Hashimoto-Torii, K. HSP70 and HSP90 in neurodegenerative diseases. Neurosci. Lett. 716, 134678 (2020).
Dickey, C. A. et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 117, 648–658 (2007).
Young, Z. T., Mok, S. A. & Gestwicki, J. E. Therapeutic strategies for restoring tau homeostasis. Cold Spring Harb. Perspect. Med. 8, a024612 (2018).
Schopf, F. H., Biebl, M. M. & Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360 (2017).
Weickert, S., Wawrzyniuk, M., John, L. H., Rüdiger, S. G. D. & Drescher, M. The mechanism of Hsp90-induced oligomerizaton of Tau. Sci. Adv. 6, eaax6999 (2020).
Ferrari, L. et al. Arginine π-stacking drives binding to fibrils of the Alzheimer protein Tau. Nat. Commun. 11, 571 (2020).
Oroz, J. et al. Structure and pro-toxic mechanism of the human Hsp90/PPIase/Tau complex. Nat. Commun. 9, 4532 (2018).
Kasza, A. et al. Dihydropyridine derivatives modulate heat shock responses and have a neuroprotective effect in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 53, 557–571 (2016).
Roe, S. M. et al. The crystal structure of the Hsp90-LA1011 complex and the mechanism by which la1011 may improve the prognosis of Alzheimer’s Disease. Biomolecules 13, 1051 (2023).
Moll, A. et al. Hsp multichaperone complex buffers pathologically modified Tau. Nat. Commun. 13, 3668 (2022).
Karagöz, G. E. et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 156, 963–974 (2014).
Trushina, N. I., Bakota, L., Mulkidjanian, A. Y. & Brandt, R. The Evolution of Tau Phosphorylation and Interactions. Front. Aging Neurosci 11, 256 (2019).
Wang, H. et al. ATP impedes the inhibitory effect of Hsp90 on Aβ40 fibrillation. J. Mol. Biol. 433, 166717 (2021).
Evans, C. G., Wisén, S. & Gestwicki, J. E. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. J. Biol. Chem. 281, 33182–33191 (2006).
Ran, X. & Gestwicki, J. E. Inhibitors of protein-protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 44, 75–86 (2018).
Martino, E. et al. Mapping, structure and modulation of PPI. Front. Chem. 9, 718405 (2021).
Bruzzoni-Giovanelli, H. et al. Interfering peptides targeting protein-protein interactions: the next generation of drugs?. Drug Discov. 23, 272–285 (2018).
Pelay-Gimeno, M. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem. Int. Ed. 54, 8896–8927 (2015).
Laxio Arenas, J. Peptides and peptidomimetics as inhibitors of protein-protein interactions involving β-sheet secondary structures. Curr. Opin. Chem. Biol. 52, 157–167 (2019).
Pellegrino, S. et al. β-Hairpin mimics containing a piperidine–pyrrolidine scaffold modulate the β-amyloid aggregation process, preserving the monomer species. Chem. Sci. 8, 1295–1302 (2017).
Tonali, N. et al. Structure-activity relationships of β-hairpin mimics as modulators of amyloid β-peptide aggregation. Eur. J. Med. Chem. 154, 280–293 (2018).
Lesma, J. et al. β-Hairpin peptide mimics decrease human islet amyloid polypeptide (hIAPP) aggregation. Front. Cell Dev. Biol. 9, 729001 (2021).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Liu, H. et al. Disclosing the mechanism of spontaneous aggregation and template-induced misfolding of the key hexapeptide (PHF6) of Tau protein based on molecular dynamics simulation. ACS Chem. Neurosci. 10, 4810–4823 (2019).
von Bergen, M. et al. Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl Acad. Sci. 97, 5129–5134 (2000).
Abriata, L. A. A simple spreadsheet program to simulate and analyze the far-UV circular dichroism spectra of proteins. J. Chem. Educ. 88, 1268–1273 (2011).
LeVine, H. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284 (1999).
Gade Malmos, K. et al. ThT 101: a primer on the use of thioflavin T to investigate amyloid formation. Amyloid 24, 1–16 (2017).
Louros, N., Schymkowitz, J. & Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 24, 912–933 (2023).
Elbaum-Garfinkle, S., Ramlall, T. & Rhoades, E. The role of the lipid bilayer in Tau aggregation. Biophys. J. 98, 2722–2730 (2010).
Li, W. & Lee, V. M.-Y. Characterization of two VQIXXK motifs for tau fibrillization in vitro. Biochemistry 45, 15692–15701 (2006).
Daebel, V. et al. β-Sheet core of tau paired helical filaments revealed by solid-state NMR. J. Am. Chem. Soc. 134, 13982–13989 (2012).
Paul, A. et al. Inhibition of tau amyloid formation and disruption of its preformed fibrils by Naphthoquinone–Dopamine hybrid. FEBS J. 288, 4267–4290 (2021).
Seideler, P. M. et al. Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176 (2018).
Zhang, W. et al. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 8, e43584 (2019).
Lövestam, S. et al. Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. eLife 11, e76494 (2022).
Huvent, I. et al. A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge. Biochem. Biophys. Res. Commun. 445, 299–303 (2014).
Chakraborty, P. et al. Co-factor-free aggregation of tau into seeding-competent RNA-sequestering amyloid fibrils. Nat. Commun. 12, 4231 (2021).
Soeda, Y. et al. Methylene blue inhibits formation of tau fibrils but not of granular Tau oligomers: a plausible key to understanding failure of a clinical trial for Alzheimer’s disease. J. Alzheimers Dis. 68, 1677–1686 (2019).
Rizzu, P. et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 64, 414–421 (1999).
Momeni, P. et al. Clinical and pathological features of an Alzheimer’s disease patient with the MAPT Delta K280 mutation. Neurobiol. Aging 30, 388–393 (2009).
Barghorn, S. et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721 (2000).
Kumar, P., Nagarajan, A. & Uchil, P. D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb. Protoc. 2018, (2018).
Riss, T. L. et al. Cell Viability Assays. In Assay Guidance Manual (eds Markossian, S. et al.) (Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda (MD), 2004).
Igaev, M. et al. A refined reaction-diffusion model of Tau-microtubule dynamics and its application in FDAP analysis. Biophys. J. 107, 2567–2578 (2014).
Pinzi, L. et al. Quantitative live cell imaging of a tauopathy model enables the identification of a polypharmacological drug candidate that restores physiological microtubule interaction. Nat. Commun. 15, 1679 (2024).
Rahman, M. H. et al. Resveratrol and neuroprotection: impact and its therapeutic potential in Alzheimer’s disease. Front. Pharmacol. 11, 619024 (2020).
Vanmechelen, E. et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci. Lett. 285, 49–52 (2000).
Tatebe, H. et al. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 12, 63 (2017).
Wijesekara, N. et al. Combination of human tau and islet amyloid polypeptide exacerbates metabolic dysfunction in transgenic mice. J. Pathol. 254, 244–253 (2021).
Chatterjee Bhowmick, D. & Jeremic, A. Functional proteasome complex is required for turnover of islet amyloid polypeptide in pancreatic β-cells. J. Biol. Chem. 293, 14210–14223 (2018).
Cummings, J. L., Tong, G. & Ballard, C. Treatment combinations for alzheimer’s disease: current and future pharmacotherapy options. J. Alzheimers Dis. JAD 67, 779–794 (2019).
Gong, C.-X., Liu, F. & Iqbal, K. Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J. Alzheimers Dis. JAD 64, S107–S117 (2018).
Brandt, R. & Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J. Biol. Chem. 268, 3414–3419 (1993).
Wallin, C. et al. The neuronal tau protein blocks in vitro fibrillation of the amyloid-β (Aβ) peptide at the oligomeric stage. J. Am. Chem. Soc. 140, 8138–8146 (2018).
Studier, F. W., Rosenberg, A. H., Dunn, J. J. & Dubendorff, J. W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89 (1990).
Taylor, A. I. P. et al. Simple, reliable protocol for high-yield solubilization of seedless amyloid-β monomer. ACS Chem. Neurosci. 14, 53–71 (2023).
Fath, T., Eidenmüller, J. & Brandt, R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J. Neurosci. 22, 9733–9741 (2002).
Conze, C. et al. Caspase-cleaved tau is senescence-associated and induces a toxic gain of function by putting the brake on axonal transport. Mol. Psychiatry 27, 3010–3023 (2022).
Conze, C. et al. Redox signaling modulates axonal microtubule organization and induces a specific phosphorylation signature of microtubule-regulating proteins. Redox Biol. 83, 103626 (2025).
Ukmar-Godec, T. et al. Proteasomal degradation of the intrinsically disordered protein tau at single-residue resolution. Sci. Adv. 6, eaba3916 (2020).
Lee, W., Tonelli, M. & Markley, J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015).
Acknowledgments
This research has received funding from the European Union’s Horizon 2020 research and innovation program H2020-MSCAITN-2019-EJD: Marie Skłodowska-Curie Innovative Training Networks (European Joint Doctorate)—Grant Agreement No: 860070—TubInTrain (DDL, NB, RB, SO). M.Z. and L.R. thank Maria-Sol Cima-Omori for the expression and purification of 2N4R 15N-Tau. M.Z. was supported by the European Research Council (ERC) under the EU Horizon 2020 research and innovation program (grant agreement numbers 787679 and 101069214). I.G. has been supported by the Erasmus+ program (Pisa University). Robert Thai (CEA Paris Saclay) is thanked for his contribution to the LC-MS analysis of pronase stability assays.
Author information
Authors and Affiliations
Contributions
D.D.L. performed the synthesis, NMR and CD analyzes, pronase stability and participate to ThT assays on Tau under the co-supervision of N.T. and S.O. and the preparation of TEM specimens and their analysis under the supervision of V.D. N.B. performed and analyzed MTT assays and quantitative live-cell imaging under the supervision of R.B. J.K. performed and analyzed the ThT assays (Wt-Tau, TauΔK280, Aβ1-42, hIAPP). L.R. performed and analyzed the NMR titrations of WT (2N4R) 15N-Tau with β-Hsp90 and β-Tau, under the supervision of M.Z. O.L. performed and analyzed the NMR titrations of Aβ1-42 15N-Tau with β-Hsp90. I.G. synthesized AcPHF6* and performed the ThT assays of AcPHF6* under the supervision of D.D.L. and N.T. J.L. produced Wt-Tau for ThT assays and TEM. Y.H. and I.E. performed and analyzed TEM imaging of Wt-Tau and Aβ1-42. V.D. designed, supervised and analyzed TEM experiments. N.S. paid and supervised D.D.L. for TEM during his post-doctoral period at Bielefeld University. M.L.G. co-supervised D.D.L. during his period at Università degli Studi di Milano. N.T. designed the compounds and co-supervised D.D.L. R.B. supervised N.B. for MTT assays and quantitative live-cell imaging. S.O. supervised D.D.L. and N.B. and managed the global project and the preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chrisostomos Prodromou, Jun-Seok Lee and the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Di Lorenzo, D., Bisi, N., Kaffy, J. et al. Synthetic chaperone based on Hsp90-Tau interaction inhibits Tau aggregation and rescues physiological Tau-Microtubule interaction. Nat Commun 16, 8756 (2025). https://doi.org/10.1038/s41467-025-63824-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63824-1
