
Abstract
The hydrogenation of CO2 to methanol is a promising route for carbon capture and utilization, however achieving high selectivity and productivity remains a challenge. This study presents a novel catalyst synthesized by pyrolyzing a zirconium-based metal-organic framework impregnated with indium, yielding ultrafine In2O3 nanoparticles uniformly embedded within a ZrO2 and carbon matrix. The resulting In2O3/ZrO2 heterojunction exhibited abundant oxygen vacancies at the interface, which is crucial for enhancing the catalytic performance. Under gas-phase conditions, the catalyst achieves an exceptional methanol selectivity of 81% with a record-high productivity of 2.64 gMeOH·gcat⁻¹·h⁻¹ at mild reaction conditions, while in liquid-phase hydrogenation, methanol selectivity reaches 96%. Comprehensive structural characterizations confirmed that oxygen vacancies and the heterointerface served as active sites, facilitating CO2 activation and methanol stabilization. Mechanistic insights from in-situ DRIFTS and ATR-IR spectroscopy revealed that methanol formation proceeds via the formate pathway, further supported by in-situ ambient-pressure X-ray photoelectron spectroscopy, demonstrating electronic structural modulation and an increased concentration of oxygen vacancies. These findings underscore the critical role of defect engineering in optimizing CO2 hydrogenation catalysts and provide a pathway for designing highly efficient systems for sustainable methanol production.
Similar content being viewed by others

Introduction
Rising CO2 levels in the atmosphere have heightened concerns about global climate change and ocean acidification1. Hydrogenating CO2 into alcohol or hydrocarbons is a key strategy for recycling atmospheric CO2 emissions from combustion processes. With growing industrial demand for methanol, significant efforts have been made to convert CO2 into methanol2. Various materials have been tested as catalysts for this conversion, and most industrial processes rely on metal nanoparticles supported on oxide substrates. Cu-based catalysts, often combined with ZnO and/or Al2O3, have been in use for over 50 years3. Although these catalysts avoid methane production, they show limited activity for CO2 hydrogenation at low temperatures (T < 500 K), leading to higher hydrogen consumption and reduced methanol yields4. Additionally, the presence of water accelerated the sintering of Cu and ZnO, causing catalyst deactivation5.
Indium-based catalysts have emerged as promising solutions for the conversion of CO2 into methanol6. In2O3 offers up to 100% selectivity for methanol under optimal CO2 hydrogenation conditions, with exceptional stability due to its oxide-based active phase7. While unsupported In2O3 has been improved by the addition of active components or promoters to enhance H2 dissociation, supported In-based catalysts are more suitable for industrial use, requiring less active materials and providing greater stability8,9,10,11. Recent research has focused on supported In-based catalysts, with monoclinic zirconia (m-ZrO2) proving to be an excellent support for achieving high CO2 conversion and methanol selectivity12. In addition, Tsoukalou et al.12 and Chen et al. 13 have concentrated on identifying the active phase of the In2O3/ZrO2 catalyst used in methanol synthesis from CO2 and H2. The In-Ov-Zr sites (Ov representing oxygen vacancies) formed by the interaction between In2O3 and ZrO2 are crucial for maximizing the activity and stability in methanol production. The effectiveness of CO2 reduction to methanol is largely influenced by the size and concentration of the In2O3 domains13. However, increasing the interfacial area between the In2O3 domains and m-ZrO2 support remains a key challenge for further enhancing the performance.
The synthesis of MOF-derived materials can be used to increase the interfacial area and oxygen vacancies. However, the design of heterojunctions with abundant interfaces in complex systems remains challenging14. Certain MOFs and MOF composites serve as ideal precursors or templates for producing heterostructured transition metal/metal oxides via a straightforward hydrothermal-calcination method15,16,17,18. These MOF-derived heterostructures typically retain the high porosity and versatile morphology of MOFs, making them highly promising for catalytic applications19,20,21. For example, CoMn-MOF-74-derived Co/MnO heterostructured nanoparticles showed improved CO2 adsorption and activation, delivering excellent catalytic performance in low-temperature CO2 methanation22. Building on this success, we developed a In2O3/ZrO2-based heterostructural catalyst using MOF precursors and evaluated its efficiency for CO2 hydrogenation to methanol.
In this study, we developed a composite catalyst featuring a heterojunction of In2O3 and ZrO2 (Deg In-Zr (3:8)), derived from an indium-impregnated Zr-BDC MOF. The Deg In-Zr (3:8) catalyst demonstrated an outstanding performance in the selective hydrogenation of CO2 to methanol in both the liquid and gas phases under ambient conditions. This catalyst achieved a high CH₃OH productivity of 2.64 gMeOH·gcat−1. h−1 in the gas phase under mild reaction conditions (220 °C, 40 bar combined H2 and CO2 pressure (H2: CO2 3:1)). Additionally, the catalyst contained a low indium content (0.6 wt. %), with 2-3 nm In2O3 nanoparticles uniformly dispersed on the surface, making it a cost-effective option for industrial use. The unique In2O3/ZrO2 heterointerface provided numerous active sites for enhanced H2 dissociation and CO2 activation, as confirmed by detailed structural analysis using XAS (XANES and EXAFS). The reaction mechanism responsible for this exceptional performance was investigated using in-situ ATR-IR for liquid-phase reactions, in-situ DRIFTS for gas-phase reactions, and quantum chemistry modelling. Furthermore, ambient pressure in situ XPS was conducted to gain insights into the active phase during CO2 hydrogenation to methanol.
Results
Synthetic route
The Zr-BDC MOF was synthesized by dissolving zirconium tetrachloride (ZrCl4) (0.053 g, 0.227 mmol) and 1,4-benzene dicarboxylic acid (H2BDC) (0.034 g, 0.227 mmol) in N,N-dimethylformamide (DMF) at room temperature23. The mixture was heated at 120 °C for 24 h to induce crystallization. After cooling, the solid was filtered, washed with DMF and methanol, and dried at 60 °C overnight. X-ray diffraction (XRD) confirmed that the synthesized MOF matched the previously reported Zr-BDC structure24. Indium was incorporated into the MOF via the incipient wetness impregnation (IWI) method to produce In-Zr BDC catalysts with various indium-to-zirconium ratios. The mixture was refluxed with indium nitrate in methanol at 70 °C, centrifuged, and then dried. The XRD pattern confirms the successful incorporation of indium. All the samples were calcined at 600 °C in nitrogen, resulting in a Deg In-Zr catalyst. During calcination, solvent molecules were lost at approximately 150 °C, and the H2BDC ligand decomposed near 350 °C. The Deg In-Zr BDC (3:8) catalyst, containing ZrO2 and In2O3 phases, exhibited the highest CO2 conversion to methanol, achieving a high efficiency with minimal indium content.
Crystalline phase and surface properties
The XRD patterns of various samples, including ZrO2, In2O3, In2O3-ZrO2/C, In2O3-ZrO2, Zr-BDC MOF, indium-impregnated Zr-BDC (In-Zr BDC), and degraded In-Zr MOF (Deg In-Zr), are shown in Fig. 1b. The Zr-BDC MOF displayed sharp peaks, indicating high crystallinity, with a pattern matching previous reports23. After thermal degradation at 600 °C, the MOF structure collapsed, altering the diffraction pattern and revealing the presence of In2O3 7and ZrO2 phases with peak shifts as compared to bare In2O3 and ZrO2, suggesting crystal distortion and a small crystal domain size. In contrast, the impregnated In2O3-ZrO2 and the as-synthesised graphitic-carbon-supported In2O3 and ZrO2 exhibited sharp crystalline peaks, indicating the presence of large crystalline phases. Moreover, the peaks corresponding to In2O3 and ZrO2 remained unchanged relative to those of the pure In2O3 and ZrO2. All the degraded MOF catalysts with different In: Zr ratios exhibited similar diffraction patterns (Fig. S1). Raman spectra of Zr-BDC (Fig. 1c) show characteristic shifts at 634 and 860 cm⁻¹ (C–H bond vibrations) and at 1140 and 1611 cm⁻¹ (C = C modes of the benzene ring). Peaks at 1434 and 1448 cm⁻¹ correspond to symmetric and asymmetric C–O2 stretching25,26. Indium impregnation retained the structural integrity of the MOF. After degradation, the Deg In-Zr (3:8) catalyst showed D and G bands at 1316 cm⁻¹ and 1590 cm⁻¹, indicating graphitic carbon formation27. Further details are available in the supplementary information (page number 19).

a Catalyst synthesis scheme, b XRD patterns, c Raman spectra of various catalysts, d In 3 d, e Zr 3 d and f O 1 s XPS of different catalysts.
X-ray Photoelectron Spectroscopy (XPS) was performed to analyze the surface composition and oxidation states of the elements in the various catalysts (Fig. 1 (d)–(f), Figs. S2, S3). The high-resolution XPS spectra for indium (Fig. 1(d)) reveal distinct peaks corresponding to In2O3 and In(OH)3 for the as-synthesized metal oxides28. In comparison, the Deg In-Zr (3:8) catalyst shows the largest binding energy shift for In3+, suggesting a strong electronic interaction between In2O3 and ZrO2 in this material10. This shift indicates significant electronic modulation, as further detailed in supplementary information. For ZrO2, (Fig. 1 (e)) the Zr 3d5/2 and 3d3/2 peaks appear at 182.01 eV and 184.34 eV29, indicating the presence of Zr4+. In the Deg In-Zr (3:8) catalyst, these peaks shift to higher binding energies (182.42 eV and 184.40 eV), likely due to surface restructuring and enhanced electronic interaction between ZrO2 and In2O330. The oxygen 1 s XPS spectra of the metal oxide catalysts (Fig. 1(f)) show three peaks at 529.62 eV, 531.2 eV, and 532.7 eV, corresponding to lattice oxygen (OA), oxygen vacancies (OB), and chemically adsorbed oxygen or C–O bond-associated oxygen (OC), respectively31,32. The Deg In-Zr (3:8) catalyst exhibits the highest oxygen vacancy concentration, as shown in Fig. 1(f) and Table S3, compared to other as-synthesized catalysts such as In2O3, ZrO2, In2O3-ZrO2, and In2O3-ZrO2/C. The higher concentration of oxygen vacancies likely contributed to the enhanced activity in CO2 hydrogenation. A detailed discussion of the varying indium and zirconium ratios in the degraded catalyst is provided in the Supplementary Information (Figure S3) (page number 29). Oxygen vacancies can catalyse the decomposition of water in the liquid phase. Recent studies have suggested that surface defects such as oxygen vacancies may facilitate water activation. For instance, Deng et al. reported that sulfur vacancies at the edges of MoS₂ act as active sites, enabling a water-mediated CO₂ hydrogenation mechanism33. In their work, the OH* and H* species in dynamic equilibrium with water served as moderate hydrogenating agents, leading to formate formation, with residual O* species subsequently reduced by hydrogen. The presence of water has been shown to boost methanol selectivity and CO₂ conversion on the In₂O₃-ZrO₂ catalyst, as previously demonstrated by Song et al. using DFT calculations34. Control experiments were performed to test the hydrogenation of CO2 in the absence of H₂ in water. HPLC analysis (Fig. S21) revealed the absence of HCOOH or methanol, which may indicate a very limited contribution from water-mediated hydrogenation under the current reaction conditions. This suggests that, while oxygen vacancies may interact with water to some extent, the dominant pathway in our system remains H₂ activation and CO₂ adsorption facilitated by the engineered In₂O₃–ZrO₂ heterojunction.
X-ray Absorption Spectroscopy (XAS) was employed to gain precise quantitative insights into the electronic behaviour and coordination environments (Figs. 2, S5 and S6). The X-ray Absorption Near Edge Structure (XANES) at the In K-edge provided information on the valence state of the absorbing atom, although the low indium density across the samples made analysis challenging. All samples showed significant deviations from metallic Zr (Fig. 2g). In In-Zr Metal-Organic Frameworks (MOFs), a minor electron transfer from indium to the Zr MOF was observed, with minimal impact on the structural integrity of the Zr framework. However, in the degraded In-Zr MOFs (Deg In-Zr (3:8)), the Zr MOF decomposed into ZrO2, displaying differences from monoclinic ZrO2 and similarities with In-modified ZrO2 in impregnated In-ZrO2 samples35.

Curve fitting results of the k3-weighted EXAFS data at Zr K-edge for Fourier transform a Zr foil b ZrO2 c Zr BDC MOF d Deg In-Zr MOF (Deg In-Zr (3:8)) e In-Zr MOF and f In-ZrO2 (impregnated indium in ZrO2) Red circles: observed; Red solid line: fitted; Blue circles: imaginary part; Blue solid line: fitted. g XANES of Zr K-edge h WT of Deg In-Zr (3:8) and (i) In-Zr BDC.
We also analyzed the extended X-ray absorption fine structure (EXAFS) spectra at the Zr K-edge to investigate the local structure around Zr atoms in the samples. At the Zr K-edge of the Zr MOF, two types of Zr-O bonds and a Zr-Zr bond were detected, indicative of the inherent structure of the Zr MOF (Fig. 2c). However, the Zr-Zr coordination number was lower than expected, suggesting distortions around the Zr atoms within the MOF framework, reducing the coordination number, as estimated by EXAFS.
EXAFS analysis at the In-K-edge for the In-Zr MOF was anticipated, but insufficient signals were obtained, suggesting either a lower-than-expected In density (as estimated by ICP) or a distorted and heterogeneous local structure around the In atoms, complicating precise EXAFS measurements. Although the ICP-MS results indicate an overall In loading of 0.6 wt%, the actual indium density in the active regions of the catalyst may be much lower because of the highly dispersed nature of the In₂O₃ nanoparticles on the ZrO₂ support. Indium species may exist in a highly disordered or amorphous environment at the In₂O₃/ZrO₂ interface, leading to weak and poorly defined oscillations in the EXAFS region. Such structural distortion can severely dampen the signal, making reliable fitting difficult, even under fluorescence detection. Nonetheless, the Zr-Zr coordination number was lower in the In-Zr MOF, indicating increased distortion around the Zr atoms owing to the incorporation of In into the framework pores. The EXAFS Fourier transform showed a peak at approximately 0.3 nm, which is shorter than that of Zr MOFs, and the presence of Zr-In bonds improved the fitting analysis. Direct Zr-In bonding at 0.299 nm ± 0.003 was observed, suggesting the formation of Zr-O-In bonds at the interface of the MOF pore walls (Fig. 2e, i)12.
At the Zr K-edge, EXAFS analysis of the monoclinic ZrO2 samples revealed Zr-O bonds at 0.215 nm and Zr-Zr bonds at 0.350 nm (Fig. 2b). In the degraded In-Zr MOF (Deg In-Zr (3:8)), the presence of indium affected the structural parameters (Fig. 2d, h), showing two distinct Zr-O bonds at 0.214 nm and 0.233 nm, compared to the single Zr-O bond at 0.215 nm in ZrO2. Zr-Zr bonding in the degraded In-Zr MOF was observed at 0.362 nm, compared to 0.350 nm in ZrO2, while Zr-In bonding at 0.299 nm was also noted, similar to that in the as-synthesized In-Zr MOF. However, the coordination number of the Zr-In bonds was low, as detailed in Tables S6–S11. The R-factor for the fitting involving Zr-In bonds (3.5%) was superior to that for Zr-Zr bonds alone (4.6%). Therefore, the ZrO2 formed from the degradation of the In-Zr MOF at 600 °C exhibited a different lattice structure compared to ZrO2, likely due to the formation of a new mixed In-Zr oxide with Zr-O-In bonds. This mixed oxide likely contains numerous oxygen defects owing to the differing valences of Zr4+ and In3+ 36.
CO2-TPD was performed to assess the CO2 adsorption behavior of various catalysts. The CO2-TPD profiles (Fig. S9), all catalysts showed a desorption peak between 50–200 °C, attributed to physically absorbed CO228,32,37,38. A second peak above 200 °C indicates CO2 chemisorption at oxygen vacancies, which is critical for CO2 activation39. The Deg In-Zr (3:8) catalyst showed a higher CO2 chemisorption intensity between 200 and 450 °C compared to bare In2O3 and ZrO2, suggesting that the heterointerfaces between In2O3 and ZrO2, along with oxygen vacancies, act as key sites for CO2 adsorption and activation13.HCOOH and CH3OH temperature-programmed desorption (TPD) experiments were conducted to examine the surface species responsible for high methanol yield during CO2 hydrogenation (Fig. S7(c), (b)). In the case of the Deg In-Zr catalyst, the HCOOH decomposition peak appeared at lower temperatures, indicating reduced stability with increased indium content, leading to its decomposition into CO2 and H2. This suggests that optimizing the indium content is key to effectively converting the HCOO* intermediates into CH3O* species. Similar results by Han et al. showed that lower indium loadings stabilized HCOO species13. The CH3OH TPD profile revealed that methanol stability decreases with increasing indium content, highlighting the importance of reducing the indium concentration to improve methanol selectivity in CO2 hydrogenation. H2-TPR analysis also suggested the formation of the highest number of oxygen vacancies in the Deg In-Zr (3:8) catalyst which is explained in the supplementary information (page number 27 and Fig. S8).
Morphological and textural properties of the catalysts
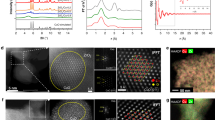
The morphologies of the materials were analyzed using TEM and HR-TEM. TEM images of Zr-BDC (Figs. 3, S10, and S11) revealed a square-like morphology, which was also maintained in the Deg In-Zr (3:8) catalyst, featuring ultra-small metal oxide nanoparticles ranging from 2 to 3 nm. As shown in Fig. 3, this catalyst contains both In2O3 and ZrO2 nanoparticles. The d-spacing of 0.417 nm, observed in In2O3, is slightly larger than the standard 0.413 nm for the In2O3 (211)40 lattice plane, likely due to crystal lattice distortion, as supported by the XRD data41. For ZrO2, the HR-TEM images display d-spacing values of 0.498 and 0.269 nm, corresponding to the (001) and (200) lattice planes of the monoclinic phase, respectively42. This increased d-spacing in the Deg In-Zr (3:8) catalyst compared to that of bare ZrO2 indicates a crystal distortion. Notably, no tetragonal ZrO2 phase was observed in HR-TEM analysis. Previous research by Muller et al. highlighted the high activity of monoclinic ZrO2 in CO2 hydrogenation to methanol, a finding supported by the absence of the tetragonal form in EXAFS analysis12. The SEAD pattern also confirms the presence of both In2O3 and ZrO2, with a diffusive pattern attributed to the ultrasmall nanoparticles in the Deg In-Zr (3:8) catalyst43. STEM images (Fig. 3(d)–(h)) and EDX spectra (Fig. S12) further demonstrated the uniform dispersion of indium and zirconium within the carbonated MOF matrix.

a–c HR-TEM of Deg In-Zr (3:8) and d–i HADDF-STEM images and corresponding elemental mapping of Deg In-Zr (3:8).
Catalytic activity and stability
CO2 catalytic conversion in the gas phase was conducted at 220 °C with a feed mixture of (1:3) CO2 and H2 at a total pressure of 30 bar over various catalysts. As shown in Fig. 4(a), (b), the Deg In-Zr BDC (3:8) catalyst demonstrated outstanding methanol productivity (2.64 gMeOH·gcat−1·h−1) after 51 h, with an exceptional gas hourly space velocity (GHSV) of 78,000 h⁻¹. This superior performance is likely due to the formation of an In2O3-incorporated ZrO2 phase, which introduces oxygen vacancies or defective sites that enhance CO2 activation and methanol production32. Previous studies, such as that by Javier et al., suggest that incorporating In2O3 into ZrO2 induces the formation of a polymorphic ZrO2 structure, which significantly improves CO2 adsorption on the catalyst surface32. Catalytic tests on bare In2O3 and ZrO2 revealed that ZrO2 mainly produced CO with a low CO2 conversion (0.09%), whereas In2O3 showed slightly higher activity with 0.9% CO2 conversion, producing methanol. The performance of the Deg In-Zr (3:8) catalyst was further compared with those of In2O3-impregnated ZrO2 (In2O3-ZrO2) and In2O3-ZrO2/C (impregnated on graphite). These conventional catalysts achieved CO2 conversion rates of 3.2% and 4.6%, respectively, but were still far less active than Deg In-Zr (3:8), which achieved 27.8% CO2 conversion after 51 h at 220 °C and 40 bar pressure. The catalytic activity was also assessed by varying the indium content of MOF-derived catalysts. Figure 4(b), (e) show that increasing the indium content initially enhanced the activity, but beyond a certain threshold, the catalytic performance declined. This is likely due to the optimal balance between In2O3 and ZrO2, both of which serve as active sites for CO2 hydrogenation to methanol32,36.

a–f Gas phase and g–i liquid-phase reactions.
The time-on-stream profile (Figs. 4(f) and S13 (a)) indicates that the Deg In-Zr (3:8) catalyst had an induction period of 24 h, during which methanol selectivity reached 81%, with a productivity of 2.61 gMeOH·gcat−1·h−1. The methanol selectivity stabilized at 81.4%, with by-products including formic acid (10.3%) and CO (1.6%). The small amount of CO is likely due to the reverse water-gas shift (RWGS) reaction, which typically requires higher activation energy and occurs at temperatures above 280 °C7. After 51 h, the methanol production remained stable at 2.64 gMeOH·gcat−1·h−1. Initially, formic acid was the major by-product, but as the reaction progressed, its selectivity decreased, while methanol selectivity increased. CO2 and H2 are adsorbed onto the catalyst surface, forming a surface-bound formate44 (HCOO −), which is then reduced to methanol through intermediates such as H2CO* and H3CO*45. Oxygen vacancies in the In2O3 species assist in this process by facilitating CO2 adsorption and activation. As the temperature increased, the methanol selectivity improved; however, ethanol formation began at temperatures above 230 °C. The time-on-stream profiles for different temperatures are shown in Fig. S13(b), (c), where CO selectivity increased with temperature due to the RWGS reaction35.
For liquid-phase CO2 hydrogenation, the Deg In-Zr (3:8) catalyst was tested in a high-pressure Parr reactor. The catalyst was highly active in converting CO2 to methanol, with reaction parameters such as temperature, pressure, and reaction time (Fig. 4(g)–(i)). Prior to the reaction, the catalyst was reduced at 300 °C under 4% H2 in Ar for 2 h. As shown in Fig. 4 (h), the highest methanol selectivity (96.8 %) was achieved at 130 °C and 30 bar pressure (3:1 H2: CO2 ratio). Increasing the temperature to 190 °C enhanced the CO2 conversion to 35.3%, but ethanol and propanol started to form at higher temperatures, likely because of methanol chain growth via C-C coupling46. The effect of pressure on the CO2 conversion and methanol selectivity was also evaluated, with pressures ranging from 20 to 70 bar (Fig. 4(g)). As the pressure increased, the CO2 conversion increased from 23.6% to 32.2%; however, the methanol selectivity decreased above 30 bar, leading to the formation of ethanol and propanol. The propanol selectivity reached 15.5% at 70 bar with an ethanol selectivity of 2.3%. The time-on-stream profile of the liquid phase over 72 h (Fig. S18) revealed that the methanol selectivity peaked at 96.8% after 5 h of reaction with 26.6% CO2 conversion. Over extended reaction times, the butanol selectivity increased, reaching 25.5% after 72 h, likely due to C-C coupling reactions46. This phenomenon aligns with the findings of Wang et al., who observed ethanol formation via C-C coupling over Co catalysts after extended reaction times. The reusability of the Deg In-Zr (3:8) catalyst (Fig. 4(i)) was tested over five cycles at 130 °C, total pressure of 30 bar, and reaction time of 5 h. The CO2 conversion showed only a slight decline from 26.6% to 25.8% and the methanol selectivity remained high (96.8–95.1%). ICP-MS (Table S12) analysis indicated minimal loss of the active metals (In and Zr), confirming the stability of the catalyst in the liquid-phase reaction medium. The reused catalysts were characterised by XRD, XPS, and TEM (Fig. S23), confirming that the catalyst maintained its structural integrity after five catalytic cycles. XPS analysis of In and Zr in the reused catalyst revealed oxidation states of +3 and +4, respectively, consistent with those observed in the fresh catalyst.
Structure performance relationship establishment via in situ AP XPS and Kinetic Study
In-situ XPS analysis was conducted on various catalysts at different temperatures and hydrogen pressures, as shown in Fig. 5. The O 1 s high-resolution XPS spectra were deconvoluted into three peaks at 529.6 eV, 531.7 eV, and 533.3 eV, corresponding to lattice oxygen, oxygen vacancies, and –OH groups, respectively47. As the hydrogen and CO2 pressures increased from 10 to 30 mTorr, the number of oxygen vacancies also increased (Fig. 5(a)). This trend was also observed with increasing temperature (Fig. 5(b)), indicating that higher temperatures and pressures promoted the generation of oxygen vacancies, which in turn created more active sites for CO2 adsorption on the catalyst surface48. Figure 5(c), (d) show a shift in the indium binding energy to higher values under increased pressure and temperature. Conversely, the binding energy of Zr shifted to lower values under the same conditions (Fig. 5(e) and (f)), suggesting a charge transfer between In and Zr, which may lead to the formation of In-Zr bimetallic sites49,50. The C 1 s XPS spectra were deconvoluted into three peaks at 284.3 eV, 285.5 eV, and 288.6 eV, corresponding to the C–C, C–O–C, and O–C = O bonds, respectively51.

a, b O 1 s XPS, c, d In 3 d XPS, e, f Zr 3 d XPS, and g, h C 1 s XPS spectra.
A kinetic study was performed using the Deg In-Zr (3:8) catalyst and compared with the conventional In2O3-ZrO2 and In2O3-ZrO2/C catalysts (Fig. S15(a)–(c)). The activation energy for CO2 conversion to methanol and the reverse water-gas shift (RWGS) reaction was determined using the Arrhenius equation by plotting the inverse of temperature (1/T) against ln(ri), where ri represents the initial rate constant. CO2 conversion to methanol was carried out at four different temperatures: 210, 220, 230, and 240 °C. Among the catalysts, Deg In-Zr (3:8) exhibited the lowest activation energy (46.2 ± 5 kJ mol⁻¹) for methanol production, significantly lower than the reported activation energy for In2O3-ZrO2 catalysts by Frei et al. The activation energy for the RWGS reaction over Deg In-Zr (3:8) was higher, at 98.6 ± 4 kJ mol⁻¹32. Due to its low activation energy barrier for methanol production, Deg In-Zr (3:8) demonstrated superior catalytic activity for CO2 conversion to methanol. For the hydrogenation of CO ₂ to methanol, the Deg In-Zr (3:8) catalyst exhibited a notably low activation energy of 46.2 kJ/mol. This reduced barrier is attributed to the formation of interfacial sites between In₂O₃ and ZrO₂ and the presence of oxygen vacancies, which were confirmed by HR-TEM, EXAFS, and XPS analyses. The In₂O₃–ZrO₂ interface introduces both geometric and electronic confinement effects52. These interfacial sites modify the local electron density and coordination environment, thereby enabling more efficient adsorption and activation of reactants52. Similar effects have been documented by Murray et al. for CeO₂-based interfaces, which also showed reduced energy barriers due to tailored interfacial interactions53. According to Sabatier’s principle, an optimal catalyst must bind intermediates with a moderate strength54,55. The temperature-programmed desorption (TPD) data for CO₂, CH₃OH, and HCOOH (Figs. S7 and S9) indicate that the binding energies at the Deg In-Zr (3:8) interface are indeed balanced and strong enough for activation, but weak enough to allow product desorption. Fluctuations in the indium concentration, whether too high or too low, can disrupt these interactions, ultimately resulting in a higher activation energy barrier.
An investigation was conducted using the Deg In-Zr (3:8) catalyst to evaluate how variations in hydrogen and carbon dioxide partial pressures affect CO₂ conversion and methanol productivity (g CH₃OH·g_cat⁻¹·h⁻¹, or STY). To ascertain the reaction order with respect to hydrogen, researchers have varied the hydrogen partial pressure while keeping a constant CO ₂ pressure. They monitored both the CO₂ conversion and methanol productivity over time at hydrogen pressures of 10, 20, 23, and 30 bar, with a fixed CO₂ pressure of 7 bar (see Fig. S16(a), (b))56. The findings revealed that as the hydrogen pressure increased from 10 to 30 bar, both CO₂ conversion and methanol productivity improved markedly. In a parallel experiment focused on carbon dioxide, the time-on-stream performance for both CO₂ conversion and methanol productivity was measured at different CO₂ partial pressures (2, 7, 10, and 20 bar) while maintaining the hydrogen pressure at 23 bar (Fig. S16(c), (d)). Here, increasing the CO₂ pressure from 7 to 30 bar yielded only a slight improvement in performance compared to the more significant enhancements observed when adjusting the hydrogen pressure. This trend is consistent with earlier results reported by Zhang et al. 57. This observation supports the notion that hydrogen activation plays a more crucial role in the conversion process than the activation of CO₂ or dissociation of the C–O bond in the methanol synthesis reaction from CO₂. In essence, for converting CO₂ to methanol, the activation of hydrogen is a key step for achieving high methanol yields. Previously, Li et al. demonstrated similar findings, where in methanol synthesis, the supply of activated hydrogen is lacking, even though there is enough activated CO₂56. Consequently, an increase in the number of hydrogen activation sites on the catalyst directly boosts methanol production. This result further reinforces both the experimental data and DFT calculations in this study, indicating that the activation of CO₂ and its conversion into formate species is not the rate-limiting step.
Furthermore, to determine the reaction orders, the natural logarithm of the partial pressure of each gas was plotted against the natural logarithm of the methanol STY (as shown in Fig. S16(g), (h)) and ref.56. The analysis yielded a reaction order of 0.18 for CO₂ and 0.92 for hydrogen, which is nearly one. These values are consistent with previous findings. Li et al. observed that, under low-pressure conditions, the reaction order for CO₂ is nearly zero or only a very small decimal value56. Moreover, Zhang et al. reported that when the hydrogen partial pressure exceeded 7 bar, the reaction order for hydrogen in methanol production ranges from 0.5 to 157.
To identify the rate-limiting step and correlate the findings from CH₃OH and HCOOH TPD analyses, we conducted two separate reactions over a Deg In-Zr (3:8) catalyst: one converting CO₂ to formic acid and another hydrogenating formic acid to methanol. The first reaction was limited to 30 min to produce formic acid in a controlled manner. Two distinct reactions were investigated: conversion of CO₂ to formic acid and subsequent transformation of formic acid into methanol. The activation energies for both reactions were determined using the Arrhenius equation by plotting ln(ri) against the inverse temperature (1/T), where ri denotes the initial reaction rate (Fig. S17(a)–(c)). CO₂ hydrogenation to methanol was performed at four different temperatures: 210, 220, 230, and 240 °C. It was observed that the hydrogenation of formic acid/formate to methanol requires a higher activation energy than the conversion of CO₂ to formic acid. For the Deg In-Zr (3:8) catalyst, the activation energy for the CO₂-to-formic acid reaction was measured as 41.4 ± 2 kJ/mol, while for the formic acid-to-methanol step, it was 84.6 ± 3 kJ/mol, indicating that the hydrogenation of formate to methanol is the rate-determining step. In the case of the In₂O₃-ZrO₂-C catalyst, the activation energies were 94.2 ± 3 kJ/mol for CO₂ to formic acid and 115.3 ± 4 kJ/mol for formic acid to methanol. Similarly, for the In₂O₃-ZrO₂ catalyst, the activation energies were 101.1 ± 3 kJ/mol and 118.3 ± 2 kJ/mol for the two steps, respectively.
In situ DRIFT for gas phase and in situ ATR-IR for liquid phase
To investigate the reaction mechanism and identify intermediate species in the gas-phase conversion of CO2 to methanol, diffuse reflectance infrared Fourier transform spectroscopy (DRIFT) was conducted on Deg In-Zr (3:8), Deg In-Zr (1:4), and In-Zr BDC catalysts at 220 °C and 10 bar with a 25% CO2 and 75% H2 feed. At the beginning of the reaction (Fig. S22), a CO2 peak at 2350 cm⁻¹ was detected for all catalysts58. After 10 min, small peaks appeared at 2121 cm⁻¹ (CO) and 1498 cm⁻¹ (monodentate carbonate, CO3-2) appeared45. The formate species (HCOO*) became prominent after 25 min over the Deg In-Zr (3:8) catalyst (Fig. S22(b)), with peaks at 1576 cm⁻¹ (asymmetric OCO stretching) and 1372 cm⁻¹ (symmetric OCO stretching)59. Additionally, the bending vibration of CH in HCOO* was detected at 1386 cm⁻¹ after 20 minutes60. Peaks corresponding to H3CO* species (2830, 2894, and 1040 cm⁻¹)61 appeared after 30 min, with the formate intensity peaking at 35 min and declining after 50 min, whereas the H3CO* intensity increased over time, reaching its maximum after 140 min. The decrease in the CO2 peak at 2350 cm⁻¹ confirmed CO2 adsorption and conversion into HCOO*, which was subsequently reduced to H3CO* and eventually formed CH3OH. These findings are consistent with those of previous studies of In-Zr-based catalysts45. DRIFT analysis was also performed on Deg In-Zr (1:4), which showed the appearance of HCOO* peaks after 35 min, although no H3CO* species were detected, indicating lower activity than that of Deg In-Zr (3:8). For the indium-impregnated Zr-BDC catalyst (In-Zr BDC), the formate peak at 1576 cm⁻¹ was weak, confirming its much lower activity (Fig. S22(a), (c)), respectively.
In situ ATR-IR spectroscopy was conducted to further investigate the CO2 hydrogenation mechanism over Zr-BDC, In-Zr BDC, and Deg In-Zr (3:8) catalysts (Fig. 6(b), (d), (f)) within a 0–180 min time frame. Initially, a doublet for adsorbed CO2 at 2350 cm⁻¹ was observed for all catalysts62,63. For the Deg In-Zr (3:8) catalyst (Fig. 6(f)), a peak at 1500 cm⁻¹ appeared after 10 min, corresponding to surface-bound zirconium bidentate carbonate, consistent with CO2 reduction studies by Katayama et al. 64. As the reaction progressed, a new peak at 1620 cm⁻¹, associated with adsorbed formate (HCOOad), emerged after 20 min. CO2 and H2 were adsorbed onto the catalyst surface, where oxygen vacancies and active H species likely combined with the adsorbed CO2 to form a formate45. After 30 min, the formate peak intensified65, and a peak at 1298 cm⁻¹ (symmetric C = O stretching in bidentate HCOOad) appeared62. After 40 min, a small peak at 1396 cm⁻¹ for OCH3 adsorbed species (OCH3ad) was detected64, indicating the hydrogenation of HCOOad to OCH3ad, following the reaction pathway CO2(ad) → HCOO(ad) → H2CO(ad) → OCH3(ad), as supported by both DFT calculations and experimental evidence44. The OCH3(ad) peak increased in intensity over time, reaching its maximum at 180 min, whereas the formate peak steadily declined, confirming its conversion to OCH3(ad) and eventually methanol. Additionally, the CO2 peak at 2350 cm⁻¹ decreased significantly, indicating CO2 reduction to CH3OH. No CO peak was observed, suggesting the absence of a reverse water-gas shift (RWGS) reaction, which typically occurs at higher temperatures66.

a Reaction mechanism over Deg In-Zr (3:8) catalyst, b, d, f time dependent in situ ATR-IR of Zr-BDC, In-Zr BDC, and Deg In-Zr (3:8) respectively; c, e, g ATR-IR of the catalysts in different gaseous environment for Zr-BDC, In-Zr BDC, and Deg In-Zr (3:8) respectively in liquid phase.
In contrast, the other two catalysts, Zr-BDC (bare MOF) (Fig. 6(b)) and In-Zr BDC (indium-impregnated Zr MOF) (Fig. 6(d)), showed no significant peaks except for CO2, indicating that they were inactive for CO2 hydrogenation to methanol during the 0–180 min reaction window.
DFT calculations
To gain mechanistic insight into the hydrogenation of CO2 to CH3OH at the interface of In2O3 and ZrO2 and the impact of oxygen vacancies, density functional theory (DFT) calculations were carried out. It was observed that the possibility of population oxygen vacancies was more likely to be exhibited at the interface of In2O3 and ZrO2, as the process of removal of an ‘O’ atom from the interface is more exothermic (−0.35 eV) than that of In2O3 (−0.12 eV) and ZrO2 surface (0.29 eV), as depicted in Fig. S24 and Table S2 supplementary information. CO2 was adsorbed on the interface of In2O3-ZrO2 in a V-shape configuration by forming In-O, O-C, and Zr-O bonds with bond lengths of 2.2 Å, 1.4 Å, and 2.3 Å, respectively (Fig. 7 (a)). At the oxygen vacant site, CO2 was adsorbed by forming an ‘In-O’ bond (2.2 Å), a ‘Zr-C’ bond (2.4 Å), and the second ‘O’ got bonded with both In (2.4 Å) and Zr (2.3 Å), as illustrated in Fig. 7 (b). The process of CO2 adsorption on the O-defect site of the interface releases an energy of −2.64 eV, which is nearly 2-fold higher than that of the pristine interface (−1.29 eV). This indicates that oxygen vacancies serve as primary adsorption sites for CO2 adsorption. Furthermore, *CO2 can be hydrogenated in the presence of H to form either a formate (*HCOO) or carboxylate (*COOH) intermediate. At the interface, the reaction energy determined for *HCOO formation (−0.5 eV) was more exothermic than that for *COOH formation (0.21 eV). In contrast, at the Ov-site, the energy released during the hydrogenation of *CO2 to *HCOO and *COOH was measured to be −1.6 eV and 0.15 eV, which were 1.1 eV and 0.6 eV more exothermic in comparison to the pristine interface. In addition to this, the formation of *HCOO from *CO2 hydrogenation was found to be more favourable with a lower activation barrier of 2.03 eV as compared to *COOH (2.33 eV), and O-vacancy played a crucial role in the formation of *HCOO intermediate by lowering the activation barrier to 1.64 eV than the pristine interface. The hydrogenation of *HCOO and *COOH intermediates was further investigated to yield *HCOOH, where the activation energies were measured to be 0.42 eV and 0.58 eV, respectively at the pristine interface and 0.45 eV and 0.83 eV at the Ov-site. This observation indicated that *CO2 was hydrogenated to form *HCOOH more likely through the formate (HCOO) pathway with higher intermediate stability67 and lower activation barrier than the carboxylate (COOH) pathway, in agreement with the findings of the *HCOO species in the in situ DRIFTS experiments. Further transformation of *HCOOH proceeds through the stepwise formation of the intermediates *H2COOH, *CH2O, *CH3O, and *CH3OH. Among these steps, the hydrogenation of *CH2O to *CH3O intermediate was found to be the rate-limiting step with the highest activation energy of 2.04 eV at the pristine interface and −1.76 eV at the Ov-site44. This observation aligns with the kinetic evaluation of the reaction and detection of *CH2O in the in situ DRIFTS analysis. The energy profile diagram (Figs. 7(c), (d), S25–S26) shows that the presence of oxygen vacancies plays a crucial role in stabilizing the intermediates generated during the hydrogenation of CO2 to methanol at the interface of In2O3-ZrO2. Additionally, these vacancies facilitate hydrogenation by lowering the activation barriers.

Geometry optimized structure of CO2 adsorption on a pristine In2O3-ZrO2 interface and b Ov-site of In2O3-ZrO2 interface color code: yellow (Indium), cyan (Zirconium), grey (Carbon), and red (Oxygen), and the energy profile diagram of CO2 hydrogenation to CH3OH at c In2O3-ZrO2 interface and d Ov-sites of In2O3-ZrO2 interface.
The influence of oxygen vacancies in the non-degraded MOF and degraded MOF (Deg In-Zr (3:8)) on the conversion of CO₂ into methanol was further confirmed by QTAIM analysis, an alternative DFT approach that is described in detail in the supplementary information (page 30-31 supplementary information). Oxygen vacancies serve as critical active sites to enhance both CO₂ activation and H₂ dissociation. QTAIM analysis revealed that at these vacancy sites, the adsorbed CO₂ adopts a bent configuration, indicative of charge polarization and activation. This polarization leads to significant electron density redistribution, particularly around the C = O bonds, which facilitates nucleophilic attack and subsequent hydrogenation steps. Mechanistically, oxygen vacancies act as Lewis basic centres that anchor CO₂ by allowing one of its oxygen atoms to fill the vacancy, whereas adjacent cationic sites (In³⁺, Zr4+) promote heterolytic H₂ dissociation. In conclusion, oxygen vacancies significantly influence both the adsorption strength of reactants and the energetics of key reaction steps. These insights, supported by QTAIM and DFT data, provide a clearer understanding of the structure–activity relationship.
Plausible reaction mechanism and structure, and performance relationship
Initially, H2 undergoes heterolytic cleavage on the defective surface of indium oxide, marking the first step in the hydrogenation of chemisorbed CO2 at oxygen vacancies (Fig. 6(a))68,69. A plausible pathway for methanol synthesis on the In2O3(111) surface follows the sequence: *HCOO → H2COO* → H3CO*. In this study, in situ DRIFTS under realistic conditions revealed that *HCOO-is the dominant surface intermediate in the CO2 hydrogenation process to methanol, which is consistent with previous reports60,69. While formate species were previously suggested as bystanders on Cu-based catalysts70, in situ DRIFTS confirmed the active involvement of *HCOO as a reaction intermediate for In-Zr catalysts. Increasing the indium content enhanced H2 splitting and promoted the formation of *HCOO, as demonstrated by the generation of *HCOO on the Deg In-Zr (3:8), Deg In-Zr (1:8), and Deg In-Zr (3:2) catalysts. Indium sites are also likely crucial for the formation of *H3CO species71. CH3OH TPD analysis shows that catalysts with a higher indium content exhibited a stronger affinity between the catalyst surface and CH3OH. Additionally, HCOOH TPD analysis indicates that bulk In2O3 has a stronger interaction with HCOOH than other catalysts, suggesting that the conversion of HCOOH to CH3O* depends on the optimal interaction with the catalyst surface, where the correct indium amount is critical for achieving high CH3OH selectivity. However, when the indium content was too high, the interaction with HCOOH became too stable, potentially inhibiting its conversion to CH3O*. The well-dispersed In2O3 on ZrO2 forms a complex interface with the ZrO2 support. At these interfacial sites, In2O3 dissociates gaseous H2 into reactive hydrogen atoms, which then interact with adsorbed CO2 to generate bidentate *HCOO. This intermediate is more likely to undergo hydrogenation given the delicate balance between the adsorption strengths of *HCOO and *H3CO. Meanwhile, the reduced interaction of *HCOO with ZrO2 inhibits CO formation as it struggles to bind effectively to the ZrO2 surface. To clarify the reaction mechanism, the decomposition of HCOOH was tested on various catalysts in the presence of CO2 and H2. ZrO2 alone primarily produced CO, while the Deg In-Zr catalysts generated CO, CO2, and H2 during steady-state HCOOH decomposition, indicating that ZrO2 plays a crucial role in CO formation during the catalytic conversion of CO2 to methanol.
Discussion
Crystal engineering of MOFs combined with pyrolysis techniques has been used to synthesize ultrafine In2O3 nanoparticles embedded in a ZrO2 matrix, forming an oxygen vacancy-incorporated heterojunction between the In2O3 and ZrO2 phases. This catalyst, obtained by pyrolyzing indium-impregnated Zr-BDC MOF with a low indium content of 0.2 wt.%, has been thoroughly characterized using EXAFS, XANES, and HR-TEM, confirming the creation of In2O3 and ZrO2 heterostructures. XPS analysis further revealed the presence of oxygen vacancies on the catalyst surface, which likely contributed to its exceptional catalytic performance in both liquid- and gas-phase CO2 hydrogenation to methanol. The active sites for methanol synthesis are believed to be the interface between ZrO2 and defective In2O3, with reaction mechanisms involving the formate pathway. Oxygen vacancies play a key role in activating CO2 and enhancing the stability of methanol on catalyst surfaces. This catalyst demonstrated remarkable stability in both liquid- and gas-phase reactions, highlighting its robust performance. In situ DRIFT spectroscopy (gas phase) and in situ ATR-IR spectroscopy (liquid phase) confirmed that methanol synthesis predominantly followed the formate pathway. CH3OH TPD and HCOOH TPD experiments showed that an optimal indium content enhances CH3OH selectivity. A higher concentration of indium stabilizes formate species on the catalyst surface, reducing their conversion to CH3O* species. Conversely, higher ZrO2 concentrations promote the decomposition of formate into CO and H2O. These TPD studies revealed the active surface species driving the reaction. In situ XPS was used to further explore the relationship between the catalyst surface structure and catalytic performance. The study confirmed that the abundance of oxygen vacancies and electronic modulation between In2O3 and ZrO2 are key factors driving high catalytic activity. A previous work by Kumari et al. also highlighted that oxygen vacancies play a critical role in CO2 activation by lowering the reaction barriers for CO2 dissociation. DFT calculations in this study reinforced these findings72. This paper presents a systematic atomic-level approach for designing multi-metal catalysts with enhanced active sites for CO2 hydrogenation to methanol. The method of optimizing indium content and controlling oxygen vacancies can be applied to other catalytic systems, providing a cost-effective and efficient route to catalyst synthesis, with indium loading as low as 0.2 wt.%. This approach offers significant potential for industrial-scale methanol production via CO2 hydrogenation. Future work will involve operando EXAFS studies to monitor structural changes in the catalyst during reactions in real time. These studies will provide deeper insights into the reaction mechanism, helping to develop more effective catalysts for CO2 hydrogenation.
Methods
Chemicals
ZrCl4, benzene dicarboxylic acid, indium (III) nitrate hydrate (In(NO3)3 · xH2O), N, N-dimethylformamide, methanol, and ZrO2 were purchased from Sigma Aldrich.
Catalyst preparation
Zr BDC MOF was prepared using a previously reported method involving the dissolution of ZrCl4 (0.053 g, 0.227 mmol) and 1,4-benzene dicarboxylic acid (H2BDC) (0.034 g, 0.227 mmol) in N, N-dimethylformamide (DMF) (24.9 g, 340 mmol) at room temperature23. The mixture was then heated in an oven at 120 °C for 24 h, allowing crystallization to occur under static conditions. After cooling to room temperature in air, the solid product was filtered, washed multiple times with DMF and methanol, and dried overnight (14 h) in an oven at 60 °C. Indium was impregnated into the Zr-BDC MOF using the incipient wetness impregnation (IWI) technique to synthesize the In-Zr BDC catalyst. Various indium-to-zirconium molar ratios (3:2, 3:4, 3:8, 1:2, 1:4, and 1:8) were used to produce indium-impregnated Zr BDC MOF. The corresponding amount of indium nitrate was added to 1 g of Zr BDC MOF in a methanol solution. The resulting solution was refluxed at 70 °C for 24 h. Subsequently, the solution was centrifuged, and the solid (In-Zr BDC) was dried in an oven at 60 °C overnight. All In-Zr BDC were thermally treated in a 600 °C tubular furnace under a N2 atmosphere for 4 h at a heating rate of 2 °C/min. This was denoted as Deg In-Zr.
Catalyst characterization
All detailed catalyst characterizations are provided in the Supplementary Information.
DFT calculations
The details of the DFT calculations are shown in the Supplementary Information.
Catalyst evaluation (gas phase and liquid phase)
The catalytic activity was assessed in a fixed-bed reactor that was 300 mm long and 7 mm in diameter, constructed from stainless steel. The bed comprised 0.4 g of catalyst (with a mesh size distribution of 20–40) and 2 g quartz particles arranged between two layers of silica wool. Initially, the reactor pressure was raised to 3.0 MPa with N2 flow, and the temperature was increased to 300 °C. Subsequently, the catalyst was reduced and activated using a stream of diluted hydrogen (10 vol. % H2/N2) at 350 °C for 1 h. The catalytic activity for methanol synthesis was evaluated under the following conditions: H2/CO2 = 3 and flow rate of 10 mL/min (30 bar pressure). The gases were depressurised to atmospheric pressure at the outlet of the reactor, and the reaction products were analysed using an online gas chromatograph (GC, Agilent 7890 A) equipped with two detectors. A flame ionization detector, along with an HP-FFAP column and H2 as the carrier gas, was used to detect CO, CH4, and CH3OH. A thermal conductivity detector equipped with MS-5A and Hayesep Q columns using He as the carrier gas was used to identify other gaseous products (including H2, CO2, N2, and CO). The carbon balance exceeded 95% in all experiments. The calculations for CO2 conversion, selectivity of C-containing products, and product yield were carried out as follows
Here, N denotes the carbon-containing species in the products, including CO, CH4, and CH3OH. The results were obtained when the reaction reached a steady state.
All CO2 hydrogenation reactions were performed in a 250 ml stainless steel Parr autoclave built with a pressure gauge setup. The typical hydrogenation reaction conditions were a temperature of 130 °C, 60 ml water, 40 mg of catalyst, oxygen pressure of 30 bar (3:1 H2: CO2), and reaction time of 5 h. After completion of the reaction, the mixture was collected, filtered, and analyzed by HPLC. C-18 column in combination with milli Q water as the mobile phase.
Data availability
Source data are present in Cartesian coordinates EXCEL file. The cartesian coordinates of atoms in the optimized structure of Deg In2O3-OV1, Deg In2O3-OV2, nonDeg In2O3-1, nonDeg In2O3-2, and nonDeg Zr BDC MOF catalyst models, calculated at PBE0/6-31 G* level of theory. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Aresta, M. Carbon dioxide as chemical feedstock. (John Wiley & Sons, 2010).
Jadhav, S. G., Vaidya, P. D., Bhanage, B. M. & Joshi, J. B. Catalytic carbon dioxide hydrogenation to methanol: a review of recent studies. Chem. Eng. Res. Des. 92, 2557–2567 (2014).
Lunkenbein, T., Schumann, J., Behrens, M., Schlögl, R. & Willinger, M. G. Formation of a ZnO overlayer in industrial Cu/ZnO/Al2O3 catalysts induced by strong metal–support interactions. Angew. Chem. 127, 4627–4631 (2015).
Inui, T., Hara, H., Takeguchi, T. & Kim, J.-B. Structure and function of Cu-based composite catalysts for highly effective synthesis of methanol by hydrogenation of CO2 and CO. Catal. today 36, 25–32 (1997).
Wu, J., Saito, M., Takeuchi, M. & Watanabe, T. The stability of Cu/ZnO-based catalysts in methanol synthesis from a CO2-rich feed and from a CO-rich feed. Appl. Catal. A Gen. 218, 235–240 (2001).
Sun, K. et al. Hydrogenation of CO2 to methanol over In2O3 catalyst. J. CO2 Utilization 12, 1–6 (2015).
Martin, O. et al. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angew. Chem. 128, 6369–6373 (2016).
Ye, J., Liu, C. -j, Mei, D. & Ge, Q. Methanol synthesis from CO2 hydrogenation over a Pd4/In2O3 model catalyst: a combined DFT and kinetic study. J. Catal. 317, 44–53 (2014).
Liu, X., Men, Y., Wang, J., He, R. & Wang, Y. Remarkable support effect on the reactivity of Pt/In2O3/MOx catalysts for methanol steam reforming. J. Power Sources 364, 341–350 (2017).
Pinheiro Araújo, T. et al. Flame-made ternary Pd-In2O3-ZrO2 catalyst with enhanced oxygen vacancy generation for CO2 hydrogenation to methanol. Nat. Commun. 13, 5610 (2022).
Vera, C. Y. R. et al. Mechanistic understanding of support effect on the activity and selectivity of indium oxide catalysts for CO2 hydrogenation. Chem. Eng. J. 426, 131767 (2021).
Tsoukalou, A. et al. Operando X-ray absorption spectroscopy identifies a monoclinic ZrO2: In solid solution as the active phase for the hydrogenation of CO2 to methanol. ACS Catal. 10, 10060–10067 (2020).
Chen, T. -y. et al. Unraveling highly tunable selectivity in CO2 hydrogenation over bimetallic In-Zr oxide catalysts. Acs Catal. 9, 8785–8797 (2019).
Lan, M. et al. Fabrication of porous Pt-doping heterojunctions by using bimetallic MOF template for photocatalytic hydrogen generation. Nano Energy 33, 238–246 (2017).
Lu, X. F., Chen, Y., Wang, S., Gao, S. & Lou, X. W. Interfacing manganese oxide and cobalt in porous graphitic carbon polyhedrons boosts oxygen electrocatalysis for Zn–air batteries. Adv. Mater. 31, 1902339 (2019).
Hu, P. et al. Improved interface compatibility of hollow H-Zr0. 1Ti0. 9O2 with UiO-66-NH2 via Zr-Ti bidirectional penetration to boost visible photocatalytic activity for acetaldehyde degradation under high humidity. Appl. Catal. B Environ. 296, 120371 (2021).
Crake, A. et al. The effect of materials architecture in TiO2/MOF composites on CO2 photoreduction and charge transfer. Small 15, 1805473 (2019).
Zhao, C. et al. Dual MOFs template-directed fabrication of hollow-structured heterojunction photocatalysts for efficient CO2 reduction. Chem. Eng. J. 416, 129155 (2021).
Cui, W. G. & Hu, T. L. Incorporation of active metal species in crystalline porous materials for highly efficient synergetic catalysis. Small 17, 2003971 (2021).
Chen, Y.-Z., Zhang, R., Jiao, L. & Jiang, H.-L. Metal–organic framework-derived porous materials for catalysis. Coord. Chem. Rev. 362, 1–23 (2018).
Liu, H. et al. Single atoms meet metal–organic frameworks: collaborative efforts for efficient photocatalysis. Energy Environ. Sci. 15, 3722–3749 (2022).
Cui, W.-G. et al. Engineering Co/MnO heterointerface inside porous graphitic carbon for boosting the low-temperature CO2methanation. Appl. Catal. B Environ. 287, 119959 (2021).
Cavka, J. H. et al. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 130, 13850–13851 (2008).
Schaate, A. et al. Modulated synthesis of Zr-based metal–organic frameworks: from nano to single crystals. Chem. A Eur. J. 17, 6643–6651 (2011).
Carneiro, A. C. R. et al. On-line micro-packed column solid-phase extraction of cadmium using metal-organic framework (MOF) UiO-66 with posterior determination by TS-FF-AAS. J. Braz. Chem. Soc. 33, 958–968 (2022).
Xu, H., Zhu, J., Cheng, Y. & Cai, D. Functionalized UIO-66@ Ag nanoparticles substrate for rapid and ultrasensitive SERS detection of di-(2-ethylhexyl) phthalate in plastics. Sens. Actuators B: Chem. 349, 130793 (2021).
Chai, L. et al. Bimetallic-MOF derived carbon with single Pt anchored C4 atomic group constructing super fuel cell with ultrahigh power density and self-change ability. Adv. Mater. 36, 2308989 (2024).
Sharma, P., Ho, P. H., Shao, J., Creaser, D. & Olsson, L. Role of ZrO2 and CeO2 support on the In2O3 catalyst activity for CO2 hydrogenation. Fuel 331, 125878 (2023).
Liu, J. et al. Low on-resistance diamond field effect transistor with high-k ZrO2 as dielectric. Sci. Rep. 4, 1–5 (2014).
Meng, C. et al. Oxygen-deficient metal oxides supported nano-intermetallic InNi3C0. 5 toward efficient CO2 hydrogenation to methanol. Sci. Adv. 7, eabi6012 (2021).
Park, H. et al. Highly conducting In2O3 nanowire network with passivating ZrO2 thin film for solution-processed field effect transistors. Adv. Electron. Mater. 2, 1600218 (2016).
Frei, M. S. et al. Role of zirconia in indium oxide-catalyzed CO2 hydrogenation to methanol. Acs Catal. 10, 1133–1145 (2019).
Wang, Z. et al. Boosting CO2 hydrogenation to formate over edge-sulfur vacancies of molybdenum disulfide. Angew. Chem. Int. Ed. 62, e202307086 (2023).
Jiang, X. et al. A combined experimental and DFT study of H2O effect on In2O3/ZrO2 catalyst for CO2 hydrogenation to methanol. J. Catal. 383, 283–296 (2020).
Pustovarenko, A. et al. Metal–organic framework-derived synthesis of cobalt indium catalysts for the hydrogenation of CO2 to methanol. ACS Catal. 10, 5064–5076 (2020).
Zhang, X. et al. Support effect and surface reconstruction in In2O3/m-ZrO2 catalyzed CO2 hydrogenation. ACS Catal. 12, 3868–3880 (2022).
Li, N. et al. CO2 hydrogenation to methanol promoted by Cu and metastable tetragonal CexZryOz interface. J. Energy Chem. 68, 771–779 (2022).
Li, Z. et al. Boosting CO2 hydrogenation efficiency for methanol synthesis over Pd/In2O3/ZrO2 catalysts by crystalline phase effect. Appl. Surf. Sci. 603, 154420 (2022).
Cai, Z. et al. Pd supported on MIL-68 (In)-derived In2O3 nanotubes as superior catalysts to boost CO2 hydrogenation to methanol. ACS Catal. 10, 13275–13289 (2020).
Zhang, C., Sun, D., Huan, Y., Wu, K. & Liao, H. Highly sensitive ZnO nanoparticles-loaded In2O3 hollow microsphere for detecting ppb-level NO2 at low working temperature. Prog. Nat. Sci. Mater. Int. 30, 469–476 (2020).
Choudhury, B. & Choudhury, A. Lattice distortion and corresponding changes in optical properties of CeO2 nanoparticles on Nd doping. Curr. Appl. Phys. 13, 217–223 (2013).
Peng, Z. et al. Heterophase-structured nanocrystals as superior supports for Ru-based catalysts in selective hydrogenation of benzene. Sci. Rep. 7, 39847 (2017).
Chen, Y., Song, B., Tang, X., Lu, L. & Xue, J. Ultrasmall Fe3O4 nanoparticle/MoS2 nanosheet composites with superior performances for lithium ion batteries. Small 10, 1536–1543 (2014).
Wang, J. et al. CO2 hydrogenation to methanol over In2O3-based catalysts: from mechanism to catalyst development. ACS Catal. 11, 1406–1423 (2021).
Tsoukalou, A. et al. Surface intermediates in in-based ZrO2-supported catalysts for hydrogenation of CO2 to methanol. J. Phys. Chem. C. 126, 1793–1799 (2022).
Wang, L. et al. Selective hydrogenation of CO2 to ethanol over cobalt catalysts. Angew. Chem. Int. Ed. 57, 6104–6108 (2018).
Kwon, H.-M. et al. A correlation between oxygen vacancies and reliability characteristics in a single zirconium oxide metal-insulator-metal capacitor. IEEE Trans. Electron Devices 61, 2619–2627 (2014).
Fu, L., Chen, H., Wang, K. & Wang, X. Oxygen-vacancy generation in MgFe2O4 by high temperature calcination and its improved photocatalytic activity for CO2 reduction. J. Alloy. Compd. 891, 161925 (2022).
Sengar, S. K., Mehta, B. & Govind, G. Size and alloying induced shift in core and valence bands of Pd-Ag and Pd-Cu nanoparticles. J. Appl. Phys. 115, 124301 (2014).
Wang, D. et al. Electronic behaviour of Au-Pt alloys and the 4f binding energy shift anomaly in Au bimetallics-X-ray spectroscopy studies. AIP Adv. 8, 065210 (2018).
Chen, X., Wang, X. & Fang, D. A review on C1s XPS-spectra for some kinds of carbon materials. Fuller. Nanotubes Carbon Nanostruct. 28, 1048–1058 (2020).
Vogt, C. & Weckhuysen, B. M. The concept of active site in heterogeneous catalysis. Nat. Rev. Chem. 6, 89–111 (2022).
Cargnello, M. et al. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 341, 771–773 (2013).
Pan, Y., Shen, X., Yao, L., Bentalib, A. & Peng, Z. Active sites in heterogeneous catalytic reaction on metal and metal oxide: theory and practice. Catalysts 8, 478 (2018).
Aguila, B. et al. Lower activation energy for catalytic reactions through host–guest cooperation within metal–organic frameworks. Angew. Chem. 130, 10264–10268 (2018).
Tang, C. et al. Insights into the selectivity determinant and rate-determining step of CO2 hydrogenation to methanol. J. Phys. Chem. C. 126, 10399–10407 (2022).
Wang, X. & Zhang, H. Kinetically relevant variation triggered by hydrogen pressure: a mechanistic case study of CO2 hydrogenation to methanol over Cu/ZnO. J. Catal. 406, 145–156 (2022).
Cui, X. et al. Improving methanol selectivity in CO2 hydrogenation by tuning the distance of Cu on catalyst. Appl. Catal. B Environ. 298, 120590 (2021).
Fehr, S. M., Nguyen, K. & Krossing, I. Realistic operando-DRIFTS studies on Cu/ZnO catalysts for CO2 hydrogenation to methanol–direct observation of mono-ionized defect sites and implications for reaction intermediates. ChemCatChem 14, e202101500 (2022).
Wang, J. et al. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 3, e1701290 (2017).
Wu, C. et al. Inverse ZrO2/Cu as a highly efficient methanol synthesis catalyst from CO2 hydrogenation. Nat. Commun. 11, 5767 (2020).
Baruch, M. F., Pander III, J. E., White, J. L. & Bocarsly, A. B. Mechanistic insights into the reduction of CO2 on tin electrodes using in situ ATR-IR spectroscopy. Acs Catal. 5, 3148–3156 (2015).
Schädle, T., Pejcic, B. & Mizaikoff, B. Monitoring dissolved carbon dioxide and methane in brine environments at high pressure using IR-ATR spectroscopy. Anal. methods 8, 756–762 (2016).
Katayama, Y. et al. An in situ surface-enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-bound and O-bound reaction intermediates. J. Phys. Chem. C. 123, 5951–5963 (2018).
Yang, X. et al. MOF-derived Cu@ Cu2O heterogeneous electrocatalyst with moderate intermediates adsorption for highly selective reduction of CO2 to methanol. Chem. Eng. J. 431, 134171 (2022).
Zhao, Y.-F. et al. Insight into methanol synthesis from CO2 hydrogenation on Cu (1 1 1): Complex reaction network and the effects of H2O. J. Catal. 281, 199–211 (2011).
Yang, C. et al. Strong electronic oxide–support interaction over In2O3/ZrO2 for highly selective CO2 hydrogenation to methanol. J. Am. Chem. Soc. 142, 19523–19531 (2020).
Frei, M. S. et al. Mechanism and microkinetics of methanol synthesis via CO2 hydrogenation on indium oxide. J. Catal. 361, 313–321 (2018).
Albani, D. et al. Semihydrogenation of acetylene on indium oxide: proposed single-ensemble catalysis. Angew. Chem. 129, 10895–10900 (2017).
Kattel, S., Yan, B., Yang, Y., Chen, J. G. & Liu, P. Optimizing binding energies of key intermediates for CO2 hydrogenation to methanol over oxide-supported copper. J. Am. Chem. Soc. 138, 12440–12450(2016).
Su, J. et al. Direct conversion of syngas into light olefins over zirconium-doped indium (III) oxide and SAPO-34 bifunctional catalysts: design of oxide component and construction of reaction network. ChemCatChem 10, 1536–1541 (2018).
Kumari, N., Haider, M. A., Agarwal, M., Sinha, N. & Basu, S. Role of reduced CeO2 (110) surface for CO2 reduction to CO and methanol. J. Phys. Chem. C. 120, 16626–16635 (2016).
Acknowledgements
All authors acknowledge the RMIT Microscopy and Microanalysis Facility (RMMF) for their scientific and technical assistance and for providing access to their research facilities. Distinguished Professor Suresh Bhargava wishes to express his gratitude towards his mentor & supervisor, the late Professor E. W. Abel, Exter UK, for giving him the power of knowledge and mentorship to produce future scientists. Prof. S.K. Bhargava and Prof. Ravindra D. Gudi acknowledge the prestigious VAJRA (Visiting Advanced Joint Research) award support from the Science and Engineering Research Board (SERB), New Delhi, India to Prof. S.K. Bhargava.
Author information
Authors and Affiliations
Contributions
P.K: Conceptualization, experimental data investigation, analysis, methodology, writing original draft, and editing. S.C.S.: Conceptualization, experimental data investigation, analysis, methodology, writing, and editing. T.Y: EXAFS analysis and data collection. D.J.: Writing and editing, H.A.M. and T.U.: EXAFS analysis and data collection. J.K. and T.H.: Theoretical calculations, writing, and editing. S.P.: Experimental data investigation, writing, review, and editing. R.D.G. and D.D.M.: Theoretical calculations, writing, and editing, Y.I.: EXAFS writing, supervision, review and editing. S.K.B.: Project administration, supervision, writing, review, and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guanghui Zhang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Koley, P., Shit, S.C., Yoshida, T. et al. Metal organic framework derived In2O3/ZrO2 heterojunctions with interfacial oxygen vacancies for highly selective CO2-to-methanol hydrogenation. Nat Commun 16, 8903 (2025). https://doi.org/10.1038/s41467-025-63932-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63932-y
