
Abstract
Prostate cancer (PCa) germline testing, while gaining momentum, is ancestry restrictive and African exclusive. Through whole genome sequencing for 217 African ancestral cases (186 southern African, 31 Pan representative), we identify 172 potentially pathogenic variants in 78 DNA damage repair or PCa related genes. Prevalence for reported (13/217, 5.99%) and cumulative predicted (24/217, 11.06%) variants of significance (11 genes) falls below that reported for non-Africans. Conversely, BRCA1, HOXB13, CDK12, MLH1, MSH2, and BRIP1 remain unimpacted. Through pathogenic ranking based on variant frequency and functionality, clinical presentation and tumour-matched biallelic inactivation, top-ranked candidates include PREX2, POLE, FAT1, BRCA2, POLQ, LRP1B and ATM. Besides notable impact of DNA polymerases, including POLG, Fanconi anaemia genes include FANCD2, FANCA, FANCG, ERCC4, FANCE and FANCI, while DNA mismatch repair genes MSH3 and PMS1 outranked known namesakes MSH6 and PMS2. This study provides insights into the spectrum of African-relevant potentially pathogenic PCa variants, highlighting much-needed gene candidates for ancestry-inclusive germline testing.
Similar content being viewed by others

Introduction
Germline testing (GT) for prostate cancer (PCa) is essential to optimise patients who benefit the most from precision medicine while predicting the risk of further malignancy for the patient and their relatives1. It encompasses testing for rare gene variants that are attributed to hereditary cancers, such as those involved in DNA repair2. With increased therapeutic implications1,3, GT is moving beyond PCa risk assessment to include management of patients and screening of healthy men, as advocated by the National Comprehensive Cancer Network (NCCN) guidelines and other health organisations2,4,5.
Besides a family history of PCa and younger age, African ancestry is a well-established risk factor for incidence, advanced disease and mortality6,7. However, guidelines for GT have almost exclusively been developed using non-African studies2,8,9. Recently, we showed that current GT panels are less optimal for rare pathogenic variants in South African patients of African ancestry10, with prevalence only half of that reported for non-African populations (5.6% vs 11.8–17.2%)11,12. Concurring with previous, yet limited, African American and West African studies13,14, we hypothesise that pathogenic variants mediating the high-mortality pattern of PCa among African ancestral men are largely unknown. Notably, the lack of African-relevant data led the 2019 Philadelphia PCa Consensus Conference to exclude men of African ancestry from the current PCa GT criteria8.
Representing globally the greatest PCa mortality rates15 and home to genetically the most diverse populations16, we initiated the Southern African Prostate Cancer Study (SAPCS), the founding study for the Health Equity Research and Outcomes Improvement Consortium Prostate Cancer Precision Health Africa1K (HEROIC PCaPH Africa1K)17. The overall aim - to generate African-relevant whole genome sequencing (WGS) data for the purpose of addressing PCa health disparities. Merging both published18 and unpublished (this study) SAPCS data with Pan Prostate Cancer Group (PPCG) derived African ancestral WGS data19 for a total of 217 cases, we use this unique data source to perform untargeted gene-wide interrogation for yet unknown potentially pathogenic variants. Here, we provide insights into potential gene candidates to establish PCa GT criteria for men of African ancestry.
Results
Genomic resources and patient characteristics
PCa cases recruited as part of the SAPCS or PPCG (Methods) from which blood-derived WGS germline data had been generated were sourced (Table S1). SAPCS data included 116 published18, and 70 additional cases, the latter generated to an average of 43.3X coverage (range 36.4 to 69.1X), for a total of 186 South Africans of African ancestry. PPCG data (n = 990) was sourced from five countries, including Canada, Germany, United Kingdom, Australia (Melbourne and Sydney18), and France or French Caribbean, of which 31 cases are African ancestral19. WGS African-representative younger aged (<50 years) no cancer control data included 49 population-matched South Africans (southern African controls, SAC) and 40 Kenyans representing both east Bantu and Nilotic ethno-linguistic diversity (east African controls, EAC). Medical Genome Reference Bank (MGRB) WGS control data was sourced from 3,209 largely European ancestral Australians (1332 male, 1877 female) \(\ge\)75 years at time of recruitment and with no known cancer, hypertension or dementia20. Irrespective of data source, single-nucleotide variants (SNVs) and small insertions and deletions (indels; <50 base pairs) were called using GATK best practices.
Using 64,654 ancestry-informative SNVs, population substructure analysis was performed (Methods), confirming African ancestries for all 217 cases (Fig. 1A). At optimal k = 3 population inference (Supplementary Data 1), non-African fractions >10% are notably scarce for SAPCS (2.7%, 5/186; per patient range 12% to 64%) compared to PPCG patients (64.5%, 20/31, range 10.4% to 69.1%). Further, k = 4 defined SAPCS patients as southern African, with 68.8% (128/186) including southern African Khoe-San heritage (range 2% to 51.3%) (Fig. 1B). Ancestries within the PPCG are primarily west African derived (range 23% to 99.9%), with overall larger non-African fractions, as expected for Caribbean and African American patients, apart from a single PPCG patient with 52.9% southern African ancestry. SAPCS patients presented on average 2 years later (mean 66.7 years; range 43-99) compared with PPCG cases (mean 64.8 years; range 45-77) and with significantly advanced International Society of Urological Pathology Grade Group (ISUP) \(\ge\)4 (53.2% vs 19.4%, Chi-squared p-value < 0.0001) disease (Table S2). As previously reported21, SAPCS men present with elevated Prostate-Specific Antigen (PSA) levels (mean 233.6 ng/mL; range 1 to 4,841) at almost 4-fold greater than PPCG Africans (mean 60.8 ng/mL; range 5 to 1150).

Admixture plot for the study cohort including 186 South African (SAPCS) and 31 PPCG African ancestral patients using k-means clustering for k = 3 (A, cross-validation error = 0.252, Supplementary Data 1) and k = 4 (B, cross-validation error = 0.255, Supplementary Data 1). Population fractions have been determined against reference controls defined as; European (CEU, n = 20), Asian (CHB, n = 20), west African or Yoruba (YRI, n = 20), African American (ASW, n = 20), San (KSGP, n = 20) and east African or Luhya (LWK, n = 20).
Potentially pathogenic variants in African ancestral PCa patients
Nearly 59 million SNVs and 10 million indels from 217 African patients were interrogated for known potentially pathogenic variants (PPVs; Fig. 2 Step 1). Using the non-African biased ClinVar database, which includes the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) guidelines22, pathogenic or likely pathogenic variants were identified and screened for all populations and African restricted minor allele frequency (MAF) using gnomAD v.4.023. Consequently, 252 low-frequency inclusive PPVs were identified in 223 genes (195 SNVs, Supplementary Data 2 and 57 indels, Supplementary Data 3; 90 missense, 86 stop gain or loss, 22 splice variants, 51 frameshifts, 5 non-coding), of which 33 PPVs are absent from current databases (defined as unknown). Focusing on rare variants (MAF < 1%) resulted in 241 PPVs in 214 genes, with further Gene Set Enrichment Analysis (GSEA)24 focused on genes associated with DNA damage repair (DDR) or PCa germline gene candidates, leaving 45 rare PPVs (11.11% unknown) in 34 genes (Table 1). Conversely, a single PPV in the DDR gene POLG p.Phe749Ser, while rare in population-wide global data (MAF = 0.0002) and absent in our African ancestral PPCG patients, presented at low frequencies in our SAPCS cases (MAF = 0.0134) and as such is classified here as a population-specific low-frequency (PSLF) PPV.

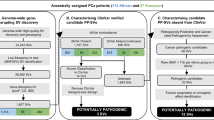
Step 1. From genome-wide small variants (SNVs, single nucleotide variants; indels, insertions or deletions <50 bases) derived from 217 African PCa cases (blue) 45 rare DNA Damage Repair (DDR) or PCa related PPVs in 34 genes (Table 1) and a single PSLF-PPV (Table 3) were identified. Step 2. Rare and low-frequency PPV candidate genes (n = 223) were further filtered for non-African representative PPVs using European-biased PCa (PPCG, orange) and healthy (MGRB, red) datasets, provided multi-ethnic validation for 22 gene candidates, genetic conservation for five genes and no further PPV candidate exclusion. Step 3. Prioritizing African-derived variants of unknown significance (VUS) for classification as POVs, as per exclusion and inclusion criteria (grey), yielded 138 rare DDR/PCa related POVs in 61 genes (Table 2) and 16 PSLF-POVs in 11 genes (10 overlapping with POV candidates, Table 3). Minor allele frequency (MAF) filtering (steps 1 and 3) was based on all population and African restricted gnomAD v4.0 data. Step 4. All class potential pathogenic variants were further filtered using population control MAFs >2% (SAC, southern African controls; EAC, east African controls) and variant allele frequency (VAF) < 30% for a total of 172 variants of pathogenic potential across 78 candidate genes.
Cross-ancestral correlations for African-relevant PPV-derived gene candidates
Focusing on the 223 genes harbouring low-frequency inclusive PPVs from African patients, we further interrogated for PPVs in 959 non-African PPCG cases and 3,209 MGRB healthy-aged controls (Fig. 2 Step 2). Here we identified 293 rare PPVs impacting 53.8% (120/223) of our African-derived gene candidates in 37.6% (361/959) non-African patients (Supplementary Data 4). Known PCa GT genes include BRCA2 (14 unique PPVs), ATM (7), CHEK2 (5), TP53 (3), RAD50 (2) and RAD54L (1). We also found PPVs in multiple African-relevant gene candidates including RECQL4 (5 unique PPVs), JAK2 (4) INO80 (3), EGFR (2), and ASPM (2), while APTX, LRP1B, FANCG, FANCD2, ERBB3, BUB1B, POLE, BLM, RAD54L, JAK3 and U2AF1 each presented with a single unique PPV each. Notable African-relevant GT candidate genes that lacked variance included TRRAP, CHD1L, ERBB4, MSH3, ROS1, PREX2, MYC, RET, CHD4, NF1, DONSON and STAG2. Additionally, 13 rare PPVs were shared between the ancestries (Table S3), including CHEK2 p.Arg283X and RAD50 p.Glu723fs, both previously known in PCa.
For the healthy European ancestral population, we identified 855 rare PPVs impacting 74% (163/223) of gene candidates in 63.4% (2,004/3,209) of MGRB participants (Supplementary Data 5). The most abundantly impacted genes, although rare, include known PCa GT panel genes ATM (12 unique PPVs) and CHEK2 (7), while African-relevant candidates included EGFR (13), CHD4 (12), ERBB4 and RECQL4 (7 each). Of the 19 PPVs shared with our African PCa patients (Table S4), two impacted African-relevant candidates RET p.Val804Met and the STAG2 splice variant rs1603095192G>T in a single individual each (MAF = 0.00016) and as such were not removed as candidates. In contrast, African-relevant PCa GT candidate genes RAD54L, ROS1, LRP1B, JAK3 and U2AF1 were highly conserved (lacked notable variance). Taken together, no genes were excluded based on the European population data.
Characterising variants of unknown significance as potentially oncogenic
Low-frequency inclusive African variants of unknown significance (VUS), that are not in ClinVar and/or defined using ACMG-AMP criteria as pathogenic/likely pathogenic or benign/likely benign were further interrogated for oncogenic potential (Fig. 2 Step 3). After exclusion for common variants (MAF > 5%) found in all population and African restricted gnomAD data, VUS were maintained based on their functional potential defined as deleterious in SIFT25, and/or damaging in PolyPhen-226, or disrupting a stop codon or splice junction, with additional oncogenic potential established using the Cancer Genome Interpreter (CGI)27 providing the definition in this study as a potentially oncogenic variant (POVs). Identifying 529 POVs in 274 genes, after exclusion for common/low-frequency POVs (MAF > 1%) left 476 rare POVs in 261 genes (Supplementary Data 6). Focusing on DDR or PCa-associated genes, 138 rare POVs (15 unknown) remained in 61 gene candidates, including seven in known PCa GT-panel genes with an additional nine previously identified as PPV-derived candidates (Table 2), leaving 45 potential POV-derived candidate genes (Supplementary Data 7), and 16 PSLF-POVs in 12 (11 overlapping with rare POV-derived) candidate genes (Table 3).
Population-matched control and CHIP-associated filtering
As southern Africans are poorly represented in population databases such as gnomAD28, we further sought to determine MAFs in healthy population-matched southern and east African controls (Fig. 2 Step 4). While three PPVs, RAD50 p.Glu723fs (known to PCa), TRRAP p.Ala505fs and the inframe deletion identified in RECQL4, presented in a single East African (EAC MAF = 0.0116279), the latter including a single Southern African (SAC MAF = 0.0102041), none were excluded from further analyses (Table 1). Absent or negligible in all population or African restricted gnomAD data, 13 POVs were found to be rare in either SACs or EACs (single subject each) and as such were not excluded (Table 2 and Supplementary Data 7). Although JAK2 p.Arg922Trp and NDRG1 p.Ala84Ser were found in two SACs each (MAF=0.0204082), due to their absence from our EACs, we elected to cautiously maintain these POVs in downstream analyses, setting our MAF threshold for exclusion at >2%. As such, three POVs ERCC6 p.Thr699Met (EAC MAF = 0.0465116), p.Ala906Gly (EAC MAF = 0.0348837) and ERCC4 p.Ala860Asp (SAC/EAC combined MAF = 0.031915) were removed, leaving 135 POVs in 61 genes for further consideration.
Rare globally (through all population analyses) yet presenting at low frequencies within our African ancestral cases, the single PSLF-PPV and 13 of the 16 PSLF-POVs (81.25%) were restricted to our SAPCS cohort (Table 3). Notably, PSLF-POVs impacting known PCa GT panel genes ATM (p.Asp44Gly) and PMS2 (p.Leu729X) presented in both our SACs and EACs (MAFs range 0.0204 to 0.0306) as did nine of the remaining PSLF-POVs and as such were removed from further analyses (Supplementary Data 8). Absent from SAC and EAC cohorts, besides the PSLF-PPV impacting the DDR gene POLG (p.Phe749Ser), the five remaining PSLF-POVs impacting the DDR-relevant oncogene PREX229 (p.Lys787Glu, p.Arg1230Trp, and rs150773140 slice donor) and DDR genes POLQ (p.Leu232Ile) and CREBBP (p.Gln2204 frameshift) warrant further consideration.
Additionally, clonal haematopoiesis of indeterminate potential (CHIP), the natural process of acquiring somatic alterations in haematopoietic stem cells as a person ages, was further considered. After visual confirmation using Integrative Genomics Viewer (IGV)30, read count was used to determine variant allele frequencies (VAFs) and in turn associated CHIP. Of our 81 rare/PSLF PPV/POV derived candidate genes, five are recognised as CHIP associated31 and include by ranking DNMT3A (1st), TET2 (2nd), PPM1D (4th), TP53 (5th), and JAK2 (7th). Falling within the CHIP associated VAF threshold, defined conservatively here as <0.332, all six DNMT3A POVs (VAF range 0.205882 to 0.257143), the single TP53 PPV (VAF = 0.209302) and one each of the three TET2 POVs (VAF = 0.24) and of the two PPM1D POVs (VAF = 0.17) were removed from further analysis. Appreciating missing PPCG VAF data, additional unknown CHIP gene-associated variants removed included both TRRAP PPVs occurring in a single 84-year-old patient, the BRCA2 p.Lys2740X PPV, the KMT2C p.Gly3170Ala POV and the single NCM8 POV p.Phe274Ile. After MAF (SAC and EAC) and VAF (CHIP) filtering 41 rare PPVs (32 genes) and 125 rare POVs (59 genes) remained. As none of the PSLF-PPV/POVs fell below the CHIP-associated VAF threshold, all 6 MAF-filtered PSLF variants remained (4 genes). A total of 172 pathogenic variants impacting 78 candidate genes were further considered (Supplementary Data 9).
Ranking variant pathogenicity and gene candidates
Providing further evidence for our focus on DDR-relevant genes, gene ontology (GO) enrichment and pathway analysis using g:profiler33 for all 473 genes harbouring low-frequency inclusive PPVs (n = 252) and POVs (n = 529) revealed DNA damage response and DNA repair as the most enriched biological processes (Fig. S1). Molecular functions were biased towards catalytic activity on DNA and ATP-dependent activity on DNA across the genes. To provide further pathogenic-value to the 172 variants across 78 genes, we developed a 9-step ranking system which provides a weighting (see Methods) for variant features, clinical presentation and when available (116 SAPCS, 31 PPCG) somatic biallelic inactivation (Fig. 3A). A half rank was removed for variants within CHIP-associated genes31, although well above the VAFs CHIP-threshold, while a full rank was gained for SAC/EAC MAFs <1%, PPV over POV status, and for variants showing potential Loss of Function (pLoF) as estimated using LOFTEE23. For clinical features at presentation, less weighting (half a rank) was applied for PSA levels, as elevated non-age-driven PSA heterogeneity has been observed for SAPCS men presenting both with and without PCa21. While presenting up to 10 years younger than the study mean, having an ISUP GG \(\ge\)4 and a family history of PCa all earned a full rank each, this was doubled for men presenting over 10 years younger than the study mean and halves for men with a family history of breast or ovarian cancer. Tumour features were defined by loss of heterozygosity (LOH), requiring overlapping somatic copy number loss or somatic SNV with allelic fractions >65% or 15% greater than the germline allele frequency34, and/or a second hit following Knudson’s two-hit hypothesis35, while a minimal value was applied for missing data (no matched tumour). While our system provides weighting for variance recurrence, gene-matched rare and PSLF PPV/POVs were ranked separately.

A Ranking system overview based on variant, clinical and tumour features. B Ranking for 24 rare PPV/POVs identified in known PCa GT genes, including previously reported (known) and not reported (unknown) variants. C The 11 known PCa GT genes ranked by weight (total ranked score), prevalence and total number of variants. D Ranking for 142 reported (known) and not reported (unknown) rare PPV/POVs impacting 66 candidate genes not included in PCa GT panels. E Ranking by weight (ranked score) for all 78 known and unknown PCa GT gene candidates, with population-specific low-frequency (PSLF) candidates assessed independently and represented as gene duplicates (stars), while providing an additional gene candidate CREBBP.
Focusing on known PCa GT genes (24 rare PPV/POVs in 11 genes), we observe a study prevalence of 11.06% (24/217), with a single PPCG patient (PPCG0019, 57% African genetic ancestry) presenting with three candidate variants in RAD54L, PMS2 and FANCA each, with the latter variant showing a 2nd hit and LOH in the patient-matched tumour. The skewing towards PPCG (25.81%, 8/31) over SAPCS patients (8.60%, 16/186), likely reflects not only the elevated non-African ancestral fractions within PPCG patients, but also the under-representation of southern Africans in PCa genetic data. The highest ranked variants include ATM (p.Arg3047X and p.Arg2832Cys), BRCA2 (p.Ile1924fs), FANCA (p.Arg504Gly) and BRCA2 (p.Trp31Arg and p.Gln2850fs) (Fig. 3B), which includes the highest ranked genes BRCA2, ATM, and FANCA, followed by RAD54L and PMS2 (Fig. 3C). For the unknown gene candidates (142 rare PPV/POVs in 66 genes), the highest ranked variants (>5.5 median) include TRRAP (p.Ala2335Gly), POLE (p.Pro99Leu), APTX (rs146487634 splice donor variant), ASPM (p.Arg1271X), POLE (p.Glu1241X), RTEL1 (p.Arg898Cys), KMT2D (p.Gln3861fs), LRP1B (p.Ala3469Thr), ERBB3 (p.Thr618Ser), and MSH3 (p.Ile537fs) (Fig. 3D). While TRRAP, POLE, APTX, RTEL1 and MSH3 are known DDR genes, more recently ASPM36, KMT2D37, LRP1B38 and ERBB339 have been defined as DDR relevant. Merged with our known and PSLF gene candidates, while PREX2 PSLF variant restricted, POLE and FAT1 outrank BRCA2, and POLQ and LRP1B outrank ATM (Fig. 3E). When combining rare and PSLF variants, POLQ outranks PREX2, while POLG ranking approaches that of ATM. In contrast to the DDR DNA poloymerase genes, POLE, POLQ and POLG, and DDR-relevant genes, PREX2 and LRP1B, FAT1 is a known PCa tumour suppressor40. Additional unknown candidate genes outranking FANCA include known DDR genes ERCC2, RECQL4, CLSPN, MSH3, FANCD2, HERC2, TRRAP and CREBBP (PSLP driven), DDR-relevant genes ROS1, ASPM, KMT2D, ERBB3, PRDM2, FGFR4, KMT2C, LEF1 and PER1, and the PCa germline associated oncogene RET (Supplementary Data 10).
Southern African patient-matched tumour mutational burden and signatures
Besides tumour features linked directly to PPV/POV ranking, having observed an overall higher tumour mutational burden (TMB, 1.197 vs 1.061 mutations/Mb, Log10-transformed t = 2.5207, P = 0.01308) and enrichment of mutational signatures of unknown significance (10 vs 1) in our SAPCS versus European-derived tumours18, we further sought to correlate biologically relevant PPV/POV status with patient-matched TMB, with a focus on the PPV/POVs impacting the DNA polymerases, and tumour enrichment for signatures known to be associated with the same or similar largely DDR-related aetiologies. Ranking TMBs for all 116 SAPCS patients, 10/20 (50%) of DNA polymerase presenting PPV/POV patients presented with a TMB above the median (1.23 mutations/Mb), ranging from 1.53 to 3.31 mutations/Mb and including a single outlier UP2113 (59.61) with associated microsatellite instability (MSI) (Table 4). Three patients presented with two POL gene PPV/POVs each, including the TMB outlier (POLE p.Pro99Leu and POLQ p.Leu232Ile), while KAL0074 (POLE p.Ser864Cys and POLG p.Arg993Cys) presented with an above median TMB (1.598). Notably, mutational signatures associated with TMB or DNA polymerase variants, such as single-base-substitution (SBS)9, SBS10 (all), SBS14 and double-base-substitution (DBS)3, were absent in our study.
While the BRCA2-associated signature SBS3 was found to be enriched in a single SAPCS patient with no DDR/known PCa-associated PPV/POV germline variant, no enrichment was observed for signatures with associated DDR-related aetiologies, including SBS6, SBS15, SBS21, SBS26 and SBS44, while the MSH6 POV carrier did not present with the gene-associated copy-number (CN)25 tumour enrichment. Conversely, 22 PPV/POV-presenting SAPCS patients harboured DDR-like mutational signatures (Table 5), including DBS7 (defective DNA mismatch repair), insertion-deletion (ID)1 and ID2 (defective DNA mismatch repair/DNA replication slippage), ID6 (homologous recombination DNA damage repair associated with BRCA2/1 mutations), ID8 (repair of DNA double strand breaks by non-homologous DNA end-joining mechanisms) and structural-variation (SV)3 (homologous recombination deficiency), of which 9/22 (40.9%) or 9/20 (45%, excluding for MAF/VAF criteria) presented with two or more PPV/POVs. Notably, two patients with POLQ POVs (p.Arg784Cys and p.Ser1618X) showed enrichment for both ID1 and SV3.
Discussion
Recent research indicates that 88% of early PCa mortality occurs in individuals with high genetic susceptibility or a family history of cancer, while only one-third of these deaths are preventable through lifestyle modification41. Additionally, outcomes for patients with DDR-specific pathogenic variants have been shown to ameliorate with adjunct hormone therapy or chemotherapy2, including a positive response to poly-(ADP ribose) polymerase (PARP) inhibitors42. Taken together, this underscores the importance of GT, which is gaining momentum4. Targeting largely DDR genes, the prevalence among men meeting NCCN screening criteria is estimated at 15–17%11,12. Focusing on 60 cancer susceptibility genes, a recent study of 1883 men undergoing tumour WGS, irrespective of clinical presentation yet biased towards metastatic disease, found 22% with a cancer driver also presented with an actionable pathogenic germline variant43. As with the latter study, current literature has almost exclusively focused on European ancestral populations. As such, detecting pathogenic variants in African populations at greatest risk for PCa-associated mortality is hindered by a paucity of data10,14.
Here we perform a comprehensive non-targeted WGS-based interrogation for African ancestral PCa patients, with a focus on the region most impacted by associated lethality—southern Africa15. Reporting a prevalence of 5.99% for PPVs in known PCa GT candidate genes (12 PPVs, 6 genes in 13 patients), restricting our analysis to men with \( > \)90% African genetic ancestry reduced the prevalence to 4.69% (9/192) and a roughly 3-fold reduction in reported PCa GT efficiency. Appreciating that African-relevant PPVs are likely underrepresented in current databases, exacerbated by European-centric guidelines, we used a previously employed method to filter VUS with a high possibility of oncogenicity10. Identifying 12 POVs in 12 patients, we increased the number of represented known PCa GT genes to 11 and a prevalence of 11.06% (24/217 all African) or 9.90% (19/192 restricted African ancestry >90%), which remains below that reported for non-African populations. While the most impactful variants defined by our ranking system were both in ATM (p.Arg3047X and p.Arg2832Cys), overall the most impacted known PCa GT gene was BRCA2. Conversely, no PPVs/POVs were identified in BRCA1, HOXB13, CDK12, MLH1, MSH2, or BRIP1.
The decreased prevalence for known PCa GT candidate-impacted genes in our African cohort, with further genetic conservation of six candidates, further highlights the potential for yet unknown African-inclusive gene candidates. Irrespective of gene candidates or function, we found notable enrichment for DDR biological processes for genome-wide PPV/POVs, providing further justification for tailored gene discovery. Aware of the under-representation of African-derived data in ClinVar and used for the development of ACMG/AMP guidelines, it was essential that we provide further clarification for VUS, which, taken together, resulted in the identification of 148 rare/PSLF PPV/POVs across 67 unknown gene candidates. Notably, PREX2, POLE and FAT1 outrank BRCA2, while POLQ and LRP1B outranks ATM. Overall, the DNA polymerases POLE, POLQ, and POLG represent the highest combined rankings, with the latter two including PSLF-POV representation. This coincides with a recent study reporting germline POLE and POLQ variants in African American PCa patients44, while the reported benefit for Durvalumab therapy in colorectal cancer patients with germline POLE mutations45 holds potential for PCa precision oncology. Additionally, we found 50% of the tumour-matched SAPCS POLE, POLQ, and POLG carriers to present with an above median TMB. While Fanconi Anemia-associated genes BRCA2, FANCA and PALB2 are known PCa GT candidates11,12, FANCD2 outranked FANCA, with FANCG, ECCR4, FANCE and FANCI (in order of ranking) potential candidates. Intriguingly, the FANCG p.Tyr213fs deletion has previously been associated with breast cancer in a South African patient46. While DNA mismatch repair genes MSH6 and PMS2 are known PCa GT candidates11,12, unknown candidates MSH3 and PMS1 out-ranked their namesake counterparts by 3.5- and 1.1-fold, respectively. Our findings are further supported by MSH3 germline rare variants having been associated with PCa in Chinese patients47, while rare PMS1 variant has been linked to hereditary breast cancer48. Two of the three DNA helicase genes RECQL4 and BLM rank 7 and 0.5 points above the study median, respectively, with RECQL4 supported by published PCa germline variants49,50. We found KMT2D, KMT2C, TRRAP and CREBBP, genes involved in chromatin remodelling, to outrank FANCA. Conversely, the epigenetic modulators DNMT3A and TET2 showed CHIP-associated VAFs for all six DNMT3A one of three TET2 variants. While DNMT3A was removed from our candidate gene list, rare TET2 variants have been reported for African American PCa patients51. Additionally, while the single PPV in the known PCa GT and highly ranked CHIP-associated gene TP53 showed evidence for non-inheritance and was as such removed, all three PPV/POVs in the highly ranked CHIP-associated gene JAK2 were retained as somatic, achieving a median ranking. Another Janus kinase (JAK) gene making the list included JAK3 (6 ranking).
Providing insights for possible African-relevant PCa GT candidate genes, it is notable that although a recent DDR-targeted study of 17,000 European PCa patients advocated for the inclusion of XRCC2, MRE11, POLK, POLH, and MSH59, only MRE11 (4.5 ranking) was identified in our study. Irrespective of ancestry, however, both studies call for focus on the DNA polymerase genes. Additionally, while NCCN guidelines2 recommend the inclusion of BARD1 (4.5 ranking) and RAD54L (5.5 ranking), these genes are largely absent from commercially available panels12. Besides the missense POVs reported here, recently we described a BARD1 pLoF large deletion in a SAPCS patient with associated somatic LOH52, emphasising the potential for overlooked inherited structural variants through our study focus on small variants. Other potential limitations include assessing for pLoF in oncogenic candidates such as RET, ROS1, FGFR4, and MYC, while FAT1 and LEF1 reported to oscillate between oncogenic and tumour suppressive behaviour. While no PPV/POVs identified in these genes showed pLoF, we are unable to determine their potential gain-of-function. Additionally, ROS1 (ranking 11 points above the median) has been shown to display DDR activity53, is a BRCA-negative breast cancer gene candidate54, and has been shown to harbour PPVs in Chinese PCa patients55. Furthermore, our highest impacted gene PREX2, a DDR-relevant oncogene29, harboured a single splice donor disrupting pLoF PSLF-POV requiring further functional clarification. While our data alludes to the benefits of our whole genome approach, we acknowledge limitations of defining true functionality, with the inevitable potential for pathogenic misclassification. Additionally, while candidate PPV/POVs in known GT genes, including BRCA2 (p.Trp31Arg) and FANCA (p.Arg504Gly) showed tumour associated DDR-like mutational signature enrichment, the 28 PPV/POVs in unknown GT-candidates showing DDR-like mutational enrichment provides further merit for consideration.
Besides unknown and overlooked gene candidates, the lack of guidelines or management plans for over 20% of current GT genes identified in PCa has limited GT application56. Increased affordability and accessibility for GT have seen a growth in uptake among men not meeting NCCN criteria57, with a caveat of poor panel coverage leading to negative results and false reassurance11, which is more likely in African populations who exhibit understudied and distinct genetic patterns8,10,18,28. Noting that numerous actionable germline variants are overlooked using current panels, a recent non-African study advocated for WGS as a cost-effective alternative58. Additional non-genomic considerations include the elevated clinical heterogeneity observed across ethno-linguistic groups from the same region within sub-Saharan Africa59,60, while defining high- or very-high-risk PCa based on European-derived NCCN PSA inclusion criteria (PSA > 20 ng/mL) for PCa GT screening, as shown for SAPCS21, requires African-specific criteria. In concordance with others6,61,62, we need to consider reduced PCa awareness in addition to cultural barriers driving later diagnosis and reduction in knowledge with regards to family history as observed for more rurally located SAPCS recruits17,63.
In conclusion, our findings underscore the complexity of designing an African-inclusive GT panel for PCa, necessitating multiple panels or a broader range of genes than those pertinent to non-African populations. Our refined set of genes and germline variants provides a much-needed framework for stratification in clinical trials and serves as a roadmap for functional validation studies. These can be utilised across African populations in precision medicine, with potential applications extending both within Africa and worldwide.
Methods
Ethics and inclusion statement
As per the HEROIC PCaPH Africa1K inter-institutional Collaborative Research Agreement (CRA) and Global Code of Conduct for research in resource-poor settings, locals have been included in all aspects of the research including study design, local primary ethics approvals and stewardship, study implementation, analysis and authorship, to intellectual property and data ownership. Capacity building across South Africa and Kenya includes (i) awarded and self-managed budget allocation, which has led to numerous employments including clinicians, scientists, nurses, field workers and administrators, (ii) sourcing infrastructure, resourcing and providing clinical training to provide much needed urology screening in under-resourced regions, (iii) co-supervision and exchanges for postgraduate students to genomic intensive partner laboratories, (iv) providing access to off-site high performance computational infrastructure, while (v) holding on-site annual training workshops in projects related topics. Through engagement and inclusion of local policy makers, consumer representatives and public health leaders, the team is committed to the dissemination of scientific data back to communities and local government.
Ethics approvals and institutional agreements
Biological male patients (verification of prostate organ) and population-representative sex/gender-unbiased controls provided informed consent to participate in the study and were recruited as part of the SAPCS (patients and controls) or East African Prostate Cancer Study (EAPCS, controls only). For the SAPCS, study approval was granted by the University of Pretoria Faculty of Human Research Ethics Committee (HREC #43/2010, including US Federal-wide Assurance FWA00002567 and IRB00002235 IORG0001762) in South Africa, with additional Institutional Review Board (IRB) approval granted by the Human Research Protection Office (HRPO) of the US Army Medical Research and Development Command (E02371.2a TARGET Africa; E03333.1a and E05986.1a HEROIC PCaPH Africa1K). For the EAPCS, study approval was granted by the Kenyatta National Hospital (KNH) and University of Nairobi (UON) Ethics Research Committee (ERC) in Kenya (KNH/UON-ERC P637/07/2019), with additional IRB approval granted by the US Army Medical Research and Development Command HRPO (E03347.1b and E05987.1a HEROIC PCaPH Africa1K). Samples (whole blood) were shipped to the University of Sydney in accordance with institutional Material Transfer Agreements (MTAs) and including for the SAPCS under a Republic of South Africa Department of Health Export Permit (National Health Act 2003; J1/2/4/2), while data sharing includes is made possible by a full-executed inter-institutional CRA between the HEROIC PCaPH Africa1K study leads including the University of Sydney (Australia), University of Pretoria (South Africa), University of Nairobi (Kenya) and University of Chicago (U.S.A.). Molecular genetic research for patients from the SAPCS bioresource was approved by the St. Vincent’s Hospital Human Research Ethics Committee in Sydney in Australia (#SVH15/227), with additional IRB approval granted by the US Army Medical Research and Development Command HRPO (E02371 TARGET Africa; E03280.1a and E05984.1a HEROIC PCaPH Africa1K). As an International Cancer Genome Consortium (ICGC) member, the PPCG collection is subject to the standards of ethical consent. Country-specific IRB approvals, which included Australian samples from Melbourne (Epworth Health 34506; Melbourne Health 2019.058) and Sydney (St Vincent’s HREC #SVH/12/231).
Participants
PCa patients
The 217 African ancestral participants were recruited either at routine, and as such non-compensated, PCa diagnosis from a participating SAPCS urology clinic in South Africa or at radical prostatectomy from a participating PPCG member site. Study inclusion was based on a histopathological confirmation of PCa defined as a Gleason score or an International Society of Urological Pathology Grade Group (ISUP) and a self-reported and/or genetically predicted African ancestry. For the SAPCS, 186 men self-identifying as African ancestral or more specifically from a southern African Bantu ethno-linguistic group, were selected for whole genome interrogation, including both published (n = 116)18 and unpublished data (n = 70). The additional PCa patients represented South Africans recruited at research hubs for the TARGET Africa and/or HEROIC PCaPH Africa1K US-DoD-funded projects, which included Dr George Mukhari Academic Hospital of the Sefako Makgatho Health Sciences University, an urban hub in the province of Gauteng, or at Tshilidzini Hospital, an approved University of Pretoria research hub, within the rural province of Limpopo. Conversely, the PPCG includes whole genome data for 959 PCa cases sourced from Canada (n = 303), Germany (n = 238), United Kingdom (n = 226)64,65, Australia (n = 143 Melbourne, 53 Sydney)18, and France (n = 25), of which 31 (3.1%), including 11 Canadians, 10 British and 10 French Caribbeans, reported African ancestry18.
African controls
The HEROIC PCaPH Africa1K has access to 49 southern Africans self-identified from one or more southern Bantu ethno-linguistic group and recruited as part of the SAPCS, and 40 east Africans self-identified from either an eastern Bantu or Nilotic ethno-linguistic group via the EAPCS. Participation as a population-matched study control included two-generational African ethno-linguistic identity, being less than 50 years of age, no PCa or any cancer diagnosis, and unlike our case cohort, representing any self-reported gender. Having undergone deep whole genome sequencing (unpublished), provided the background for targeted candidate gene interrogation for population-relevant MAFs.
Healthy controls
The MGRB samples were gathered from 3,209 European ancestral Australian individuals aged 75 years or older with no known metabolic illnesses including hypertension, cancer, or dementia20. WGS of the samples was performed on Illumina HiSeq X sequencers, generating a median coverage of 37.31X (range 21.95 to 44.12X). Mapping was built on GRCh37 and variant calling was performed following GATK best practices as previously described20.
Whole genome sequencing and variant calling
As previously described for the SAPCS18, DNA was extracted from whole blood (Qiagen kits) from treatment-naïve patients and 2 x 150 cycle paired-end whole genomes were sequenced (Illumina HiSeq X Ten or NovaSeq) to an average of 45X coverage (range, 30 to 71X) and aligned to the GRCh38 reference. SNVs and small insertions and deletions (indels; <50 base pairs) were called using the Genome Analysis Toolkit (GATK v4.1.2.0, Broad Institute)66 and variant data made available through the SAPCS Data Access Committee (DAC), with data deposited for 116 published genomes at the European Genome-phenome Archive (Table S1). Another 70 Southern African PCa patients were deep sequenced using the Illumina NovaSeq Plus (University of New South Wales Ramaciotti Genomics Facility) to an average of 43.3X coverage (range 36.4 to 69.1X), with SNVs and indels called using the Sydney Informatics Hub quality control (QC) and germline-ShortV joint-calling (see Code Availability). PPCG whole genome data have been generated by each participating country, as previously described19, with data sourced from Australia, including Sydney’s Garvan/St Vincent’s PCa Database18 and Melbourne Research group, Canadian PCa Genome Network, French ICGC PCa group, Germany ICGC PCa group, and CRUK-ICGC Prostate Group, UK. Apart from the Australian Sydney variant data called using the SAPCS pipeline18, all remaining PPCG variants were called using a single GRCh37-referenced liftover19.
Genetic ancestral fractions
Further clarification of African ancestry and population substructure was performed for all 217 cases. Representative control populations from the Human Genome Diversity Project (HGDP) and 1000 Genomes Project (1KGP) and incorporated within the gnomAD v3.1. The database included 20 individuals each representing East African (Luhya, LWK), West African (Yoruba, YRI), African American (ASW), European (CEU) and Asian (Han Chinese, CHB) ancestries23. The 20 southern African KhoeSan were derived from the KhoeSan Genome Project (KSGP, unpublished data Hayes Lab). Using a set of 77,369 linkage disequilibrium (LD)-pruned exomic single nucleotide variants (SNVs), previously used to characterise the major substructure between African regions28, after filtering for variants that were not fixed in the current dataset, a total of 64,654 SNVs were used for ADMIXTURE v1.3.067 analysis and tested for k = 1 to 10 with five-fold cross-validation (CV) and 10 replications each. While k = 3 generated the lowest mean CV error at 0.2525 (10/10 replicates in concordance), k = 4 had slightly higher mean CV error at 0.255 (10/10 replicates in concordance) and could distinguish Southern African ancestry from West African ancestry, which was used to further refine patient ancestral population substructure.
Variant pathogenicity prediction and classification
Following the identification of pathogenic/likely pathogenic variants in the Clinvar database, which includes the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines, variants with a population minor allele frequency (MAF) < 5%, as defined using gnomAD v.4.023, were recorded here as potentially pathogenic variant (PPVs). Genes whose link to DDR was more recently discovered, as well as genes with evidence of reported germline variants in PCa were also included. Genes harbouring PPVs among the African PCa patients were interrogated for variant pathogenicity among the PPCG European PCa patients and the MGRB healthy controls. The genes were excluded from the African-relevant list if the overall MAF of the PPVs were higher in these populations compared with the African patients. For all the remaining variants, those reported as deleterious or damaging using the SIFT25 and PolyPhen-226 prediction tools, respectively, that resulted in a stop codon or splice junction disruption were further selected. Variants were removed if they were reported as benign/likely benign in ClinVar or by the ACMG/AMP guidelines or had an MAF > 5% from all population-defined gnomAD data. Finally, variants were described as potentially oncogenic variant (POVs) if they were reported as an oncogenic driver in the Cancer Genome Interpreter (CGI)27. These variants were further refined to include those involved in DDR24, those with evidence of germline variants in PCa (according to the same standards), and MAF < 1%. All candidate PPVs and POVs were visually confirmed through allele frequencies using Integrative Genomics Viewer (IGV)30.
Candidate gene ranking
To confirm that the variants in our candidate gene list were inherited rather than resulting from CHIP, we analysed read counts to ascertain variant allele frequencies, removing variants with VAF < 30%32. For our 9-step ranking system, variant feature weighting included (i) CHIP-associated gene (−0.5), (ii) SAC/EAC MAF < 1% (+1), (iii) PPV over POV (+1), and (iv) pLoF (+1), clinical features of patients at diagnosis/surgery with weighting included (v) age up to 10 years younger (+1) or over 10 years younger (+2) than cohort mean (mean 67 years for SAPCS and 65 years for PPCG patients), (vi) ISUP GG = 3 (+0.5) and \(\ge\)4 (+1), (vii) PSA > 60 ng/mL (+1), which is based on the more conservative PPCG cohort mean, and (viii) family history (1st or 2nd-degree relatives) of PCa (+1) or breast and/or ovarian cancer (+0.5), and lastly (ix) tumour features including gene-matched LOH and/or second somatic hit (+1), while factoring for samples where tumour was not available (+0.5).
Statistics and reproducibility
Sample size was determined by the availability of recruited patients and/or whole genome data meeting the study criteria, African ancestral patients with a clinicopathological diagnosis of PCa. As such, no statistical method was used to predetermine sample size and after meeting inclusion criteria, no patient/data were excluded from the analyses. While the experiments were not randomised, for both initial SAPCS and PPCG data generation and analyses, investigators were blinded to patient ancestry. After genetic testing, men of confirmed African ancestry were selected for downstream analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Access to published whole genome sequence data published in Jaratlerdsiri et al18 was made available via Data Access Committee (DAC) approval as outlined under the European Genome-Phenome Archive (EGA) [https://ega-archive.org] project-specific access policies under overarching study EGAS00001006425, which includes the Southern African Prostate Cancer Study (SAPCS) Dataset at EGAD00001009067 and as part of the PPCG cohort the Garvan/St Vincent’s Prostate Cancer or Sydney Database at EGAD00001009066, while additional PPCG Datasets are summarised in Table S1 and include Canadian PCa Genome Network [https://ega-archive.org/datasets/EGAD00001004170], CRUK-ICGC Prostate Group UK [https://ega-archive.org/datasets/EGAC00001000852], French/Caribbean ICGC PCa Group [https://ega-archive.org/datasets/EGAD00001003835], Germany ICGC PCa Group [https://ega-archive.org/datasets/EGAD00001005997], and Melbourne Research Group Australia [https://ega-archive.org/datasets/EGAD00001004182]. MGRB data is available as defined by study EGAS00001003511 and dataset EGAD00001005228. The additional 70 SAPCS germline whole genome data has been deposited under the overarching study EGAS50000001132 [https://submission.ega-archive.org/submissions/EGA50000001053] and dataset EGAD50000001626 [https://submission.ega-archive.org/submissions/EGA50000001053/datasets]. Additional variant and annotation data for the African PCa patients, European PPCG patients, African and healthy control populations study are available within the main text and supplementary information.
Access to the additional SAPCS sequencing data generated in this study may be requested via the SAPCS DAC and will be made available to researchers with appropriate feasibility and corresponding ethics approvals to ensure the safeguarding of patient genomic information (contact V.M.H. or M.S.R.B.). Restrictions include (i) No transfer to third parties allowed, (ii) acknowledgment of the SAPCS in publications/presentations, (iii) a report of the results of the research to be provided to DAC after completion (or when requested), (iv) researchers cannot utilise the data for commercial purposes or any other purposes not approved by the DAC, and (v) approval will not be given that excludes other researchers from accessing data. Data currently being used for capacity building in under-resourced studies across Sub-Saharan Africa will be given priority and at times may be granted time-limited exclusive rights for no more than a two-year period.
SNVs and indels data supporting the findings of this study are available within the main text and Supplementary information. Previously published SNV and indel sites and their minor allele frequencies are available in the dbSNP [https://www.ncbi.nlm.nih.gov/snp/]68, and gnomAD databases [https://gnomad.broadinstitute.org/]23. Gene regions are available in the ENSEMBL database [https://www.ensembl.org]69, and DDR gene list is available at GSEA24.
Code availability
Software and scripts for DNA sequence read data collection, and the scripts for sequence read alignment and quality control are available on GitHub (https://github.com/Sydney-Informatics-Hub/Bioinformatics).
References
Stadler, Z. K. et al. Therapeutic implications of germline testing in patients with advanced cancers. J. Clin. Oncol. 39, 2698–2709 (2021).
National Comprehensive Cancer Network NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer, Version 1.2023 (2023).
Giri, V. N. et al. Genetic testing in prostate cancer management: Considerations informing primary care. CA Cancer J. Clin. 72, 360–371 (2022).
National Comprehensive Cancer Network NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection, Version 1.2023 (2023).
Wei, J. T. et al. Early detection of prostate cancer: AUA/SUO guideline part I: prostate cancer screening. J. Urol. 210, 46–53 (2023).
Nyame, Y. A. et al. Deconstructing, addressing, and eliminating racial and ethnic inequities in prostate cancer care. Eur. Urol. 82, 341–351 (2022).
Fletcher, S. A. et al. Geographic distribution of racial differences in prostate cancer mortality. JAMA Netw. Open 3, e201839 (2020).
Giri, V. N. et al. Implementation of germline testing for prostate cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J. Clin. Oncol. 38, 2798–2811 (2020).
Darst, B. F. et al. Germline sequencing analysis to inform clinical gene panel testing for aggressive prostate cancer. JAMA Oncol. 9, 1514–1524 (2023).
Gheybi, K. et al. Evaluating germline testing panels in southern african males with advanced prostate cancer. J. Natl. Compr. Canc. Netw 21, 289–296.e3 (2023).
Nicolosi, P. et al. Prevalence of germline variants in prostate cancer and implications for current genetic testing guidelines. JAMA Oncol 5, 523–528 (2019).
Russo, J. & Giri, V. N. Germline testing and genetic counselling in prostate cancer. Nat. Rev. Urol. 19, 331–343 (2022).
Giri, V. N., Hartman, R., Pritzlaff, M., Horton, C. & Keith, S. W. Germline variant spectrum among african american men undergoing prostate cancer germline testing: need for equity in genetic testing. JCO Precis. Oncol. 6, e2200234 (2022).
Matejcic, M. et al. Pathogenic variants in cancer predisposition genes and prostate cancer risk in men of African ancestry. JCO Precis. Oncol. 4, 32–43 (2020).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Pereira, L., Mutesa, L., Tindana, P. & Ramsay, M. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genet. 22, 284–306 (2021).
Hayes, V. M. et al. Health equity research outcomes and improvement consortium prostate cancer health precision Africa1k: closing the health equity gap through rural community inclusion. J. Urol. Oncol. 22, 144–149 (2024).
Jaratlerdsiri, W. et al. African-specific molecular taxonomy of prostate cancer. Nature 609, 552–559 (2022).
Burns, D. et al. Rare germline variants are associated with rapid biochemical recurrence after radical prostate cancer treatment: A Pan Prostate Cancer Group Study. Eur. Urol. 82, 201–211 (2022).
Pinese, M. et al. The Medical Genome Reference Bank contains whole genome and phenotype data of 2570 healthy elderly. Nat. Commun. 11, 435 (2020).
Tindall, E. A. et al. Clinical presentation of prostate cancer in black South Africans. Prostate 74, 880–891 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods. 7, 248–249 (2010).
Tamborero, D. et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 10, 25 (2018).
Soh, P. et al. Prostate cancer genetic risk and associated aggressive disease in men of African ancestry. Nat. Commun. 14, 8037 (2023).
Li, M. et al. PREX2 contributes to radiation resistance by inhibiting radiotherapy-induced tumor immunogenicity via cGAS/STING/IFNs pathway in colorectal cancer. BMC Med. 22, 154 (2024).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Kessler, M. D. et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612, 301–309 (2022).
Vlasschaert, C. et al. A practical approach to curate clonal hematopoiesis of indeterminate potential in human genetic data sets. Blood. 141, 2214–2223 (2023).
Reimand, J. et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 14, 482–517 (2019).
Carvalho, N. D. A. D. et al. Prevalence and clinical implications of germline pathogenic variants in cancer predisposing genes in young patients across sarcoma subtypes. J. Med. Genet. 61, 61 (2024).
Knudson, A. G. Jr Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA. 68, 820–823 (1971).
Tsai, K. K., Bae, B.-I., Hsu, C.-C., Cheng, L.-H. & Shaked, Y. Oncogenic ASPM Is a regulatory hub of developmental and stemness signaling in cancers. Cancer Res 83, 2993–3000 (2023).
Liu, W. et al. Histone-methyltransferase KMT2D deficiency impairs the Fanconi anemia/BRCA pathway upon glycolytic inhibition in squamous cell carcinoma. Nat. Commun. 15, 6755 (2024).
Gu, W., Zhuang, W., Zhuang, M., He, M. & Li, Z. DNA damage response and repair gene mutations are associated with tumor mutational burden and outcomes to platinum-based chemotherapy/immunotherapy in advanced NSCLC patients. Diagn. Pathol. 18, 119 (2023).
Park, C. M., Kawasaki, Y., Refaat, A. & Sakurai, H. Mechanisms for DNA-damaging agent-induced inactivation of ErbB2 and ErbB3 via the ERK and p38 signaling pathways. Oncol. Lett. 15, 1758–1762 (2018).
Lu, Y. et al. Expression of the fat-1 gene diminishes prostate cancer growth in vivo through enhancing apoptosis and inhibiting GSK-3 beta phosphorylation. Mol. Cancer Ther 7, 3203–3211 (2008).
Plym, A. et al. Early prostate cancer deaths among men with higher vs lower genetic risk. JAMA Network Open 7, e2420034–e2420034 (2024).
Lozano, R. et al. Genetic aberrations in DNA repair pathways: a cornerstone of precision oncology in prostate cancer. Br. J. Cancer 124, 552–563 (2021).
Truong, H. et al. Gene-based confirmatory germline testing following tumor-only sequencing of prostate cancer. Eur. Urol. 83, 29–38 (2023).
Yadav, S. et al. Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 39, 4299–4311 (2020).
Oh, C. R. et al. Phase II study of durvalumab monotherapy in patients with previously treated microsatellite instability-high/mismatch repair-deficient or POLE-mutated metastatic or unresectable colorectal cancer. Int. J. Cancer 150, 2038–2045 (2022).
Eygelaar, D., van Rensburg, E. J. & Joubert, F. Germline sequence variants contributing to cancer susceptibility in South African breast cancer patients of African ancestry. Sci. Rep. 12, 802 (2022).
Wu, J. et al. Prevalence of comprehensive DNA damage repair gene germline mutations in Chinese prostate cancer patients. Int. J. Cancer 148, 673–681 (2021).
Landry, K. K. et al. Investigation of discordant sibling pairs from hereditary breast cancer families and analysis of a rare PMS1 variant. Cancer Genet 260-261, 30–36 (2022).
Nientiedt, C. et al. High prevalence of DNA damage repair gene defects and TP53 alterations in men with treatment-naïve metastatic prostate cancer –Results from a prospective pilot study using a 37 gene panel. Urol. Oncol. 38, 637.e617–637.e627 (2020).
Paulo, P. et al. Targeted next generation sequencing identifies functionally deleterious germline mutations in novel genes in early-onset/familial prostate cancer. PLoS Genet. 14, e1007355 (2018).
Koboldt, D. C. et al. Rare variation in TET2 is associated with clinically relevant prostate carcinoma in African Americans. Cancer Epidemiol. Biomarkers Prev 25, 1456–1463 (2016).
Gong, T. et al. Rare pathogenic structural variants show potential to enhance prostate cancer germline testing for African men. Nat. Commun. 16, 2400 (2025).
Srinivas, U. S., Tan, B. W. Q., Vellayappan, B. A. & Jeyasekharan, A. D. ROS and the DNA damage response in cancer. Redox. Biol. 25, 101084 (2019).
Isidori, F. et al. RASAL1 and ROS1 gene variants in hereditary breast cancer. Cancers (Basel) 12, 2539 (2020).
Liang, Y. et al. Whole-exome sequencing reveals a comprehensive germline mutation landscape and identifies twelve novel predisposition genes in Chinese prostate cancer patients. PLoS Genet. 18, e1010373 (2022).
Samadder, N. J. et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol 7, 230–237 (2021).
Kurian, A. W. et al. Germline genetic testing after cancer diagnosis. JAMA 330, 43–51 (2023).
Davidson, A. L. et al. The clinical utility and costs of whole-genome sequencing to detect cancer susceptibility variants—a multi-site prospective cohort study. Genome Med 15, 74 (2023).
Korir, A. et al. Cancer risks in Nairobi (2000–2014) by ethnic group. Int. J. Cancer 140, 788–797 (2017).
Gheybi, K. et al. Linking African ancestral substructure to prostate cancer health disparities. Sci. Rep. 13, 20909 (2023).
Mbugua, R. G., Karanja, S. & Oluchina, S. Effectiveness of a community health worker-led intervention on knowledge, perception, and prostate cancer screening among men in Rural Kenya. Adv. Prev. Med. 8, 4621446 (2022).
Maladze, N., Maphula, A., Maluleke, M. & Makhado, L. Knowledge and Attitudes towards Prostate Cancer and Screening among Males in Limpopo Province, South Africa. Int. J. Environ. Res. Public Health 20, 5220 (2023).
Hayes, V. M. & Bornman, M. S. R. Prostate cancer in Southern Africa: does africa hold untapped potential to add value to the current understanding of a common disease?. J. Glob. Oncol. 4, 1–7 (2018).
Woodcock, D. J. et al. Genomic evolution shapes prostate cancer disease type. Cell Genomics 4, 100511 (2024).
Wedge, D. C. et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 50, 682–692 (2018).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics. 43, 11.10.11–11.10.33 (2013).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Harrison, P. W. et al. Ensembl 2024. Nucleic Acids Res. 52, D891–D899 (2024).
Acknowledgements
The authors are forever grateful to the patients, their families, respective clinical staff and bioresource managers who have who have contributed to the data within each Consortium represented in this study. Specifically, we acknowledge the contributions of the SAPCS participants and health care workers from South Africa who have contributed both published24, and additional data. Furthermore, we thank the Ramaciotti Centre for Genomics, University of New South Wales, Sydney, for data generation, as well as the Sydney Informatics Hub at the University of Sydney and the National Computational Institute (NCI) in Canberra for providing the high-performance computational infrastructure Artemis and Gaddi used in this study, respectively. We also acknowledge the support of the research staff in the PPCG contributing teams who so carefully curated the samples and the follow-up data. Genomic sequencing and interrogation of SAPCS data was supported by the Ancestry and Health Genomics Laboratory at the University of Sydney through National Health and Medical Research Council (NHMRC) of Australia funding (2018/GNT1165762, 2020/GNT2001098 and 2021/GNT2010551 to V.M.H.), and USA Congressionally Directed Medical Research Programs (CDMRP) Prostate Cancer Research Program (PCRP) funding, including an Idea Development Award (PC200390, TARGET Africa to V.M.H.) and HEROIC Consortium Award (PC210168 and PC230673, HEROIC PCaPH Africa1K to V.M.H., M.S.R.B., P.M.N. and G.S.P.), the latter including data generation for African control data. For PPCG curation, management and analysis (to R.A.E. and Z.K-J.) we acknowledge support from Cancer Research UK (C5047/A14835/A22530/A17528, C309/A11566, C368/A6743, A368/A7990, C14303/A17197), Prostate Cancer UK (MA-TIA23-002), Dallaglio Foundation (CR-UK Prostate Cancer ICGC Project and Pan Prostate Cancer Group), PC-UK/Movember, the NIHR support to The Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. Further support for SAPCS and PPCG analysis was provided by the USA National Institute of Health (NIH) National Cancer Institute (NCI) Award (1R01CA285772-01 to V.M.H.) and a USA Prostate Cancer Foundation (PCF) Challenge Award (2023CHAL4150 to V.M.H., M.S.R.B. and G.S.P.). V.M.H. is further supported by the Petre Foundation via the University of Sydney Foundation (Australia) and M.M.H. by a Sydney Cancer Partners pilot grant (Australia).
Author information
Authors and Affiliations
Consortia
Contributions
Conception and design: V.M.H. Administrative support: J.J., T.M.N.M., M.T.L., S.M.P., G.S.P. and K.D.S. Provision of study materials or patients (SAPCS and controls): R.A.C., M.B.R., M.N., Muv.O., Mar.O., S.B.A.M., M.S.R.B. and V.M.H., and (EAPCS controls): W.M.O., M.O.O. and P.M.N. Pathology review: Mel.L. and Mas.L. Sample processing and preparation (70 SAPCS genomes): M.M.H. Variant calling and data uploads (70 SAPCS genomes): J.J. Collection and assembly of data: K.G., P.X.Y.S., J.J., D.B., P.M., D.K., W.J., D.C.W., R.G.B., D.S.B., C.S.C., J.R., G.C-T., O.C., C.M.H., N.M.C., P.D.S., T.S., J.W., S.B.A.M., P.M.N., D.M.T., Z.K-J., R.A.E., M.S.R.B. and V.M.H. Ancestry fraction determination: P.X.Y.S. Data analysis and interpretation: K.G., P.X.Y.S., J.J., D.B., P.M., D.K., J.W., D.M.T., Z.K-J., R.A.E. and V.M.H. Manuscript writing and Figure generation: K.G., P.X.Y.S. and V.M.H. Final approval and review of manuscript: All authors
Corresponding author
Ethics declarations
Competing interests
Member of Active Surveillance Movember Committee (V.M.H., R.A.E.). Member of external expert committee to Astra Zeneca UK (R.A.E.). Honoraria from GU-ASCO, Janssen, University of Chicago, Dana Farber Cancer Institute USA as a speaker (R.A.E.). Educational honorarium from Bayer and Ipsen (R.A.E.). Member of the SAB of Our Future Health (R.A.E.). Undertakes private practice as a sole trader at The Royal Marsden NHS Foundation Trust and 90 Sloane Street SW1X 9PQ and 280 Kings Road SW3 4NX, London, UK (R.A.E.). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qian (Janie) Qin, Solomon Rotimi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gheybi, K., Soh, P.X.Y., Jiang, J. et al. Pathogenic variants reveal candidate genes for prostate cancer germline testing for men of African ancestry. Nat Commun 16, 8799 (2025). https://doi.org/10.1038/s41467-025-63865-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63865-6
